

 Thirty-five macamide analogues were synthesised by modifying the initial molecular structure.
Thirty-five macamide analogues were synthesised by modifying the initial molecular structure.
Abstract
Thirty-five macamide analogues were synthesised by modifying the initial molecular structure. The resulting structures were confirmed using NMR and MS. Cytotoxicity and the anti-inflammatory activity of these synthetic macamides were evaluated in the THP-1 cell line. Preliminary biological evaluation indicated that most of these synthetic macamides did not present cytotoxicity (MTT assay) in the tested cell line with respect to the control (actinomycin D). Regarding the anti-inflammatory activity, several analogues had a greater potential for inhibition of TNF-α than natural macamides. Synthetic macamide 4a was the most active (IC50 = 0.009 ± 0.001 μM) compared to the C87 (control). Through looking at the link between the chemical structure and the activity, our study proves that changes made to natural macamides at the level of the alkyl chain, the benzyl position, the amide bond, and the addition of two methyl groups to the aromatic ring (meta position) lead us to obtaining new macamides with greater anti-inflammatory activity.
1. Introduction
Our immune system is an incredible network of cells that protects us from all kinds of diseases. However, sometimes the system can be faulty and misinterpret the signals, causing our immune system to not recognise its own body and start attacking itself.1 The functioning of the immune system is regulated by the activity of pro and anti-inflammatory mediators. The imbalance of any of these mediators can lead to the development of increased susceptibility to infections, autoimmune diseases, and chronic inflammatory conditions.2 Currently, therapeutic investigations have focused on suppressing the production of pro-inflammatory mediators and on inhibiting the activation of the immune response, using modulation of TNF-α, NF-kB, STAT3 and IFN-γ signalling pathways.3
In particular, the use of inhibitors against the production and biological activity of the tumour necrosis factor alpha (TNF-α) has been widely described and discussed.4 This is because this factor plays an important role in inflammatory disorders related to diseases with various etymologies. These diseases can come from an infectious origin (meningitis, tuberculosis, acquired immunodeficiency syndrome), a tumour (prostate cancer, lung cancer, breast cancer) or have an autoimmune origin (inflammatory bowel disease, rheumatoid arthritis, lupus).5 Several drugs (e.g. dexamethasone) commonly used as immune-suppressants have been used to modulate the TNF-α activity, although their effects are associated with considerable toxicity.6
In the search for new compounds that involve the inhibition of TNF-α and its activity in cells, while presenting less toxicity, with greater potency and, thus, offering better clinical alternatives, our research suggests a series of chemical modifications to natural macamides which have been reported as having anti-inflammatory potential (TNF-α inhibitors).7
Macamides are secondary metabolites of the species Lepidium meyenii “Maca”8 and Tropaeolum tuberosum “Mashua”9 that grow exposed to extreme climatic conditions (poor soils with a slightly acidic pH between 5–6 and exposure to ultraviolet radiation at altitudes of 3.800 meters above sea level), producing this type of compounds considered as biomarkers of these species. Chemically, macamides consist of a residue of a benzylamine (product of the hydrolysis of glucosinolates by the enzyme myrosinase) and a fatty acid residue (product of the hydrolysis of membrane lipids).10
Recent pharmacological studies have shown that macamides have also other activities such as antioxidants, analgesics and antidepressants;11 anti-tuberculosis;12 anti-cholesterolemics;13 antifibrogenics;14 anticonvulsants;15 neuroprotectives16 and anticancer,17 being interesting molecules for chemical studies due to their wide pharmacological spectrum use.
The objective of the present work is to conclude on how the anti-inflammatory potential of macamides can be improved through the use of synthesised analogues. For this study, we first determined the cytotoxicity of the synthetic macamides, as their effect may hide anti-inflammatory activity. Subsequently, we evaluated their inhibitory activity on TNF-α in the THP-1 cell line.
2. Results and discussion
In order to improve the pharmacological profile of macamides as potential anti-inflammatory agents (TNF-α inhibition), we proceeded to carry out a chemical synthesis of structural analogues, by means of various modifications in the positions considered of greatest importance (Fig. 1).
Fig. 1. General structure of macamide and positions explored on this study.

The synthetic pathway was performed using an amidation reaction (modification of the Steglich reaction18,19) between the corresponding carboxylic acid and the benzylamine with N,N′-dicyclohexylcarbodiimide (DCC), as shown in Scheme 1.
Scheme 1. Synthesis of macamide derivatives by means of the amidation reaction.

Concerning the alkyl chain, it was observed that its elongation (derivatives 1a → 1d) does not generate a change in cytotoxicity (MTT) in the THP-1 cell line. However, synthetic macamide 1a (four-carbon alkyl chain) did not show a relevant inhibition on TNF-α, it exhibited the same value as the free carboxylic acid (oleic acid). This effect is due to the increase in the number of carbon atoms present in the chain, leading to an increase in the biological activity.20 In this sense, derivative 1d showed TNF-α inhibition with an IC50 of 16.6 ± 1.59 μM. Analysing the n-octanol/water partition coefficients (clog P) of derivatives 1a → 1d, a relationship between lipophilicity and biological activity was observed, and this is justified by the fact that increases in lipophilicity lead to an improvement in biological activity (Table 1). Free oleic acid was tested as negative control to determine if it was responsible for the inhibitory activity. Yet, we saw that free carboxylic acid did not produce significant activity on the target due to its low liposolubility (clog P = 3.47) compared to derivative 1d (clog P = 7.68).
