

Abstract
Ultraviolet (UV) radiation is among the most prevalent environmental factors that influence human health and disease. Even one hour of UV irradiation extensively damages the genome. To cope with resulting deleterious DNA lesions, cells activate a multitude of DNA damage response pathways, including DNA repair. Strikingly, UV-induced DNA damage formation and repair are affected by chromatin state. When cells enter S phase with these lesions, a distinct mutation signature is created via error-prone translesion synthesis. Chronic UV exposure leads to high mutation burden in skin and consequently the development of skin cancer, the most common cancer in the United States. Intriguingly, UV-induced oxidative stress has opposing effects on carcinogenesis. Elucidating the molecular mechanisms of UV-induced DNA damage responses will be useful for preventing and treating skin cancer with greater precision. Excitingly, recent studies have uncovered substantial depth of novel findings regarding the molecular and cellular consequences of UV irradiation. In this review, we will discuss updated mechanisms of UV-induced DNA damage responses including the ATR pathway, which maintains genome integrity following UV irradiation. We will also present current strategies for preventing and treating nonmelanoma skin cancer, including ATR pathway inhibition for prevention and photodynamic therapy for treatment.
Graphical Abstract

Ultraviolet (UV) radiation is an extremely prevalent environmental factor that extensively damages the genome. Cells respond to UV-induced DNA damage by activating multiple biological processes: DNA damage checkpoint for cell cycle arrest, DNA repair that is affected by chromatin state, and DNA damage tolerance that may promote survival with mutations. Chronic UV exposure increases mutation burden, leading to skin cancer development. This process is facilitated by reactive oxygen species (ROS), inflammation, and immunosuppression. Recent studies have elucidated precise molecular and cellular consequences of UV irradiation. Targeting UV-induced DNA damage responses is an effective means to prevent and treat skin cancer.
Introduction
Ultraviolet (UV) radiation is one of the most common environmental exposures to humans and other organisms. Its role in influencing human health and disease was first suggested in the late 19th and early 20th centuries (1–4). Since then, the response of the human body to UV, and more recently the cellular and molecular responses to UV, have been continually elucidated in incrementally greater detail.
UV exposure primarily affects the skin in humans (Fig. 1). Molecular mechanisms have been identified for UV radiation in skin erythema and vasodilation (5,6), inflammation (5,7,8), sunburn and suntan (9,10), epidermal hyperplasia (11), wrinkle formation and photoaging (also known as dermatoheliosis) (12,13), and immunosuppression (14,15). UV exposure can also induce production of the endogenous opioid β-endorphin in the skin and create addictive behavior to UV light (16,10). As a result of this addiction, people may seek excessive UV exposure. However, chronic UV irradiation increases the risk of skin carcinogenesis (17–19,3,1,20). Furthermore, the burden of skin cancer increases with age (21,22). Three major types of skin cancer are basal cell carcinoma (BCC), cutaneous squamous cell carcinoma (cSCC), and melanoma. Among these, BCC and cSCC are categorized as nonmelanoma skin cancer (NMSC, also known as keratinocyte carcinoma (23)) and are the most common cancers in the United States (24). The annual incidence of NMSC is 5.4 million in the United States (24).
Figure 1.

Time course of UV-induced responses in the skin and skin cancer evolution. UV irradiation instantaneously generates DNA lesions that are potentially mutagenic and carcinogenic. To survive UV damage, cells cope with these deleterious lesions by activating a variety of signaling pathways including ATR (DNA damage response). UV also generates reactive oxygen species (ROS) that promote inflammation and carcinogenesis. Translesion synthesis enables DNA synthesis in the presence of UV-induced DNA lesions but may incorporate mutations. In the days and weeks following UV exposure, the skin exhibits tissue-level responses such as erythema, tanning, angiogenesis, epidermal hyperplasia, and immunosuppression. Chronic UV exposure leads to accumulation of genetic and epigenetic alterations. As a result, aberrant cell signaling drives transformation of normal skin cells to premalignant lesions (e.g., actinic keratosis (AK) or benign melanocytic nevi). With further increase in the burden of genetic and epigenetic alterations, cells may evolve into invasive and metastatic skin cancer. Chronic UV irradiation also promotes premature skin aging (photoaging), characterized by wrinkle formation and reduced elasticity.
Given the fact that humans are diurnal and exposed to sunlight throughout their lifetimes, UV irradiation creates a tremendous burden of damage in an extremely large number of skin cells in the human body. The human body has 37 trillion cells in total, of which approximately 2 trillion cells are in the skin (including 176 billion epidermal cells) (25). Because up to 41% of body surface area is typically exposed to the sun (26), up to 820 billion skin cells (72 billion epidermal cells) per person are theoretically exposed to UV. Among these, 3.5% of epidermal cells are typically in S phase (27), meaning that approximately 2.5 billion epidermal cells need to replicate DNA with UV-induced DNA damage at any time, increasing the likelihood of mutation incorporation. The risk of skin cancer increases over time with accumulated mutations. In humans, approximately 70,000 hours of lifetime sun exposure appears to be a threshold for increased risk of cSCC (19). Strikingly, with long-term UV exposure, the mutation burden even in normal skin approximates the mutation burden present in cancers of internal organs (not exposed to UV) (28). Thus, understanding the molecular mechanisms of how cells cope with UV-induced damage is of great interest. These fundamental mechanisms of cellular responses to environmental factors will provide insights into effective strategies for preventing and treating skin cancer with greater precision.
In this review, we will summarize recent discoveries in UV-induced damage responses, including formation of DNA damage, ATR signaling pathway, transcriptional changes, DNA repair, oxidative stress, and mutagenesis. This review will also describe strategies for prevention and treatment of nonmelanoma skin cancer, including ATR as a target for skin cancer prevention and photodynamic therapies that were pioneered by Dr. Thomas J. Dougherty in the 1970s.
Updated models of UV-induced DNA damage responses
UV-induced DNA damage formation
UV irradiation generates two major types of lesions in DNA through direct photochemical reactions within dipyrimidine sites (two adjacent cytosine or thymine residues), forming dimers on the same strand (29). The most common UV-induced DNA lesion is cyclobutane pyrimidine dimers (CPDs), most frequently at two adjacent thymines (cyclobutane thymine dimers) (30–32). The second most common UV-induced DNA lesion is pyrimidine (6-4) pyrimidone photoproducts (6–4PPs), which are generated via covalent bond formation between the carbons 6 and 4 of adjacent pyrimidines. 6–4PPs are 6-fold less abundant than CPDs with simulated sunlight (30). One hour of sunlight generates a surprisingly high number of DNA lesions; we estimate that 100,000 to 200,000 DNA lesions are formed in human diploid cells with 6 billion base pairs (520 lesions per 106 normal bases per J/cm2 UVB in the whole skin (32); UV index is 3 in October in New York (41°N) (33), and one hour of sunlight at UV index 3 reaches the minimal erythemal dose in type II skin, typically 0.025 J/cm2 UVB (34)).
Sunlight contains three different wavelength ranges of UV light, termed UVA (315–400 nm), UVB (280–315 nm), and UVC (100–280 nm). UVC is entirely absorbed by the ozone layer of the Earth, leaving UVA and UVB as the main UV components of terrestrial sunlight. The number and types of DNA lesions generated by UV depend on UV wavelengths (30,35). Shorter wavelengths generate DNA lesions more efficiently; UVC is ~100-fold more efficient to generate CPDs than UVB, and UVB is ~1,000-fold more efficient than UVA. UVC and UVB generate both CPDs and 6–4PPs, but UVA generates only CPDs.
UVA and UVB have varying effects on the skin. UVA causes photoaging, but both UVA and UVB cause mutations and cancer (13,36,37). UVB is known to be the main contributor to UV-induced skin carcinogenesis. Specifically, 315 nm (borderline wavelength between UVA and UVB) radiation has been found to induce more mutations and cancer than other UV wavelengths (38–40). UVA also causes oxidative DNA damage that may lead to mutations (41).
Direct formation of CPDs by UV is very rapid; CPDs form within one picosecond after UV exposure (42). Additionally, in melanocytes, CPDs form for 3 hours after ceasing UV exposure (43). These delayed, ‘dark CPDs’ are generated via UV-induced reactive oxygen and nitrogen species that react with melanin. Dark CPDs include cytosine-containing lesions relevant to UV signature C>T mutations. Thus, post-UV time (up to 3 hours) may be a therapeutic window to limit the formation of cytosine-containing CPDs that are potentially mutagenic. This work suggests that melanin plays a role in UV-induced mutagenesis. However, melanin has been shown to have a protective role for DNA from UV damage. In cell culture, melanin reduces the rate of CPD and 6–4PP formation (44). Furthermore, CPD formation is considerably reduced in highly pigmented skin than in less pigmented skin in humans (45).
The question of where in the genome UV-induced DNA lesions are preferentially created has only recently been addressed. The use of next-generation sequencing (NGS) has enabled the precise localization of UV-induced DNA lesions in the genome aligned with chromatin information. One study revealed genome-wide distribution of UV-induced CPD lesions at single-nucleotide resolution (‘CPD-seq’) and compared with known nucleosome and transcription factor binding positions (46). Intriguingly, the formation of CPDs within each nucleosome (147 bp of DNA double helix wrapped around a histone octamer) was affected by the distance of dipyrimidines to the histone core (Fig. 2). Due to the nature of the DNA double helix, the position of nucleotides can be slightly close (‘inner’) or distant (‘outer’) to the histone core periodically (~10 bp per turn) within a nucleosome. When dipyrimidines were close to the histone core within a nucleosome, fewer CPD lesions were generated due to relatively constrained flexibility of the nucleotides at these inner positions (46). Also, this study showed that CPD formation is largely inhibited at binding sites of yeast transcription factors Abf1 and Reb1. However, binding sites for the E-twenty-six (ETS) transcription factor family have been found with increased CPD formation due to changes in DNA conformation after ETS binding (47,48). Another study shows that transcription factor binding can inhibit, stimulate, or have no effect on UV-induced lesion formation depending on the specific transcription factor (49). Furthermore, whole-genome mapping of UV-induced DNA lesions prior to DNA repair recently revealed that UV-induced lesions are generated at higher frequencies in relatively inaccessible chromosomal regions (heterochromatin), whereas euchromatic regions characterized by active transcription and DNase hypersensitivity are protected from UV damage (50). Intriguingly, long interspersed nuclear elements (LINEs) – heterochromatic regions present at the nuclear lamina at the nuclear periphery – were among the genomic regions most susceptible to UV-induced DNA damage formation. Such genomic elements have been proposed to provide a ‘sink’ to prevent UV-induced DNA damage in active genes, thereby protecting genome integrity (50). In addition to chromatin state, UV-induced CPD lesions are preferentially formed at dipyrimidine sites with cytosine methylation (51). It has been suggested that cytosine methylation within TC dipyrimidines induces structural changes at the nucleotide level that increase the risk of CPD formation, but not 6–4PP formation (52).
Figure 2.

UV-induced DNA lesion formation affected by nucleotide positioning in nucleosome. Within an individual nucleosome, due to the rotation of nucleotides within the DNA double helix, nucleotides are periodically positioned close to (‘inner’) or far from (‘outer’) the histone core every ~10 bp. Compared to the ‘outer’ positions, nucleotides at the ‘inner’ positions have constrained flexibility. Thus, UV induces dimer formation more frequently at ‘outer’ dipyrimidines where DNA bending is more flexible.
ATR and ATM pathways to cope with UV-induced DNA lesions
DNA damage checkpoints are a surveillance mechanism to ensure genome integrity and respond to genotoxic stress by activating DNA damage response pathways. These pathways are essential for surviving DNA damage in all cells (53–55). DNA damage response is regulated by two major kinases that respond to various types of genotoxic stress: ataxia telangiectasia mutated (ATM) primarily responsible for DNA double-strand breaks and ataxia telangiectasia and Rad3-related (ATR) primarily responsible for replication stress. In response to replication stress (e.g., UV that generates replication-blocking lesions, and hydroxyurea that depletes deoxynucleotides), ATR plays a key role in maintaining genome integrity by inducing cell cycle arrest, inhibiting DNA replication origin firing, and stabilizing stalled replication forks (54) (Fig. 3A).
Figure 3.

UV-induced DNA damage responses to maintain genome integrity. (A) ATR activation by replication blockage. During DNA replication, DNA polymerase stalls at UV-induced DNA lesions while MCM helicase continues to unwind duplex DNA. This uncoupling of helicase and polymerase activities generates a long stretch of single-stranded DNA (ssDNA) that is rapidly coated with RPA. RPA-coated ssDNA recruits the ATR-ATRIP complex, TopBP1, RHINO, and ETAA1, resulting in ATR activation. ATR phosphorylates many downstream targets to induce cell cycle arrest, inhibit DNA replication origin firing, and prevent replication fork collapse. (B) ATM activation by transcription blockage. In noncycling cells, UV-induced DNA lesions stall transcription and displace spliceosomes. This leads to hybridization of pre-mRNA with the template DNA strand, leaving the nontemplate strand as ssDNA and forming an R-loop structure that recruits ATM. Subsequently, ATM induces alternative splicing of pre-mRNA, leading to altered gene expression in UV-irradiated cells. (C) Nucleotide excision repair (NER) for UV-induced DNA lesions. NER consists of global genome repair (GGR), which recognizes lesions throughout the genome independently of transcription or replication, and transcription-coupled repair (TCR), which recognizes DNA lesions on transcribed strands of active genes during transcription. In both GGR and TCR, the damaged DNA strand is cleaved at both sides of the DNA lesion, generating a lesion-containing product (~30 nt). The resulting ssDNA gap is filled by DNA polymerases δ/ε. (D) EXO1-mediated ATR activation and response to excessive lesions. In noncycling cells, EXO1 competes with NER and extends the NER-generated ssDNA gap, making it long enough to activate the DNA damage checkpoint. Compared to low doses, high doses of UV irradiation generate lesions more frequently, and thus there is a greater chance that two DNA lesions are closely positioned on opposing strands (closely opposing lesions, COLs). One lesion is removed by NER, but the other lesion remains in the resulting ssDNA gap. NER gap filling by DNA polymerases δ/ε stalls at this lesion, and EXO1 extends the ssDNA gap. Translesion synthesis (TLS) polymerases fill the lesion-containing gap and displace EXO1. If TLS polymerases are deficient, EXO1 will continue to extend the ssDNA gap, leading to double-strand breaks and cell death. (E) Translesion synthesis (TLS) as DNA damage tolerance. When cells enter S phase with UV-induced DNA lesions, replicative DNA polymerases stall at the lesion. To resume DNA replication, repriming occurs downstream of the lesion. The resulting ssDNA gap will be filled by TLS polymerases that can synthesize DNA across the lesion but may incorporate mutations. TLS occurs in an error-free or error-prone manner.
The ATR activation mechanism has been extensively studied (54,53). UV generates DNA lesions that can stall replication forks by blocking replicative DNA polymerase; meanwhile, DNA helicase continues to unwind duplex DNA (56). This uncoupling of helicase and DNA polymerase activities generates a long stretch of single-stranded DNA (ssDNA) to which replication protein A (RPA) is recruited (57). ATR exists in a stable complex with ATR-interacting protein (ATRIP), and the ATR-ATRIP complex is recruited to this RPA-coated ssDNA structure (58,54). ATR has kinase activity that is stimulated by many mediator proteins that are recruited to the same RPA-ssDNA structure. One of these mediators is DNA topoisomerase II binding protein 1 (TopBP1), which localizes to DNA damage sites or arrested replication forks (59,60). TopBP1 recruitment to RPA-coated ssDNA is required for full activation of ATR kinase activity (61). RHINO is also recruited to sites of DNA damage and interacts with TopBP1 to fully activate ATR (62). At RPA-ssDNA, ATR autophosphorylates itself at Thr1989 to promote TopBP1-ATR interaction, stimulating ATR kinase activity (63). More recently, Ewing’s tumor-associated antigen 1 (ETAA1) has been identified as an RPA-binding protein to directly activate ATR independently of TopBP1 (64).
Activated ATR phosphorylates various downstream targets (65,66) in order to elicit cell cycle checkpoints (G1-S (67), intra-S (68,69), S-G2 (70), and G2-M (71)) (Fig. 4). After UV generates DNA damage, p53 is phosphorylated at Ser15, and checkpoint kinase 1 (CHK1) is phosphorylated at Ser345. These phosphorylation events are mediated by ATR, but not by ATM (72). In turn, phosphorylated p53 (Ser15) is prevented from degradation, allowing for cell cycle arrest or apoptosis (73). Phosphorylated, activated CHK1 (Ser345) triggers degradation of the CDC25A phosphatase that is required for cyclin-dependent kinase 2 (CDK2)-mediated G1-S transition, and sequestration of the CDC25C phosphatase that is required for CDK1-mediated G2-M transition (54,71). During S phase, stalled replication forks must be stabilized to prevent fork collapse, and the firing of new DNA replication origins must be inhibited until replication blockage is resolved. DNA replication origin firing requires phosphorylation of Treslin by CDK (74). It is plausible that replication stress-induced CHK1 activation decreases the function of CDK, thereby inhibiting Treslin and DNA replication origin firing (74). ATR phosphorylates SMARCAL1 to prevent collapse of stalled replication forks (75). By these processes during replication stress, ATR activation allows time for DNA repair and maintains genome integrity. ATR also plays a role in unperturbed conditions; ATR regulates the frequency of replication origin firing in the absence of DNA damage (76). During mitosis, ATR promotes faithful chromosome segregation independently of DNA damage (77). Collectively, the ATR pathway plays a major role in ensuring faithful DNA replication and maintaining genome integrity (54,70,77).
