

Abstract
Simple Summary
Germline and somatic variant testing of the BRCA1 and BRCA2 genes are important to predict treatment response to PARP inhibitors in ovarian cancer patients. However, germline variants in other genes besides BRCA1 and BRCA2 are associated with ovarian cancer predisposition, which would be missed by a genetic testing aimed only at treatment decision. We aimed to evaluate the yield of clinically actionable germline variants using next-generation sequencing of a customized panel of 10 genes for the analysis of pathology samples of ovarian carcinomas. We identified clinically actionable germline variants in a significantly higher proportion of ovarian cancer patients when compared with genetic testing focused only on BRCA1 and BRCA2. This strategy increases the chance to make available genetic counseling, presymptomatic genetic testing, and gynecological cancer prophylaxis to female relatives who turn out to be healthy carriers of deleterious germline variants.
Abstract
Since the approval of PARP inhibitors for the treatment of high-grade serous ovarian cancer, in addition to cancer risk assessment, BRCA1 and BRCA2 genetic testing also has therapeutic implications (germline and somatic variants) and should be offered to these patients at diagnosis, irrespective of family history. However, variants in other genes besides BRCA1 and BRCA2 are associated with ovarian cancer predisposition, which would be missed by a genetic testing aimed only at indication for PARP inhibitor treatment. In this study, we aimed to evaluate the yield of clinically actionable germline variants using next-generation sequencing of a customized panel of 10 genes for the analysis of formalin-fixed paraffin-embedded samples from 96 ovarian carcinomas, a strategy that allows the detection of both somatic and germline variants in a single test. In addition to 13.7% of deleterious germline BRCA1/BRCA2 carriers, we identified 7.4% additional patients with pathogenic germline variants in other genes predisposing for ovarian cancer, namely RAD51C, RAD51D, and MSH6, representing 35% of all pathogenic germline variants. We conclude that the strategy of reflex gene-panel tumor testing enables the identification of clinically actionable germline variants in a significantly higher proportion of ovarian cancer patients, which may be valuable information in patients with advanced disease that have run out of approved therapeutic options. Furthermore, this approach increases the chance to make available genetic counseling, presymptomatic genetic testing, and gynecological cancer prophylaxis to female relatives who turn out to be healthy carriers of deleterious germline variants.
Keywords: ovarian cancer, tumor testing, next generation sequencing, multi-gene panel, genetic predisposition, clinically actionable alterations
1. Introduction
Ovarian cancer (OC) is the third most common malignant disease among patients with gynecological neoplasia, being the eighth most common cancer and the eighth most common cause of cancer-related death in women [1]. The lack of specific symptoms and effective early detection strategies contributes to the high mortality rate, as most OC are diagnosed at an advanced stage [2]. Epithelial ovarian cancer (EOC) is recognized as a heterogeneous disease mainly represented by five histological subtypes (high-grade serous, low-grade serous, clear cell, endometrioid, and mucinous) that are characterized by distinct clinical and molecular features. Despite this knowledge, the initial standard treatment has typically been maximal effort cytoreductive surgery, with platinum-paclitaxel chemotherapy being the standard of care treatment [3,4]. Despite the usually good initial response, about 70–80% of patients will relapse within the subsequent three years and have limited treatment options thereafter [5,6].
The first poly(ADP-ribose) polymerase (PARP)-inhibitor (PARPi) received the approval from the European Medicines Agency (EMA) in 2014 for use as a maintenance therapy in relapsed high-grade serous ovarian cancer (HGSOC) patients with platinum-sensitivity and with BRCA1/BRCA2 somatic or germline variants [7,8]. The same treatment was approved later for first-line maintenance therapy in BRCA mutated advanced EOC [9]. The mechanism underlying the efficacy of PARPi is synthetic lethality, which occurs in cells with deficiency in homologous recombination (HR) repair, where BRCA1/BRCA2 proteins play an essential role. When base excision repair (BER) is blocked by PARPi and thereby inhibits single strand break repair, the combined effect with HR results in accumulation of DNA aberrations and cell death [10]. Since PARPi approval, BRCA status became a relevant criterion to select patients who may benefit from this therapy or to identify the main responders to this class of drugs. Consequently, OC patients have been referred for BRCA1 and BRCA2 genetic testing (germline and somatic) to guide treatment decisions in addition to familial risk assessment regardless of family history of breast or OC [11]. This seems to be the best approach as some studies showed that more than 30% of women with germline variants had no family history of breast or OC [12].
Although most are sporadic, it has been reported that about 23% of OC are associated with hereditary predisposition [12], and a large proportion of these cases are due to inherited variants in BRCA1 and BRCA2 genes. Women with germline variants in these genes are at higher risk of developing OC, when compared with women in the general population, with the lifetime risk being 44% and 17% for BRCA1 and BRCA2, respectively [13]. The use of next-generation sequencing (NGS) with multi-gene panels may allow the identification of other genes associated with inherited predisposition to OC [12]. Recent studies with blood samples showed that 18–24% of OC patients carry germline variants in genes associated with OC risk and, of these, 16–18% had germline variants in BRCA1 or BRCA2 genes and 4–6% had variants in other genes such as BRIP1, RAD51C, and RAD51D [12,14]. The identification of carriers of deleterious germline variants in OC predisposition genes allows the use of prophylactic surgery for a disease that currently has no effective screening allowing early diagnosis.
Many laboratories are now performing BRCA1/BRCA2 testing in formalin-fixed paraffin-embedded (FFPE) tumor samples to detect germline and somatic variants to identify, in a single test, those patients with somatic and germline variants who are most likely to benefit from PARPi therapy. Using this strategy, there is a risk that patients without BRCA1/BRCA2 variants are not evaluated for other genes predisposing to OC, thereby missing the opportunity for presymptomatic testing and prophylactic measures. Eventually, they might subsequently be referred for germline NGS testing of other genes, which increases the sequencing costs [11]. In this study, we designed a customized NGS gene panel that includes all known OC cancer predisposing genes for the detection of pathogenic germline variants in a single test using FFPE tumor samples. We show that this strategy significantly increases the yield of clinically actionable pathogenic germline variants in a single NGS test, which can subsequently be evaluated in a peripheral blood sample by Sanger sequencing for its eventual constitutional origin after genetic counseling.