Table 1. Influence of the length of the alkyl chain, degree of unsaturation in the alkyl chain and structural homology of the synthetic macamides on TNF-α.
| No. | Derivatives | Viability (CC50 μM) THP-1 cells | TNF-α inhibition (IC50 μM) THP-1 cells | clog P a |
| Oleic acid |
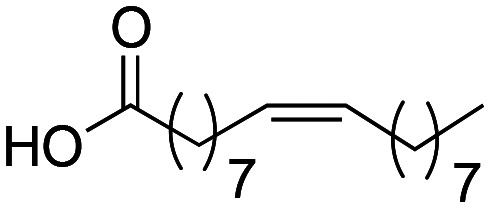
|
100 ± 1.99 | 79.0 ± 1.78 | 3.47 |
| 1a |
|
100 ± 1.71 | 78.4 ± 1.19 | 1.70 |
| 1b |
|
100 ± 1.66 | 58.7 ± 1.61 | 4.01 |
| 1c |
|
100 ± 1.98 | 43.1 ± 1.77 | 6.30 |
| 1d |
|
100 ± 1.83 | 16.6 ± 1.59 | 7.68 |
| 1e |
|
97.3 ± 1.18 | 15.2 ± 1.51 | 8.31 |
| 1f |
|
98.4 ± 1.43 | 14.2 ± 1.47 | 7.96 |
| 1g |
|
97.8 ± 1.22 | 21.0 ± 1.45 | 7.82 |
| 1h |
|
88.15 ± 1.20 | 15.7 ± 1.69 | 8.28 |
| 1i |
|
80.3 ± 1.31 | 8.3 ± 0.49 | 8.60 |
aValues of clog P were calculated by DataWarrior.27
This result can be justified by the lipophilicity effect, a physicochemical parameter that determines the ability of a substance to cross the biological membrane.21,22 Therefore, molecules with a longer chain length can more easily cross the cell barrier by diffusion.
After confirming that the aliphatic chain of C18 is the most suitable, the next step was to study the influence of the double bond. The oleic and linoleic fatty acids were used as precursors in order to obtain natural macamides23 through an amidation reaction.
The results reveal that the presence of double bonds improves TNF-α inhibition, due to the increased lipophilic capacity of derivatives 1e and 1f with respect to derivative 1d.24 The difference between derivatives 1e and 1f was not statistically significant (nsp > 0.999) (Table 1). Consequently, it was decided to use derivative 1f for subsequent modifications, since this macamide is found in a greater proportion in nature25 and because it showed considerable biological activity (14.2 ± 1.47 μM).
After the promising results, it was decided to continue the pharmacomodulation study with the synthesis of homologues (through the addition or subtraction of methylene groups) on the benzylic position. We started from (9Z,12Z)-octadeca-9,12-dienoyl chloride and used different amines, from aniline (n = 0) to 3-phenyl-1-propylamine (n = 3), obtaining the corresponding synthetic macamides (1g → 1i) (Scheme 2).
Scheme 2. Synthesis of 1g → 1i macamide derivatives.

Analysing the results, it was observed that as the number of methylenes increased, their lipophilic capacity increased and, in turn, their biological activity was higher. In the case of derivative 1h, the variation with respect to derivative 1f was not statistically significant (nsp > 0.999), but, after the introduction of a second methylene group (1i), the effect was more pronounced (IC50 = 8.3 ± 0.49 μM). However, derivative 1i showed a cytotoxic effect (CC50 = 80.3 ± 1.31 μM) on THP-1 cells and the difference was statistically significant with respect to derivative 1f (***p < 0.001) (Table 1).
These data can be justified based on electronic distribution, in which nitrogen, being a strong activator, modifies the electronic distribution of the aromatic ring and, consequently, its interaction26 as observed when comparing derivatives 1f and 1i.
The next phase of the investigation of the structure–activity relationship was to study the influence of substituents on the benzylic position. Due to the decrease in yield when the alkyl chain length is increased using an amidation reaction, (9Z,12Z)-octadeca-9,12-dienoyl chloride was used as a synthesis precursor instead of oleic acid, following Scheme 3.
Scheme 3. Synthesis of 2a → 2e macamide derivatives.

To understand how this position affects activity, the following substitutions in the benzyl position have been tested: hydrazide residue (2a), methyl residue (2b), ethyl residue (2c), hydroxyethyl residue (2d) and gem-dimethyl residue (2e). The data revealed that the substitution at the benzylic position affected biological activity. Synthetic macamide 2a, which presents a hydrazide bond instead of amide, improved biological activity, exhibiting an IC50 of 8.9 ± 0.98 μM. Also, substitution in the benzylic position by a methyl group (2b), significantly improved the activity (IC50 of 5.2 ± 0.81 μM). This result is due to the greater lipophilic capacity of derivative 2b (clog P = 8.20) compared to derivative 2a (clog P = 7.40) (Table 2).