Figure 4.

Cell cycle checkpoints elicited by UV-induced ATR activation. G1-S checkpoint: Following UV-induced DNA damage in G1 phase, activated ATR phosphorylates p53 at Ser15 and CHK1 at Ser345. Phospho-CHK1 inactivates CDC25A, preventing dephosphorylation of CDK2 and inducing G1 arrest. The p53-p21 pathway also inhibits CDK2. Intra-S checkpoint: UV activates the ATR-CHK1 pathway in S phase, decreasing the function of CDK. DNA replication origin firing requires phosphorylation of Treslin by CDK, and thus it is plausible that replication stress prevents origin firing via inhibiting Treslin phosphorylation. S-G2 checkpoint: ATR senses ongoing DNA replication during unperturbed S phase, inhibiting CDK1 activity and preventing cell cycle progression to G2 phase. When DNA replication is completed, ATR activity is diminished, allowing CDK1 to phosphorylate FOXM1 for S-G2 transition. G2-M checkpoint: UV-induced ATR-CHK1 activation inhibits CDC25C and CDK1 activities, preventing G2-M transition. WEE1 phosphorylates and inhibits CDK1, also inducing G2 arrest.
Most cancer cells are defective in the p53 pathway that induces G1 arrest in response to DNA damage. These p53-defective cells rely on the ATR pathway to survive DNA damage. Thus, targeting the ATR pathway can selectively sensitize p53-defective or G1 checkpoint-defective cells to DNA damage-induced death (78–81). Paradoxically, in noncycling cells, ATR inhibition decreases apoptosis in response to DNA damage induced by UV or UV mimetic N-acetoxy-2-acetylaminofluorene (AAAF), an agent that generates bulky lesions on guanines in DNA and blocks RNA polymerase II progression (82). This suggests that ATR mediates pro-apoptotic signaling in noncycling cells. Thus, ATR inhibition can selectively kill proliferating, cancerous cells while sparing nonproliferating, normal cells.
UV irradiation primarily activates the ATR pathway, but in noncycling cells, the ATM pathway is also activated by UV. A recent study revealed a non-canonical, RNA-mediated mechanism for the activation of the ATM pathway in noncycling cells (83) (Fig. 3B). UV-induced DNA damage blocks transcription, leading to displacement of spliceosomes which normally process pre-mRNA into mature transcripts. The resulting displacement of spliceosomes facilitates formation of R-loops with pre-mRNA, i.e., the hybridization of pre-mRNA with the template DNA strand that leaves the nontemplate strand as ssDNA. These R-loops activate ATM, which then contribute to alternative splicing of pre-mRNA (83). As a result, ATM significantly contributes to UV-induced gene expression changes (83).
Transcriptional changes following acute UV exposure
The gene expression profile is altered within hours after acute UV exposure. In one study of human epidermis, 12% of 5,380 analyzed genes had at least 2-fold altered gene expression following UV exposure: 246 upregulated and 373 downregulated (84). Among these 619 genes, 21%, 53%, and 26% of differentially expressed genes were found at 2, 24, and 72 hours after UV exposure, respectively (84). UV-upregulated genes encoded histones, interferons, matrix metalloproteinases, glutathione peroxidases, and S100 calcium-binding proteins. After 72 hours, half of these UV-regulated genes returned to pre-exposure baseline expression levels, indicating the transient nature of acute responses to UV exposure (84). Transcription recovery depends on the length of genes; after UV exposure, transcription of long genes (>20 kb) is preferentially inhibited, and transcription recovery is delayed (85). Thus, short genes exhibited higher levels of expression than long genes in the acute period after UV exposure (85). UV irradiation increases transcription of XPC, a key protein in nucleotide excision repair (NER), in a p53-dependent manner within 24 hours (86). In human keratinocytes, within 1 hour after UV exposure, a variety of cytokeratins were upregulated; 6 hours after UV exposure, genes related to growth, apoptosis, DNA repair, cytokines, and cell adhesion exhibited altered expression (87). Importantly, gene expression of human epidermis in vivo and that of cultured keratinocytes in vitro are different with and without UV exposure, reflecting differences in cellular environments (88,84).
Acute UV exposure alters expression of not only mRNA but also microRNA (miRNA; ~22 nt noncoding RNA molecules which regulate gene expression). After 5 J/cm2 of UVA irradiation, expression of miRNAs miR-21 and miR-203 are increased; miR-21 is known to have oncogenic properties, and miR-203 antagonizes the expression of p63, which is a transcription factor maintaining the “stemness” of skin cells (89). However, 30 mJ/cm2 of UVB irradiation had no effect on miR-21 expression and increased miR-203 expression, suggesting different effects of UVA and UVB on different miRNA molecules. cSCC is associated with increased miR-21 expression, whereas miR-203 expression is decreased in well differentiated cSCC and further decreased in moderately differentiated cSCC (89). Further investigations are needed to elucidate the roles of these miRNAs in cSCC pathogenesis. UV exposure also affects expression of long noncoding RNAs (lncRNA; >200 nt). A total of 660 lncRNAs were up- or downregulated by UVA and 3,559 lncRNAs by UVB (90). Intriguingly, the majority of lncRNAs were only affected either by UVA or UVB, and only few lncRNAs were up- or downregulated by both UVA and UVB, suggesting differences in cellular response to UVA and UVB irradiation (90). Although the specific roles of most of these lncRNAs have not been elucidated, lncRNAs have been broadly implicated in disease via regulation of protein-coding genes.
Repair of UV-induced DNA lesions and the effect of chromatin state
DNA repair is essential for any organism whose genome is damaged by UV from the sun (91). Bacteria, plants, and animals (except for placental mammals) have light-activated DNA repair enzymes known as photolyases that repair UV-induced DNA damage, either CPD or 6–4PP, upon illumination by visible light (92). Photolyase acts very rapidly; CPD-photolyase (also known simply as photolyase) from Escherichia coli repairs CPDs within one nanosecond (93), and 6–4PP-photolyase (also known as (6-4) photolyase) from Arabidopsis thaliana repairs 6–4PPs within tens of nanoseconds (94). Humans do not have photolyases (92,95); however, humans and other organisms have the nucleotide excision repair (NER) mechanism, which can repair UV-induced DNA damage (both CPD and 6–4PP) without light (96,97) (Fig. 3C). NER consists of two subpathways that differ in initial damage recognition: global genome repair (GGR) and transcription-coupled repair (TCR) (96,98). GGR is initiated by recognition of DNA lesions throughout the genome independent of transcription or replication, whereas TCR recognizes DNA lesions on transcribed strands of active genes during transcription (96,98). In GGR, UV-damaged DNA-binding protein (UV-DDB) directly binds to DNA lesions to initiate the DNA repair process. After damage recognition in both GGR and TCR, DNA repair proteins are recruited for dual incision in the damaged DNA strand at both sides of the lesion, creating a lesion-containing product (26–27 nt) and leaving the other strand single-stranded. To fill the single-stranded DNA (ssDNA) gap, DNA polymerases δ/ε are recruited and synthesize DNA using the intact strand as a template in an error-free manner (96). Remodeling of chromatin assists the NER machinery with access to lesions (96). Interestingly, RNA-binding protein DGCR8, which is involved in miRNA processing, has been found to facilitate TCR independently of RNA processing (99). This novel function of DGCR8 requires UV-induced phosphorylation of DGCR8 at Ser153 (99).
In noncycling cells, exonuclease 1 (EXO1) competes with the gap filling process of NER and extends the NER-generated ssDNA gap, making it long enough to activate DNA damage checkpoints (100) (Fig. 3D). When cells receive an excessive dose of UV, many DNA lesions are generated to the extent that two DNA lesions are likely to be close to each other and on two opposing strands (closely opposing lesions, COLs). These COLs are problematic for NER. Removal of one of two COLs by NER leaves the second lesion in the complementary strand in an ssDNA configuration. Because the ssDNA gap contains a lesion, DNA polymerases δ/ε cannot synthesize DNA using the damaged strand as a template, and EXO1 starts extending the ssDNA gap. This promotes the recruitment of specialized DNA polymerases, which can synthesize DNA across a lesion (translesion synthesis, TLS), to the lesion-containing ssDNA gap in order to fill the gap, preventing further action of EXO1 (101). If TLS polymerases are deficient, EXO1 will continue to extend the ssDNA gap while DNA polymerases δ/ε remain stalled, resulting in DNA double-strand breaks and cell death (101).
Xeroderma pigmentosum (XP) and Cockayne syndrome (CS) are rare, autosomal recessive disorders of DNA repair. XP is characterized by increased sun sensitivity and higher skin cancer risk (98). XP is caused by mutations in the XP genes (XPA through XPG), which are necessary for NER (98,96,102). Compared to normal cells, DNA repair deficiency by XP mutations leads to ~4-fold higher mutation frequency after acute UV exposure (5–7 days after one dose of 0.5 mJ/cm2 UVC) (103). XP patients carry a 10,000-fold increased risk of NMSC and may develop early-onset NMSC if their skin is not protected from the sun (98). There is another XP subgroup, XP variant (XPV), that exhibits normal NER but defective TLS due to lack of DNA polymerase η (104). The incidence of XP is estimated to be 1 in 22,000 in Japan, 1 in 250,000 in the U.S., and 1 in 435,000 live births in Western Europe (105,106). However, the prevalence of XP in the U.S. may be higher than clinically reported, and thus unsuspected XP mutations may contribute to high frequency of skin cancer in the general U.S. population (107). An immunohistochemistry analysis of a tissue microarray revealed that XP protein XPC, the main damage-recognition protein for GGR, is absent in 49% of invasive cSCCs from immunocompetent non-XP patients and 59% of invasive cSCCs from immunosuppressed non-XP patients (108). This high frequency of XPC inactivation in cSCC may be explained by a recent study demonstrating that XPC silencing drives malignant transformation (109). Accumulation of unrepaired DNA following XPC silencing in normal keratinocytes increases DNA-dependent protein kinase (DNA-PK) activity, which leads to production of reactive oxygen species (ROS), alters cellular metabolism, and drives malignant transformation (109).
CS is characterized by increased sun sensitivity, progressive neurological degeneration, and premature aging (98,110). CS is caused by mutations in the CS genes (CSA and CSB), which are responsible for TCR (98,96). Intriguingly, CS is not associated with increased risk of any cancer (110,103). Compared to normal cells, UV induces an increased number of mutations in XP and CS cells, both of which are DNA repair-deficient (103). However, types of mutations generated in XP and CS cells differ; UV-induced mutations in XP predominantly have UV signature (C>T), whereas the majority of mutations in CS have oxidative damage signature (G>T) (103). This suggests that CS proteins may be more important for repair of oxidative DNA damage (8-hydroxy-2’-deoxyguanosine, 8-OH-dG) than for repair of direct UV-induced DNA damage (CPD and 6–4PP).
The efficiency of NER varies throughout the genome and is affected by chromatin state (Fig. 5A). Two recent studies analyzed the kinetics of NER and its relationship with chromatin accessibility using the novel technology ‘eXcision Repair-sequencing’ (XR-seq), which measures repair of CPDs and 6–4PPs genome-wide at single-nucleotide resolution (111,112). At the early time points (1–8 hours) following UV irradiation, CPDs were repaired primarily by TCR due to the high abundance of CPDs and thus higher likelihood of encountering CPDs during transcription (111,112). Repair of CPDs on nontranscribed strands and intergenic regions persisted up to 48 hours after UV irradiation (112). In contrast, 6–4PPs were repaired predominantly by GGR due to their highly DNA-distorting nature, presumably making them easily detectable throughout the genome (111,113). Active and open chromatin regions were repaired more rapidly than heterochromatin for both CPD and 6–4PP lesions (112). Intriguingly, regions with delayed repair (heterochromatin) had a higher frequency of cancer-related mutations (112). This high frequency of mutations in heterochromatin may be attributable to slow repair in heterochromatin (112) and more frequent generation of UV-induced DNA lesions in heterochromatin (50). Consistently, chronic low-dose UVB (CLUV) irradiation leads to the accumulation of residual CPD lesions that persist in heterochromatin even in DNA repair-proficient cells (114). These residual CPDs that are refractory to repair are diluted by semiconservative DNA replication (114).
Figure 5.
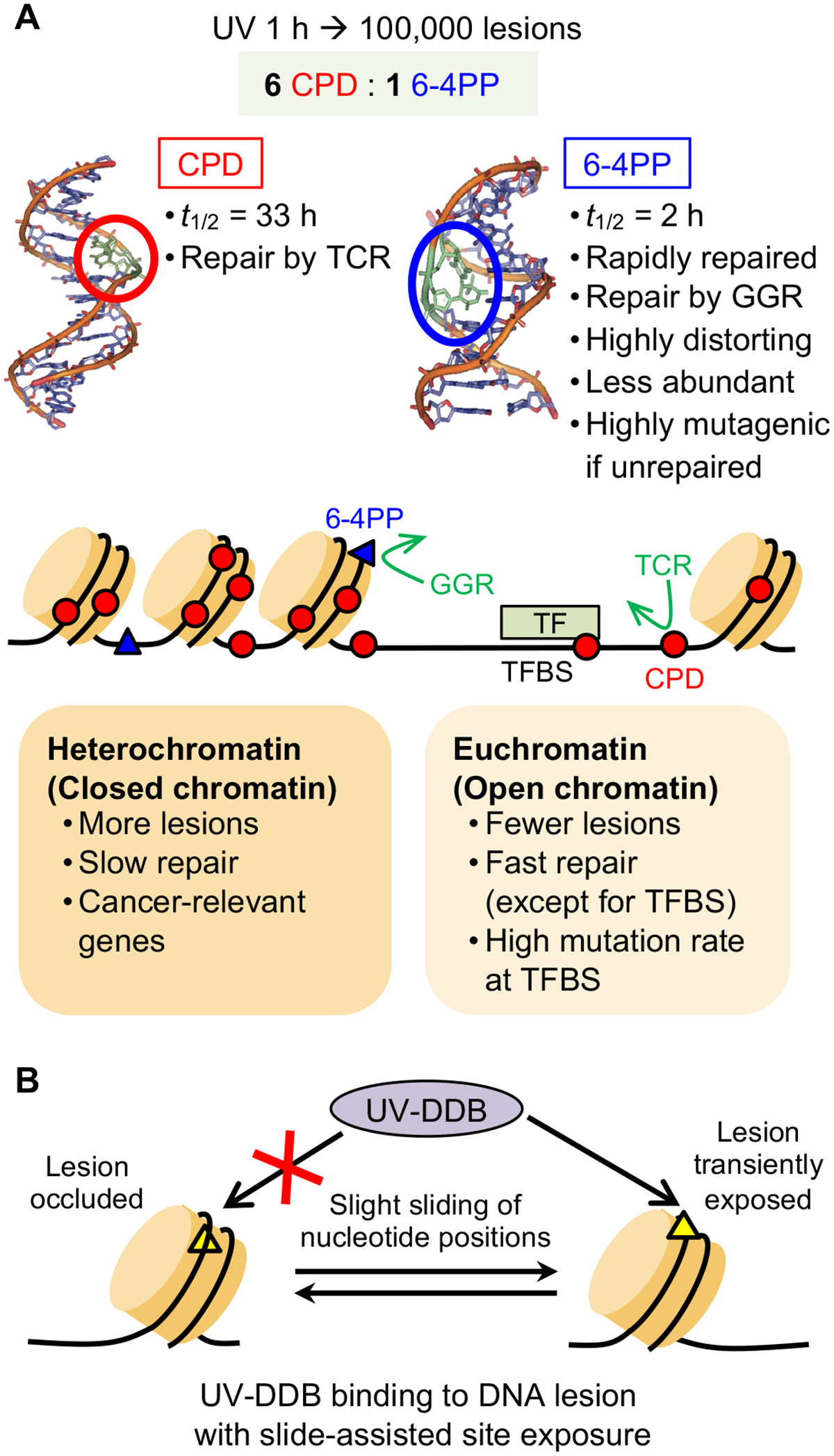
Repair of UV-induced DNA lesions depends on chromatin state and sliding of nucleotide positions. (A) UV irradiation forms dimers at dipyrimidine sites on the same strand, distorting the DNA double helix. The two major types of DNA lesions are cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (6–4PPs), which differ in terms of their abundance, degree of DNA distortion, and the primary mode of repair (global genome repair (GGR) or transcription-coupled repair (TCR)). Chromatin state influences formation and repair of UV-induced DNA lesion; lesions form more frequently and are repaired more slowly in heterochromatin than in euchromatin. Transcription factor binding sites (TFBS) have variable frequency of lesion formation and slow lesion repair that leads to high mutation rate. The structures of DNA double helix with CPD and 6–4PP lesions are adapted with permission from Rastogi, R. P. et al. J Nucleic Acids (2010) 2010:592980. (B) UV-induced DNA lesions that are occluded within a nucleosome undergo ‘slide-assisted site exposure’: slight sliding of nucleotide positions relative to the histone core, without affecting the overall nucleosome architecture. This process transiently exposes occluded lesions to UV-damaged DNA-binding protein (UV-DDB), which recognizes UV damage and initiates global genome repair (GGR).
An important question related to slow DNA repair in heterochromatin is how UV-DDB detects occluded DNA lesions in nucleosomes to initiate GGR. A recent study determined the mechanism by which UV-induced DNA lesions are recognized in chromatin, in which nucleosomes restrict access to DNA (115) (Fig. 5B). Occluded nucleosomal lesions that face the histone core become exposed and accessible to UV-DDB after slight sliding of nucleotide positions that shifts the contacts between the histone core and occluded DNA lesions (‘slide-assisted site exposure’). This lesion exposure process does not require histone octamer disassembly or looping off of nucleosomal DNA.
