

 Ghrelin O-acyltransferase (GOAT) inhibitors hold promise in treating many diseases like obesity, diabetes and NAFLD. In this article we review the current drug-discovery landscape of GOAT inhibitors since the discovery of the enzyme.
Ghrelin O-acyltransferase (GOAT) inhibitors hold promise in treating many diseases like obesity, diabetes and NAFLD. In this article we review the current drug-discovery landscape of GOAT inhibitors since the discovery of the enzyme.
Abstract
Ghrelin is a stomach-derived peptide hormone which stimulates appetite. For ghrelin to exert its orexigenic effect, octanoylation on the serine-3 residue of this gut–brain peptide is essential. The octanoylation of ghrelin is mediated by a unique acyltransferase enzyme known as ghrelin O-acyltransferase (GOAT). Thus modulating this enzyme offers viable approaches to alter feeding behaviors. Over the past decade, several small-molecule based approaches have appeared dealing with the discovery of compounds able to modulate this enzyme for the treatment of obesity and type 2 diabetes. Drug discovery efforts from academic groups and several pharmaceutical companies have fielded compounds having efficacy in altering acylated ghrelin levels in animal models but to date, compounds modulating the activity of the GOAT enzyme do not yet represent clinical options. This mini-review covers the drug discovery approaches of the last decade since the discovery of the GOAT enzyme.
Introduction
Diabesity is a growing public health concern worldwide.1,2 In the United States, the incidence of type II diabetes and obesity have been increasing rapidly.3 Therapeutic interventions in addition to lifestyle alterations and surgical options are urgently needed. Ghrelin or acyl ghrelin is a peptide hormone comprised of 28-amino acids, that is synthesized mainly in the stomach X/A cells. Ghrelin was discovered by Kojima and colleagues in 1999,4 and it is implicated in many physiological processes related to appetite stimulation, insulin sensitivity and energy homeostasis through regulation of body fat.5,6
Ghrelin in known to appear in two forms: the unacylated ghrelin (UAG) which is the most common form in the plasma, and acylated ghrelin (AG). The amino acid sequence of mammalian acyl ghrelin is highly conserved, differing only at two residues between humans and rodents.7 Modification of serine-3 residue post-translationally by an eight carbon-fatty acid octanoyl group results in acylated ghrelin (commonly referred to as ‘ghrelin’) (Fig. 1). This octanoylation probably renders the peptide brain-penetrant and enables the acylated form of ghrelin to bind to the growth hormone-secretagogue receptor 1a (GHS-R1a) in brain to exert its physiological effects related to appetite and growth hormone secretion. The discovery of liver-expressed antimicrobial peptide 2 (LEAP2) as a potent, endogenous antagonist of GHS-R1a has been reported.8 The secretion of LEAP2 mainly in the liver and small intestine, is suppressed by fasting. LEAP2 fully inhibits GHS-R1a activation (IC50 = 6 nM) by ghrelin and blocks the major effects of ghrelin in vivo, including food intake, GH release, and maintenance of viable glucose levels during chronic caloric restriction.8,9
Fig. 1. Generation of ‘active ghrelin’.
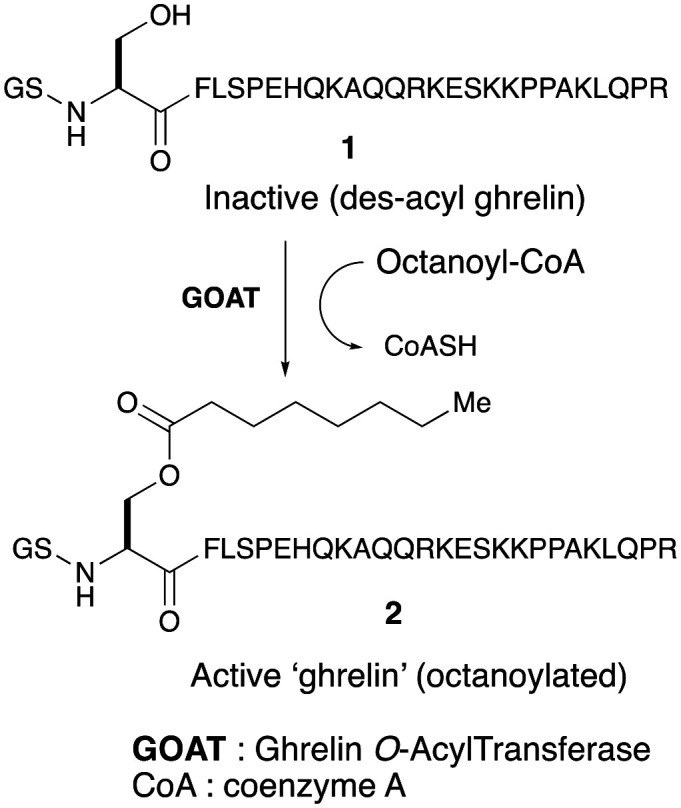
The octanoylation of ghrelin is an important regulatory process mediated by an acyltransferase enzyme known as ghrelin O-acyltransferase (GOAT). The molecular identity of GOAT was identified by two groups concurrently and reported in 2008.10,11 GOAT is a 45 kDa transmembrane protein belonging to the family of membrane-bound O-acyltransferases (MBOAT). In the search for acyltransferase responsible for octanoylation of ghrelin, Yang and co-workers identified the MBOAT family of enzymes as possible mediators. They screened sixteen MBOATs encoded by the mouse genome for the ability to acylate ghrelin. Using rat insulinoma (INS-1) cell line, each MBOAT was expressed alongside ghrelin followed by the addition of octanoic acid to the culture medium and analyzed for the presence of octanoyl ghrelin.10 It was discovered that only treatment with MBOAT4 produced acyl ghrelin. MBOAT4 was thus termed ghrelin O-acyltransferase (GOAT). Octanoyl coenzyme A (CoA) was later identified as the required acyl donor in the transformation. With transfection assays, it was also found that GOAT attached an octanoyl group specifically to serine-3 in ghrelin and the attachment required two amino acids in the enzyme (ASP-307 and HIS-338) that are highly conserved in MBOATs.10,12,13
Previous studies have shown that GOAT mRNA expression was high in the stomach and gut,11 and more recently its expression was also reported in adrenal cortex, breast, right and left colon, duodenum, jejunum, ileum, fat, fallopian tube, gallbladder, lymph node, lymphocyte cell line, kidney, liver, lung, muscle, myocardium, pituitary, oesophagus, pancreas, ovary, placenta, prostate, testis, spleen and thyroid.14 GOAT expression could hence correspond to the widespread role of ghrelin in autocrine and paracrine effects.