Table 2. Influence of the substitution in the benzylic position and modified analogues at the amide bond of the synthetic macamides on TNF-α.
| No. | Derivatives | Viability (CC50 μM) THP-1 cells | TNF-α inhibition (IC50 μM) THP-1 cells | clog P a |
| 2a |
|
98.1 ± 1.84 | 8.9 ± 0.98 | 7.40 |
| 2b |
|
95.8 ± 1.97 | 5.2 ± 0.81 | 8.20 |
| 2c |
|
96.5 ± 1.79 | 19.7 ± 1.94 | 8.29 |
| 2d |
|
98.0 ± 1.46 | 56.8 ± 1.75 | 7.29 |
| 2e |
|
97.3 ± 1.94 | 13.1 ± 1.74 | 8.33 |
| 2f |
|
89.1 ± 1.23 | 23.8 ± 1.44 | 8.11 |
| 2g |
|
95.4 ± 1.88 | 6.4 ± 0.91 | 8.10 |
| 2h |
|
84.3 ± 1.12 | 5.3 ± 0.75 | 8.17 |
| 2i |
|
98.7 ± 1.97 | 1.6 ± 0.07 | 8.30 |
| 2j |
|
95.0 ± 1.82 | 0.97 ± 0.02 | 8.31 |
aValues of clog P were calculated by DataWarrior.27
In the case of derivatives 2c and 2e, although they have a greater lipophilic capacity than derivative 2b, they showed less biological activity. This effect can be justified by the fact that its substituents have a higher volume that prevents correct interaction with the target. Regarding the low biological activity of the derivative 2d, this result is due to its lower lipophilic capacity with respect to derivative 2a.
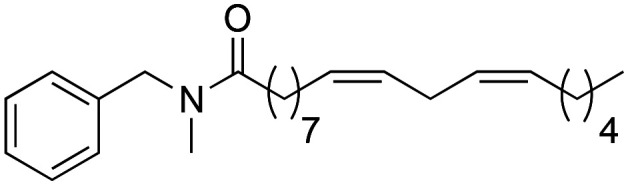
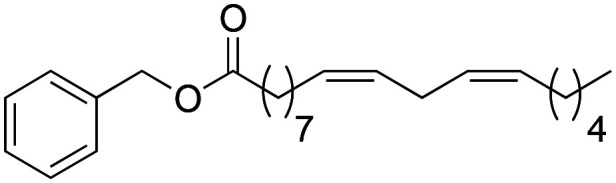
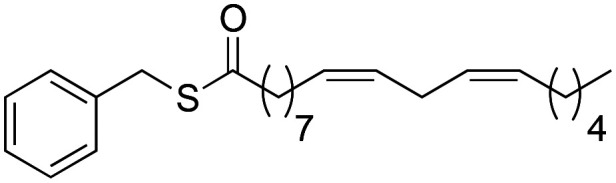
Once the structure–activity study on the benzylic position was completed, we proceeded to analyse how the amide bond influences biological activity. At this point, changes were made to the amide bond. For this, the nitrogen-methylated derivative (2f), the benzyl ester (2g) and the benzyl thioester (2h) were synthesised (Scheme 4).
Scheme 4. Synthesis of 2f → 2h macamide derivatives.

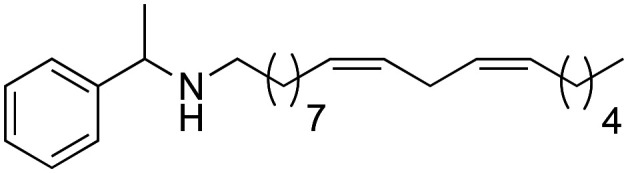
The synthetic macamide 2i was obtained reducing derivative 2b with LiAlH4 in a 90% yield (Scheme 5).
Scheme 5. Synthesis of 2i macamide derivative.

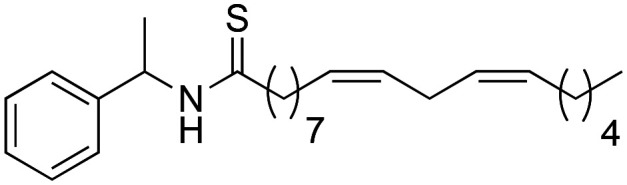
The synthetic macamide 2j was quantitatively synthesised from the reaction of the derivative 2b and Lawesson's reagent (Scheme 6).
Scheme 6. Synthesis of 2j macamide derivative.

The data revealed that the methylated nitrogen derivative 2f showed a lower biological activity (IC50 of 23.8 ± 1.44 μM) compared to derivative 1f (IC50 of 14.2 ± 1.47 μM). This effect is due to the absence of hydrogen in the amide bond that allows the macamide to form a hydrogen bond with the target. Similarly, it was observed that derivative 2f did not show cytotoxicity (≥90%).
Through the substitution of N–H in the amide bond by other heteroatoms such as oxygen and sulphur, derivatives 2g and 2h had a favourable effect on the inhibition of TNF-α, exhibiting IC50 values of 6.4 ± 0.91 and 5.3 ± 0.75 μM similar to those of derivative 2b (IC50 = 5.2 ± 0.81 μM), yet the difference was not statistically significant between them (nsp > 0.999). Likewise, the similarity between these derivatives with respect to their activity is due to their lipophilic capacity in the range of 8.10–8.20 (Table 2).