UV-induced DNA lesions have been thought to be repaired solely by NER. However, a recent study demonstrated the existence of an NER-independent repair mechanism for removing UV-induced DNA damage in human cells (116). An anti-diabetic drug, acetohexamide, was recently identified as an agent that alleviates the UV sensitivity of NER-deficient cells. Acetohexamide inhibited expression of DNA glycosylase MUTYH, which is known to be involved in base excision repair (BER). Surprisingly, NER-deficient (XPA-deficient) cells treated with acetohexamide were able to repair CPDs, indicating the existence of an NER-independent repair mechanism that is normally masked by MUTYH.
Methylated RNA as an early responder to UV-induced DNA damage
Due to its dynamic and reversible nature, post-transcriptionally modified RNA has great potential to participate in many non-hereditary, regulatory roles (117). N6-methyladenosine (m6A), the most prevalent posttranscriptional modification of mRNA, is enriched around stop codons and participates in pre-mRNA processing, mRNA nuclear export, translation, and mRNA decay (118). One recent study uncovered a novel function of RNA m6A modification in the UV-induced DNA damage response (119). After UV irradiation, RNA m6A modification was rapidly induced at sites of DNA damage, peaking at 2 minutes, and decreased to background levels at 10 minutes post-UV (119). Also, DNA polymerase κ, known to be involved in NER and TLS, was rapidly recruited to DNA damage sites, and its immediate localization required the catalytic activity of the methyltransferase METTL3. Downregulation of METTL3 resulted in reduced m6A RNA at sites of UV-induced DNA damage, reduced recruitment of DNA polymerase κ to these sites, and delayed repair of UV-induced CPD lesions. Rapid induction of m6A RNA was uniquely detected after UV irradiation, but not after γ irradiation or DNA-damaging chemicals. This induction occurred in S/G2/M-phase cells, but not in G1-phase cells. This study demonstrated that rapid METTL3-mediated RNA m6A methylation and recruitment of DNA polymerase κ facilitate DNA repair after UV exposure (119). DNA polymerase κ might act as a TLS polymerase to facilitate lesion bypass during S phase, allowing repair of the lesion subsequently. However, it remains to be determined how the rapid increase and decrease in RNA m6A methylation within only 10 minutes after UV irradiation contributes to the relatively slower process of DNA repair.
Mechanisms of UV mutagenesis
Compared to instantaneous generation of DNA lesions by sunlight, DNA repair kinetics is relatively slow, which may lead to cells entering S phase before completing DNA repair. During DNA replication in the presence of UV-induced DNA lesions, mutations may be incorporated opposite of these highly DNA-distorting lesions. Skin that is chronically sun-exposed over decades accumulates a very large number of mutations, approaching the mutation frequency present in cancers of internal organs (not exposed to UV) (28). The gradual acquisition of cancer-related mutations that drive malignant transformation leads to skin cancer.
DNA lesions generated from all three wavelength ranges of UV radiation most commonly lead to C>T mutations at dipyrimidine sites, termed the UV signature mutation (120,36,41). These C>T mutations occur when cells undergo error-prone DNA replication across unrepaired DNA lesions. This process is known as error-prone translesion synthesis (TLS) and is one mechanism of DNA damage tolerance that allows cells to bypass replication-blocking lesions without actually removing the lesions (121) (Fig. 3E). Specifically, replicative DNA polymerases δ/ε stall at a DNA-distorting, UV-induced lesion. To continue DNA synthesis across the lesion, specialized TLS polymerases are recruited to the lesion. These TLS polymerases are capable of accommodating bulky chemical groups (e.g., UV-induced DNA lesions) on the template strand, allowing DNA synthesis on the lesion-containing template strand; however, TLS polymerases have high error rates (122). Posttranslational modification of proliferating cell nuclear antigen (PCNA) is important for DNA damage tolerance; PCNA normally forms a ring around double-stranded DNA and tethers replicative DNA polymerases to their DNA template during DNA replication. Helicase-like transcription factor (HLTF) promotes monoubiquitination of PCNA and facilitates recruitment of TLS polymerase η to the DNA lesion (123). A unique TLS polymerase is also recruited to the lesion: PrimPol, which contains both DNA polymerase and DNA primase activities (124–127). PrimPol not only performs TLS but also creates DNA primers that are necessary for other DNA polymerases to reinitiate DNA synthesis downstream of the lesion (124–127). TLS polymerases (error rate >10–2) have low fidelity compared to replicative polymerases δ/ε (error rate 10–5–10–7) (128). TLS polymerases occasionally incorporate incorrect nucleotides opposite of the UV-induced lesion (e.g., adenine opposite of a cytosine-containing lesion), leading to a mismatched base pair (Fig. 6). During the next round of DNA replication using the incorrect nucleotide-containing strand as the template, a complementary nucleotide is incorporated opposite of the incorrect nucleotide (T incorporated opposite of A), thus generating a mutation (net change C>T mutation). This final step is known as mutation fixation. Another mechanism for generating C>T mutation is mediated by deamination independently of error-prone TLS. Predominantly at CPDs and on transcribed strands, significant proportions of cytosines are deaminated to uracil. This uracil is complemented by adenine on the opposite strand via error-free TLS (129,130,41). The next round of DNA replication incorporates thymine in place of uracil, resulting in C>T mutation as the net effect.
Figure 6.
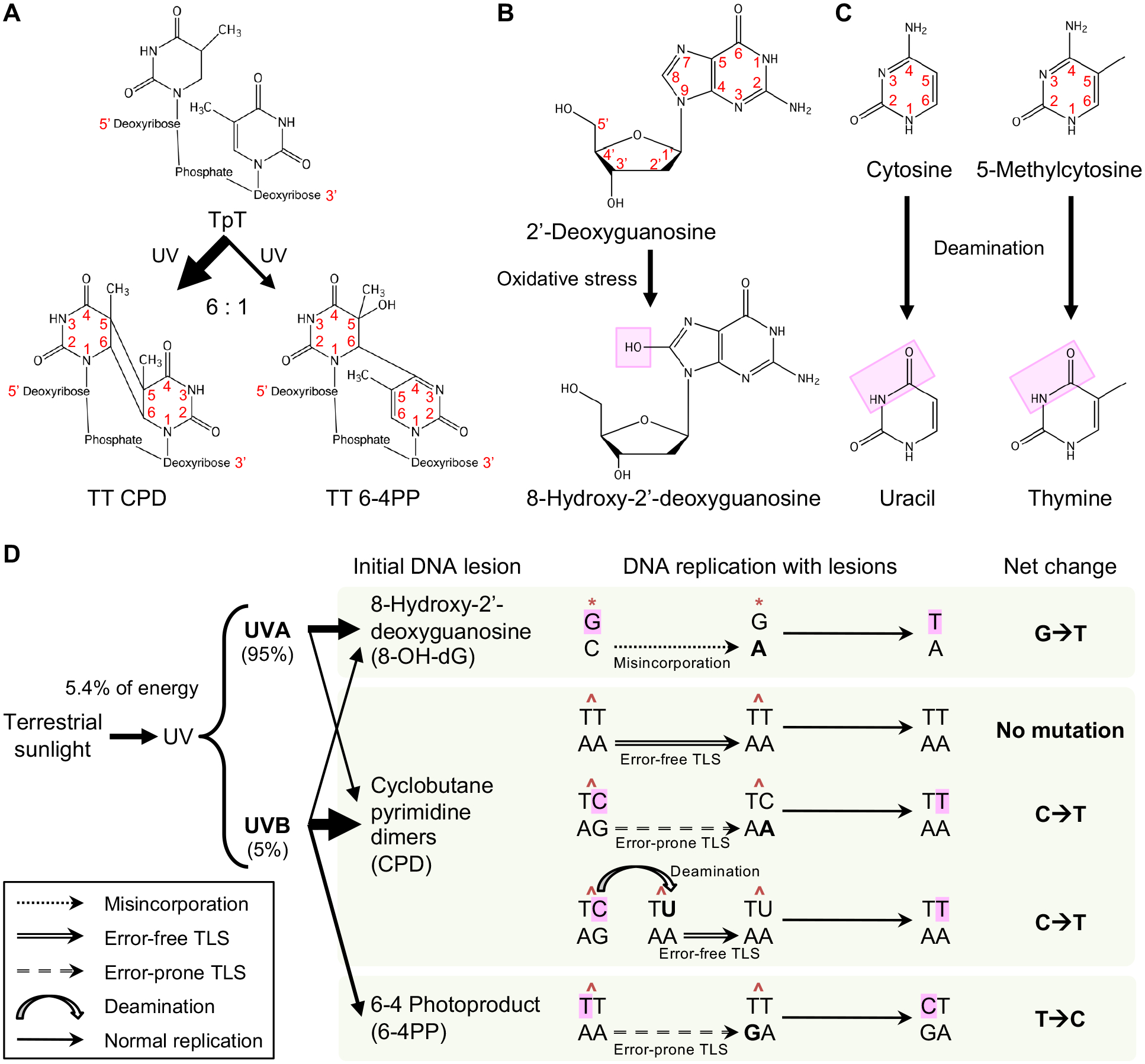
Chemical structures of UV-induced DNA lesions and mechanisms of UV-induced mutagenesis. (A) Cyclobutane pyrimidine dimer (CPD) and 6–4 photoproduct (6–4PP) lesions are UV-induced dimers formed at dipyrimidine sites, e.g., two adjacent thymines (TpT). CPDs are generated by UV from simulated sunlight 6 times more frequently than 6–4PPs. (B) Oxidative stress can be induced by UVA rather than UVB and damages 2’-deoxyguanosine, converting it into the DNA lesion 8-hydroxy-2’-deoxyguanosine (8-OH-dG). (C) Cytosine and 5-methylcytosine (5-mC) can be deaminated into uracil and thymine, respectively. Deamination occurs more frequently when cytosine or 5-mC is part of a CPD on a transcribed strand. (D) UVA and UVB compose 95% and 5% of UV radiation from terrestrial sunlight, respectively. UVA and UVB irradiation generate 8-OH-dG, CPD, and 6–4PP DNA lesions. Deamination may occur at CPDs. During the first round of DNA replication after lesion formation, correct or incorrect nucleotides are incorporated opposite of the lesion via different mechanisms including misincorporation and error-free and error-prone translesion synthesis (TLS). During the second round of DNA replication, complementary nucleotides are incorporated opposite of the correct or incorrect nucleotides that were from the first round. This results in mutation fixation. Due to the abundance and slow repair of CPDs, the resulting C>T mutations are the most prevalent UV-induced mutations and are known as UV signature mutations. Thickness of each arrow leading from UVA and UVB to DNA lesion types in the panel indicates the relative contributions of UVA and UVB to formation of each lesion type.
On a per-lesion basis, 6–4PPs are more mutagenic than CPDs. To induce a 1 bp mutation in DNA repair-proficient mammalian cells, 159 CPDs are required, whereas 53 6–4PPs are required (131). However, CPDs are identified as the principal cause of UV-associated skin cancer, presumably due to high abundance and slow repair. When CPDs were removed by CPD-photolyase, skin cancer development following chronic UV irradiation was prevented in a mouse model (132).
The advancement of massively parallel sequencing technology has revealed the mutational landscapes of UV-associated skin cancer on a large scale. In 2007, more than 1,000 somatic mutations were identified in the coding exons of 518 protein kinase genes in 210 diverse human cancers (133). Mutations found in melanoma were predominantly the UV signature (C>T) mutation, indicative of past UV exposure. In 2010, a cancer genome sequence was reported for the first time using the patient-derived melanoma cell line COLO-829, comprehensively cataloging somatic mutations across the genome (134). More than 33,000 somatic base substitutions were identified. Among these, 187 mutations (0.6%) were nonsynonymous substitutions in protein-coding sequences, some of which may be cancer driver mutations. Consistent with earlier studies, the predominant mutations detected were the UV signature (C>T) mutation. This study also found that these C>T mutations were more frequent on nontranscribed strands than on transcribed strands, likely due to TCR that removes UV-induced DNA lesions only on transcribed strands.
The mutagenic nature of UV is also observed in the genome of even normal skin. A recent study examined the mutation burden present in normal, sun-exposed epidermis (28). Consistent with previous studies, C>T mutations were most frequent. Surprisingly, many cancer-associated gene mutations were present in normal, sun-exposed skin at a frequency similar to that of internal cancers. Approximately 25% of normal skin cells harbored cancer-causing mutations at a density of ~140 driver mutations/cm2. A major difference between cSCC and normal, sun-exposed skin was the frequency of mutations; the number of driver point mutations per cell was 10-fold higher in cSCC cells (average 2.7 mutations per cell) compared to that in normal cells (average 0.27 mutations per cell). However, normal cells could carry 2–3 driver mutations without malignant transformation, indicating that merely having multiple driver mutations is not sufficient for acquiring malignant potential (28). These findings suggest that malignant transformation may require specific combinations of cancer driver mutations.
UV induces genetic heterogeneity within a single tumor, which affects the immune response and tumor growth of melanoma in vivo. A recent study demonstrated that UV irradiation increases genetic heterogeneity in a cell population from a melanoma cell line, as determined by the variant allele frequency (135). Compared to highly heterogeneous tumors, tumors of low heterogeneity elicited stronger immune responses and grew at a dramatically reduced rate in mice. In humans, lower tumor heterogeneity has been linked to stronger immune response and better patient survival (135).
UV-induced reactive oxygen species (ROS) and carcinogenesis
Reactive oxygen species (ROS) are unstable molecules that contain oxygen and readily react with other molecules in cells. ROS are generated in cells as a result of normal cellular metabolism, but ROS are also generated by exogeneous agents including UV irradiation (136). Interestingly, visible light (400–700 nm wavelength) also produces ROS (137). ROS include superoxide (O2•–), hydrogen peroxide (H2O2), and hydroxyl radical (•OH). ROS are eliminated by intracellular antioxidants such as glutathione (GSH) (136). UV irradiation produces excessive amounts of ROS that overwhelm these antioxidant molecules (136). In melanocytes, the melanosome is proposed to be a major source of ROS (138). Also, the extracellular matrix (ECM) of skin is a potentially significant photosensitizer to produce ROS upon UV exposure (139). When dermal fibroblasts were treated with ECM proteins (collagen and elastin) that were pre-irradiated with UV, these cells exhibited oxidative stress responses (139). This suggests that ECM proteins act as photosensitizers to generate ROS upon UV irradiation in skin. These high levels of ROS contribute to abnormal cell proliferation signaling, epigenetic alterations, and DNA lesion formation, leading to disease states including cancer (136,138). In particular, UV-induced ROS oxidatively damage the nucleoside 2’-deoxyguanosine, converting it to 8-hydroxy-2’-deoxyguanosine (8-OH-dG; tautomer 8-oxo-7,8-dihydro-2’-deoxyguanosine, 8-oxo-dG) (140,41) (Fig. 6). In DNA, 8-OH-dG lesions are repaired by base excision repair (BER), but if unrepaired, these lesions result in G>T mutations (141,41). A carcinogenic role of ROS is demonstrated in XPC deficiency; XPC loss causes ROS-mediated aberrations in cellular metabolism, driving malignant transformation of keratinocytes (109).
ROS generated by UV activate a number of signaling pathways that are related to inflammation and carcinogenesis (136). Examples of oncogenic signaling activated by UV-induced ROS include the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway (142,136), which is overactive in many cancers (143), epidermal growth factor receptor (EGFR) (144), p38 MAPK (142), activating protein-1 (AP-1) (136), and NF-κB (136). Activation of NRF2, a transcription factor that induces antioxidant defense, decreases ROS levels and prevents ROS-induced mutations following UV irradiation, thereby inhibiting skin carcinogenesis (145). Although UV is a poor activator of NRF2, the basal expression level of NRF2 is high in epidermal keratinocytes, preventing skin from UV damage (145). Intriguingly, excessive NRF2 may be detrimental; in transgenic mice, overexpression of a constitutively active NRF2 mutant leads to epidermal thickening, corneocyte fragility, impaired desquamation, and increased immune infiltration into the skin at the early ages of 1–3 months (145).
Although mutagenic and carcinogenic roles of UV-induced ROS have been demonstrated (41,136,109), a recent report intriguingly demonstrates that oxidative stress inhibits metastasis of melanoma in a mouse model; in other words, reducing oxidative stress promotes metastasis (146). In this study, melanoma cells circulating in blood or in visceral metastatic nodules experienced high levels of ROS. Successfully metastasized melanoma cells had increased expression of NADPH-generating enzymes that are involved in antioxidant production. Exogenous administration of antioxidants promoted distant metastasis of melanoma cells, whereas inhibition of NADPH-generating enzymes suppressed metastatic dissemination. As opposed to ROS-mediated oncogenic signaling, this study suggests that oxidative stress in melanoma cells inhibits metastasis.
Prevention and treatment of nonmelanoma skin cancer
Incidence and progression of skin cancer
All types of skin cancer are highly associated with UV exposure (17–19,3,1,20). Consistent with high prevalence of UV exposure, skin cancer is the most common cancer in the United States (24). Despite declining incidence and mortality of cancer in general, the incidence of skin cancer has been escalating (147,148,24,149–151), demonstrating an urgent need for effective and efficient strategies for prevention and treatment. The annual incidence of nonmelanoma skin cancer (NMSC), comprising basal cell carcinoma (BCC) and cutaneous squamous cell carcinoma (cSCC), in the United States was 3.5 million in 2006 (148) and increased to 5.4 million in 2012 (24). This exceeds the incidence of all other cancers combined, which is 1.8 million in 2019 (147). For cSCC, estimated new cases are 1 million in 2012 in the United States, and estimated deaths are 15,000 (152). For melanoma, estimated new cases are 96,480 in 2019 in the United States, and estimated deaths are 7,230 (147). Melanoma incidence is predicted to increase to 116,000 annual new cases in 2026–2031 among U.S. whites (151).