2. Results
2.1. Variant Analysis
Of 96 FFPE OC samples, one (OC65) was excluded from further analysis because it did not pass quality filters. Among the ten genes included in the NGS panel (BRCA1, BRCA2, BRIP1, MLH1, MSH2, MSH6, PMS2, RAD51C, RAD51D, and TP53), a total of 3457 variants was annotated and classified among the remaining 95 tumor samples. After variant filtering 97 variants remained, of which 57 were classified as pathogenic/likely pathogenic in ClinVar. Among the variants not classified in ClinVar, two variants were nonsense, seven were frameshift variants, two were canonical +1 and −2 splice site variants, and one was an inframe variant, all of which were considered pathogenic since there is strong evidence of pathogenicity [15]. Additionally, there were 28 missense variants not classified, with unknown significance or conflicting interpretations of pathogenicity. The potential pathogenicity of these missense variants was evaluated through in silico prediction tools, and 19 of these variants were predicted to have a deleterious impact on the protein. We, therefore, retained 88 variants across seven genes, of which 69 (77.3%) were pathogenic and 19 (22.7%) were missense variants of uncertain significance (VUS). Of 95 tumor samples analyzed, variants in the genes under study were identified in 66 (69.5%).
2.2. Germline Variants
After variant filtering, all variants of the genes of interest identified by NGS in the tumor of the 66 patients were tested in peripheral blood samples, and only those which were confirmed to be germline are included in this report. Of the 88 variants detected, 26 variants were confirmed to be germline across seven of the 10 genes included in the panel. Out of the 26 germline variants identified, 20 (76.9%) were pathogenic variants and six (23.1%) were VUS. In 69 of the 95 (73.7%) patients included in the study, no germline variants among the 10 genes tested were identified. Pathogenic variants were found in 20 patients (21.1%): seven in BRCA1, six in BRCA2, four in RAD51D, two in RAD51C, and one in MSH6. Germline VUS were identified in six patients (6.3%) in BRCA2, MSH6, TP53, and BRIP1 (Figure 1 and Table 1).
Figure 1.

Spectrum of germline variants (pathogenic and variants of uncertain significance (VUS)).
Table 1.
Germline variants identified among 95 ovarian cancer patients with next generation sequencing of a 10-gene panel.
| Patient | Gene | HGVS Coding | HGVS Protein | VAF | ClinVar | MetaLR 1 | MetaSVM 1 | C-Score 1 | Varsome | Additional Tests | Personal History (Age 2) | Family History |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OC09 | BRCA1 | c.2037delinsCC | p.(Lys679AsnfsTer4) | 60% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (46) | 4 × OC, 2 × PaC, 1 × GC |
| OC11 | BRIP1 | c.2477A>G | p.(Asn826Ser) | 74% | VUS | D | D | 26.3 | VUS | N/A | OC (39) | 1 × CRC, 1 × L |
| OC12 | BRCA1 | c.211A>G | p.(Arg71Gly) | 81% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (58) | 2 × BC, 1 × PC |
| OC16 | RAD51C | c.709C>T | p.(Arg237Ter) | 81% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (57) | 1 × OC |
| OC17 * | RAD51D | c.748del | p.(His250ThrfsTer2) | 68% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (57) | 2 × OC, 2 × BC, 1 × CRC |
| OC19 | BRCA1 | c.3331_3334del | p.(Gln1111AsnfsTer5) | 81% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (61) | 4 × BC, 1 × OC |
| OC23 | BRCA1 | c.3817C>T | p.(Gln1273Ter) | 92% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (46) | 6 × BC, 1 × CRC |
| OC24 | MSH6 | c.3848_3862del | p.(Ile1283_Tyr1287del) | 45% | N/A | N/A | N/A | N/A | N/A | IHC: loss of expression | OC (51), UC (51) | 2 × BC, 1 × CRC,1 × LC |
| OC29 | BRCA2 | c.5073dup | p.(Trp1692MetfsTer3) | 81% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (48), BC (48) | 3 × BC, 3 × PC, 3 × CRC |
| OC30 * | RAD51D | c.748del | p.(His250ThrfsTer2) | 81% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (73) | 2 × OC, 2 × BC, 1 × CRC |
| OC31 | RAD51C | c.709C>T | p.(Arg237Ter) | 77% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (62) | 1 × BC |
| OC36 | BRCA2 | c.8036A>G | p.(Asp2679Gly) | 36% | CIP | D | D | 31 | LP | N/A | OC (59) | 1 × CRC, 1 × |
| OC37 | MSH6 | c.1729C>T | p.(Arg577Cys) | 43% | VUS | D | D | 34 | VUS | IHC: normal; MSI: normal | OC (68) | 1 × LC |
| OC39 | BRCA1 | c.3331_3334del | p.(Gln1111AsnfsTer5) | 69% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (63) | 2 × BC, 1 × CRC |
| OC40 | BRCA1 | c.3331_3334del | p.(Gln1111AsnfsTer5) | 58% | Pathogenic | N/A | N/A | N/A | N/A | N/A | BC (49), OC (50) | --------- |
| OC50 | BRCA2 | c.9364G>C | p.(Ala3122Pro) | 88% | VUS | D | D | 31 | LP | N/A | OC (62) | 1 × CRC, 1 × BC, 1 × PC |
| OC52 | BRCA1 | c.2037_2038insC | p.(Lys680GlnfsTer3) | 54% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (52) | 2 × BC, 1 × PC |
| OC54 | RAD51D | c.748del | p.(His250ThrfsTer2) | 93% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (48) | 2 × BC, 1 × PC, 3 × CRC |
| OC55 * | RAD51D | c.748del | p.(His250ThrfsTer2) | 56% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (54) | 2 × OC, 2 × BC, 1 × CRC |
| OC60 | BRCA2 | c.7975A>G | p.(Arg2659Gly) | 62% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (56) | 3 × BC, 1 × PaC, 1 × CRC |
| OC72 | BRCA2 | c.5073dup | p.(Trp1692MetfsTer3) | 56% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (60) | 1 × PaC, 1 × PC |
| OC73 | BRCA2 | c.5073dup | p.(Trp1692MetfsTer3) | 86% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (44) | 2 × BC, 1 × CRC |
| OC79 | MSH6 | c.3182T>C | p.(Leu1061Pro) | 48% | VUS | D | D | 24.8 | VUS | IHC: loss of expression | OC (49) | 1 × CRC |
| OC88 | BRCA2 | c.2T>G | p.Met1? | 93% | Pathogenic | N/A | N/A | N/A | N/A | N/A | BC (59), OC (69) | 1 × BC, 1 × PC |
| OC91 | TP53 | c.845G>A | p.(Arg282Gln) | 47% | VUS | D | D | 35 | LP | N/A | OC (58), TC (60), PaC (60) | 1 × GC, 1 × CRC, BC |
| OC93 | BRCA2 | c.8488-1G>A | --------- | 57% | Pathogenic | N/A | N/A | N/A | N/A | N/A | OC (65) | 1 × BC, 1 × PaC |
BC: Breast cancer; CIP: conflicting interpretations of pathogenicity; CRC: Colorectal cancer; GC: gastric cancer; IHC: Immunohistochemistry; L: Lymphoma; LC: Lung cancer; LP: Likely pathogenic; MSI: Microsatellite instability; N/A: not available; OC: ovarian cancer; PaC: Pancreatic cancer; PC: Prostate cancer; UC: uterine cancer; VAF: variant allele frequency; VUS: variant of unknown significance. * OC17, OC30, and OC55 belong to the same family. 1 Missense variants were retained if they were predicted to be damaging (D) by MetaLR, MetaSVM, and CADD-score (>15). 2 Age at diagnosis.