To observe the influence of the amide bond on the biological activity, the carbonyl group of derivative 2b was modified obtaining the synthetic macamides (2j) and (2i). In both cases, there was an increase in TNF-α inhibition, with the value of derivative 2j standing out (IC50 = 0.97 ± 0.02 μM). These results denote that alteration in the amide bond is key to biological activity. Likewise, it was observed that the increase in the biological activity of derivatives 2j and 2i was due to their greater lipophilic capacity with respect to derivative 2b (Table 2).
To end the first step of our study on the structure–activity relationship, we investigated the influence of the aromatic ring on the inhibition of TNF-α. Changes were made to the position of the aromatic ring. For this, it was investigated if its presence was influential and how and if the introduction of substituents was relevant. In this sense, two types of derivatives were prepared: those in which the aromatic ring was replaced by alkyl groups (3a → 3c), and those in which the aromatic ring had different types of substituents (3d → 3j). In all cases, the synthetic methodology followed Scheme 7.
Scheme 7. Synthesis of 3a → 3j macamide derivatives.

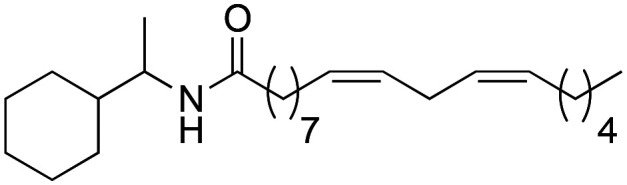
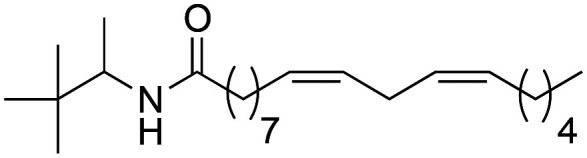
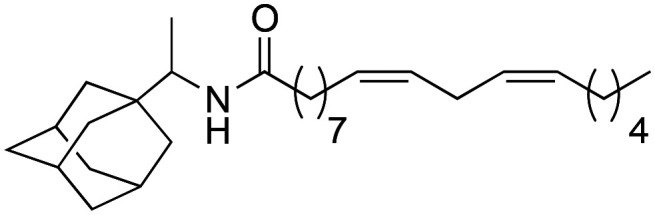
As it can be seen in Table 3, the changes made to the aromatic ring have a very favourable effect on the activity. When the benzene ring is replaced by a cyclohexyl group (3a), a tert-butyl group (3b), or an adamantyl group (3c), an improvement in biological activity occurs, showing IC50 values of 0.71 ± 0.03, 1.00 ± 0.03 and 0.78 ± 0.02 μM, respectively, with respect to derivative 2b (5.2 ± 0.81 μM). Likewise, derivatives 3a and 3c showed a greater lipophilic capacity with respect to derivative 2b, determining their greater biological activity. These data demonstrate that aromaticity is not a key effect for activity.
Table 3. Influence of the aromatic ring and the position of methyl on the aromatic ring of the synthetic macamides on TNF-α.
| No. | Derivatives | Viability (CC50 μM) THP-1 cells | TNF-α inhibition (IC50 μM) THP-1 cells | clog P a |
| 3a |
|
93.2 ± 1.76 | 0.71 ± 0.03 | 8.64 |
| 3b |
|
94.7 ± 1.25 | 1.00 ± 0.03 | 8.07 |
| 3c |
|
90.9 ± 1.22 | 0.78 ± 0.02 | 8.45 |
| 3d |
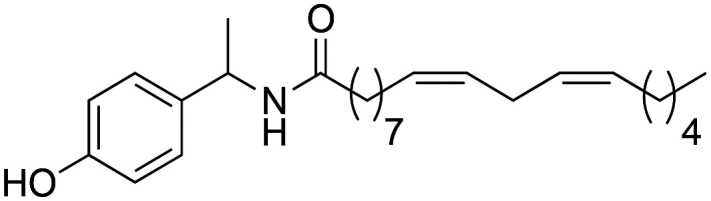
|
93.5 ± 1.58 | 1.84 ± 0.03 | 8.00 |
| 3e |
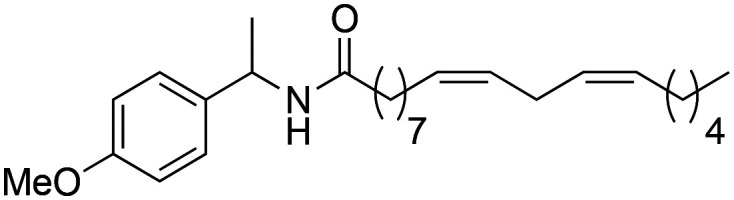