Actinic keratosis (AK) is a precursor to cSCC. The risk of progression of AK to invasive cSCC is ~8% per year (153). cSCC has a good prognosis if it is completely resected at an early stage. However, one in 50 cSCCs metastasizes, resulting in limited treatment options and poor prognosis (154,155). Notably, although blacks are 70 times less likely to develop skin cancer than Caucasians (156), people of color often present with skin cancer at a more advanced stage, leading to poor prognosis (157). The most common skin cancer type, BCC, rarely metastasizes, and patients with BCC have excellent prognosis.
UV exposure avoidance for skin cancer prevention
Avoidance of excessive UV exposure is a cornerstone of skin cancer prevention. Chronic UV exposure from the sun is a significant risk factor for skin cancer among the general population (19). Although UV exposure had been known to contribute to skin carcinogenesis for decades (158), it was only in 2002 that UV exposure was first listed as a known carcinogen by the U.S. Department of Health and Human Services (159,160). Although it is impossible to eliminate this carcinogen for most people, UV exposure can be mitigated sufficiently to prevent skin cancer, particularly in susceptible populations. For skin cancer prevention, the “SunSmart” mass media campaign to “slip [on a shirt], slop [on sunscreen], slap [on a hat], seek [shade], and slide [on sunglasses]” has been in place in Australia since the 1980s to encourage the public to avoid excess UV exposure (161). In 2007, the slogan was updated to add “seek [shade] and slide [on sunglasses]” to emphasize two additional sun protection measures. This program has potentially contributed to recent declines in melanoma incidence in Australia (151), one of the countries with the highest burdens of melanoma and NMSC in the world (21,20). Lifetime sunlight exposures of approximately 10,000 hours and beyond 70,000 hours are associated with elevated risks of BCC and cSCC, respectively (19). In mice, topical application of Sun Protection Factor (SPF)-15 sunscreen before UV irradiation nearly completely protected skin from generating p53 mutations (162). Thus, the long-term avoidance of UV exposure and the use of sunscreen are crucial components of skin cancer prevention.
Molecular targets for skin cancer prevention
In the face of rising skin cancer incidence and the challenge of treating metastatic cancer, the development of novel preventive and therapeutic measures against skin cancer is critically important. Targeting UV-induced signaling may be effective in preventing skin cancer (163,164) (Fig. 7). Common dietary compounds that can prevent skin cancer are of great interest; caffeine in coffee and the tea polyphenol (–)-epigallocatechin-3-gallate (EGCG) have been shown to inhibit UV-induced skin carcinogenesis (165–169).
Figure 7.
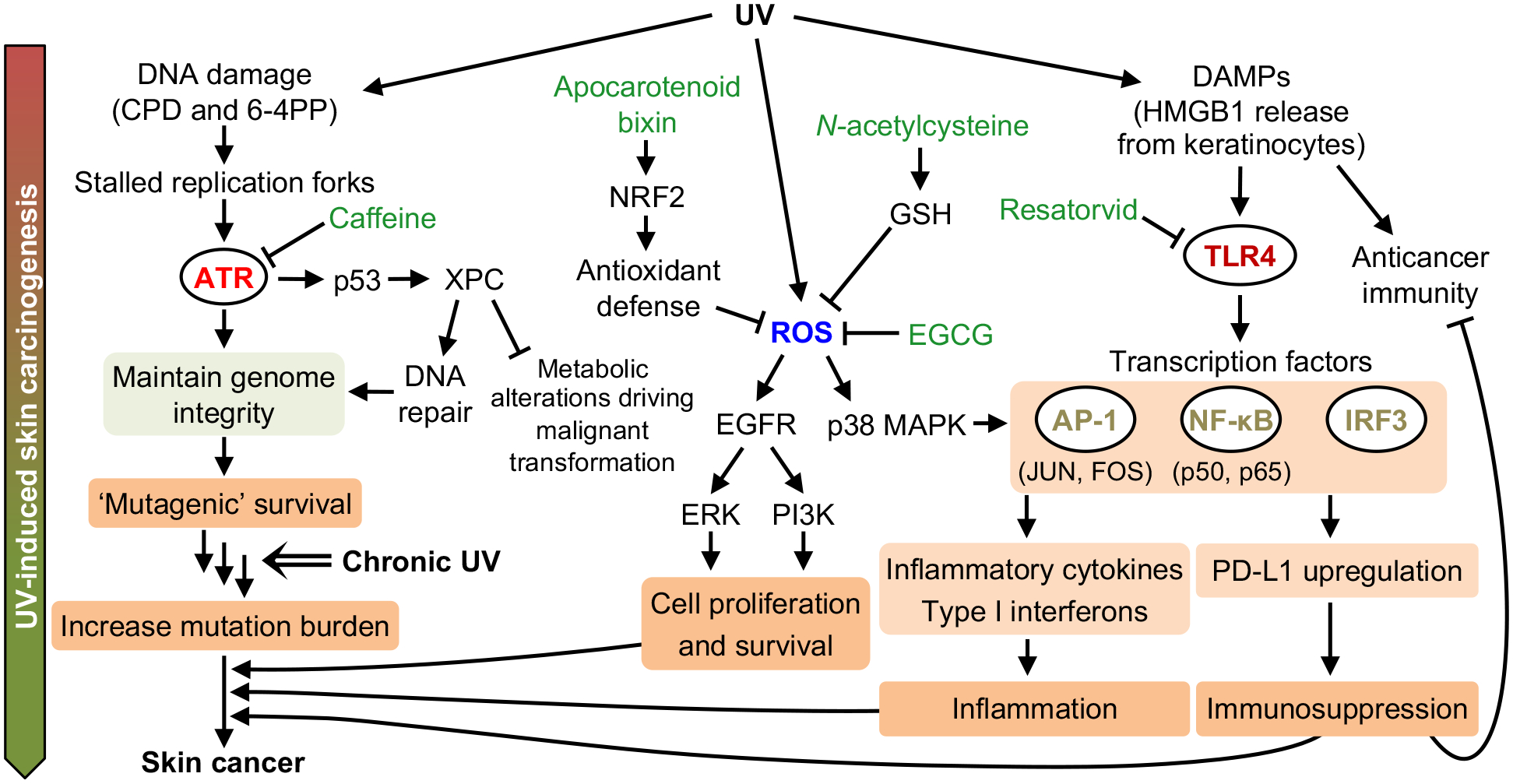
Targeting UV-induced signaling pathways to suppress skin carcinogenesis. UV-induced DNA lesions stall DNA replication forks and activate the ATR pathway. The ATR pathway maintains genome integrity, allowing cells to survive UV-induced DNA damage but possibly with unrepaired lesions that become mutations (‘mutagenic survival’). Thus, chronic UV exposure will increase mutation burden, accumulating cancer driver mutations that may promote skin cancer development. UV-induced p53 upregulates XPC, which facilitates DNA repair and prevents metabolic alterations that drive cancer. UV-induced reactive oxygen species (ROS) activate the EGFR and p38 MAPK pathways, contributing to skin cancer development. HMGB1 released from UV-damaged keratinocytes activates the TLR4 pathway, which can drive skin cancer via inflammation and immunosuppression. Inhibiting ATR (via caffeine), ROS (via the tea polyphenol EGCG), or TLR4 (via resatorvid) has been demonstrated to prevent UV-induced skin carcinogenesis in vivo.
UV-induced inflammation is important for skin carcinogenesis (8). UV-damaged epidermal keratinocytes release high mobility group box 1 (HMGB1), which activates Toll-like receptor 4 (TLR4) to trigger inflammation (7,8). The TLR4 antagonist resatorvid inhibited UV-induced AP-1 and NF-κB signaling, thereby suppressing UV-induced inflammation and skin tumorigenesis in SKH-1 hairless mice (170). Also, UV induces autophagy in the epidermis, and the autophagy gene Atg7 regulates UV-induced cytokine expression and secretion. Deletion of Atg7 suppressed UV-induced inflammation and skin tumorigenesis (171). Thus, autophagy is another potential target for skin cancer prevention. Furthermore, the apocarotenoid bixin is an FDA-approved food additive that activates NRF2, which is a transcription factor that induces antioxidant defense. Bixin reduces oxidative damage and inflammatory response after acute UV exposure (172), thus potentially attenuating UV-induced skin carcinogenesis.
Caffeine and ATR inhibition for skin cancer prevention
Several epidemiological studies have reported that caffeine consumption is associated with reduced risks of NMSC and melanoma. A large study of approximately 77,000 Caucasian women enrolled in the Women’s Health Initiative Observational Study reported a 10.8% lower prevalence of NMSC in participants who consume caffeinated coffee on a daily basis, compared with nondrinkers (173). Strikingly, caffeinated coffee consumption of ≥6 cups per day was associated with a 30% reduction in prevalence of NMSC. Decaffeinated coffee had no preventive effect. This study suggests that caffeine consumption reduces the risk of NMSC in a dose-dependent manner. For both men and women, caffeine consumption reduced the risk of BCC in a dose-dependent manner where approximately 600 mg of daily caffeine consumption led to a 16% reduced risk of BCC (174). In a large study of middle-aged and elderly Chinese in Singapore, coffee reduced the risks of both BCC and cSCC in a dose-dependent manner; coffee consumption of ≥3 cups per day led to a 46% reduced risk of BCC and a 67% reduced risk of cSCC (175). Caffeinated coffee consumption of ≥4 cups per day demonstrated a 25% reduction in the risk of melanoma in non-Hispanic whites (176).
In parallel to human epidemiological studies, mouse experiments have been performed to investigate the preventive effect of caffeine on UV-induced skin carcinogenesis, elucidate molecular mechanisms by which caffeine prevents skin cancer, and identify targets for skin cancer prevention (169,167). Topical application and oral administration of caffeine following chronic UV exposure have been shown to augment apoptosis (166,169), diminish photodamage (177), and suppress the onset of UV-induced skin cancer in mice (178,166,169). Caffeine has multiple molecular targets and is a nonspecific inhibitor of ATR kinase (179), which is activated in response to UV to induce cell cycle arrest. Because ATR is crucial to UV-induced DNA damage responses, studies have been performed to investigate whether ATR is the relevant target of caffeine for preventing UV-induced skin cancer. In primary human keratinocytes, caffeine augmented UV-induced apoptosis by more than 2-fold; ATR inhibition via siRNA increased UV-induced apoptosis to the same degree, but addition of caffeine to these cells had no additive effect on apoptosis (180). These findings suggest that caffeine augments UV-induced apoptosis via ATR inhibition. This augmentation of apoptosis may contribute to elimination of UV-damaged cells that are at increased risk of malignant transformation, thereby inhibiting skin cancer development (Fig. 8A). Indeed, mice with genetic inhibition of ATR in skin exhibited augmented UV-induced apoptosis and reduced incidence of UV-induced skin cancer (181).
Figure 8.
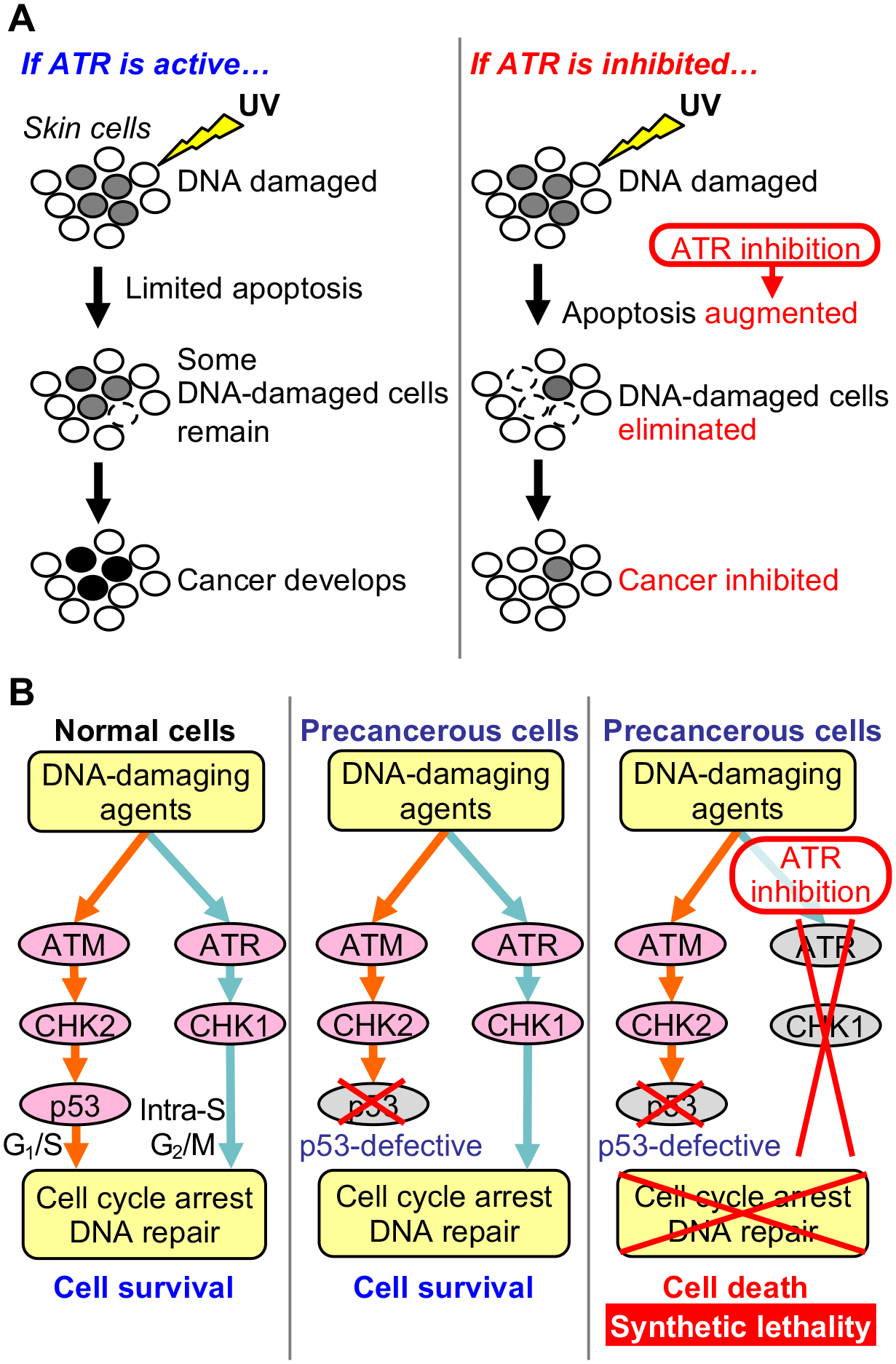
Targeting the ATR pathway to prevent cancer development. (A) ATR inhibition prevents UV-induced skin cancer development. After UV exposure, ATR induces cell cycle arrest that allows time for DNA repair, resulting in limited apoptosis. However, when DNA replication is ongoing before completion of DNA repair, mutations may be incorporated via DNA damage tolerance mechanisms (‘mutagenic survival’), leading to skin cancer development. In contrast, inhibition of the ATR pathway that is essential for surviving UV damage leads to augmented apoptosis. This contributes to elimination of DNA-damaged cells that are at increased risk of malignant transformation, thereby inhibiting skin cancer development. (B) ATR inhibition sensitizes p53-defective cells to DNA-damaging agents. Normal cells have two major pathways that respond to DNA damage: the ATM-CHK2-p53 and ATR-CHK1 pathways, which induce cell cycle arrest and DNA repair. However, precancerous cells are often defective in p53 and thus rely on the ATR-CHK1 pathway to survive DNA damage. When p53-defective precancerous cells are treated with an ATR inhibitor in the presence of a DNA-damaging agent (e.g., UV or chemotherapeutic drug), these cells will have no intact DNA damage response pathway to survive the DNA damage (synthetic lethality of defective p53 and ATR inhibition). As a result, precancerous cells will undergo apoptosis, and cancer development will be suppressed. This synthetic lethality provides the molecular basis for the use of ATR inhibitors to prevent cancer development from p53-defective cells.
Human cancers frequently harbor mutations in TP53, which encodes the p53 tumor suppressor, leading to the loss of wild-type p53 functions and conferring resistance to apoptosis (182–184). TP53 mutations are found in 94.9% of aggressive cSCCs (185) and even in 4% of normal, sun-exposed epidermal cells (28,186). Apoptosis-resistant, p53-deficient cells can be sensitized by caffeine or ATR inhibition to DNA damage-induced death, including death induced by UV and chemotherapeutic drugs (187,79). This finding suggests that ATR inhibition and loss of p53 function lead to synthetic lethality in the presence of DNA damage (Fig. 8B). Taken together, these studies support a model in which ATR inhibition or caffeine sensitizes UV-damaged, premalignant cells to death, thereby preventing and suppressing UV-induced skin carcinogenesis.
Conventional treatments for NMSCs
As is true in general for cancer therapy, treatment of NMSCs depends on the clinical stages of cancers (188,189). Complete surgical excision, destruction with cryotherapy, or topical agent is the first-line treatment for skin cancer when a lesion has not yet invaded deeper structures (in situ) (190). Alternatively, radiotherapy may be used for cSCCs when surgery is contraindicated, when cSCC is metastasized, or when surgical excision was unable to completely clear marginal tissue of carcinoma (190). However, radiotherapy is contraindicated when cSCC is found at a previously irradiated, traumatized, or poorly vascularized site, or when advanced lesions invade bones, joints, or tendons (190). When cSCC is contraindicated for surgery or radiotherapy or is unresolved after surgery or radiotherapy, chemotherapy may be indicated. 5-fluorouracil (5-FU) has been used with some success in topical and oral forms (190).