Identification of GOAT expression and distribution in human tissues is important as it will facilitate the search for GOAT inhibitors as an attractive approach to reduce the actions of ghrelin such as modulating insulin secretion, decreasing food intake and reducing adiposity. This may hence become a promising therapeutic target because the action of GOAT and thus its inhibition is very specific to ghrelin.
Details on ghrelin physiology has been extensively reviewed.15 While the role of acyl ghrelin in metabolic function is obvious, this peptide-hormone has also been implicated in areas related to cognition, learning and memory, as well as appetitive behavior16,17 and gastrointestinal (GI) motility.18 Ghrelin dysregulation is also responsible for metabolic effects seen in Prader–Willi syndrome and elevated levels of ghrelin in infancy can be consequential for developing obesity related disorders in adulthood.19 Ghrelin's role has also been linked to modulating alcohol and drug intake.20–22 Ghrelin deletion has been reported to be protective against non-alcoholic steatohepatitis (NASH).23 Studies on direct phenotypic effect of GOAT modulation have also appeared recently. Genetic deletion studies have suggested that GOAT is important for survival in severe starvation.24 It has been found that GOAT knockout mice fed with a sucrose supplemented high fat or medium chain triglyceride diet were measurably more resistant to weight gain than WT mice.25 Recent data suggested that plasma GOAT levels could represent a new diagnostic biomarker for prostate cancer (PCa).26
Recently the first structural model of a eukaryotic membrane-bound O-acyltransferase (MBOAT), ghrelin O-acyltransferase (GOAT) was reported.13 The human GOAT (hGOAT) structure was generated in the absence of any experimental data and is highly consistent with a recently reported crystal structure for the bacterial MBOAT homolog d-alanyltransferase DltB.27 The reported model revealed the design for transmembrane protein acylation with catalysis occurring in an internal channel connecting the endoplasmic reticulum (ER) lumen and cytoplasm. It was postulated that ghrelin and octanoyl-CoA enter the GOAT internal channel from the ER lumenal pore and cytoplasmic acyl donor binding sites, respectively, followed by acyl transfer to the ghrelin serine-3 side chain hydroxyl group. Octanoylated ghrelin dissociates to the ER lumen resulting in the octanoyl chain transiting through the GOAT interior, and coenzyme A is released back to the cytoplasm.7 In the absence of X-ray or cryo-EM structure, the exact location and nature of the active site and substrate-binding sites within GOAT remain unknown, however several conserved residues have been shown to be required for enzyme function.28 Among these residues as discussed previously asparagine 307 and histidine 338 are highly conserved within the MBOAT family and mutation of either residue has been shown to lead to a loss of acylation activity of GOAT.
While structural and mechanistic basis for GOAT-catalyzed ghrelin octanoylation has expanded rapidly in the last decade along with a vast array of non-radioactive, fluorescence-based tools for assaying GOAT activity,7,29–32 many aspects of GOAT enzymatic function remain to be determined. Hence there is a need for generating selective, high affinity inhibitors and pharmacological tools for studying this enzyme. The past decade has seen active research from various laboratories in the design and development of GOAT inhibitors. This article is focused on small-molecule based drug discovery work from the literature as well as patent disclosures encompassing the past decade on inhibiting GOAT for therapeutic gain.
Discovery of GOAT inhibitors
Shortly after the discovery of GOAT in ghrelin pathway, Yang et al. reported that the enzyme was inhibited by its own reaction product, the octanoyl ghrelin (2) with a modest IC50 (7 μM).12 It was shown that when Ser-3 in ghrelin was replaced with DAP ((S)-2,3-diaminopropionic acid) creating an octanoyl-amide in lieu of ester both the 28-mer 3 and the truncated 5-mer acyl ghrelin 5 were potent GOAT inhibitors with IC50 values of 0.2 and 1 μM, respectively. The octanoyl truncated analog 4, in contrast was much less potent with an IC50 value of 45 μM. It is likely that strong product inhibition of GOAT by these compounds is a function of hydrolytic stability of the amide linkage over the ester functionality. Most importantly, this work also showed that GOAT can catalyse acylation of truncated peptides derived from N-terminal ghrelin up to pentapeptide length. Utilizing modifications of this inhibitor with different length acyl chains, Darling et al.33 demonstrated that an eight-carbon acyl group is required for maximum potency against GOAT (Fig. 2).
Fig. 2. Chemical structures of earliest peptide-based GOAT inhibitors.