|
93.6 ± 1.53 | 1.71 ± 0.03 | 8.14 |
| 3f |
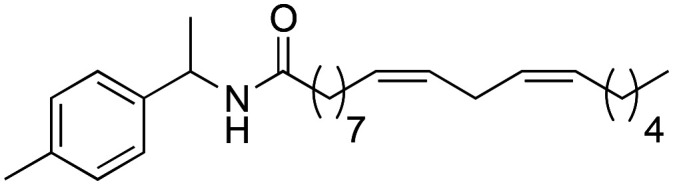
|
95.0 ± 1.98 | 0.62 ± 0.03 | 8.31 |
| 3g |
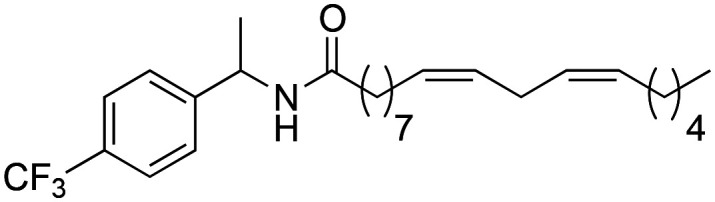
|
82.3 ± 1.73 | 1.52 ± 0.04 | 7.99 |
| 3h |
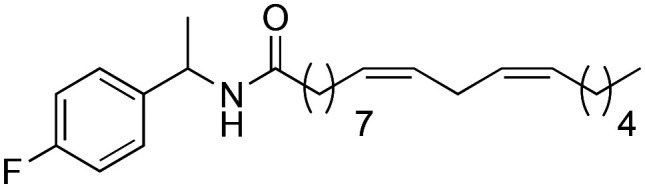
|
82.5 ± 1.86 | 0.78 ± 0.03 | 8.04 |
| 3i |
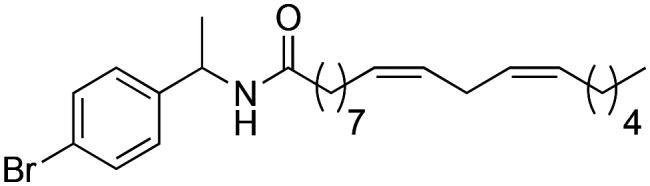
|
81.4 ± 1.80 | 0.74 ± 0.03 | 8.37 |
| 3j |
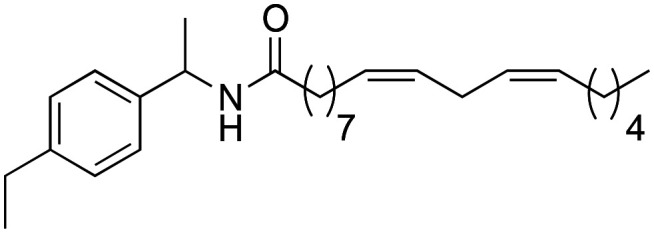
|
89.8 ± 1.30 | 1.21 ± 0.04 | 8.52 |
| 3k |
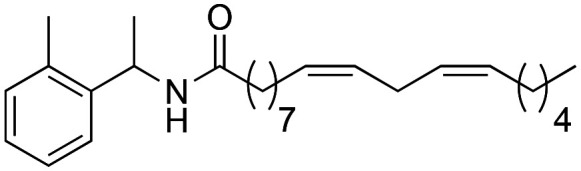
|
92.5 ± 1.64 | 0.54 ± 0.03 | 8.20 |
| 3l |
|
91.7 ± 1.19 | 0.33 ± 0.02 | 8.31 |
| 3m |
|
93.2 ± 1.53 | 0.08 ± 0.001 | 8.50 |
aValues of clog P were calculated by DataWarrior.27
Subsequently, it was decided to introduce electron donating groups (EDG) and electron withdrawing groups (EWG) in the aromatic ring to observe its effect on the biological activity. Although all substitutions improved activity with respect to derivative 2b, the trend shows that substitutions with strong EDG and EWG28 (3d and 3j) generate worse results with respect to derivatives that have weak EDG and EWG29,30 (3f, 3h and 3i). This effect is due to the greater lipophilic capacity of these derivatives (3f = 8.14; 3h = 8.04 and 3i = 8.37) (Table 3).
Regarding the cytotoxicity of this series of derivatives, the synthetic macamides 3g, 3h and 3i showed a CC50 of 82.3 ± 1.73; 82.5 ± 1.86 and 81.4 ± 1.80 μM, being cytotoxic (≥90%) on THP-1 cells. In the case of derivative 3j, its CC50 of 89.8 ± 1.30 μM was not statistically significant (nsp > 0.999) with respect to the negative control (untreated cells).
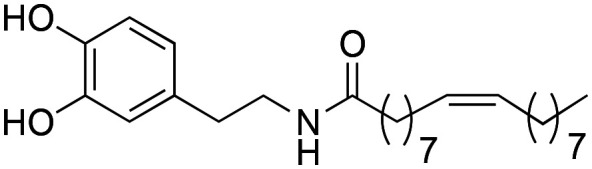
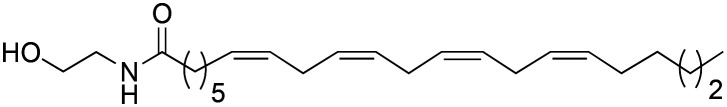
Moreover, analysing previous works, we observed that the natural macamide N-oleoyldopamine (IC50 = 3.54 ± 0.02 μM) showed a lower activity with respect to the natural macamide N-(2-hydroxyethyl)-7Z,10Z,13Z,16Z-docosatetraenamide (IC50 = 1.77 ± 0.07 μM).9 This effect is due to the fact that the second macamide showed a greater length of the alkyl chain with a greater number of double bonds. In our work, synthetic macamide 3d is an improved derivative of natural macamide N-oleoyldopamine. From a chemical point of view, the increase in the activity of the 3d derivative with respect to N-oleoyldopamine is due to the presence of an additional double bond, decrease of methylene groups in the benzyl position and addition of a methyl residue in the benzyl position.