Molecularly targeted therapies for skin cancer
Molecularly targeted therapies are a current topic of investigation for NMSCs as single agents, combination therapy, and adjuvant and neoadjuvant treatments (191,192). For locally advanced or metastatic BCCs, targeted inhibition of the Hedgehog pathway is approved for clinical use (vismodegib and sonidegib) (193–195). Molecularly targeted therapies have been explored in small-scale trials for advanced cSCC (191). In one clinical trial of patients with unresectable cSCC, cetuximab, a monoclonal antibody that inhibits EGFR, was tested as a first-line single-drug therapy and achieved 69% disease control rate (196). However, more patient participation in clinical trials is needed to recommend EGFR inhibitors for treatment of advanced cSCC. Also, inhibition of the oncogenic Wnt/β-catenin pathway has been proposed as a therapeutic strategy for cSCC (197).
The MAPK (RAS-RAF-MEK-ERK) pathway plays a key role in cell proliferation and survival and is an important target for melanoma treatment (Fig. 9). RAF inhibitors are effective against melanomas with BRAFV600E mutations but may induce cSCCs in these melanoma patients. Intriguingly, two studies identified RAS mutations in cSCCs that developed in melanoma patients treated with RAF inhibitors (vemurafenib or sorafenib) (198,199). These therapy-induced cSCCs harbored activating HRAS mutations. This may be explained by that BRAF inhibitors paradoxically activate the MAPK pathway and accelerate the progression of preexisting RAS-mutant lesions (198). Thus, simultaneously inhibiting multiple targets in the signaling network essential for cancer may eradicate cancer effectively (200). Indeed, patients with BRAF-mutated melanoma benefit from combination therapy with BRAF inhibitor (dabrafenib) and MEK inhibitor (trametinib); cSCC occurred in 1% of melanoma patients treated with this combination therapy, compared to 18% of those treated with BRAF inhibitor monotherapy (201).
Figure 9.
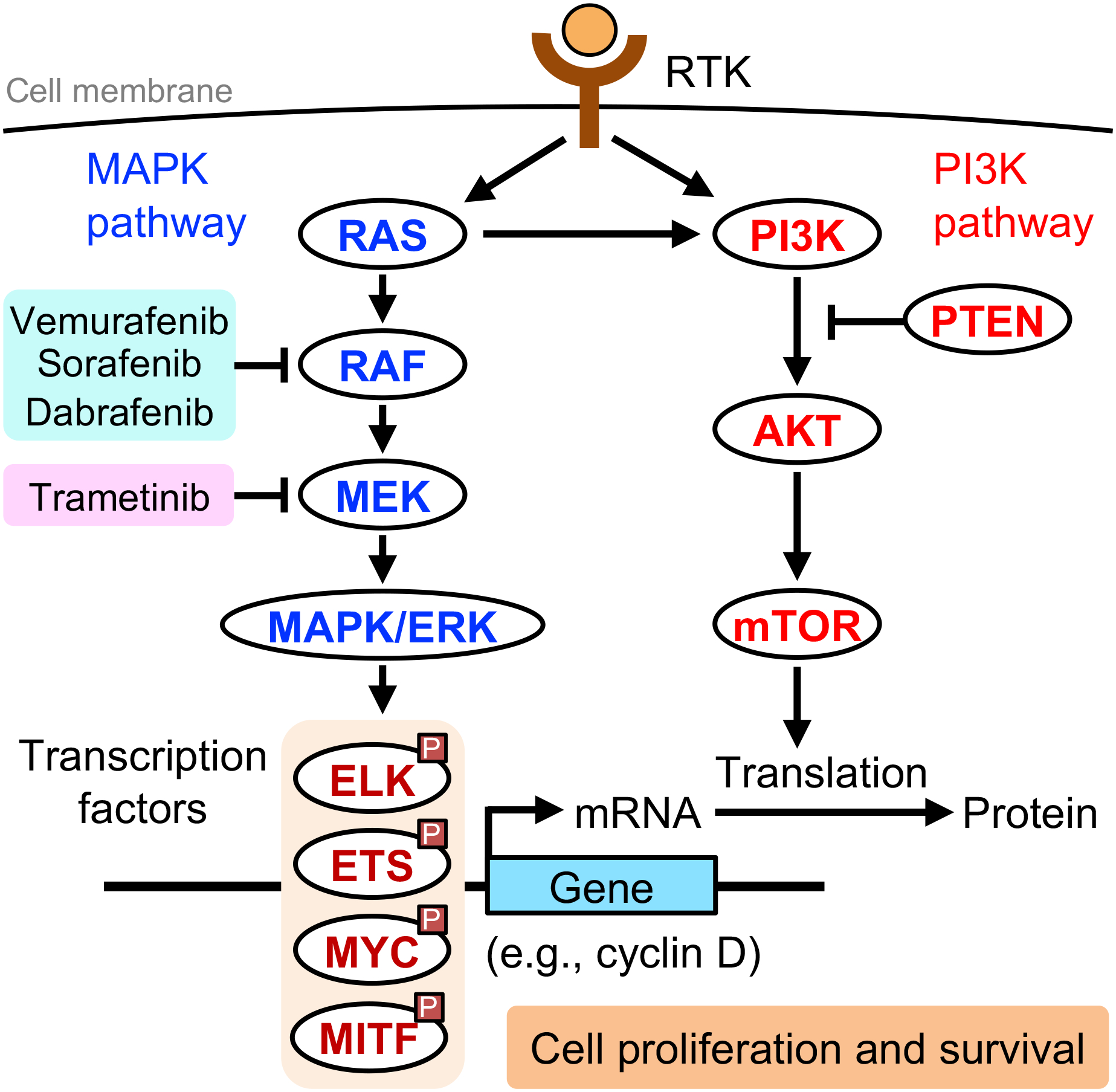
Targeting oncogenic MAPK and PI3K pathways in skin cancer. Receptor tyrosine kinases (RTKs) activate the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway and the phosphoinositide 3-kinase (PI3K) pathway, which are frequently aberrant in skin cancer. MAPK/ERK phosphorylates transcription factors and upregulates expression of genes important for cell proliferation and survival. The mammalian target of rapamycin (mTOR) upregulates mRNA translation of these genes. Aberrant activation of these pathways (e.g., activating BRAFV600E mutation) leads to increased proliferation and survival. RAF inhibitors, such as vemurafenib, sorafenib, and dabrafenib, and the MEK inhibitor trametinib are currently used in melanoma treatment. However, RAF inhibitor monotherapy for melanoma can accelerate the progression of preexisting RAS-mutant skin lesions into cutaneous squamous cell carcinoma (cSCC). Combination therapy with dabrafenib and trametinib, each targeting a different kinase within the MAPK pathway, reduces the incidence of cSCC in melanoma patients treated with a RAF inhibitor.
Many of the cancer-driving pathways in cSCC and melanoma are commonly altered in cancers of other organ systems (202,185). These commonalities may allow for targeted therapies developed for other cancers to be applied to skin cancer, and vice versa. The molecular profile of cSCC is similar to that found in other carcinogen-driven SCCs (i.e., alcohol- or tobacco-driven), especially head and neck SCC, although virus-driven SCC (i.e., human papillomavirus-driven cervical SCC) is dissimilar to cSCC (203,185). Thus, treatments for these carcinogen-driven SCCs may be developed using cSCC, which is the most common SCC and an accessible model (203).
Immunotherapy for cSCC with immune checkpoint inhibition
Concurrently with the evolution of molecularly targeted therapies, immunotherapy has rapidly advanced and is now being applied to melanoma and cSCC (204–206). Immune checkpoints are inhibitory pathways that normally maintain self-tolerance but are dysregulated in tumors, resulting in resistance to the anticancer immune response. The interaction between PD-1 in T cells and PD-L1 in cancer cells elicits immune checkpoints, suppressing T cell-mediated cytotoxicity against cancer cells. Thus, immune checkpoint blockade (e.g., via inhibitors of PD-1 and CTLA-4) has been an attractive strategy to combat cancer. UV radiation influences immune checkpoints; UV radiation upregulates PD-L1 via HMGB1-activated IRF3 and NF-κB, contributing to UV-induced immunosuppression in the skin (15) (Fig. 7). For immunotherapy for advanced cSCC, the PD-1 inhibitor cemiplimab is the first FDA-approved drug (approved in September 2018). Cemiplimab induces a therapeutic response in approximately half of patients with advanced cSCC (207). Other PD-1 inhibitors and CTLA-4 inhibitors have also shown promise, although they are not yet approved for cSCC (208). Although immunotherapy is effective for some patients, this treatment strategy is not suitable for all skin cancer patients. With cemiplimab treatment, half of advanced cSCC patients still do not benefit from immunotherapy. Furthermore, organ transplant patients, who have a 100-fold higher risk of developing cSCC (209), pose special challenges because immune activation by PD-1 blockade has been shown to trigger allograft rejection (210). Combining multiple therapies (e.g., immunotherapy and molecularly targeted therapy) may improve the efficacy of these drugs (211).
Photodynamic therapy for treatment of skin cancer
Photodynamic therapy (PDT) is a form of minimally invasive treatment that uses visible light to selectively kill cells that contain an introduced photosensitizer – a nontoxic small-molecule compound that becomes toxic when it is excited by light (Fig. 10). After absorbing light, this photosensitizer produces ROS, leading to apoptosis, necrosis, autophagy, inflammation, and a systemic immune response against tumor tissues (212–214). This ROS-based tumor destruction mechanism is similar to the mechanism of action of widely used chemotherapeutics such as bleomycin (215). PDT also leads to destruction of tumor vasculature, causing persistent tumor hypoxia and contributing to long-term tumor control (214). Widely used prodrugs for PDT are 5-aminolevulinic acid (5-ALA) and its relatively lipophilic derivative methyl aminolevulinate (MAL). These prodrugs are metabolized into the photosensitizer protoporphyrin IX (PpIX) and can be preferentially distributed to tumors (216,214). The mechanisms involved in the preferential distribution of photosensitizers in tumors are not fully understood. Research in PDT continues to develop novel photosensitizers for improved tissue selectivity and light absorbance (214,217).
Figure 10.
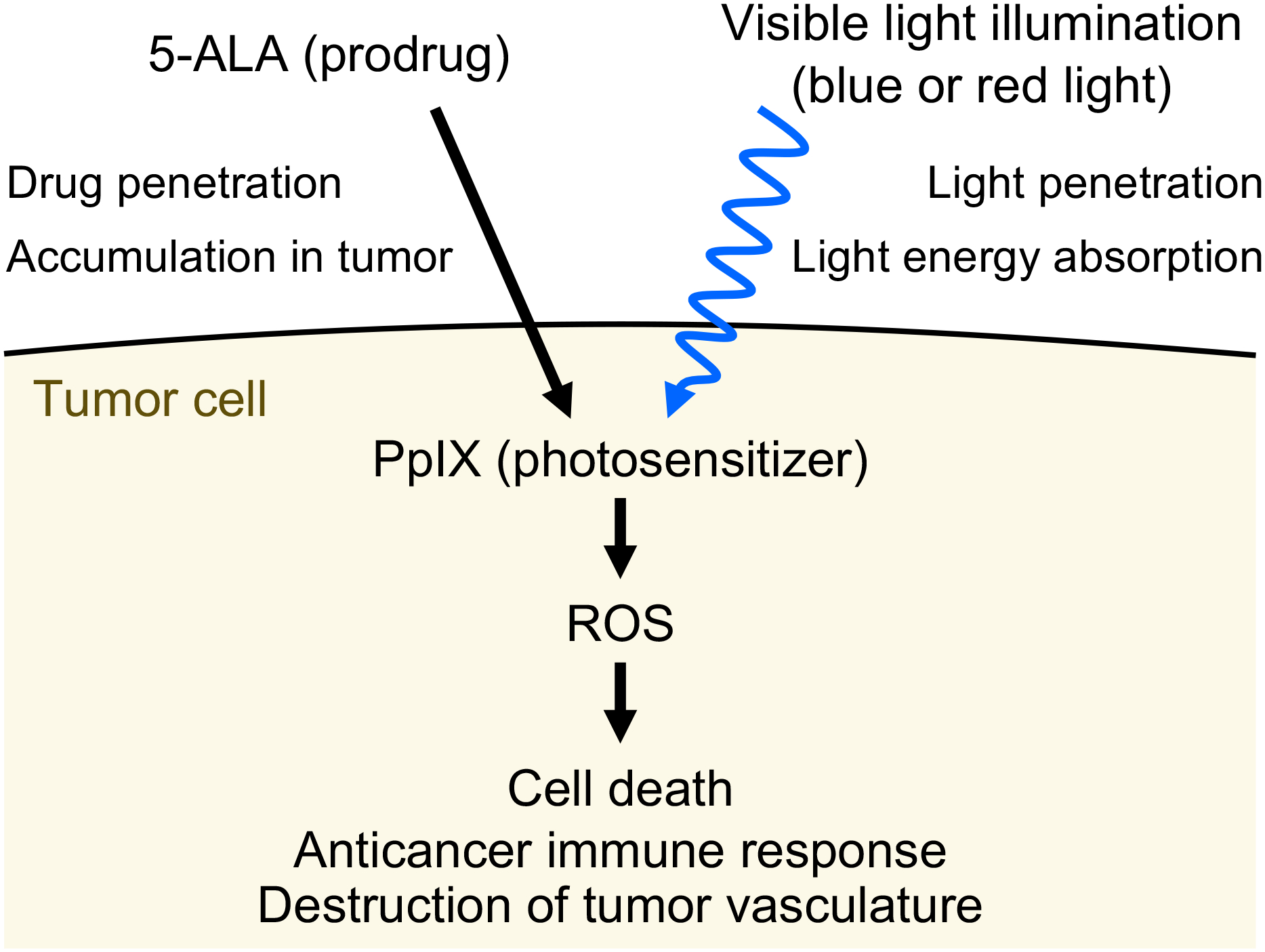
Mechanism of photodynamic therapy (PDT) for cancer. The prodrug 5-aminolevulinic acid (5-ALA) is preferentially distributed to tumor tissue in a patient. Tumor cells metabolize 5-ALA into the photosensitizer protoporphyrin IX (PpIX). Subsequently, target cells are exposed to visible light (either 400 nm or 635 nm) that is absorbed by PpIX. Excited PpIX generates reactive oxygen species (ROS) that lead to tumor cell death, anticancer immune response, and destruction of tumor vasculature. Visible light illumination and the prodrug 5-ALA have limited penetration into deeper tumor tissue, reducing the efficacy of PDT for larger tumors.
PDT has been trialed or implemented for a variety of cancers including NMSCs (218). In 1975, Dr. Thomas J. Dougherty at the Roswell Park Memorial Institute (now Roswell Park Comprehensive Cancer Center) showed for the first time that PDT can selectively treat tumors in mouse and rat models (219). Three years later, he demonstrated the efficacy of PDT for tumors in humans (220). In 1994, PDT was FDA-approved for the first time for esophageal cancer. PDT with blue light and 5-ALA was FDA-approved to treat non-hyperkeratotic, mild- and moderate-thickness AKs of the face and scalp in 1999. PDT with blue light is also approved for BCC and cSCC in situ (Bowen’s disease) in Europe. Assessment of PDT for these diseases continue; PDT has been shown to be effective in treatment of BCC, AK, Bowen’s disease, and invasive cSCC (217,213,221,214). PDT has also been used for treating cancers of the lung, gastrointestinal tract, and genitourinary tract (214,222). For melanoma, PDT has had limited success, and resistance to PDT has been observed (223). A recent study demonstrated that flavin mononucleotide, which is a water-soluble form of riboflavin (vitamin B2), selectively accumulated in melanoma cells and suppressed tumor growth in a mouse xenograft model (224).
To increase the efficacy of PDT, efforts are being made to enhance light and drug penetration into deeper tissue and energy absorption by photosensitizers (225). Typically, PDT has been used with one wavelength of either 400 nm (blue light) or 635 nm (red light) to excite a photosensitizer. To determine the optimal wavelength for treating a tumor, the characteristics of different wavelengths must be considered. Compared to shorter wavelengths, longer wavelengths may have greater penetration into deeply invaded tumors but have less efficient light absorption by photosensitizers. The widely used photosensitizer PpIX, which is metabolized from a prodrug such as 5-ALA, has five absorption peaks at 410 nm, 510 nm, 545 nm, 580 nm, and 630 nm, some of which may be simultaneously targeted for more energy absorption. Indeed, a combination of 405 nm and 505 nm wavelengths generated more ROS than 405 nm alone in cells treated with 5-ALA (226). In a mouse xenograft model, this dual-wavelength PDT suppressed tumor growth more effectively than 405 nm single-wavelength PDT (226).