Earliest reports of inhibiting GOAT to modulate serum acylated ghrelin were claimed in the patent from Johns Hopkins University.34,35 These inhibitors were designed as bi-substrate analogs of the enzymatic reaction and were coupled with one of the peptides able to cross cell membranes, the 11-mer Tat peptide. The patent filed had compounds shown in general formula 6 where GS is the amino acid glycine and serine, Dap is 1,2-diamino propanoic acid, which is a serine isostere replacing serine at the 3 position, Ahx is the amino-hexanoyl linker which connects the R amino acid residue with Tat sequence which is abbreviated for trans-acting activator of transcription. ‘Tat’ is a 11-amino acid/peptide:YGRKKRRQRRR. The idea of a bi-substrate inhibitor is to covalently combine two substrates to increase potency and specificity. The main design feature of the inhibitor also included a coenzyme A and an octanoyl ghrelin-based peptide (Fig. 3).
Fig. 3. In vivo active GOAT inhibitors from Johns Hopkins group.

Consistent with earlier work, five residues of ghrelin were sufficient for inhibition but three residues were not. Inclusion of 10 or more residues increased potency, with a ceiling effect of ∼75% inhibition of acylation seen. Amongst many variations, a compound named GO-CoA-Tat 7 was potent (100% inhibition at 100 nM), selective and effective in vitro in cell culture and in vivo in mice in inhibiting GOAT and lowering acyl ghrelin levels. After intraperitoneal administration, it enhanced insulin response to a glucose load and led to a statistically significant weight loss in mouse fed with a high fat diet. Treatment with this compound also led to selective reduction of fat versus lean mass. The GO-CoA-Tat inhibitor affected the weight and glucose levels in wild-type but not ghrelin knock-out mice confirming the specific targeting of ghrelin pathway.36 Photoactivatable GO-CoA-Tat analogs that can bind and chemically crosslink GOAT have also been claimed in this filing. To analyze GO-CoA-Tat's inhibition of GOAT, a direct binding assay based on photo-crosslinking on GOAT was developed. Two chemically modified versions of the bisubstrate inhibitor, namely, GO-CoA-Tat-F4BP and GO-CoA-Tat-L5BP, in which Phe4 or Leu5, is replaced with a photoreactive amino acid benzoyl-phenylalanine and each was tagged with a biotin group.36 The chemical modification showed that this compound could covalently crosslink to recombinant solubilized or microsomal GOAT and the crosslinking could be blocked by an excess of unlabeled GO-CoA-Tat. This technique provided unequivocal evidence for specificity and direct binding of GO-CoA-Tat to GOAT. This compound, has largely been useful as a pharmacological tool37,38 and had limited utility as a pharmaceutical because of its unfavorable druggable profile as seen in its high molecular weight (∼3700 Da) and propensity for proteolytic degradation.
Around the same time as the Johns Hopkins patent application, Patrick Harran in collaboration with University of Texas also filed their invention for inhibiting GOAT. Their inhibitor design was based on tight binding but non-reactive mimic of putative transition state geometry of GOAT catalysis.39 They envisioned that their compounds would be able to bind GOAT in a manner to the tetrahedral oxyanion in 8 and the synthesized compounds will have a stable tetrahedral geometry seen in 9. The molecules also contained appropriate hydrophobic tail for GOAT specific octanoylation and functionality for stabilization of the enzyme/inhibitor complex. It was rationalized that replacement of the oxygen with carbon and selecting a poor leaving group for R3 will achieve the transition state mimetics involved in octanoyl/acyl transfer. The invention proposed compounds of the broad formula 10. Synthetic variation yielded compounds with GOAT inhibiting activity in the low micromolar range. Two compounds PH1147 (IC50 = 90 μM) and BK1114 (IC50 = 50 μM) which differed by a methyl group at the quaternary carbon showed promising GOAT inhibition (Fig. 4).
Fig. 4. Mechanistic insight of the catalytic domain and peptidomimetic GOAT inhibitors from Harran and co-workers.

Harran's group had an additional disclosure where they improved their earlier work to develop yet another series of potent and selective GOAT inhibitors with negligible cellular toxicity. The compounds in this invention were represented by the general formula 13 and many compounds bearing a thioamido-group which were tested showed GOAT inhibition in the low nanomolar to micromolar range.40 A thiosarcocine derived peptidomimetic was screened for GOAT-inhibiting activity using membrane fractions prepared from Sf9 cells infected with baculovirus encoding mouse GOAT as a source of acyltransferase. Compound 1 (14) designated as lead inhibitor inhibited GOAT in vitro in a dose-dependent manner (IC50 ∼30 nM). A similar truncated analog which was termed as negative control was inactive at a much higher concentration. Compound 1 was administered IP to mice (80 mg kg–1) showed that the circulating acyl ghrelin levels were undetectable within minutes of the inhibitor administration. The compound's physicochemical properties were deemed to be in the range for an orally bioavailable compound (%F = 10–15%).
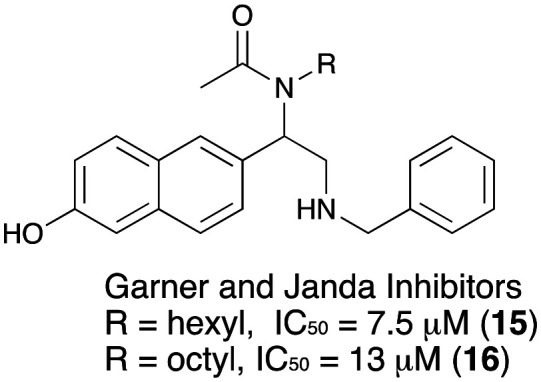
While the initial design of GOAT inhibitors were largely peptide mimetics, Janda et al. reported naphthyl glycine benzamide analogs active against GOAT in an ELISA based assay. Two promising compounds had a GOAT inhibitory IC50 of 7.5 μM and 13 μM.41,42 Further optimization of these compounds has not appeared (Fig. 5).
Fig. 5. Napthalene-based small-molecule GOAT inhibitors from Garner and Janda.
Takeda Pharmaceuticals have patented a class of aromatic/heterocycle-based GOAT inhibitors. They reported several compounds having general structure 17 that fully inhibit GOAT activity at 10 μM in vitro.43 Recently they have reported a detailed evaluation of two compounds: compound B, (4-chloro-6-{[2-methyl-6-(trifluoromethyl)pyridin-3-yl]methoxy}-1-benzothiophen-3-ylacetic acid) was synthesized and exhibited potent GOAT inhibition with oral bioavailability.30 The representative hit compound (compound A, 18) and lead compound (compound B, 19) showed reduced IC50 values at high concentrations of octanoyl-CoA. Meanwhile, neither compound caused a shift at high concentrations of des-acyl ghrelin. These results indicated that compounds possessed competitive inhibitory activity against octanoyl-CoA and noncompetitive inhibitory activity against des-acyl ghrelin. Takeda team successfully developed a homogeneous assay system and identified small-molecule inhibitors of GOAT that showed activity both in vitro and in vivo. Moreover, these two compounds decreased acyl ghrelin production in the stomach of mice after their oral administration. Compounds from Takeda has been currently licensed by Rhythm Pharmaceuticals and compound RM-853 (T-3525770, structure unknown) is undergoing clinical development (Fig. 6).
Fig. 6. Takeda GOAT inhibitors.