The substituent that exhibited the best biological activity was derivative 3f, which has a weak EDG (methyl residue) at the para position of the aromatic ring, with an IC50 of 0.62 ± 0.03 μM. Based on this result, it was verified how the introduction of an ethyl group in the para position (3j) affects the activity, and it was observed that the introduction of one more carbon atom generates a decrease in biological activity (IC50 of 1.21 ± 0.04 μM).
In view of the data, it was decided to analyse how the position of methyl on the aromatic ring influences biological activity. The variation of the position within the ring causes a considerable improvement in the biological activity; this effect is especially relevant when the substitution is made in the meta position (3l), where the corresponding amide had an IC50 of 0.33 ± 0.02 μM. Based on this value, it was decided to study the di-substitution at the meta position, obtaining the best derivative substituted in the aromatic ring (3m), presenting an IC50 of 0.08 ± 0.001 μM. These data demonstrate the strong dependence between the geometry of the synthetic macamide and the biological activity, resulting in the modification on the aromatic ring, showing that this is the position of the most influential pharmacophore (Table 3).
Accounting for these results, it was decided to analyse how the position of the methyl in the aromatic ring influences biological activity. The variation of the position within the ring caused a considerable improvement in the biological activity. This effect was especially relevant when the substitution was made in the meta position, where the derivative 3l presented an IC50 of 0.33 ± 0.02 μM. Based on this observation, we decided to study the di-substitution in the meta position, obtaining the derivative 3m, with an IC50 of 0.08 ± 0.001 μM. Likewise, derivative 3m showed a greater lipophilic capacity with respect to derivative 2b. These data demonstrate the strong dependence between the geometry of the synthetic macamide and the biological activity, confirming once more that this is the position of the most influential pharmacophore (Table 3).
Finally, compiling the information on the influence of the alkyl chain (aliphatic chain lengthening, 1f), the position of the benzyl (introduction of a methyl residue, 2b), the modification of the amide bond (ester, 2g; amine, 2i and thioamide, 2j) and the introduction of substituents on the aromatic ring (two methyl groups at the meta position, 3m), it was decided to synthesise analogues with the optimal substituents at each position to analyse their biological activity. The synthetic methodology used to obtain the synthetic macamide 4a was the same as that used to synthesise derivative 2g, reacting the acid chloride with an alcohol. In the case of the synthetic macamide 4b, it was obtained starting from the reduction of the derivative 3m with LiAlH4. Finally, to obtain synthetic macamide 4c, derivative 3m was reacted with Lawesson's reagent.
The introduction of the thiocarbonyl group (4c) achieved a slight improvement in activity with an IC50 of 0.06 ± 0.001 μM. In the case of synthetic macamide 4b, as expected, in view of the previous results, the effect was more pronounced with an IC50 of 0.05 ± 0.001 μM. Finally, synthetic macamide 4a showed the highest inhibitory effect with an IC50 of 0.009 ± 0.001 μM. This effect could be justified on the basis of the difference between the amide bond and the ester bond, since the latter has less rigidity than the amide bond and could be adapted to the active centre of the target. Similarly, it was observed that, in this series of derivatives, synthetic macamide 4c showed a higher cytotoxicity (71.5 ± 1.13 μM) and synthetic macamide 3b showed a moderate cytotoxicity (80.9 ± 1.96 μM) with respect to untreated cells (negative control) (Table 4).
Table 4. Comparative study of the optimal synthetic macamides against natural macamides and the positive control C87 on TNF-α.
| No. | Derivatives | Viability (CC50 μM) THP-1 cells | TNF-α inhibition (IC50 μM) THP-1 cells | clog P a |
| 4a |
|
90.1 ± 1.27 | 0.009 ± 0.001 | 9.09 |
| 4b |
|
80.9 ± 1.96 | 0.05 ± 0.001 | 9.19 |
| 4c |
|
71.5 ± 1.13 | 0.06 ± 0.001 | 8.75 |
| 1 |
|
>100 | 3.54 ± 0.02 | 8.79 |
| 2 |
|
>100 | 1.77 ± 0.07 | 6.58 |
| C87 |
|
10.81 ± 1.23 | 0.11 ± 0.01 | 4.57 |
aValues of clog P were calculated by DataWarrior.271 = N-oleoyldopamine; 2 = N-(2-hydroxyethyl)-7Z,10Z,13Z,16Z-docosatetraenamide.