Discussion and future directions
UV radiation has a profound impact on the biology of skin, including photoaging and carcinogenesis. Although the reasons behind the increasing incidence of skin cancer remain unclear, genetic alterations following UV exposure have been extensively studied in order to understand the evolution of UV-exposed normal skin into cancer. Genome-wide mapping of UV-induced DNA lesions and NER revealed how chromatin state affects lesion formation and repair. Lesions are generated more frequently and repaired more slowly in heterochromatin, likely contributing to cancer development. Error-prone TLS is a DNA damage tolerance process that allows for cell survival under genotoxic stress, but it enables the survival of both normal and cancer cells. The question of how to target this mechanism to selectively kill cancer cells while sparing normal cells should be investigated. Given the abundance of UV-induced mutations even in normal skin, identifying critical cancer driver mutations may pose a challenge, particularly when these driver mutations occur in noncoding genomic regions. For cancer prevention, ATR is an attractive target because ATR inhibition selectively sensitizes p53-deficient cells to genotoxic agents including UV. This is consistent with human epidemiological studies that demonstrate decreased skin cancer risk with consumption of caffeine, a nonspecific ATR inhibitor. As our understanding of molecular mechanisms of UV-induced phenomena becomes more precise and comprehensive, approaches in preventing and treating skin cancer will be increasingly driven by identifying and utilizing specific molecular targets such as ATR. With the development of targeted strategies for precision medicine, cancer therapy will likely be more effective, selective, and durable. Over the past several decades, photodynamic therapy has shown benefit in a wide variety of cancer types, although with limited penetration into tumors. Further improvement of photosensitizers and light penetration holds promise for application of photodynamic therapy to larger, more aggressive tumors.
Acknowledgements
This work is dedicated to the memory of Dr. Kazuhiko Yamane (University of Washington) who participated in early discussions. This work was supported by Dermatology Foundation Research Career Development Award (to M.K.), Dermatology Foundation Diversity Research Supplement Award (to M.K.), Washington Research Foundation Project Grant (to M.K.), Fred Hutch/University of Washington Cancer Consortium Cancer Center Support Grant (CCSG) New Investigator Support (NIH/NCI P30 CA015704) (to M.K.), University of Washington Medical Scientist Training Program (MSTP) (NIH/NIGMS T32 GM007266) (to J.W.L.), American Skin Association Hambrick Medical Student Grant (to J.W.L.), and Institute of Translational Health Sciences (ITHS) Early Investigator Catalyst Award (NIH/NCATS UL1 TR002319) (to K.R.). The authors thank Dr. Paul Nghiem for critical reading of the manuscript.
Footnotes
This article is part of a Special Issue dedicated to Dr. Thomas Dougherty.
Competing Interests: The authors declare no competing interests.
References
- 1.Kwa RE, Campana K and Moy RL (1992) Biology of cutaneous squamous cell carcinoma. J Am Acad Dermatol 26, 1–26. [DOI] [PubMed] [Google Scholar]
- 2.Albert MR and Ostheimer KG (2002) The evolution of current medical and popular attitudes toward ultraviolet light exposure: part 1. J Am Acad Dermatol 47, 930–937. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong BK, Kricker A and English DR (1997) Sun exposure and skin cancer. Australas J Dermatol 38 Suppl 1, S1–6. [DOI] [PubMed] [Google Scholar]
- 4.Wacker M and Holick MF (2013) Sunlight and Vitamin D: A global perspective for health. Dermatoendocrinol 5, 51–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clydesdale GJ, Dandie GW and Muller HK (2001) Ultraviolet light induced injury: immunological and inflammatory effects. Immunol Cell Biol 79, 547–568. [DOI] [PubMed] [Google Scholar]
- 6.Warren JB (1994) Nitric oxide and human skin blood flow responses to acetylcholine and ultraviolet light. FASEB J 8, 247–251. [DOI] [PubMed] [Google Scholar]
- 7.Johnson KE, Wulff BC, Oberyszyn TM and Wilgus TA (2013) Ultraviolet light exposure stimulates HMGB1 release by keratinocytes. Arch Dermatol Res 305, 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, Kohlmeyer J, Riesenberg S, van den Boorn-Konijnenberg D, Homig-Holzel C, Reuten R, Schadow B, Weighardt H, Wenzel D, Helfrich I, Schadendorf D, Bloch W, Bianchi ME, Lugassy C, Barnhill RL, Koch M, Fleischmann BK, Forster I, Kastenmuller W, Kolanus W, Holzel M, Gaffal E and Tuting T (2014) Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 507, 109–113. [DOI] [PubMed] [Google Scholar]
- 9.D’Orazio J, Jarrett S, Amaro-Ortiz A and Scott T (2013) UV radiation and the skin. Int J Mol Sci 14, 12222–12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen NT and Fisher DE (2019) MITF and UV responses in skin: From pigmentation to addiction. Pigment Cell Melanoma Res 32, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Abaseri TB, Putta S and Hansen LA (2006) Ultraviolet irradiation induces keratinocyte proliferation and epidermal hyperplasia through the activation of the epidermal growth factor receptor. Carcinogenesis 27, 225–231. [DOI] [PubMed] [Google Scholar]
- 12.Imokawa G and Ishida K (2015) Biological mechanisms underlying the ultraviolet radiation-induced formation of skin wrinkling and sagging I: reduced skin elasticity, highly associated with enhanced dermal elastase activity, triggers wrinkling and sagging. Int J Mol Sci 16, 7753–7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon JR and Brieva JC (2012) Images in clinical medicine. Unilateral dermatoheliosis. N Engl J Med 366, e25. [DOI] [PubMed] [Google Scholar]
- 14.Schwarz T (2005) Mechanisms of UV-induced immunosuppression. Keio J Med 54, 165–171. [DOI] [PubMed] [Google Scholar]
- 15.Wang W, Chapman NM, Zhang B, Li M, Fan M, Laribee RN, Zaidi MR, Pfeffer LM, Chi H and Wu ZH (2019) Upregulation of PD-L1 via HMGB1-Activated IRF3 and NF-kappaB Contributes to UV Radiation-Induced Immune Suppression. Cancer Res 79, 2909–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fell GL, Robinson KC, Mao J, Woolf CJ and Fisher DE (2014) Skin beta-endorphin mediates addiction to UV light. Cell 157, 1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilchrest BA, Eller MS, Geller AC and Yaar M (1999) The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med 340, 1341–1348. [DOI] [PubMed] [Google Scholar]
- 18.Narayanan DL, Saladi RN and Fox JL (2010) Ultraviolet radiation and skin cancer. Int J Dermatol 49, 978–986. [DOI] [PubMed] [Google Scholar]
- 19.Rosso S, Zanetti R, Martinez C, Tormo MJ, Schraub S, Sancho-Garnier H, Franceschi S, Gafa L, Perea E, Navarro C, Laurent R, Schrameck C, Talamini R, Tumino R and Wechsler J (1996) The multicentre south European study ‘Helios’. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer 73, 1447–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob JJ, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A, Roesch A and Hauschild A (2015) Melanoma. Nat Rev Dis Primers 1, 15003. [DOI] [PubMed] [Google Scholar]
- 21.Karimkhani C, Dellavalle RP, Coffeng LE, Flohr C, Hay RJ, Langan SM, Nsoesie EO, Ferrari AJ, Erskine HE, Silverberg JI, Vos T and Naghavi M (2017) Global Skin Disease Morbidity and Mortality: An Update From the Global Burden of Disease Study 2013. JAMA Dermatol 153, 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hay RJ, Johns NE, Williams HC, Bolliger IW, Dellavalle RP, Margolis DJ, Marks R, Naldi L, Weinstock MA, Wulf SK, Michaud C, C JLM and Naghavi M (2014) The global burden of skin disease in 2010: an analysis of the prevalence and impact of skin conditions. J Invest Dermatol 134, 1527–1534. [DOI] [PubMed] [Google Scholar]
- 23.Karimkhani C, Boyers LN, Dellavalle RP and Weinstock MA (2015) It’s time for “keratinocyte carcinoma” to replace the term “nonmelanoma skin cancer”. J Am Acad Dermatol 72, 186–187. [DOI] [PubMed] [Google Scholar]
- 24.Rogers HW, Weinstock MA, Feldman SR and Coldiron BM (2015) Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol 151, 1081–1086. [DOI] [PubMed] [Google Scholar]
- 25.Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, Vitale L, Pelleri MC, Tassani S, Piva F, Perez-Amodio S, Strippoli P and Canaider S (2013) An estimation of the number of cells in the human body. Ann Hum Biol 40, 463–471. [DOI] [PubMed] [Google Scholar]
- 26.Barger-Lux MJ and Heaney RP (2002) Effects of above average summer sun exposure on serum 25-hydroxyvitamin D and calcium absorption. J Clin Endocrinol Metab 87, 4952–4956. [DOI] [PubMed] [Google Scholar]
- 27.van Erp PE, Boezeman JB and Brons PP (1996) Cell cycle kinetics in normal human skin by in vivo administration of iododeoxyuridine and application of a differentiation marker--implications for cell cycle kinetics in psoriatic skin. Anal Cell Pathol 11, 43–54. [PubMed] [Google Scholar]
- 28.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH and Campbell PJ (2015) Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawasumi M and Nghiem P (2012) Chapter 112 Ultraviolet Radiation Carcinogenesis In Fitzpatrick’s Dermatology in General Medicine. (Edited by Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Wolff K and Leffell DJ), pp. 1251–1260. McGraw-Hill, United States. [Google Scholar]
- 30.Perdiz D, Grof P, Mezzina M, Nikaido O, Moustacchi E and Sage E (2000) Distribution and repair of bipyrimidine photoproducts in solar UV-irradiated mammalian cells. Possible role of Dewar photoproducts in solar mutagenesis. J Biol Chem 275, 26732–26742. [DOI] [PubMed] [Google Scholar]
- 31.Ravanat JL, Douki T and Cadet J (2001) Direct and indirect effects of UV radiation on DNA and its components. J Photochem Photobiol B 63, 88–102. [DOI] [PubMed] [Google Scholar]
- 32.Mouret S, Baudouin C, Charveron M, Favier A, Cadet J and Douki T (2006) Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci U S A 103, 13765–13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.World Health Organization (2019) Ultraviolet radiation and the INTERSUN Programme. Available at: https://www.who.int/uv/intersunprogramme/activities/uv_index/en/index3.html. Accessed on September 29, 2019.
- 34.Vanicek K, Frei T, Litynska Z and Schmalwieser A (1999) UV-Index for the Public: A guide for publication and interpretation of solar UV Index forecasts for the public prepared by the Working Group 4 of the COST-713 Action “UVB Forecasting”. Available at: https://www.researchgate.net/publication/288523666_UV-Index_for_the_Public. Accessed on September 29, 2019.
- 35.Besaratinia A, Yoon JI, Schroeder C, Bradforth SE, Cockburn M and Pfeifer GP (2011) Wavelength dependence of ultraviolet radiation-induced DNA damage as determined by laser irradiation suggests that cyclobutane pyrimidine dimers are the principal DNA lesions produced by terrestrial sunlight. FASEB J 25, 3079–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfeifer GP, You YH and Besaratinia A (2005) Mutations induced by ultraviolet light. Mutat Res 571, 19–31. [DOI] [PubMed] [Google Scholar]
- 37.Runger TM and Kappes UP (2008) Mechanisms of mutation formation with long-wave ultraviolet light (UVA). Photodermatol Photoimmunol Photomed 24, 2–10. [DOI] [PubMed] [Google Scholar]
- 38.Ikehata H, Higashi S, Nakamura S, Daigaku Y, Furusawa Y, Kamei Y, Watanabe M, Yamamoto K, Hieda K, Munakata N and Ono T (2013) Action spectrum analysis of UVR genotoxicity for skin: the border wavelengths between UVA and UVB can bring serious mutation loads to skin. J Invest Dermatol 133, 1850–1856. [DOI] [PubMed] [Google Scholar]
- 39.Kunisada M, Kumimoto H, Ishizaki K, Sakumi K, Nakabeppu Y and Nishigori C (2007) Narrow-band UVB induces more carcinogenic skin tumors than broad-band UVB through the formation of cyclobutane pyrimidine dimer. J Invest Dermatol 127, 2865–2871. [DOI] [PubMed] [Google Scholar]
- 40.Ikehata H, Mori T, Douki T, Cadet J and Yamamoto M (2018) Quantitative analysis of UV photolesions suggests that cyclobutane pyrimidine dimers produced in mouse skin by UVB are more mutagenic than those produced by UVC. Photochem Photobiol Sci 17, 404–413. [DOI] [PubMed] [Google Scholar]
- 41.Ikehata H and Ono T (2011) The mechanisms of UV mutagenesis. J Radiat Res 52, 115–125. [DOI] [PubMed] [Google Scholar]
- 42.Schreier WJ, Schrader TE, Koller FO, Gilch P, Crespo-Hernandez CE, Swaminathan VN, Carell T, Zinth W and Kohler B (2007) Thymine dimerization in DNA is an ultrafast photoreaction. Science 315, 625–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Premi S, Wallisch S, Mano CM, Weiner AB, Bacchiocchi A, Wakamatsu K, Bechara EJ, Halaban R, Douki T and Brash DE (2015) Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 347, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi N, Muramatsu T, Yamashina Y, Shirai T, Ohnishi T and Mori T (1993) Melanin reduces ultraviolet-induced DNA damage formation and killing rate in cultured human melanoma cells. J Invest Dermatol 101, 685–689. [DOI] [PubMed] [Google Scholar]
- 45.Brenner M and Hearing VJ (2008) The protective role of melanin against UV damage in human skin. Photochem Photobiol 84, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao P, Smerdon MJ, Roberts SA and Wyrick JJ (2016) Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc Natl Acad Sci U S A 113, 9057–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao P, Brown AJ, Esaki S, Lockwood S, Poon GMK, Smerdon MJ, Roberts SA and Wyrick JJ (2018) ETS transcription factors induce a unique UV damage signature that drives recurrent mutagenesis in melanoma. Nat Commun 9, 2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elliott K, Bostrom M, Filges S, Lindberg M, Van den Eynden J, Stahlberg A, Clausen AR and Larsson E (2018) Elevated pyrimidine dimer formation at distinct genomic bases underlies promoter mutation hotspots in UV-exposed cancers. PLoS Genet 14, e1007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu J, Adebali O, Adar S and Sancar A (2017) Dynamic maps of UV damage formation and repair for the human genome. Proc Natl Acad Sci U S A 114, 6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Nieto PE, Schwartz EK, King DA, Paulsen J, Collas P, Herrera RE and Morrison AJ (2017) Carcinogen susceptibility is regulated by genome architecture and predicts cancer mutagenesis. EMBO J 36, 2829–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tommasi S, Denissenko MF and Pfeifer GP (1997) Sunlight induces pyrimidine dimers preferentially at 5-methylcytosine bases. Cancer Res 57, 4727–4730. [PubMed] [Google Scholar]
- 52.Martinez-Fernandez L, Banyasz A, Esposito L, Markovitsi D and Improta R (2017) UV-induced damage to DNA: effect of cytosine methylation on pyrimidine dimerization. Signal Transduct Target Ther 2, 17021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciccia A and Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saldivar JC, Cortez D and Cimprich KA (2017) The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pilie PG, Tang C, Mills GB and Yap TA (2019) State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 16, 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Byun TS, Pacek M, Yee MC, Walter JC and Cimprich KA (2005) Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19, 1040–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.MacDougall CA, Byun TS, Van C, Yee MC and Cimprich KA (2007) The structural determinants of checkpoint activation. Genes Dev 21, 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zou L and Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
- 59.Yamane K, Wu X and Chen J (2002) A DNA damage-regulated BRCT-containing protein, TopBP1, is required for cell survival. Mol Cell Biol 22, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumagai A, Lee J, Yoo HY and Dunphy WG (2006) TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955. [DOI] [PubMed] [Google Scholar]
- 61.Choi JH, Lindsey-Boltz LA, Kemp M, Mason AC, Wold MS and Sancar A (2010) Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling. Proc Natl Acad Sci U S A 107, 13660–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cotta-Ramusino C, McDonald ER 3rd, Hurov K, Sowa ME, Harper JW and Elledge SJ (2011) A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH and Zou L (2011) ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell 43, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Raschle M and Mailand N (2016) Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol 18, 1196–1207. [DOI] [PubMed] [Google Scholar]
- 65.Cimprich KA and Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stokes MP, Rush J, Macneill J, Ren JM, Sprott K, Nardone J, Yang V, Beausoleil SA, Gygi SP, Livingstone M, Zhang H, Polakiewicz RD and Comb MJ (2007) Profiling of UV-induced ATM/ATR signaling pathways. Proc Natl Acad Sci U S A 104, 19855–19860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bertoli C, Skotheim JM and de Bruin RA (2013) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M and Kaufmann WK (2002) An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol 22, 8552–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Friedel AM, Pike BL and Gasser SM (2009) ATR/Mec1: coordinating fork stability and repair. Curr Opin Cell Biol 21, 237–244. [DOI] [PubMed] [Google Scholar]