Hougland and co-workers have been involved in extensive research identifying new scaffolds that have activity against GOAT.33 In their early efforts, they identified and patented GOAT inhibitors using a triazole linkage to incorporate aromatic and alkyl substituents to mimic the natural octanoyl group attached to ghrelin.44 The series of “click-chemistry”-derived triazoles utilize the same pentapeptide skeleton screened by Yang et al.12 They demonstrate the enzyme's ability to tolerate modification of the octanoate side chain. Inhibitors of the general formula 20 include a triazole portion, an alkyl linker, and a hydrophobic aromatic group on a side chain. The hydrophobic aromatic group included various length alkyl linkers. Three GOAT inhibitors with linkers n = 1, 2 and 3 were synthesized and tested in vitro for activity using a fluorescence-based assay. The compound with a chain length n = 3 had better GOAT IC50 inhibitory activity of 0.7 μM (Fig. 7).
Fig. 7. ‘Click-chemistry’ utilization of triazole-containing GOAT inhibitors.

In a recent report of small molecule GOAT inhibitors from Hougland's group, a class of molecules derived from synthetic triterpenoids was found to inhibit the human GOAT (hGOAT) ortholog. A derivative of 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO-EA, 22) was identified as hGOAT inhibitor with IC50 value of 8 μM as assessed by a microsomal hGOAT activity assay.45 In another filing, Hougland and coworkers disclosed cyanosteroid compounds that efficiently inhibit GOAT.46 The compounds were based on a steroid scaffold (23) with α,β-unsaturated ketone in the A ring, which could also bear a cyano group. The most potent compound in this series was cyanoenone 24 (IC50 = 8 μM) which can covalently modify nucleophilic thiols in the binding pocket. Studies demonstrated that these cyanoenone molecules acted as covalent, reversible inhibitors of GOAT. The requirement of α,β-unsaturation and Michael acceptor in the A ring and the increased activity of α-cyanoenone suggest that the compound could block hGOAT activity by alkylating a nucleophilic cysteine residue involved in hGOAT catalysis.47 While the compounds helped shed light on the mechanistic determinants for GOAT catalysis, the druggable parameters of these compounds are unknown (Fig. 8).
Fig. 8. Utilization of steroidal scaffold to generate covalent GOAT inhibitors.

Recently, the same group has also filed invention disclosure for imaging agents that can bind to GOAT without binding to GHS-R1a that will allow the specific detection and imaging of GOAT in various tissues.48 The imaging agent shown in 25 consists of GOAT recognition element that is coupled via an amino acid linker to a chemical fluorescent moiety, radiolabel or a metal chelator to allow imaging as seen in 26. One of the compounds in this invention had a high selectivity against GOAT over GSH-R1a (6 nM vs. 100 μM). A fluorescently labeled version of this compound SulfoCy5-3 27, had an in vitro GOAT IC50 of 17 nM and it was demonstrated that this ligand selectively labeled PC3 prostate cancer cells with no binding to HEK293 cells (Fig. 9).
Fig. 9. GOAT inhibitor based fluorescent labels for cancer imaging.