2.2.1. BRCA1 and BRCA2 Germline Variants
Of all patients included in this study, 91 (95.8%) were previously tested for BRCA1 and BRCA2 variants during clinical testing with BRCA Tumor MASTRTM Plus Dx, and 50 (52.1%) were part of the cohort of a previously published study [16]. Of these 91 patients, 15 had BRCA1/BRCA2 germline variants, and all these variants were detected in this study using the custom panel (Table S1). Among the 95 OC patients included in this study, we identified 15 germline variants in BRCA1/BRCA2 (seven in BRCA1 and eight in BRCA2). All seven variants found in BRCA1 were pathogenic, five frameshift, one nonsense, and one missense variant. Of the eight variants found in BRCA2, three were frameshift, one was a splice site variant at the canonical site -1, and four were missense variants. Of the four missense variants found in BRCA2, two were classified as pathogenic, and two were VUS (c.8036A>G and c.9364G>C). The BRCA2 germline variant c.8036A>G, p.(Asp2679Gly), was identified in patient OC36, diagnosed with HGSOC at 56 years. This variant is a missense VUS predicted to have a deleterious impact on protein by in silico tools and is considered as likely pathogenic in Varsome. However, there is no family history of either breast or OC. Furthermore, this variant was included in a study aimed to assess the clinical relevance of BRCA2 VUS missense variants and was classified as benign [17]. The variant BRCA2 c.9364G>C, p.(Ala3122Pro), was identified in patient OC50, diagnosed with HGSOC at 62 years. This variant is predicted to have a deleterious effect on protein by in silico tools and is classified as likely pathogenic in Varsome. In the patient’s family there are four relatives who have been affected with cancer, namely breast, prostate, and colorectal cancer. However, this variant was classified as likely benign in a recent study aimed to classify missense VUS variants in BRCA1 and BRCA2 using a multifactorial likelihood model [18].
2.2.2. RAD51C and RAD51D Germline Variants
The nonsense variant c.709C>T, p.(Arg327Ter), was detected in RAD51C in a proband diagnosed with HGSOC at age of 57 (OC16), who had a first cousin diagnosed with OC (Figure 2a). This variant was also identified in patient OC31, whose family has a history of other cancer types, such as breast, gastric, throat, and leukemia (Figure 2b).
Figure 2.
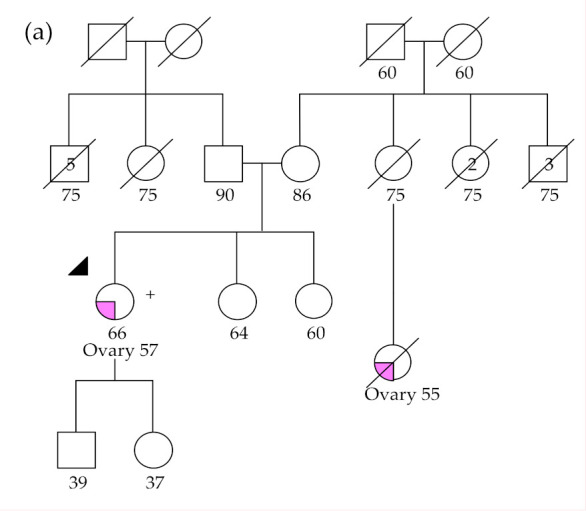

Pedigrees of patients with the RAD51C c.709C>T germline variant: (a) OC16 and (b) OC31. The index patients are indicated by an arrow.
The RAD51D frameshift variant c. 748del, p.(His250ThrfsTer2), was found in patient OC54 with a family history of colorectal, breast, prostate, and gastric cancers (Figure 3a). This variant was also detected in three patients (OC17, OC30, and OC55) belonging to the same family (seemingly unrelated to the previous), in which there is a history of breast and colorectal cancer (Figure 3b).
Figure 3.

Pedigrees of patients with RAD51D c.748del germline variant. Family of patient OC54 (a) and OC17, OC30, and OC55 (b). The index patient is indicated by an arrow. Plus (+) and minus (−) signals indicate a positive and a negative result for germline genetic testing, respectively.
2.2.3. MSH6 Germline Variants
The MSH6 inframe variant c.3848_3862del, p.(Ile1283_Tyr1287del), was identified in patient OC24, a woman diagnosed with endometrioid OC and uterine cancer at 51 years old, whose family has a history of colorectal, breast, and liver cancer (Figure 4). There is no classification for this variant in ClinVar, but it is described as a causal variant in the UMD database (www.umd.be). Immunohistochemistry analysis was performed in the ovarian tumor of the patient and showed loss of MSH6 expression. The somatic variant MSH6 c.4001+1G>T was also identified in this patient, presumably representing the second-hit leading to biallelic inactivation typical of most cancer predisposition tumor suppressor genes.
Figure 4.

Pedigree of patient OC24 with the likely pathogenic MSH6 c.3848_3862del germline variant. The index patient is indicated by an arrow.