- 70.Saldivar JC, Hamperl S, Bocek MJ, Chung M, Bass TE, Cisneros-Soberanis F, Samejima K, Xie L, Paulson JR, Earnshaw WC, Cortez D, Meyer T and Cimprich KA (2018) An intrinsic S/G2 checkpoint enforced by ATR. Science 361, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin AB, McNeely SC and Beckmann RP (2017) Achieving Precision Death with Cell-Cycle Inhibitors that Target DNA Replication and Repair. Clin Cancer Res 23, 3232–3240. [DOI] [PubMed] [Google Scholar]
- 72.Helt CE, Cliby WA, Keng PC, Bambara RA and O’Reilly MA (2005) Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem 280, 1186–1192. [DOI] [PubMed] [Google Scholar]
- 73.Hafner A, Bulyk ML, Jambhekar A and Lahav G (2019) The multiple mechanisms that regulate p53 activity and cell fate. Nat Rev Mol Cell Biol 20, 199–210. [DOI] [PubMed] [Google Scholar]
- 74.Boos D, Sanchez-Pulido L, Rappas M, Pearl LH, Oliver AW, Ponting CP and Diffley JF (2011) Regulation of DNA replication through Sld3-Dpb11 interaction is conserved from yeast to humans. Curr Biol 21, 1152–1157. [DOI] [PubMed] [Google Scholar]
- 75.Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA and Cortez D (2013) ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moiseeva TN, Yin Y, Calderon MJ, Qian C, Schamus-Haynes S, Sugitani N, Osmanbeyoglu HU, Rothenberg E, Watkins SC and Bakkenist CJ (2019) An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc Natl Acad Sci U S A 116, 13374–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kabeche L, Nguyen HD, Buisson R and Zou L (2018) A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 359, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nghiem P, Park PK, Kim Y, Vaziri C and Schreiber SL (2001) ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc Natl Acad Sci U S A 98, 9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kawasumi M, Bradner JE, Tolliday N, Thibodeau R, Sloan H, Brummond KM and Nghiem P (2014) Identification of ATR-Chk1 pathway inhibitors that selectively target p53-deficient cells without directly suppressing ATR catalytic activity. Cancer Res 74, 7534–7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM and Pollard JR (2011) Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 7, 428–430. [DOI] [PubMed] [Google Scholar]
- 81.Lecona E and Fernandez-Capetillo O (2018) Targeting ATR in cancer. Nat Rev Cancer 18, 586–595. [DOI] [PubMed] [Google Scholar]
- 82.Kemp MG and Sancar A (2016) ATR Kinase Inhibition Protects Non-cycling Cells from the Lethal Effects of DNA Damage and Transcription Stress. J Biol Chem 291, 9330–9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tresini M, Warmerdam DO, Kolovos P, Snijder L, Vrouwe MG, Demmers JA, van IWF, Grosveld FG, Medema RH, Hoeijmakers JH, Mullenders LH, Vermeulen W and Marteijn JA (2015) The core spliceosome as target and effector of non-canonical ATM signalling. Nature 523, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Enk CD, Jacob-Hirsch J, Gal H, Verbovetski I, Amariglio N, Mevorach D, Ingber A, Givol D, Rechavi G and Hochberg M (2006) The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes. Oncogene 25, 2601–2614. [DOI] [PubMed] [Google Scholar]
- 85.Andrade-Lima LC, Veloso A, Paulsen MT, Menck CF and Ljungman M (2015) DNA repair and recovery of RNA synthesis following exposure to ultraviolet light are delayed in long genes. Nucleic Acids Res 43, 2744–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Adimoolam S and Ford JM (2002) p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci U S A 99, 12985–12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Murakami T, Fujimoto M, Ohtsuki M and Nakagawa H (2001) Expression profiling of cancer-related genes in human keratinocytes following non-lethal ultraviolet B irradiation. J Dermatol Sci 27, 121–129. [DOI] [PubMed] [Google Scholar]
- 88.Katayama S, Skoog T, Jouhilahti EM, Siitonen HA, Nuutila K, Tervaniemi MH, Vuola J, Johnsson A, Lonnerberg P, Linnarsson S, Elomaa O, Kankuri E and Kere J (2015) Gene expression analysis of skin grafts and cultured keratinocytes using synthetic RNA normalization reveals insights into differentiation and growth control. BMC Genomics 16, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dziunycz P, Iotzova-Weiss G, Eloranta JJ, Lauchli S, Hafner J, French LE and Hofbauer GF (2010) Squamous cell carcinoma of the skin shows a distinct microRNA profile modulated by UV radiation. J Invest Dermatol 130, 2686–2689. [DOI] [PubMed] [Google Scholar]
- 90.Yo K and Runger TM (2017) UVA and UVB Induce Different Sets of Long Noncoding RNAs. J Invest Dermatol 137, 769–772. [DOI] [PubMed] [Google Scholar]
- 91.Sancar A (2016) Mechanisms of DNA Repair by Photolyase and Excision Nuclease (Nobel Lecture). Angew Chem Int Ed Engl 55, 8502–8527. [DOI] [PubMed] [Google Scholar]
- 92.Sancar A (2003) Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem Rev 103, 2203–2237. [DOI] [PubMed] [Google Scholar]
- 93.Kao YT, Saxena C, Wang L, Sancar A and Zhong D (2005) Direct observation of thymine dimer repair in DNA by photolyase. Proc Natl Acad Sci U S A 102, 16128–16132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li J, Liu Z, Tan C, Guo X, Wang L, Sancar A and Zhong D (2010) Dynamics and mechanism of repair of ultraviolet-induced (6–4) photoproduct by photolyase. Nature 466, 887–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lucas-Lledo JI and Lynch M (2009) Evolution of mutation rates: phylogenomic analysis of the photolyase/cryptochrome family. Mol Biol Evol 26, 1143–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marteijn JA, Lans H, Vermeulen W and Hoeijmakers JH (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15, 465–481. [DOI] [PubMed] [Google Scholar]
- 97.Eisen JA and Hanawalt PC (1999) A phylogenomic study of DNA repair genes, proteins, and processes. Mutat Res 435, 171–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.DiGiovanna JJ and Kraemer KH (2012) Shining a light on xeroderma pigmentosum. J Invest Dermatol 132, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Calses PC, Dhillon KK, Tucker N, Chi Y, Huang JW, Kawasumi M, Nghiem P, Wang Y, Clurman BE, Jacquemont C, Gafken PR, Sugasawa K, Saijo M and Taniguchi T (2017) DGCR8 Mediates Repair of UV-Induced DNA Damage Independently of RNA Processing. Cell Rep 19, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Giannattasio M, Follonier C, Tourriere H, Puddu F, Lazzaro F, Pasero P, Lopes M, Plevani P and Muzi-Falconi M (2010) Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol Cell 40, 50–62. [DOI] [PubMed] [Google Scholar]
- 101.Sertic S, Mollica A, Campus I, Roma S, Tumini E, Aguilera A and Muzi-Falconi M (2018) Coordinated Activity of Y Family TLS Polymerases and EXO1 Protects Non-S Phase Cells from UV-Induced Cytotoxic Lesions. Mol Cell 70, 34–47.e4. [DOI] [PubMed] [Google Scholar]
- 102.Sugasawa K (2008) Xeroderma pigmentosum genes: functions inside and outside DNA repair. Carcinogenesis 29, 455–465. [DOI] [PubMed] [Google Scholar]
- 103.Reid-Bayliss KS, Arron ST, Loeb LA, Bezrookove V and Cleaver JE (2016) Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc Natl Acad Sci U S A 113, 10151–10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K and Hanaoka F (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 399, 700–704. [DOI] [PubMed] [Google Scholar]
- 105.Hirai Y, Kodama Y, Moriwaki S, Noda A, Cullings HM, Macphee DG, Kodama K, Mabuchi K, Kraemer KH, Land CE and Nakamura N (2006) Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res 601, 171–178. [DOI] [PubMed] [Google Scholar]
- 106.Lehmann AR, McGibbon D and Stefanini M (2011) Xeroderma pigmentosum. Orphanet J Rare Dis 6, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pugh J, Khan SG, Tamura D, Goldstein AM, Landi MT, DiGiovanna JJ and Kraemer KH (2019) Use of Big Data to Estimate Prevalence of Defective DNA Repair Variants in the US Population. JAMA Dermatol 155, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.de Feraudy S, Ridd K, Richards LM, Kwok PY, Revet I, Oh D, Feeney L and Cleaver JE (2010) The DNA damage-binding protein XPC is a frequent target for inactivation in squamous cell carcinomas. Am J Pathol 177, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rezvani HR, Kim AL, Rossignol R, Ali N, Daly M, Mahfouf W, Bellance N, Taieb A, de Verneuil H, Mazurier F and Bickers DR (2011) XPC silencing in normal human keratinocytes triggers metabolic alterations that drive the formation of squamous cell carcinomas. J Clin Invest 121, 195–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Spivak G (2004) The many faces of Cockayne syndrome. Proc Natl Acad Sci U S A 101, 15273–15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hu J, Adar S, Selby CP, Lieb JD and Sancar A (2015) Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev 29, 948–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Adar S, Hu J, Lieb JD and Sancar A (2016) Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc Natl Acad Sci U S A 113, E2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rastogi RP, Richa, Kumar A, Tyagi MB and Sinha RP (2010) Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids 2010, 592980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Berube R, Drigeard Desgarnier MC, Douki T, Lechasseur A and Rochette PJ (2018) Persistence and Tolerance of DNA Damage Induced by Chronic UVB Irradiation of the Human Genome. J Invest Dermatol 138, 405–412. [DOI] [PubMed] [Google Scholar]
- 115.Matsumoto S, Cavadini S, Bunker RD, Grand RS, Potenza A, Rabl J, Yamamoto J, Schenk AD, Schubeler D, Iwai S, Sugasawa K, Kurumizaka H and Thoma NH (2019) DNA damage detection in nucleosomes involves DNA register shifting. Nature 571, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mazouzi A, Battistini F, Moser SC, Ferreira da Silva J, Wiedner M, Owusu M, Lardeau CH, Ringler A, Weil B, Neesen J, Orozco M, Kubicek S and Loizou JI (2017) Repair of UV-Induced DNA Damage Independent of Nucleotide Excision Repair Is Masked by MUTYH. Mol Cell 68, 797–807.e7. [DOI] [PubMed] [Google Scholar]
- 117.Zhang J (2017) Brothers in arms: emerging roles of RNA epigenetics in DNA damage repair. Cell Biosci 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao BS, Roundtree IA and He C (2017) Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, Ling D, Hsu PH, Zou L, Jambhekar A, He C and Shi Y (2017) RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brash DE (2015) UV signature mutations. Photochem Photobiol 91, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chang DJ and Cimprich KA (2009) DNA damage tolerance: when it’s OK to make mistakes. Nat Chem Biol 5, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Friedberg EC (2005) Suffering in silence: the tolerance of DNA damage. Nat Rev Mol Cell Biol 6, 943–953. [DOI] [PubMed] [Google Scholar]
- 123.Lin JR, Zeman MK, Chen JY, Yee MC and Cimprich KA (2011) SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol Cell 42, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ and Blanco L (2013) PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rudd SG, Glover L, Jozwiakowski SK, Horn D and Doherty AJ (2013) PPL2 translesion polymerase is essential for the completion of chromosomal DNA replication in the African trypanosome. Mol Cell 52, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD and Doherty AJ (2013) PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52, 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L and Mendez J (2013) Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20, 1383–1389. [DOI] [PubMed] [Google Scholar]
- 128.Loeb LA and Monnat RJ Jr. (2008) DNA polymerases and human disease. Nat Rev Genet 9, 594–604. [DOI] [PubMed] [Google Scholar]
- 129.Hendriks G, Calleja F, Besaratinia A, Vrieling H, Pfeifer GP, Mullenders LH, Jansen JG and de Wind N (2010) Transcription-dependent cytosine deamination is a novel mechanism in ultraviolet light-induced mutagenesis. Curr Biol 20, 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tu Y, Dammann R and Pfeifer GP (1998) Sequence and time-dependent deamination of cytosine bases in UVB-induced cyclobutane pyrimidine dimers in vivo. J Mol Biol 284, 297–311. [DOI] [PubMed] [Google Scholar]
- 131.Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Gali H, Hendel A, Johansson F, Erixon K, Livneh Z, Mullenders LH, Haracska L and de Wind N (2009) Separate domains of Rev1 mediate two modes of DNA damage bypass in mammalian cells. Mol Cell Biol 29, 3113–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jans J, Schul W, Sert YG, Rijksen Y, Rebel H, Eker AP, Nakajima S, van Steeg H, de Gruijl FR, Yasui A, Hoeijmakers JH and van der Horst GT (2005) Powerful skin cancer protection by a CPD-photolyase transgene. Curr Biol 15, 105–115. [DOI] [PubMed] [Google Scholar]
- 133.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA and Stratton MR (2007) Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A, Mudie LJ, Ning Z, Royce T, Schulz-Trieglaff OB, Spiridou A, Stebbings LA, Szajkowski L, Teague J, Williamson D, Chin L, Ross MT, Campbell PJ, Bentley DR, Futreal PA and Stratton MR (2010) A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, Levy R, Jimenez-Sanchez A, Trabish S, Lee JS, Karathia H, Barnea E, Day CP, Cinnamon E, Stein I, Solomon A, Bitton L, Perez-Guijarro E, Dubovik T, Shen-Orr SS, Miller ML, Merlino G, Levin Y, Pikarsky E, Eisenbach L, Admon A, Swanton C, Ruppin E and Samuels Y (2019) UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 179, 219–235.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bickers DR and Athar M (2006) Oxidative stress in the pathogenesis of skin disease. J Invest Dermatol 126, 2565–2575. [DOI] [PubMed] [Google Scholar]
- 137.Liebel F, Kaur S, Ruvolo E, Kollias N and Southall MD (2012) Irradiation of skin with visible light induces reactive oxygen species and matrix-degrading enzymes. J Invest Dermatol 132, 1901–1907. [DOI] [PubMed] [Google Scholar]
- 138.Venza M, Visalli M, Beninati C, De Gaetano GV, Teti D and Venza I (2015) Cellular Mechanisms of Oxidative Stress and Action in Melanoma. Oxid Med Cell Longev 2015, 481782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wondrak GT, Roberts MJ, Cervantes-Laurean D, Jacobson MK and Jacobson EL (2003) Proteins of the extracellular matrix are sensitizers of photo-oxidative stress in human skin cells. J Invest Dermatol 121, 578–586. [DOI] [PubMed] [Google Scholar]
- 140.Hattori Y, Nishigori C, Tanaka T, Uchida K, Nikaido O, Osawa T, Hiai H, Imamura S and Toyokuni S (1996) 8-hydroxy-2’-deoxyguanosine is increased in epidermal cells of hairless mice after chronic ultraviolet B exposure. J Invest Dermatol 107, 733–737. [DOI] [PubMed] [Google Scholar]
- 141.David SS, O’Shea VL and Kundu S (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Peus D, Vasa RA, Beyerle A, Meves A, Krautmacher C and Pittelkow MR (1999) UVB activates ERK1/2 and p38 signaling pathways via reactive oxygen species in cultured keratinocytes. J Invest Dermatol 112, 751–756. [DOI] [PubMed] [Google Scholar]
- 143.Liu F, Yang X, Geng M and Huang M (2018) Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm Sin B 8, 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Knebel A, Rahmsdorf HJ, Ullrich A and Herrlich P (1996) Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J 15, 5314–5325. [PMC free article] [PubMed] [Google Scholar]
- 145.Ikehata H and Yamamoto M (2018) Roles of the KEAP1-NRF2 system in mammalian skin exposed to UV radiation. Toxicol Appl Pharmacol 360, 69–77. [DOI] [PubMed] [Google Scholar]
- 146.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ and Morrison SJ (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Siegel RL, Miller KD and Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69, 7–34. [DOI] [PubMed] [Google Scholar]
- 148.Rogers HW, Weinstock MA, Harris AR, Hinckley MR, Feldman SR, Fleischer AB and Coldiron BM (2010) Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol 146, 283–287. [DOI] [PubMed] [Google Scholar]
- 149.Mohan SV and Chang AL (2014) Advanced Basal Cell Carcinoma: Epidemiology and Therapeutic Innovations. Curr Dermatol Rep 3, 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Guy GP Jr., Thomas CC, Thompson T, Watson M, Massetti GM, Richardson LC and Centers for Disease and Prevention (2015) Vital signs: melanoma incidence and mortality trends and projections - United States, 1982–2030. MMWR Morb Mortal Wkly Rep 64, 591–596. [PMC free article] [PubMed] [Google Scholar]
- 151.Whiteman DC, Green AC and Olsen CM (2016) The Growing Burden of Invasive Melanoma: Projections of Incidence Rates and Numbers of New Cases in Six Susceptible Populations through 2031. J Invest Dermatol 136, 1161–1171. [DOI] [PubMed] [Google Scholar]
- 152.Mansouri B and Housewright CD (2017) The Treatment of Actinic Keratoses-The Rule Rather Than the Exception. JAMA Dermatol 153, 1200. [DOI] [PubMed] [Google Scholar]
- 153.Glogau RG (2000) The risk of progression to invasive disease. J Am Acad Dermatol 42, 23–24. [DOI] [PubMed] [Google Scholar]