Martinez Grau from Eli Lilly, Madrid disclosed a singular GOAT inhibitor 29 and its enantiomers in an initial filing.49 The compound had a hGOAT inhibitory activity IC50 =192 nM. Administration of the compound of example 2 (29) in the patent for 3 days decreased plasma acylated ghrelin (AG) by –1%, 5%, 45%, and 48%, and increased unacylated ghrelin (UAG) by 2.24, 2.82. 2.53, and 2.9-fold, respectively at 0.3, 1, 3, and 10 mg kg–1. Administration at 0.3, 1, 3, and 10 mg kg–1 resulted in 39, 54, 71 and 77% reduction respectively in AG to total ghrelin ratio when compared to the vehicle-treated control animals. Similarly, compound 66 (30) at 18 mg kg–1 showed 86% reduction in AG to total ghrelin ratio when compared to the vehicle-treated control animals. These results demonstrate that the compound suppressed AG production and elevates the UAG in circulation, similar to the GOAT knock-out mouse, in vivo. In a related filing trifluoromethyl pyrimidyl-substituted compounds were evaluated with different R groups.50 Compound 2 (31) inhibited purified GOAT enzyme activity in vitro with an IC50 of 69.5 nM. Administration of this compound at 0.1, 0.3, 1, 3, and 10 mg kg–1 resulted in 57, 62, 79, 82 and 82% reduction respectively in AG to total ghrelin ratio when compared to the vehicle-treated control animals. In yet another filing from the Eli Lilly team, with expansion of the analogs belonging to the formula 28, the claims were broadened and many compounds were investigated.51 Representative compounds 31–34, had promising IC50 values in hGOAT assays. Compounds were also investigated for acyl and des-acyl ghrelin level after administration in C57BL/6 for 3 days at different doses. Compound 71 (34) in the filing showed a 66% reduction in plasma acyl ghrelin levels after 3 mg kg–1 dose and a 2.9-fold increase in unacylated ghrelin levels. Sustained decrease in plasma acyl ghrelin levels were observed after 3 mg kg–1 dose (82% reduction) and a 2.8-fold increase in unacylated ghrelin levels. The results from Eli Lilly demonstrated that the GOAT inhibitors developed by them suppressed AG production and elevated unacylated ghrelin levels in circulation and supported the phenotype see in GOAT knock-out mice in vivo. Optimization of these inhibitors to improve potency and ADMET properties led to the identification of LY3073084 (structure unknown), a potent and selective GOAT inhibitor that demonstrated robust in vivo pharmacodynamic effects. More details on the efficacy of this compound have not yet appeared (Fig. 10).
Fig. 10. Pyrimidine-substituted GOAT inhibitors from Eli Lilly.

In the last few years there has been a series of patent filings from Boehringer Ingelheim, Germany on compounds having potent inhibitory activity on GOAT. In an initial filing compounds of the general formula 35 belonging to the oxadiazolopyridine scaffold were initially reported for use as GOAT inhibitors by Godbout et al.52 Synthesis and characterization of over 160 compounds were reported in this detailed filing. Pharmacological testing and GOAT activity of analogs in HEK293 cells were reported as IC50 values in this filing. Many of the compounds showed potent GOAT inhibition exemplified by compound 36 had an IC50 of 0.02 nM. In a related genus, benzyl-, (pyridine-3-yl)methyl- or (pyridine-4-yl)methyl substituted oxadiazolopyridine derivatives 37 were discovered as highly potent GOAT inhibitors.53 Many compounds (e.g.38) in this broad invention disclosure had sub-nanomolar to low picomolar inhibition for GOAT in HEK293 cells. In vivo effects, ADMET properties or other druggable parameters of the compounds in this series were not reported. Further, pyrazole and indazole-substituted oxadiazolopyridine derivatives of the general structure 39 were also disclosed to inhibit GOAT in the subnanomolar/picomolar range.54 In yet another filing from Boehringer Ingelheim, heterocyclyl-substituted oxadiazolopyridine derivatives of the general formula 41, were reported as having inhibitory activity on GOAT (Fig. 11).55
Fig. 11. Disclosure of broad-based scaffolds for GOAT inhibition from Boehringer Ingelheim.

Compounds with various substitution patterns were synthesized and tested for GOAT inhibition. Pharmacological testing revealed many compounds consistently exhibited sub-nanomolar GOAT inhibition (e.g.42). In a departure from oxadiazolopyridine scaffold, triazolopyrimidine derivatives of the formula 43 were also disclosed as GOAT inhibitors by the Boehringer Ingelheim team.56 In this broad set of claims, compounds with a wide-ranging substitution pattern were synthesized and tested for GOAT activity. Many compounds again exhibited sub-nanomolar IC50 for hGOAT inhibition in HEK293 cells. Compound 44 was disclosed to have an IC50 of 0.13 nM. In vivo effects of the potent compounds in this series has yet to be revealed.
Recent invention disclosure from a global GSK team revealed potent, substituted benzothiophenyl acetic acid compounds of the general formula 45 as GOAT inhibitors.57 In a relatively narrow set of compounds synthesized under the scope of invention, racemic compounds were separated by chiral prep-HPLC and it is understood that the S-enantiomers of the compounds exhibited potentially favorable GOAT inhibition over R-enantiomer. One compound designated as compound 1a (46) had an invitro IC50 value < 50 nM. This compound was evaluated in vivo in three preclinical species by assessing the level of reduction in circulating acyl ghrelin levels post-treatment. A significant reduction of acyl ghrelin levels was seen in mice (1 and 10 mg kg–1) and rats (10 mg kg–1) upon treatment with 46. A single dose PK/PD study was conducted in cynomolgus monkeys and a 10 mg kg–1 dose showed a 51% reduction in acyl ghrelin levels over the first 24 h after treatment. Acyl ghrelin levels were also investigated in a high fat-high carbohydrate (HFHC) mouse model in conjunction with rimonabant (CB1R antagonist) treatment. At the end of the seven day period, mice on compound 1a (46) showed a 28% reduction in acyl ghrelin levels and a statistically significantly elevated level of des-acyl ghrelin. Rimonabant alone caused an increase in des-acyl ghrelin levels but not to the extent seen with compound 1a treatment. This filing is one of the first reports of efficacy studies with a GOAT inhibitor in higher mammalian species (Fig. 12).
Fig. 12. GOAT inhibitor disclosure from Glaxo SmithKline.

Filings from China revealed preparation of various benzothiophene compounds for activity against GOAT. In a series of substituted filings Wang claimed substituted (2-((furan-2-yl)oxy)benzo[b]thiophen-6-yl isoxazole-5-carboxylate) (47–50) and as GOAT inhibitor for treatment of obesity and diabetes.58 The prepared compounds were reported to have good inhibitory activity against GOAT at 10 μM (Fig. 13).
Fig. 13. GOAT inhibitor structures revealed in patent filings from China.