Two MSH6 missense VUS were detected in two patients. The MSH6 c.1729C>T, p.(Arg577Cys), was identified in patient OC37 diagnosed with an ovarian carcinosarcoma at age 68. This variant was reported in ClinVar and in Varsome as uncertain significance, but it is predicted to be damaging by in silico tools. Immunohistochemistry and microsatellite instability analysis were performed in the tumor of the patient, and normal MSH6 expression and no microsatellite instability were observed, thus reducing the likelihood of the variant being associated with an increased risk for OC. The MSH6 c.3182T>C, p.(Leu1061Pro) variant, was found in patient OC79, who was diagnosed with endometrioid OC at 49 years old and has two relatives affected by colorectal and skin cancer. In ClinVar, there is no classification for this variant, while in VarSome, it is classified as uncertain significance. However, immunohistochemistry analysis performed in the tumor of the proband showed loss of expression for MSH6, suggesting that this variant has a deleterious effect on the protein. Furthermore, we also identified in the OC the somatic variant MSH6 c.1100A>G, p.(His367Arg), which is classified as pathogenic in ClinVar (reviewed by an expert panel) and could represent the second-hit leading to biallelic inactivation, but the scarce available data currently do not warrant the classification of the germline variant MSH6 c.3182T>C other than a VUS.
2.2.4. BRIP1 Germline Variant
The BRIP1 missense variant c.2477A>G, p.(Asn826Ser), was identified in patient OC11, diagnosed with HGSOC at the age of 39 years and family history of colon cancer and lymphoma. This variant is classified as VUS in ClinVar and in Varsome. However, it is predicted to be damaging by in silico tools
2.2.5. TP53 Germline Variant
The TP53 missense variant c.845G>A, p.(Arg282Gln), was identified in patient OC91, diagnosed with pancreatic and thyroid cancer in addition to OC at age of 48. There is history of other cancers in the patient’s family, such as stomach, colorectal, and bone cancer. This variant has conflicting interpretations of pathogenicity in ClinVar (five uncertain significance and two likely pathogenic classifications). However, it was predicted to be damaging by in silico tools and is classified as likely pathogenic in VarSome.
3. Discussion
In this study, we aimed to evaluate the yield of germline variants using an NGS customized multi-gene panel for the analysis of ovarian carcinomas. Among the 95 OC patients analyzed, we found that 26 (27.4%) were carriers of rare germline variants in seven genes associated with genetic predisposition to OC (BRCA1, BRCA2, BRIP1, MSH6, RAD51C, RAD51D, and TP53). Of these 26 patients, 20 (77%) had pathogenic variants and six (23%) had missense VUS predicted to have a deleterious impact on the protein by in silico tools. Almost all patients included in this study were also tested for BRCA1 and BRCA2 somatic and germline variants using a clinically validated method [16], and this study was able to identify all 15 patients known to be carriers of deleterious BRCA1/BRCA2 germline variants, which allowed validation of the custom panel we designed for those genes. The frequency of pathogenic BRCA1 and BRCA2 germline variants in this unselected series of OC patients was 13.7%, which is in concordance with previous studies that reported frequencies between 13% and 18% [12,16,19,20,21]. Furthermore, BRCA1/BRCA2 tumor sequencing allows the detection of both germline and somatic variants in a single test, the latter having a frequency ranging from 4% to 7% [16,22,23]. Thus, about 20% of OC patients are likely to harbor germline or somatic BRCA1/BRCA2 deleterious variants, which are predictive of response to PARPi. To initiate genetic testing of OC patients using a FFPE tumor sample is a strategy that has the potential to significant decrease time and sequencing cost [11], as any clinically actionable variant detected in the initial NGS test can subsequently be evaluated by Sanger sequencing in a peripheral blood sample for its eventual constitutional origin after genetic counseling.
RAD51C and RAD51D are implicated in HR repair of DNA double-strand breaks, playing a key role in replication fork maintenance [24]. Germline variants in RAD51C and RAD51D are known to confer susceptibility to OC [25,26]. Studies on the prevalence of germline variants in these genes in OC vary widely, ranging between 0.57% and 2.5% for RAD51C and 0.16% and 0.57% for RAD51D, among patients unselected for age and family history [12,14,27,28,29]. For OC patients selected for family history, the prevalence of germline variants was found to be 1.27% and 0.88% for RAD51C and RAD51D, respectively [25,26]. The estimated relative risk for OC in carriers of germline variants in the RAD51C range from 5.88 to 7.55 [30,31] and from 6.30 to 7.60 for RAD51D germline variant carriers [26,31]. The National Comprehensive Cancer Network (NCCN) guidelines for breast and OC recommend that risk-reducing salpingo-oophorectomy (RRSO) should be considered beginning at age 40 to 50 years in women carriers of germline variants in these genes [32]. In this study, we identified two patients (2.1%) with a pathogenic variant in RAD51C and four patients (4.2%) with a pathogenic germline variant in RAD51D, together representing 30% of all pathogenic germline variants identified. The prevalence of germline variants in RAD51C in our cohort (2.1%) is in concordance with previous studies [12,28], while the observed frequency for RAD51D (4.2%) is higher than previously reported [26,29]. However, three of the patients with the germline variant in RAD51D turned out to be relatives, which may have spuriously increased the contribution of this gene in our study. Genetic counseling has already been offered to healthy relatives of this family, which so far allowed us to identify four female carriers of the germline variant, one of them already submitted to RRSO.
Germline variants in MMR genes (MLH1, MSH2, MSH6, and PMS2) are associated with Lynch Syndrome (LS) [33], which is characterized by predisposition to a spectrum of cancers such as stomach, ovary, pancreas, upper urologic tract, and brain tumors, in addition to the most prevalent colorectal and endometrial cancers [34]. A cumulative incidence of 13% at 75 years was observed for OC in carriers of MSH6 germline variants [35], and the lifetime time risk was estimated to be up to 13.5% by age 70 [36,37]. We identified in one patient with both endometrial and ovarian cancer a MSH6 germline inframe deletion, which is not described in the literature at the time of writing. Although this variant is predicted to lead to an inframe deletion of five aminoacids at the protein level, we observed loss of MSH6 expression in the tumor by immunohistochemistry and detected a deleterious somatic MSH6 mutation, presumably representing the second-hit leading to biallelic inactivation. Furthermore, this germline variant has been previously identified in our laboratory in another six index patients presenting clinical criteria for Lynch Syndrome, who also showed consistent MSH6 loss of expression in their tumors, allowing the classification of this variant as likely pathogenic. Because of this genetic diagnosis, the index patient was offered screening for additional tumors associated with Lynch syndrome. Furthermore, genetic counseling and presymptomatic genetic testing were offered to relatives, and carriers will be offered cancer surveillance recommended for Lynch syndrome, with consideration of bilateral salpingo-oophorectomy and hysterectomy after completed childbearing.