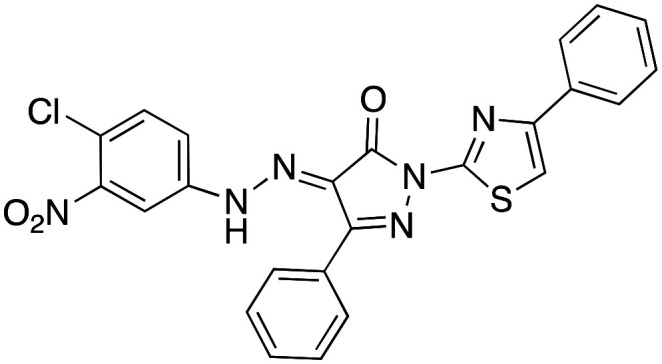
Our results show that the synthesised macamides and especially derivative 4a have a pharmacological potency between 200–400 times better than the natural N-(2-hydroxyethyl)-7Z,10Z,13Z,16Z-docosatetraenamide and N-oleoyldopamine macamides that inhibit TNF-α in THP-1 cells with IC50 of 1.77 ± 0.07 and 3.54 ± 0.02 μM, respectively,7 and 12 times better than the positive control (C87) with an IC50 of 0.11 ± 0.01 μM (Table 4).
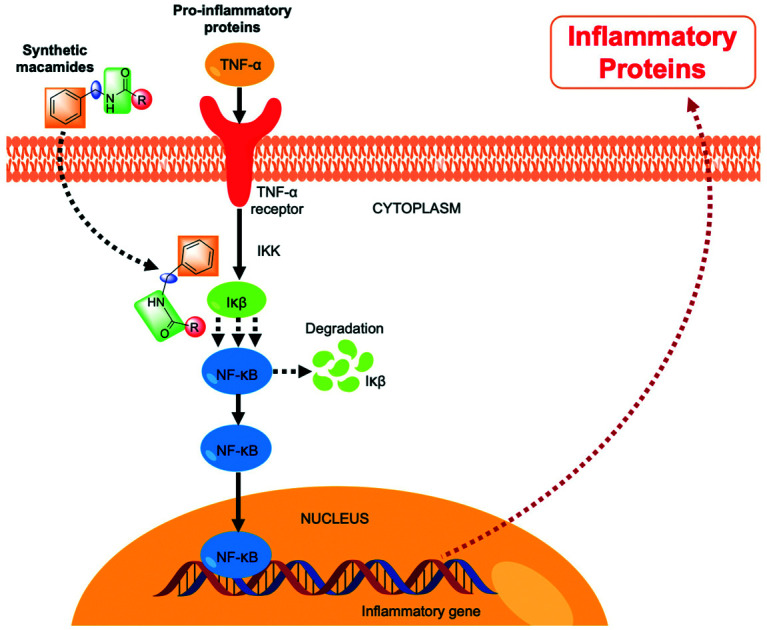
Previous works showed that the anti-inflammatory activity of natural macamides is due to the inhibitory capacity of NF-κB activation induced by TNF-α, through the direct inhibition of the IKKβ subunit and, to a smaller extent, of the IKKα subunit of the κB kinase inhibitor complex.7,25 In this sense, we can indicate that the derivatives synthesised in this work lead to direct inhibition of IκB, preventing its degradation and the subsequent inhibition of NF-κB activation.7,31 These results open the way to the development of new anti-inflammatory drugs with a better safety profile compared to the drugs available on the market.
3. Material and methods
3.1. General experimental procedures
NMR spectra were performed on a Bruker Avance AV-I operating at 300 MHz (1H) or 75 MHz (13C). The chemical shifts (δ) are indicated in ppm relative to the residual signals of deuterated solvent: δH 7.26 and δC 77.2 for CDCl3. 13C NMR spectra were acquired in a proton decoupled broadband system. The following abbreviations were used to describe the peak patterns: s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sext (sextuplet), sept (septuplet), m (multiplet) and br (broad signal). High resolution mass spectrometry (HRMS) was recorded on a quadrupole QSTAR XL spectrometer with TOF, electrospray (ESI+). In each case, the solvent and concentration used (mg mL–1) were indicated. TLC was performed using Merck silica gel 60-F254 plates. Developed chromatograms were visualised by UV absorbance (254 nm) or through application of heat to a plate stained with phosphomolybdic acid (H3PMo12O40). The manual chromatography column was performed with silica gel (60 μm) and with the indicated eluent in accordance with standard techniques. The solvents and reagents used were purchased from commercial sources and used without prior purification.
3.2. General procedure for the acylation of benzylamine with fatty acids
The corresponding carboxylic acid (1 eq.) was added in a round bottom flask and dissolved in dry CH2Cl2 (1 mL mmol–1 carboxylic acid). Subsequently, the DCC (1.2 eq.) was added. The mixture was kept under stirring for 15 minutes, after which a mixture of DMAP (10% mol) and benzylamine (1 eq.) dissolved in dry CH2Cl2 (1 mL mmol–1 benzylamine) was added. The reaction mixture was kept stirred for 12 h at room temperature. At the end of this time, the white solid formed was filtered through a filter plate and the solvent was removed from the filtrate by evaporation under reduced pressure. The crude reaction was purified by flash chromatography using Hex–AcOEt as eluent.