Studies and invention disclosures have also appeared on methods for diagnosing and/or predicting bladder and colon cancer in a patient by quantifying levels of In1-ghrelin expression in a plasma sample isolated from an individual and comparing the levels obtained with the levels of In1-ghrelin (splicing variant of ghrelin) expression, GOAT and/or GHS-R1b in tissue samples in a reference sample.59 Patients who show In1-ghrelin expression levels that are significantly higher than the levels of the reference sample, have a higher risk of developing cancer. This invention also included small interfering RNA (siRNA) useful in a cancer treatment method.
Conclusions and future perspective
From 2010 to 2019, many publications on GOAT inhibitors have emerged. Many of these inhibitors have been disclosed in the patent literature, where detailed publications on these compounds are pending. The patents from the pharma cover varied classes of compounds. In absence of computational tools for structure-based drug design, efforts have been focused on screening strategies using enzymatic assays by various groups. Initial peptide-based inhibitors and pharmacological tools paved the way for larger small-molecule based drug discovery campaigns in this realm. Common structural features like pyrimidine core, substituted thiophene acetic acids and steroidal scaffolds were chosen as starting points to expand the chemical space and understanding of the structural biology of GOAT. While a non-experimental, structural model for GOAT catalytic domain mechanism has been postulated, experiment-based, ligand-bound structural determination either by X-ray or cryo-EM will elucidate the exact molecular determinants for developing novel inhibitors for this enzyme. Development of tighter binding and covalent inhibitors can further improve the understanding of transition-state mechanism of the catalytic domain of this enzyme. It is conceivable that the revelation of the physiological roles and endogenous substrates of the GOAT could lead to new therapeutic indications by inhibiting GOAT activity. Utility of GOAT inhibitors in clinic may be revealed as compounds start entering clinical trials for indications related to obesity and insulin resistance. The mutual specificity of GOAT, octanoate and key acylation localized in the periphery (rather than CNS) suggest that a small molecule inhibitor of GOAT would not need to penetrate the brain, and hence mechanism-based toxicity from its inhibition is limited.
Besides obesity and diabetes, it is conceivable that other promising therapeutic applications of GOAT inhibitors can include disorders related to food and drug intake particularly alcohol and other drugs of abuse. While GOAT attenuation is shown to be beneficial in non-alcoholic fatty liver disease (NAFLD),38 the role of GOAT if any, in liver fibrosis leading to hepatocellular carcinoma is unknown. With the widespread expression of GOAT, the role of GOAT–ghrelin axis in inflammatory and fibrotic disorders may become important. The role of GOAT as a diagnostic marker in prostate cancer and neuroendocrine tumor has been documented and patented. However the detailed understanding of its effects in other cancer and tumor microenvironment remains unknown. Further the cross-talk between LEAP2 and GOAT can be significant in mediating ghrelin actions.60,61 In view of the emerging patent landscape Elli Lilly, Boehringer Ingelheim and GSK, have promising compounds. Significant work by Cole36 and Hougland45,47 have advanced the understanding of basic mechanistic aspects of the GOAT enzyme. Despite the emergence of these GOAT inhibitors, challenges remain related to efficacy, potency, PK/PD and other druggable properties that underscore the feasibility for development as a therapeutic. Thus, inhibition of GOAT could yet prove to be a valuable target because of its insofar unique action specific to ghrelin. The discovery of GOAT thus offers a new a pathway for exploiting the ghrelin–GOAT axis in managing obesity, diabetes and its related metabolic complications. The scope for the generation of inhibitors selective to GOAT and evaluating its potential in clinic will certainly form the crux of research in many academic and pharmaceutical research labs in the near future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
The authors would like to acknowledge the National Institute on Alcohol Abuse and Alcoholism NIAAA Intramural Research Program at the National Institutes of Health for funding support.
Biographies

Malliga Iyer
Malliga Iyer leads the Medicinal Chemistry Core program at the National Institute on Alcohol Abuse and Alcoholism (NIAAA/NIH). She obtained her MSc. in Organic Chemistry from the Indian Institute of Technology in Mumbai, India and her PhD in Chemistry from the Pennsylvania State University. She completed her post-doctoral training with Dr. Kenner Rice at National Institute on Drug Abuse (NIDA) developing opioid therapeutics for treating pain and addiction. She subsequently joined the Laboratory of Physiologic Studies in NIAAA headed by Dr. George Kunos. Her research interests include polypharmacology-based drug design and discovery for fibrosis and metabolic disorders.

Casey Wood
Casey Wood received her Bachelor of Science (2019) in Biomedical Sciences from the University of South Florida-Tampa where she conducted research at the Florida Center for Drug Discovery and Innovation under the direction of James Leahy PhD. Her research involved development of a 5-aminolevullinic acid synthase inhibitor for the treatment of porphyria. She is currently a Post-Baccalaureate IRTA fellow at the National Institutes of Health, National Institute of Alcohol Abuse and Alcoholism working in the Lab of Physiologic Studies – Medicinal Chemistry Core.