One of the side-effects of using larger NGS gene panels is the increased likelihood of finding VUS. On the other hand, it is important to report all additional information to improve variant classification, as some of the VUS may in fact be pathogenic and others may be benign, but with insufficient data to allow their classification either way. As shown in the Results section, we combined in silico, additional somatic mutation, protein expression, and segregation data to classify several germline missense variants, but six of them must remain classified as VUS with the information currently available. Of the two BRCA2 VUS and two MSH6 VUS we report here, the evidence favors the pathogenicity of one MSH6 variant and the benign nature of the remaining three variants. The remaining two VUS were found in other cancer predisposing genes, namely BRIP1 and TP53. BRIP1 binds directly to the BRCT repeats of BRCA1, having an important role in the repair mechanism of double-strand DNA breaks [38]. Deleterious BRIP1 germline variants have been described as conferring a moderately increased risk for OC [39], with an estimated relative risk of 3.41 [40], showing frequencies ranging between 0.9% and 1.36% [12,14,40]. Finally, TP53 germline variants cause Li-Fraumeni syndrome, a relatively heterogeneous cancer predisposition syndrome characterized by increased risk of four core cancers (sarcoma, breast carcinoma, brain tumor, and adrenocortical carcinoma) [41], but known to increase the risk for other tumors, including OC [42]. The data we present here on these six VUS may contribute to improving their classification in the future and to clarify their eventual relevance for OC inherited predisposition.
The strategy we propose here using a 10 gene panel for routine OC NGS genetic testing on FFPE samples allowed us to identify 7.4% patients with pathogenic germline variants in genes other than BRCA1 and BRCA2, namely RAD51C, RAD51D, and MSH6, significantly improving the yield of deleterious variants in OC predisposition genes to 21% of the patients tested, without increasing the sequencing costs. Besides the identification of women with inherited cancer predisposition, the use of the NGS multi-gene panel is also useful for the detection of pathogenic variants that are predictive biomarkers for targeted therapies. HR deficient tumors due to deleterious variants in genes other than BRCA1 and BRCA2 may be sensitive to PARPi [43,44,45]. In fact, one study showed that RAD51C-deficient cancer cells were highly sensitivity to Olaparib, significantly inhibiting tumor growth in a xenograft model [43], whereas Loveday and colleagues [26] showed that cells deficient in RAD51D were sensitive to Olaparib in a magnitude similar to the observed by silencing BRCA2. Furthermore, the Food and Drug Administration (FDA) recently approved Olaparib for the treatment of metastatic castration-resistant prostate cancer (mCRPC) patients with pathogenic germline or somatic variants in HR repair genes, including RAD51C and RAD51D [46]. However, the evidence that RAD51C and RAD51D variants are good predictive biomarkers of response to PARPi is still scarce. On the other hand, tumors with MMR deficiency display a phenotype of microsatellite instability, which is a predictive biomarker for immunotherapy [47], allowing the FDA approval of the use of pembrolizumab, an anti-PD1 therapy, for the treatment of solid tumors with high microsatellite instability (MSI-H) or MMR deficiency [48]. The use of multi-gene panel testing that allows the simultaneous detection of both somatic and germline variants in FFPE tumor samples may also be beneficial for the selection of patients to be allocated to clinical trials or for the use of off-label therapy, especially in those who experienced several disease relapses and have limited therapeutic options. Furthermore, since this panel has a customizable design, new genes can easily be incorporated when new clinically actionable information becomes available.
4. Materials and Methods
4.1. Patients and Samples
The study cohort consisted of 96 women diagnosed with OC at the Portuguese Oncology Institute of Porto (IPO-Porto) from June 2016 to July 2019. The study was approved by the institutional review board (CI-IPOP-35-2016), and written informed consent was obtained from all patients. A FFPE tumor sample was obtained from each patient, after evaluation by a pathologist, who delimited areas with >50% cancer cells. Of all patients included in this study, 91 (95.8%) were previously tested in the clinical routine for BRCA1 and BRCA2 variants in tumor and blood using CE-IVD marked methodologies specific for those genes, and 50 patients (52.1%) were part of a study from our group that aimed to test the frequency of germline and somatic variants in a consecutive series of non-mucinous OC patients [16]. The clinicopathological features of OC patients included in the study are presented in Table 2. Patients were diagnosed with OC between 27 and 80 years (median 57 years of age), mostly at advanced stage (78.1% stage III-IV). All patients included in the study were recruited regardless of family history. However, three patients (OC17, OC30, and OC55) recruited at different times were known to belong to the same family when analyzing the results.
Table 2.
Clinical features of the 96 patients included in the study.
| Clinical Features | N (%) |
|
|---|---|---|
| Age, median (range years) | 57 (27–80) | |
| Figo Stage | Figo stage I | 14 (14.6) |
| Figo stage II | 7 (7.3) | |
| Figo stage III | 55 (57.3) | |
| Figo stage IV | 20 (20.8) | |
| Histological subtype | High-grade serous | 72 (71.9) |
| Low-grade serous | 8 (8.3) | |
| Clear cell | 6 (6.3) | |
| Endometrioid | 7 (7.3) | |
| Mucinous | 1 (1.0) | |
| Mixed | 3 (3.1) | |
| Carcinosarcoma | 2 (2.1) |
4.2. DNA Extraction from FFPE Tumor Samples
DNA extraction from FFPE samples was performed using the Cobas® DNA Sample Preparation Kit (Roche Diagnostics, Basel, Switzerland) according to the manufacturer’s protocol. Quality of DNA samples was measured with a Qubit® Fluorometer with the Qubit dsDNA HS assay kit (Thermo Fisher Scientific, Waltham, MA, USA).