- 154.Ratushny V, Gober MD, Hick R, Ridky TW and Seykora JT (2012) From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J Clin Invest 122, 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Karia PS, Han J and Schmults CD (2013) Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J Am Acad Dermatol 68, 957–966. [DOI] [PubMed] [Google Scholar]
- 156.Gloster HM Jr. and Neal K (2006) Skin cancer in skin of color. J Am Acad Dermatol 55, 741–760; quiz 761–744. [DOI] [PubMed] [Google Scholar]
- 157.Agbai ON, Buster K, Sanchez M, Hernandez C, Kundu RV, Chiu M, Roberts WE, Draelos ZD, Bhushan R, Taylor SC and Lim HW (2014) Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol 70, 748–762. [DOI] [PubMed] [Google Scholar]
- 158.Council on Scientific Affairs (1989) Harmful effects of ultraviolet radiation. JAMA 262, 380–384. [PubMed] [Google Scholar]
- 159.National Toxicology Program (2002) Report on carcinogens, tenth edition. Carcinogen profiles 2002. Rep Carcinog 10, i–vi, 1–263, 1–44 passim. [PubMed] [Google Scholar]
- 160.El Ghissassi F, Baan R, Straif K, Grosse Y, Secretan B, Bouvard V, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V and WHO International Agency for Research on Cancer Monograph Working Group (2009) A review of human carcinogens–Part D: radiation. Lancet Oncol 10, 751–752. [DOI] [PubMed] [Google Scholar]
- 161.Montague M, Borland R and Sinclair C (2001) Slip! Slop! Slap! and SunSmart, 1980–2000: Skin cancer control and 20 years of population-based campaigning. Health Educ Behav 28, 290–305. [DOI] [PubMed] [Google Scholar]
- 162.Ananthaswamy HN, Loughlin SM, Cox P, Evans RL, Ullrich SE and Kripke ML (1997) Sunlight and skin cancer: inhibition of p53 mutations in UV-irradiated mouse skin by sunscreens. Nat Med 3, 510–514. [DOI] [PubMed] [Google Scholar]
- 163.Francis SO, Mahlberg MJ, Johnson KR, Ming ME and Dellavalle RP (2006) Melanoma chemoprevention. J Am Acad Dermatol 55, 849–861. [DOI] [PubMed] [Google Scholar]
- 164.Bowden GT (2004) Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat Rev Cancer 4, 23–35. [DOI] [PubMed] [Google Scholar]
- 165.Wang ZY, Huang MT, Lou YR, Xie JG, Reuhl KR, Newmark HL, Ho CT, Yang CS and Conney AH (1994) Inhibitory effects of black tea, green tea, decaffeinated black tea, and decaffeinated green tea on ultraviolet B light-induced skin carcinogenesis in 7,12-dimethylbenz[a]anthracene-initiated SKH-1 mice. Cancer Res 54, 3428–3435. [PubMed] [Google Scholar]
- 166.Lu YP, Lou YR, Xie JG, Peng QY, Liao J, Yang CS, Huang MT and Conney AH (2002) Topical applications of caffeine or (−)-epigallocatechin gallate (EGCG) inhibit carcinogenesis and selectively increase apoptosis in UVB-induced skin tumors in mice. Proc Natl Acad Sci U S A 99, 12455–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Conney AH, Zhou S, Lee MJ, Xie JG, Yang CS, Lou YR and Lu Y (2007) Stimulatory effect of oral administration of tea, coffee or caffeine on UVB-induced apoptosis in the epidermis of SKH-1 mice. Toxicol Appl Pharmacol 224, 209–213. [DOI] [PubMed] [Google Scholar]
- 168.Yang CS, Wang X, Lu G and Picinich SC (2009) Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer 9, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Conney AH, Lu YP, Lou YR, Kawasumi M and Nghiem P (2013) Mechanisms of Caffeine-Induced Inhibition of UVB Carcinogenesis. Front Oncol 3, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Blohm-Mangone K, Burkett NB, Tahsin S, Myrdal PB, Aodah A, Ho B, Janda J, McComas M, Saboda K, Roe DJ, Dong Z, Bode AM, Petricoin EF 3rd, Calvert VS, Curiel-Lewandrowski C, Alberts DS, Wondrak GT and Dickinson SE (2018) Pharmacological TLR4 Antagonism Using Topical Resatorvid Blocks Solar UV-Induced Skin Tumorigenesis in SKH-1 Mice. Cancer Prev Res (Phila) 11, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Qiang L, Sample A, Shea CR, Soltani K, Macleod KF and He YY (2017) Autophagy gene ATG7 regulates ultraviolet radiation-induced inflammation and skin tumorigenesis. Autophagy 13, 2086–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tao S, Park SL, Rojo de la Vega M, Zhang DD and Wondrak GT (2015) Systemic administration of the apocarotenoid bixin protects skin against solar UV-induced damage through activation of NRF2. Free Radic Biol Med 89, 690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Abel EL, Hendrix SO, McNeeley SG, Johnson KC, Rosenberg CA, Mossavar-Rahmani Y, Vitolins M and Kruger M (2007) Daily coffee consumption and prevalence of nonmelanoma skin cancer in Caucasian women. Eur J Cancer Prev 16, 446–452. [DOI] [PubMed] [Google Scholar]
- 174.Song F, Qureshi AA and Han J (2012) Increased caffeine intake is associated with reduced risk of basal cell carcinoma of the skin. Cancer Res 72, 3282–3289. [DOI] [PubMed] [Google Scholar]
- 175.Oh CC, Jin A, Yuan JM and Koh WP (2019) Coffee, tea, caffeine, and risk of nonmelanoma skin cancer in a Chinese population: The Singapore Chinese Health Study. J Am Acad Dermatol 81, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Loftfield E, Freedman ND, Graubard BI, Hollenbeck AR, Shebl FM, Mayne ST and Sinha R (2015) Coffee drinking and cutaneous melanoma risk in the NIH-AARP diet and health study. J Natl Cancer Inst 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Koo SW, Hirakawa S, Fujii S, Kawasumi M and Nghiem P (2007) Protection from photodamage by topical application of caffeine after ultraviolet irradiation. Br J Dermatol 156, 957–964. [DOI] [PubMed] [Google Scholar]
- 178.Huang MT, Xie JG, Wang ZY, Ho CT, Lou YR, Wang CX, Hard GC and Conney AH (1997) Effects of tea, decaffeinated tea, and caffeine on UVB light-induced complete carcinogenesis in SKH-1 mice: demonstration of caffeine as a biologically important constituent of tea. Cancer Res 57, 2623–2629. [PubMed] [Google Scholar]
- 179.Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM and Abraham RT (1999) Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 59, 4375–4382. [PubMed] [Google Scholar]
- 180.Heffernan TP, Kawasumi M, Blasina A, Anderes K, Conney AH and Nghiem P (2009) ATR-Chk1 pathway inhibition promotes apoptosis after UV treatment in primary human keratinocytes: potential basis for the UV protective effects of caffeine. J Invest Dermatol 129, 1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kawasumi M, Lemos B, Bradner JE, Thibodeau R, Kim YS, Schmidt M, Higgins E, Koo SW, Angle-Zahn A, Chen A, Levine D, Nguyen L, Heffernan TP, Longo I, Mandinova A, Lu YP, Conney AH and Nghiem P (2011) Protection from UV-induced skin carcinogenesis by genetic inhibition of the ataxia telangiectasia and Rad3-related (ATR) kinase. Proc Natl Acad Sci U S A 108, 13716–13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ and Ding L (2013) Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Olivier M, Hollstein M and Hainaut P (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2, a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mantovani F, Collavin L and Del Sal G (2019) Mutant p53 as a guardian of the cancer cell. Cell Death Differ 26, 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, Tsai KY, Curry JL, Tetzlaff MT, Lai SY, Yu J, Muzny DM, Doddapaneni H, Shinbrot E, Covington KR, Zhang J, Seth S, Caulin C, Clayman GL, El-Naggar AK, Gibbs RA, Weber RS, Myers JN, Wheeler DA and Frederick MJ (2014) Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res 20, 6582–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Leffell DJ, Tarone RE and Brash DE (1996) Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci U S A 93, 14025–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lu YP, Lou YR, Peng QY, Xie JG and Conney AH (2004) Stimulatory effect of topical application of caffeine on UVB-induced apoptosis in the epidermis of p53 and Bax knockout mice. Cancer Res 64, 5020–5027. [DOI] [PubMed] [Google Scholar]
- 188.Miller SJ, Alam M, Andersen J, Berg D, Bichakjian CK, Bowen G, Cheney RT, Glass LF, Grekin RC, Kessinger A, Lee NY, Liegeois N, Lydiatt DD, Michalski J, Morrison WH, Nehal KS, Nelson KC, Nghiem P, Olencki T, Perlis CS, Rosenberg EW, Shaha AR, Urist MM, Wang LC and Zic JA (2010) Basal cell and squamous cell skin cancers. J Natl Compr Canc Netw 8, 836–864. [DOI] [PubMed] [Google Scholar]
- 189.Bichakjian CK, Olencki T, Aasi SZ, Alam M, Andersen JS, Berg D, Bowen GM, Cheney RT, Daniels GA, Glass LF, Grekin RC, Grossman K, Higgins SA, Ho AL, Lewis KD, Lydiatt DD, Nehal KS, Nghiem P, Olsen EA, Schmults CD, Sekulic A, Shaha AR, Thorstad WL, Tuli M, Urist MM, Wang TS, Wong SL, Zic JA, Hoffmann KG and Engh A (2016) Basal Cell Skin Cancer, Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 14, 574–597. [DOI] [PubMed] [Google Scholar]
- 190.Potenza C, Bernardini N, Balduzzi V, Losco L, Mambrin A, Marchesiello A, Tolino E, Zuber S, Skroza N and Proietti I (2018) A Review of the Literature of Surgical and Nonsurgical Treatments of Invasive Squamous Cells Carcinoma. Biomed Res Int 2018, 9489163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tanese K, Nakamura Y, Hirai I and Funakoshi T (2019) Updates on the Systemic Treatment of Advanced Non-melanoma Skin Cancer. Front Med (Lausanne) 6, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sacco AG and Daniels GA (2019) Adjuvant and Neoadjuvant Treatment of Skin Cancer. Facial Plast Surg Clin North Am 27, 139–150. [DOI] [PubMed] [Google Scholar]
- 193.Sekulic A and Von Hoff D (2016) Hedgehog Pathway Inhibition. Cell 164, 831. [DOI] [PubMed] [Google Scholar]
- 194.Epstein EH (2008) Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer 8, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Guha M (2012) Hedgehog inhibitor gets landmark skin cancer approval, but questions remain for wider potential. Nat Rev Drug Discov 11, 257–258. [DOI] [PubMed] [Google Scholar]
- 196.Maubec E, Petrow P, Scheer-Senyarich I, Duvillard P, Lacroix L, Gelly J, Certain A, Duval X, Crickx B, Buffard V, Basset-Seguin N, Saez P, Duval-Modeste AB, Adamski H, Mansard S, Grange F, Dompmartin A, Faivre S, Mentre F and Avril MF (2011) Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J Clin Oncol 29, 3419–3426. [DOI] [PubMed] [Google Scholar]
- 197.Sherwood V and Leigh IM (2016) WNT Signaling in Cutaneous Squamous Cell Carcinoma: A Future Treatment Strategy? J Invest Dermatol 136, 1760–1767. [DOI] [PubMed] [Google Scholar]
- 198.Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, Chapman PB, Kim MJ, Hayward R, Martin M, Yang H, Wang Q, Hilton H, Hang JS, Noe J, Lambros M, Geyer F, Dhomen N, Niculescu-Duvaz I, Zambon A, Niculescu-Duvaz D, Preece N, Robert L, Otte NJ, Mok S, Kee D, Ma Y, Zhang C, Habets G, Burton EA, Wong B, Nguyen H, Kockx M, Andries L, Lestini B, Nolop KB, Lee RJ, Joe AK, Troy JL, Gonzalez R, Hutson TE, Puzanov I, Chmielowski B, Springer CJ, McArthur GA, Sosman JA, Lo RS, Ribas A and Marais R (2012) RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Oberholzer PA, Kee D, Dziunycz P, Sucker A, Kamsukom N, Jones R, Roden C, Chalk CJ, Ardlie K, Palescandolo E, Piris A, MacConaill LE, Robert C, Hofbauer GF, McArthur GA, Schadendorf D and Garraway LA (2012) RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol 30, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Knight ZA, Lin H and Shokat KM (2010) Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 10, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM and Schadendorf D (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372, 30–39. [DOI] [PubMed] [Google Scholar]
- 202.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, Chakravarty D, Daian F, Gao Q, Bailey MH, Liang WW, Foltz SM, Shmulevich I, Ding L, Heins Z, Ochoa A, Gross B, Gao J, Zhang H, Kundra R, Kandoth C, Bahceci I, Dervishi L, Dogrusoz U, Zhou W, Shen H, Laird PW, Way GP, Greene CS, Liang H, Xiao Y, Wang C, Iavarone A, Berger AH, Bivona TG, Lazar AJ, Hammer GD, Giordano T, Kwong LN, McArthur G, Huang C, Tward AD, Frederick MJ, McCormick F, Meyerson M, Cancer N Genome Atlas Research, Van Allen EM, Cherniack AD, Ciriello G, Sander C and Schultz N (2018) Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chitsazzadeh V, Coarfa C, Drummond JA, Nguyen T, Joseph A, Chilukuri S, Charpiot E, Adelmann CH, Ching G, Nguyen TN, Nicholas C, Thomas VD, Migden M, MacFarlane D, Thompson E, Shen J, Takata Y, McNiece K, Polansky MA, Abbas HA, Rajapakshe K, Gower A, Spira A, Covington KR, Xiao W, Gunaratne P, Pickering C, Frederick M, Myers JN, Shen L, Yao H, Su X, Rapini RP, Wheeler DA, Hawk ET, Flores ER and Tsai KY (2016) Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat Commun 7, 12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Luke JJ, Flaherty KT, Ribas A and Long GV (2017) Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol 14, 463–482. [DOI] [PubMed] [Google Scholar]
- 205.Wei SC, Duffy CR and Allison JP (2018) Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086. [DOI] [PubMed] [Google Scholar]
- 206.Paulson KG, Lahman MC, Chapuis AG and Brownell I (2019) Immunotherapy for skin cancer. Int Immunol 31, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, Chung CH, Hernandez-Aya L, Lim AM, Chang ALS, Rabinowits G, Thai AA, Dunn LA, Hughes BGM, Khushalani NI, Modi B, Schadendorf D, Gao B, Seebach F, Li S, Li J, Mathias M, Booth J, Mohan K, Stankevich E, Babiker HM, Brana I, Gil-Martin M, Homsi J, Johnson ML, Moreno V, Niu J, Owonikoko TK, Papadopoulos KP, Yancopoulos GD, Lowy I and Fury MG (2018) PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N Engl J Med 379, 341–351. [DOI] [PubMed] [Google Scholar]
- 208.Ogata D and Tsuchida T (2019) Systemic Immunotherapy for Advanced Cutaneous Squamous Cell Carcinoma. Curr Treat Options Oncol 20, 30. [DOI] [PubMed] [Google Scholar]
- 209.Lindelof B, Sigurgeirsson B, Gabel H and Stern RS (2000) Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol 143, 513–519. [PubMed] [Google Scholar]
- 210.Goldman JW, Abdalla B, Mendenhall MA, Sisk A, Hunt J, Danovitch GM and Lum EL (2018) PD 1 checkpoint inhibition in solid organ transplants: 2 sides of a coin - case report. BMC Nephrol 19, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr., Carlino MS, Fisher R, Long GV, Hodi FS, Tsoi J, Grasso CS, Mookerjee B, Zhao Q, Ghori R, Moreno BH, Ibrahim N and Hamid O (2019) Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 25, 936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Castano AP, Mroz P and Hamblin MR (2006) Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer 6, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tampa M, Sarbu MI, Matei C, Mitran CI, Mitran MI, Caruntu C, Constantin C, Neagu M and Georgescu SR (2019) Photodynamic therapy: A hot topic in dermato-oncology. Oncol Lett 17, 4085–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J and Peng Q (1998) Photodynamic therapy. J Natl Cancer Inst 90, 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wondrak GT (2009) Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal 11, 3013–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Morton CA, Szeimies RM, Sidoroff A and Braathen LR (2013) European guidelines for topical photodynamic therapy part 1: treatment delivery and current indications - actinic keratoses, Bowen’s disease, basal cell carcinoma. J Eur Acad Dermatol Venereol 27, 536–544. [DOI] [PubMed] [Google Scholar]
- 217.de Albuquerque IO, Nunes J, Figueiro Longo JP, Muehlmann LA and Azevedo RB (2019) Photodynamic therapy in superficial basal cell carcinoma treatment. Photodiagnosis Photodyn Ther 27, 428–432. [DOI] [PubMed] [Google Scholar]
- 218.Dolmans DE, Fukumura D and Jain RK (2003) Photodynamic therapy for cancer. Nat Rev Cancer 3, 380–387. [DOI] [PubMed] [Google Scholar]
- 219.Dougherty TJ, Grindey GB, Fiel R, Weishaupt KR and Boyle DG (1975) Photoradiation therapy. II. Cure of animal tumors with hematoporphyrin and light. J Natl Cancer Inst 55, 115–121. [DOI] [PubMed] [Google Scholar]
- 220.Dougherty TJ, Kaufman JE, Goldfarb A, Weishaupt KR, Boyle D and Mittleman A (1978) Photoradiation therapy for the treatment of malignant tumors. Cancer Res 38, 2628–2635. [PubMed] [Google Scholar]
- 221.Keyal U, Bhatta AK, Zhang G and Wang XL (2019) Present and future perspectives of photodynamic therapy for cutaneous squamous cell carcinoma. J Am Acad Dermatol 80, 765–773. [DOI] [PubMed] [Google Scholar]
- 222.Yanovsky RL, Bartenstein DW, Rogers GS, Isakoff SJ and Chen ST (2019) Photodynamic therapy for solid tumors: A review of the literature. Photodermatol Photoimmunol Photomed 35, 295–303. [DOI] [PubMed] [Google Scholar]
- 223.Huang YY, Vecchio D, Avci P, Yin R, Garcia-Diaz M and Hamblin MR (2013) Melanoma resistance to photodynamic therapy: new insights. Biol Chem 394, 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Akasov RA, Sholina NV, Khochenkov DA, Alova AV, Gorelkin PV, Erofeev AS, Generalova AN and Khaydukov EV (2019) Photodynamic therapy of melanoma by blue-light photoactivation of flavin mononucleotide. Sci Rep 9, 9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Champeau M, Vignoud S, Mortier L and Mordon S (2019) Photodynamic therapy for skin cancer: How to enhance drug penetration? J Photochem Photobiol B 197, 111544. [DOI] [PubMed] [Google Scholar]
- 226.Masuda H, Kimura M, Nishioka A, Kato H and Morita A (2019) Dual wavelength 5-aminolevulinic acid photodynamic therapy using a novel flexible light-emitting diode unit. J Dermatol Sci 93, 109–115. [DOI] [PubMed] [Google Scholar]