George Kunos
George Kunos received his MD degree from Semmelweis University in Budapest and a PhD in pharmacology from McGill University in Montreal. He stayed and rose through the ranks at McGill, then served as Professor and Chair of the Department of Pharmacology and Toxicology at Virginia Commonwealth University before joining the NIH in 2000 in his current position as Scientific Director of the National Institute on Alcohol Abuse and Alcoholism. His research over the last 25 years has focused on the biology of the endocannabinoid system, with special emphasis on its role in regulating metabolism, cardiovascular functions and addictive behavior.
References
- Farag Y. M. K., Gaballa M. R. Nephrol., Dial., Transplant. 2011;26:28–35. doi: 10.1093/ndt/gfq576. [DOI] [PubMed] [Google Scholar]
- Kalra S. JPMA, J. Pak. Med. Assoc. 2013;63:532–534. [PubMed] [Google Scholar]
- Abid A., Ahmad S., Waheed A. Clin. Med. Insights: Endocrinol. Diabetes. 2016;9:19–22. doi: 10.4137/CMED.S38247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M., Hosoda H., Date Y., Nakazato M., Matsuo H., Kangawa K. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- Pradhan G., Samson S. L., Sun Y. Curr. Opin. Clin. Nutr. Metab. Care. 2013;16:619–624. doi: 10.1097/MCO.0b013e328365b9be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi S., Sato T., Kangawa K., Nakazato M. Cell Metab. 2018;27:786–804. doi: 10.1016/j.cmet.2018.02.008. [DOI] [PubMed] [Google Scholar]
- Taylor M. S., Hwang Y., Hsiao P.-Y., Boeke J. D., Cole P. A. Methods Enzymol. 2012;514:205–228. doi: 10.1016/B978-0-12-381272-8.00013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X., Yang H., Bednarek M. A., Galon-Tilleman H., Chen P., Chen M., Lichtman J. S., Wang Y., Dalmas O., Yin Y., Tian H., Jermutus L., Grimsby J., Rondinone C. M., Konkar A., Kaplan D. D. Cell Metab. 2018;27:461–469.e6. doi: 10.1016/j.cmet.2017.10.016. [DOI] [PubMed] [Google Scholar]
- Schalla M. A., Stengel A. Nat. Rev. Gastroenterol. Hepatol. 2019;16:711–712. doi: 10.1038/s41575-019-0224-9. [DOI] [PubMed] [Google Scholar]
- Yang J., Brown M. S., Liang G., Grishin N. V., Goldstein J. L. Cell. 2008;132:387–396. doi: 10.1016/j.cell.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Gutierrez J. A., Solenberg P. J., Perkins D. R., Willency J. A., Knierman M. D., Jin Z., Witcher D. R., Luo S., Onyia J. E., Hale J. E. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6320–6325. doi: 10.1073/pnas.0800708105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Zhao T.-J., Goldstein J. L., Brown M. S. Proc. Natl. Acad. Sci. U. S. A. 2008;105:10750–10755. doi: 10.1073/pnas.0805353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campaña M. B., Irudayanathan F. J., Davis T. R., McGovern-Gooch K. R., Loftus R., Ashkar M., Escoffery N., Navarro M., Sieburg M. A., Nangia S., Hougland J. L. J. Biol. Chem. 2019;294:14166–14174. doi: 10.1074/jbc.AC119.009749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. T., Kola B., Grossman A., Korbonits M. Endocr. J. 2011;58:707–710. doi: 10.1507/endocrj.k11e-117. [DOI] [PubMed] [Google Scholar]
- Castañeda T. R., Tong J., Datta R., Culler M., Tschöp M. H. Front. Neuroendocrinol. 2010;31:44–60. doi: 10.1016/j.yfrne.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Carlini V. P., Monzón M. E., Varas M. M., Cragnolini A. B., Schiöth H. B., Scimonelli T. N., de Barioglio S. R. Biochem. Biophys. Res. Commun. 2002;299:739–743. doi: 10.1016/s0006-291x(02)02740-7. [DOI] [PubMed] [Google Scholar]
- Diano S., Farr S. A., Benoit S. C., McNay E. C., da Silva I., Horvath B., Gaskin F. S., Nonaka N., Jaeger L. B., Banks W. A., Morley J. E., Pinto S., Sherwin R. S., Xu L., Yamada K. A., Sleeman M. W., Tschöp M. H., Horvath T. L. Nat. Neurosci. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- Dornonville de la Cour C., Lindström E., Norlén P., Håkanson R. Regul. Pept. 2004;120:23–32. doi: 10.1016/j.regpep.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Haqq A. M., Grambow S. C., Muehlbauer M., Newgard C. B., Svetkey L. P., Carrel A. L., Yanovski J. A., Purnell J. Q., Freemark M. J. Clin. Endocrinol. Metab. 2008;69:911–920. doi: 10.1111/j.1365-2265.2008.03385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallar L. J., Farokhnia M., Tunstall B. J., Vendruscolo L. F., Leggio L. Int. Rev. Neurobiol. 2017;136:89–119. doi: 10.1016/bs.irn.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Farokhnia M., Grodin E. N., Lee M. R., Oot E. N., Blackburn A. N., Stangl B. L., Schwandt M. L., Farinelli L. A., Momenan R., Ramchandani V. A., Leggio L. Mol. Psychiatry. 2018;23:2029–2038. doi: 10.1038/mp.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski G., Cinar R., Coffey N. J., Liu J., Jourdan T., Mukhopadhyay B., Chedester L., Liu Z., Osei-Hyiaman D., Iyer M. R., Park J. K., Smith R. G., Iwakura H., Kunos G. Cell Metab. 2019;29:1320–1333.e8. doi: 10.1016/j.cmet.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillory B., Jawanmardi N., Iakova P., Anderson B., Zang P., Timchenko N. A., Garcia J. M. Aging Cell. 2018;17:e12688. doi: 10.1111/acel.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T.-J., Liang G., Li R. L., Xie X., Sleeman M. W., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., Goldstein J. L., Brown M. S. Proc. Natl. Acad. Sci. U. S. A. 2010;107:7467–7472. doi: 10.1073/pnas.1002271107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouno T., Akiyama N., Ito T., Okuda T., Nanchi I., Notoya M., Oka S., Yukioka H. J. Endocrinol. 2016;228:115–125. doi: 10.1530/JOE-15-0330. [DOI] [PubMed] [Google Scholar]