4.3. Next-Generation Sequencing and Bioinformatic Analyses
NGS was performed in 96 tumor samples using a customized QIASeq Targeted DNA Panel containing ten genes (BRCA1, BRCA2, BRIP1, MLH1, MSH2, MSH6, PMS2, RAD51C, RAD51D, and TP53) (QIAGEN, Antwerp, Belgium). These genes were selected according to international guidelines for OC screening and/or previously description in the literature as conferring a risk for the development of OC. Library preparation was performed according to the manufacturer’s instructions, and final libraries were quantified on a 4200 TapeStation System (Agilent Technologies Inc, Santa Clara, CA, USA). Sequencing was carried out using a high-output kit in the NextSeq 550 System (Illumina, Inc., San Diego, CA, USA) in a 2 × 151 bp paired-end run. Sequencing alignment and variant calling were performed using smCounter2 (QIAGEN, Antwerp, Belgium) [49], and the resulting .vcf files were imported to GeneticistAssistantTM software (SoftGenetics, LLC, State College, PA, USA) for variant annotation. All variants with minor allele frequency (MAF) greater than 1% and synonymous variants were excluded. For MAF filtering, data were obtained from the 1000 Genomes Project (1000G), Genome Aggregation Database (gnomAD), and Exome Aggregation Consortium (ExAC) databases. To screen germline variants, we used the variant allele frequency (VAF) as recommend by ASCO/AMP/CAP guidelines [15] using VAF above 30% as the cutoff, which was shown to be reliable in a previous study of our group [16]. In fact, since the present study is focused on testing tumor suppressor genes, which very often show loss of heterozygosity in the tumor, the VAFs of deleterious germline variants are actually expected to be higher than 50%, which is supported by our earlier finding of mean VAFs of 80% and 69% for BRCA1 and BRCA2 pathogenic germline variants, respectively, with the lowest VAF being 49% [16].
4.4. Variant Classification
All variants remaining after variant filtering that had been described as pathogenic/likely pathogenic in ClinVar or having literature evidence supporting their pathogenicity were retained as deleterious. Nonsense, frameshift, inframe deletion and/or insertion, as well as canonical splice site variants, were also retained since they are considered to have very strong evidence of pathogenicity [15]. The potential pathogenicity of missense VUS was evaluated using MetaLR and MetaSVM, which combine 10 in silico prediction tools (SIFT, PolyPhen-2 HDIV, PolyPhen-2 HVAR, GERP++, MutationTaster, Mutation Assessor, FATHMM, LRT, SiPhy, and PhyloP) and the maximum minor allele frequency (MMAF) from the 1000G project [50]. We also used Combined Annotation-Dependent Depletion (CADD), which is an integrative annotation built from more than 60 genomic features for scoring the deleteriousness of single nucleotide variants (SNVs) and insertion/deletion variants [51]. Missense VUS were retained only if they were predicted to be damaging by MetaLR, MetaSVM, and CADD. The potential clinical significance of germline VUS was also evaluated using the VarSome approach [52], which is a bioinformatic tool for clinical interpretation of variants according to the American College of Medical Genetics and Genomic (ACMG) and the Association for Molecular Pathology (AMP) guidelines. Variants described in ClinVar as benign/likely benign were classified as benign and discarded.
4.5. Germline Variants Validation
Variants suspected to be germline were confirmed by Sanger sequencing in peripheral blood samples. DNA was extracted from blood samples using a standard protocol. Sanger sequencing was performed in a 3500 Genetic Analyzer (Applied Biosystems Foster City, CA, USA) using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), following the manufacturer’s instructions.
4.6. Microsatellite Instability and Immunohistochemical Analysis
Microsatellite instability analysis was performed using the Bethesda panel of markers (BAT25, BAT26, D2S123, D5S346, and D17S250) and according to the 1997 National Cancer Institute Guidelines, as previously described [53,54]. Assessment of MSH6 protein immunoexpression was performed as previously described [55].
5. Conclusions
We conclude that the strategy of reflex gene-panel tumor testing enables the identification of clinically actionable germline variants in a significantly higher proportion of OC patients, while at the same time testing for somatic variants in the same genes, which may be valuable information in patients with advanced disease that have run out of approved therapeutic options. Furthermore, this approach increases the chance to make available genetic counseling, presymptomatic genetic testing, and gynecological cancer prophylaxis to female relatives who turn out to be healthy carriers of deleterious germline variants.
Supplementary Materials
The following are available online at https://www.mdpi.com/2072-6694/12/10/2834/s1, Table S1: comparison of results obtained by the custom panel and by a clinically validated test regarding the BRCA1/BRCA2 deleterious variants.
Author Contributions
Conceptualization, A.B. and M.R.T.; methodology, A.B., P.P., A.P., J.G., M.P., C.S., C.P. and C.E.; software, A.B. and P.P.; validation, A.B., P.P., A.P. and M.R.T.; formal analysis, A.B.; data curation, A.B.; resources: C.B., A.P. and D.P.; supervision, M.R.T.; project administration, M.R.T.; funding acquisition, M.R.T.; writing—original draft preparation, A.B.; writing—review and editing, A.B., P.P., A.P., J.G., M.P., C.S., C.P., C.B., A.P., D.P., J.S. and M.R.T. All authors have read and agreed to the published version of the manuscript.
Funding
Ana Barbosa is a research fellow supported by the project NORTE-01-0145-FEDER-000027, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF). Pedro Pinto, Joana Guerra, and Manuela Pinheiro are research fellows of the Fundação para a Ciência e Tecnologia (UID/DTP/0076/POCI-01-0145-FEDER-006868, SFRH/BD/138670/2018 and SFRH/BPD/113014/2015, respectively).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Guppy A., Nathan P., Rustin G. Epithelial Ovarian Cancer: A Review of Current Management. Clin. Oncol. 2005;17:399–411. doi: 10.1016/j.clon.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Santaballa A., Barretina P., Casado A., García Y.G., Martín A.G., Guerra E., Lainez N., Martínez J., Redondo A., Romero I. SEOM Clinical Guideline in ovarian cancer. Clin. Transl. Oncol. 2016;18:1206–1212. doi: 10.1007/s12094-016-1588-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan R.J., Armstrong D.K., Alvarez R.D., Bakkum-Gamez J.N., Behbakht K., Chen L.-M., Copeland L., Crispens M.A., DeRosa M., Dorigo O., et al. Ovarian Cancer, Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016;14:1134–1163. doi: 10.6004/jnccn.2016.0122. [DOI] [PubMed] [Google Scholar]
- 5.Pignata S., Cecere S.C., Du Bois A., Harter P., Heitz F. Treatment of recurrent ovarian cancer. Ann. Oncol. 2017;28:viii51–viii56. doi: 10.1093/annonc/mdx441. [DOI] [PubMed] [Google Scholar]
- 6.Ledermann J., Raja F., Fotopoulou C., Gonzalez-Martin A., Colombo N., Sessa C. ESMO Guidelines Working Group Corrections to “Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up”. Ann. Oncol. 2018;29:iv259. doi: 10.1093/annonc/mdy157. [DOI] [PubMed] [Google Scholar]
- 7.Ledermann J.A., Harter P., Gourley C., Friedlander M., Vergote I., Rustin G., Scott C.L., Meier W., Shapira-Frommer R., Safra T., et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852–861. doi: 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
- 8.Ledermann J.A., Harter P., Gourley C., Friedlander M., Vergote I., Rustin G., Scott C., Meier W., Shapira-Frommer R., Safra T., et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012;366:1382–1392. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 9.Moore K., Colombo N., Scambia G., Kim B.-G., Oaknin A., Friedlander M., Lisyanskaya A., Floquet A., Leary A., Sonke G.S., et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018;379:2495–2505. doi: 10.1056/NEJMoa1810858. [DOI] [PubMed] [Google Scholar]
- 10.Ashworth A. A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J. Clin. Oncol. 2008;26:3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 11.Capoluongo E., Ellison G., Lopez J.A.-G., Penault F.-L., Ligtenberg M.J.L., Banerjee S., Singer C., Friedman E., Markefka B., Schirmacher P., et al. Guidance Statement on BRCA1/2 Tumor Testing in Ovarian Cancer Patients. Semin. Oncol. 2017;44:187–197. doi: 10.1053/j.seminoncol.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Walsh T., Casadei S., Lee M.K., Pennil C.C., Nord A.S., Thornton A.M., Roeb W., Agnew K.J., Stray S.M., Wickramanayake A., et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuchenbaecker K.B., Hopper J.L., Barnes D.R., Phillips K.-A., Mooij T.M., Roos-Blom M.-J., Jervis S., Van Leeuwen F.E., Milne R.L., Andrieu N., et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA. 2017;317:2402–2416. doi: 10.1001/jama.2017.7112. [DOI] [PubMed] [Google Scholar]
- 14.Norquist B., Harrell M.I., Brady M.F., Walsh T., Lee M.K., Gulsuner S., Bernards S.S., Casadei S., Yi Q., Burger R.A., et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016;2:482. doi: 10.1001/jamaoncol.2015.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M.M., Datto M., Duncavage E.J., Kulkarni S., Lindeman N.I., Roy S., Tsimberidou A., Vnencak-Jones C.L., Wolff D.J., Younes A., et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017;19:4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peixoto A., Pinto P., Guerra J., Pinheiro M., Santos C., Pinto C., Santos R., Escudeiro C., Bartosch C., Canario R., et al. Tumor Testing for Somatic and Germline BRCA1/BRCA2 Variants in Ovarian Cancer Patients in the Context of Strong Founder Effects. Front. Oncol. 2020;10:1318. doi: 10.3389/fonc.2020.01318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guidugli L., Shimelis H., Masica D.L., Pankratz V.S., Lipton G.B., Singh N., Hu C., Monteiro A.N.A., Lindor N.M., Goldgar D.E., et al. Assessment of the Clinical Relevance of BRCA2 Missense Variants by Functional and Computational Approaches. Am. J. Hum. Genet. 2018;102:233–248. doi: 10.1016/j.ajhg.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parsons M.T., Tudini E., Li H., Hahnen E., Wappenschmidt B., Feliubadaló L., Aalfs C.M., Agata S., Aittomäki K., Alducci E., et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum. Mutat. 2019;40:1557–1578. doi: 10.1002/humu.23818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morgan R.D., Burghel G.J., Flaum N., Bulman M., Clamp A.R., Hasan J., Mitchell C.L., Schlecht H., Woodward E.R., Lallo F.I., et al. Prevalence of germline pathogenic BRCA1/2 variants in sequential epithelial ovarian cancer cases. J. Med. Genet. 2019;56:301–307. doi: 10.1136/jmedgenet-2018-105792. [DOI] [PubMed] [Google Scholar]
- 20.The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S., Royer R., Li S., McLaughlin J.R., Rosen B., Risch H.A., Fan I., Bradley L., Shaw P.A., Narod S.A. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011;121:353–357. doi: 10.1016/j.ygyno.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 22.Pennington K.P., Walsh T., Harrell M.I., Lee M.K., Pennil C.C., Rendi M.H., Thornton A., Norquist B., Casadei S., Nord A.S., et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2013;20:764–775. doi: 10.1158/1078-0432.CCR-13-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cunningham J.M., Cicek M.S., Larson N.B., Davila J., Wang C., Larson M.C., Song H., Dicks E.M., Harrington P., Wick M., et al. Clinical Characteristics of Ovarian Cancer Classified by BRCA1, BRCA2 and RAD51C Status. Sci. Rep. 2014;4:4026. doi: 10.1038/srep04026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto Y., Chaudhuri A.R., Lopes M., Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meindl A., Hellebrand H., Wiek C., Erven V., Wappenschmidt B., Niederacher D., Freund M., Lichtner P., Hartmann L., Schaal H., et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010;42:410–414. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 26.Loveday C., Breast Cancer Susceptibility Collaboration (UK) Turnbull C., Ramsay E., Hughes D., Ruark E., Frankum J.R., Bowden G., Kalmyrzaev B., Warren-Perry M., et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011;43:879–882. doi: 10.1038/ng.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song H., Dicks E., Ramus S.J., Tyrer J.P., Intermaggio M.P., Hayward J., Edlund C.K., Conti D., Harrington P., Fraser L., et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015;33:2901–2907. doi: 10.1200/JCO.2015.61.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harter P., Hauke J., Heitz F., Reuss A., Kommoss S., Marme F., Heimbach A., Prieske K., Richtees L., Burges A., et al. Prevalence of deleterious germline variants in risk genes including BRCA1/2 in consecutive ovarian cancer patients (AGO-TR-1) PLoS ONE. 2017;12:e0186043. doi: 10.1371/journal.pone.0186043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Konstanta I., Fostira F.F., Apostolou P., Stratikos E., Kalfakakou D., Pampanos A., Kollia P., Papadimitriou C., Konstantopoulou I., Yannoukakos D. Contribution of RAD51D germline mutations in breast and ovarian cancer in Greece. J. Hum. Genet. 2018;63:1149–1158. doi: 10.1038/s10038-018-0498-8. [DOI] [PubMed] [Google Scholar]