- Gómez-Gómez E., Jiménez-Vacas J. M., Carrasco-Valiente J., Herrero-Aguayo V., Blanca-Pedregosa A. M., León-González A. J., Valero-Rosa J., Fernández-Rueda J. L., González-Serrano T., López-Miranda J., Gahete M. D., Castaño J. P., Requena-Tapia M. J., Luque R. M. J. Cell. Mol. Med. 2018;22:5688–5697. doi: 10.1111/jcmm.13845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D., Wang Z., Merrikh C. N., Lang K. S., Lu P., Li X., Merrikh H., Rao Z., Xu W. Nature. 2018;562:286–290. doi: 10.1038/s41586-018-0568-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K. Trends Biochem. Sci. 2000;25:111–112. doi: 10.1016/s0968-0004(99)01539-x. [DOI] [PubMed] [Google Scholar]
- Sieburg M. A., Cleverdon E. R., Hougland J. L. Methods Mol. Biol. 2019;2009:227–241. doi: 10.1007/978-1-4939-9532-5_18. [DOI] [PubMed] [Google Scholar]
- Yoneyama-Hirozane M., Deguchi K., Hirakawa T., Ishii T., Odani T., Matsui J., Nakano Y., Imahashi K., Takakura N., Chisaki I., Takekawa S., Sakamoto J. SLAS Discovery. 2018;23:154–163. doi: 10.1177/2472555217727097. [DOI] [PubMed] [Google Scholar]
- Garner A. L., Janda K. D. Angew. Chem., Int. Ed. 2010;49:9630–9634. doi: 10.1002/anie.201003387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling J. E., Prybolsky E. P., Sieburg M., Hougland J. L. Anal. Biochem. 2013;437:68–76. doi: 10.1016/j.ab.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Darling J. E., Zhao F., Loftus R. J., Patton L. M., Gibbs R. A., Hougland J. L. Biochemistry. 2015;54:1100–1110. doi: 10.1021/bi5010359. [DOI] [PubMed] [Google Scholar]
- The Johns Hopkins University, WO2010039461A2, 2010.
- The Johns Hopkins University, US2011257086A1, 2011.
- Barnett B. P., Hwang Y., Taylor M. S., Kirchner H., Pfluger P. T., Bernard V., Lin Y., Bowers E. M., Mukherjee C., Song W.-J., Longo P. A., Leahy D. J., Hussain M. A., Tschöp M. H., Boeke J. D., Cole P. A. Science. 2010;330:1689–1692. doi: 10.1126/science.1196154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuffel P., Wang L., Prinz P., Goebel-Stengel M., Scharner S., Kobelt P., Hofmann T., Rose M., Klapp B. F., Reeve J. R., Stengel A. J. Physiol. Pharmacol. 2015;66:493–503. [PubMed] [Google Scholar]
- Zhang S., Mao Y., Fan X. Drug Des., Dev. Ther. 2018;12:873–885. doi: 10.2147/DDDT.S158985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- University of Texas, Board of Regents, US20100086955A1, 2010.
- The Regents of the University of California, US20170275249A1, 2017.
- Garner A. L., Janda K. D. Chem. Commun. 2011;47:7512–7514. doi: 10.1039/c1cc11817j. [DOI] [PubMed] [Google Scholar]
- Garner A. L., Janda K. D. ChemBioChem. 2011;12:523–525. doi: 10.1002/cbic.201000777. [DOI] [PubMed] [Google Scholar]
- Takeda Pharmaceutical Co [JP/JP], Aromatic Ring Compound, WO2013125732Al, 2013.
- Syracuse University, US2015018520A1, 2015.
- McGovern-Gooch K. R., Mahajani N. S., Garagozzo A., Schramm A. J., Hannah L. G., Sieburg M. A., Chisholm J. D., Hougland J. L. Biochemistry. 2017;56:919–931. doi: 10.1021/acs.biochem.6b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougland J. L. and Chisholm J. D., WO2018035079A1, 2018.
- Cleverdon E. R., Davis T. R., Hougland J. L. Bioorg. Chem. 2018;79:98–106. doi: 10.1016/j.bioorg.2018.04.009. [DOI] [PubMed] [Google Scholar]
- Syracuse University, US2020102365A1, 2020.
- Eli Lilly and Co., US20150133474A1, 2015.
- Eli Lilly and Co., WO2016168222A1, 2016.
- Eli Lilly and Co., WO2016168225A1, 2016.
- Boehringer Ingelheim International GmbH, WO2018024653A1, 2018.
- Boehringer Ingelheim International GmbH, WO2019149657A1, 2019.
- Boehringer Ingelheim Int., WO2019149658A1, 2019.
- Boehringer Ingelheim Int., WO2019149659A1, 2019.
- Boehringer Ingelheim Int., WO2019149660A1, 2019.
- GlaxoSmithKline Ip Dev Ltd., WO2019149959A1, 2019.
- (a) Peop. Rep. China, CN108610337A, 2018.; (b) Peop. Rep. China, CN108516972A, 2018.
- Universidad De Córdoba, Use of In1-Ghrelin for the Diagnosis of Prostate Cancer, EP3557258A1, 2019.
- Al-Massadi O., Müller T., Tschöp M., Diéguez C., Nogueiras R. Trends Pharmacol. Sci. 2018;39:685–694. doi: 10.1016/j.tips.2018.06.004. [DOI] [PubMed] [Google Scholar]
- Abizaid A., Hougland J. L. Trends Endocrinol. Metab. 2020;31:107–117. doi: 10.1016/j.tem.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]