- 30.Loveday C., Turnbull C., Ruark E., Xicola R.M.M., Ramsay E., Hughes D., Warren-Perry M., Snape K., Breast Cancer Susceptibility Collaboration (UK) Eccles D.M., et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012;44:475–476. doi: 10.1038/ng.2224. [DOI] [PubMed] [Google Scholar]
- 31.Yang X., Song H., Leslie G., Engel C., Hahnen E., Auber B., Horváth J., Kast K., Niederacher D., Turnbull C., et al. Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. J. Natl. Cancer Inst. 2020 doi: 10.1093/jnci/djaa030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daly M.B., Pilarski R., Berry M., Buys S.S., Farmer M., Friedman S., Garber J.E., Kauff N.D., Khan S., Klein C., et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Cancer Netw. 2016;15:9–20. doi: 10.6004/jnccn.2017.0003. [DOI] [PubMed] [Google Scholar]
- 33.Lynch H.T., Snyder C.L., Shaw T.G., Heinen C.D., Hitchins M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer. 2015;15:181–194. doi: 10.1038/nrc3878. [DOI] [PubMed] [Google Scholar]
- 34.Watson P., Vasen H.F.A., Mecklin J.-P., Bernstein I., Aarnio M., Järvinen H.J., Myrhøj T., Sunde L., Wijnen J.T., Lynch H.T. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int. J. Cancer. 2008;123:444–449. doi: 10.1002/ijc.23508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Møller P., Seppälä T.T., Bernstein I., Holinski-Feder E., Sala P., Evans D.G., Lindblom A., Macrae F., Blanco I., Sijmons R.H., et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut. 2017;67:1306–1316. doi: 10.1136/gutjnl-2017-314057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramsoekh D., Wagner A., Van Leerdam M.E., Dooijes D., Tops C.M., Steyerberg E.W., Kuipers E.J. Cancer risk in MLH1, MSH2 and MSH6 mutation carriers; different risk profiles may influence clinical management. Hered. Cancer Clin. Pract. 2009;7:17. doi: 10.1186/1897-4287-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonadona V., Bonaiti B., Olschwang S., Grandjouan S., Huiart L., Longy M., Guimbaud R., Buecher B., Bignon Y.-J., Caron O., et al. Cancer Risks Associated With Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA. 2011;305:2304–2310. doi: 10.1001/jama.2011.743. [DOI] [PubMed] [Google Scholar]
- 38.Bridge W.L., Vandenberg C.J., Franklin R.J., Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat. Genet. 2005;37:953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 39.Rafnar T., Gudbjartsson D.F., Sulem P., Jonasdottir A., Sigurdsson A., Jonasdottir A., Besenbacher S., Lundin P., Stacey S.N., Gudmundsson J., et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011;43:1104–1107. doi: 10.1038/ng.955. [DOI] [PubMed] [Google Scholar]
- 40.Ramus S.J., Song H., Dicks E., Tyrer J.P., Rosenthal A.N., Intermaggio M.P., Fraser L., Gentry-Maharaj A., Hayward J., Philpott S., et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015;107 doi: 10.1093/jnci/djv214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li F.P., Fraumeni J.F. Soft-Tissue Sarcomas, Breast Cancer, and Other Neoplasms. Ann. Intern. Med. 1969;71:747–752. doi: 10.7326/0003-4819-71-4-747. [DOI] [PubMed] [Google Scholar]
- 42.Sorrell A.D., Espenschied C.R., Culver J.O., Weitzel J.N. Tumor protein p53 (TP53) testing and Li-Fraumeni syndrome: Current status of clinical applications and future directions. Mol. Diagn. Ther. 2013;17:31–47. doi: 10.1007/s40291-013-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Min A., Im S.-A., Yoon Y.-K., Song S.-H., Nam H.-J., Hur H.-S., Kim H.-P., Lee K.-H., Han S.-W., Oh D.-Y., et al. RAD51C-Deficient Cancer Cells Are Highly Sensitive to the PARP Inhibitor Olaparib. Mol. Cancer Ther. 2013;12:865–877. doi: 10.1158/1535-7163.MCT-12-0950. [DOI] [PubMed] [Google Scholar]
- 44.AlHilli M.M., Becker M.A., Weroha S.J., Flatten K.S., Hurley R.M., Harrell M.I., Oberg A.L., Maurer M.J., Hawthorne K.M., Hou X., et al. In vivo anti-tumor activity of the PARP inhibitor niraparib in homologous recombination deficient and proficient ovarian carcinoma. Gynecol. Oncol. 2016;143:379–388. doi: 10.1016/j.ygyno.2016.08.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCabe N., Turner N.C., Lord C.J., Kluzek K., Bialkowska A., Swift S., Giavara S., O’Connor M.J., Tutt A., Zdzienicka M.Z., et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 46.De Bono J.S., Mateo J., Fizazi K., Saad F., Shore N., Sandhu S., Chi K.N., Sartor O., Agarwal N., Olmos D., et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020;382:2091–2102. doi: 10.1056/NEJMoa1911440. [DOI] [PubMed] [Google Scholar]
- 47.Le D.T., Uram J.N., Wang H., Bartlett B.R., Kemberling H., Eyring A.D., Skora A.D., Luber B.S., Azad N.S., Laheru D., et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marcus L., Lemery S.J., Keegan P., Pazdur R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019;25:3753–3758. doi: 10.1158/1078-0432.CCR-18-4070. [DOI] [PubMed] [Google Scholar]
- 49.Xu C., Gu X., Padmanabhan R., Wu Z., Peng Q., Dicarlo J., Wang Y. smCounter2: An accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics. 2018;35:1299–1309. doi: 10.1093/bioinformatics/bty790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong C., Wei P., Jian X., Gibbs R., Boerwinkle E., Wang K., Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2014;24:2125–2137. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kopanos C., Tsiolkas V., Kouris A., Chapple C.E., Aguilera M.A., Meyer R., Massouras A. VarSome: The human genomic variant search engine. Bioinformatics. 2019;35:1978–1980. doi: 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boland C.R., Thibodeau S.N., Hamilton S.R., Sidransky D., Eshleman J.R., Burt R.W., Meltzer S.J., A Rodriguez-Bigas M., Fodde R., Ranzani G.N., et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 54.Pinheiro M., Veiga I., Pinto C., Afonso L., Sousa O., Fragoso M., Santos L.L., Lopes P., Pais I., Lopes C., et al. Mitochondrial genome alterations in rectal and sigmoid carcinomas. Cancer Lett. 2009;280:38–43. doi: 10.1016/j.canlet.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 55.Bartosch C., Pires-Luís A.S., Meireles C., Baptista M., Gouveia A., Pinto C., Shannon K.M., Jerónimo C., Teixeira M.R., Lopes J.M., et al. Pathologic Findings in Prophylactic and Nonprophylactic Hysterectomy Specimens of Patients With Lynch Syndrome. Am. J. Surg. Pathol. 2016;40:1177–1191. doi: 10.1097/PAS.0000000000000684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.