3.3. Synthesis of (9Z,12Z)-N′-phenyloctadeca-9,12-dienehydrazide
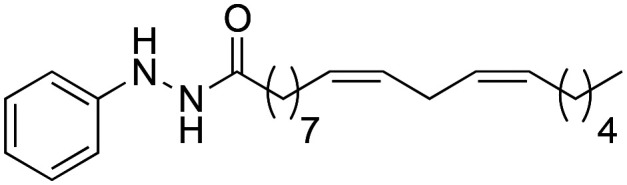
In a solution of phenylhydrazine (215 mg, 2 mmol) in dry CH2Cl2 (3 mL mmol–1), Et3N (201 mg, 2 mmol) was added and stirred at 0 °C. Next, (9Z,12Z)-octadeca-9,12-dienoyl chloride (500 mg, 1.66 mmol) was added dropwise. After 10 minutes, the reaction was observed to be complete by TLC. Water was added and extraction was carried out with CH2Cl2 2 × 15 mL. The organic phase was dried with anhydrous MgSO4, filtered and the solvent was removed under reduced pressure. (9Z,12Z)-N′-phenyloctadeca-9,12-dienehydrazide (2a) was purified by flash chromatography with a mixture of Hex–AcOEt (2 : 1) as eluent; it was obtained as a colourless liquid with a yield of 70%.
3.4. General procedure for the synthesis of (9Z,12Z)-octadeca-9,12-dienamide, (9Z,12Z)-octadeca-9,12-dienoate derivatives and (9Z,12Z)-octadeca-9,12-dienethioate derivatives
In a solution of the corresponding amine, alcohol or thiol (1.0 eq.) in dry CH2Cl2 (2 mL mmol–1), Et3N (1.2 eq.) was added and stirred at 0 °C. Next, the (9Z,12Z)-octadeca-9,12-dienoyl chloride was added dropwise. After 10 minutes, the reaction was observed to have been completed by TLC. HCl (10%) was added and the aqueous phase 2 × 15 mL of CH2Cl2 was extracted. The organic phase was dried over anhydrous MgSO4, filtered, and the solvent was removed under reduced pressure. The crude reaction was purified by flash chromatography using Hex–AcOEt as eluent.
3.5. General procedure for amide reduction
LiAlH4 (5 eq.) was added to a flask with dry THF at –78 °C, and, subsequently, the corresponding amide (1 eq.) was added, drop by drop, dissolved in dry THF. Stirring was maintained for 12 h. After the reaction was complete, hydrated Na2SO4 was added and allowed to react for 15 minutes until the formation of hydrogen was no longer observed. The solution was filtered and the solvent was removed under reduced pressure; the crude reaction was purified by flash chromatography using Hex–AcOEt as eluent.
3.6. General procedure for obtaining thioamides
In a solution of the corresponding amide (1.0 eq.) in dry THF, Lawesson's reagent (1.5 eq.) was added and it was stirred at reflux for 12 h. After completion of the reaction, the solvent was removed under reduced pressure and the reaction crude was purified by flash chromatography using Hex–AcOEt as the eluent.
3.7. Spectroscopic data
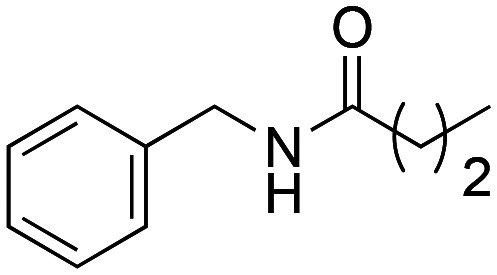
N-Benzylbutyramide (1a)
N-Benzylbutyramide (1a) was obtained from butanoic acid (300 mg, 3.41 mmol) and benzylamine (3.41 mmol). 508 mg of the synthetic macamide 1a (90% yield) were obtained as a white solid. The product was chromatographed with a Hex–AcOEt mixture (3 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.94 (t, 3H, J = 7.4 Hz), 1.66 (s, 2H, J = 7.4 Hz), 2.17 (t, 2H, J = 7.5 Hz), 4.40 (d, 2H, J = 5.7 Hz), 6.19 (bs, 1H), 7.29–7.16 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 13.9, 19.3, 38.7, 43.5, 127.5, 127.8, 128.7, 138.5, 173.1.
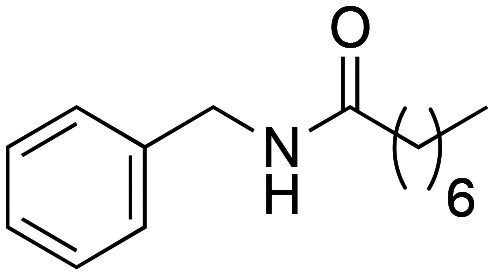
N-Benzyloctanamide (1b)
N-Benzyloctanamide (1b) was obtained from octanoic acid (443 mg, 3.07 mmol) and benzylamine (329 mg, 3.07 mmol). 620 mg of the synthetic macamide 1b (81% yield) were obtained as a white solid. The product was chromatographed with a Hex–AcOEt mixture (3 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.85 (t, 3H, J = 7.1 Hz), 1.26–1.17 (m, 8H), 1.63 (q, 2H, J = 7.4 Hz) 2.18 (t, 2H, J = 7.7 Hz), 4.41 (d, 2H, J = 5.7 Hz), 5.80 (bs, 1H), 7.28–7.19 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.2, 22.7, 25.9, 29.1, 29.4, 31.8, 36.9, 43.7, 127.5, 127.9, 128.8, 138.5, 173.1.
N-Benzylhexadecanamide (1c)
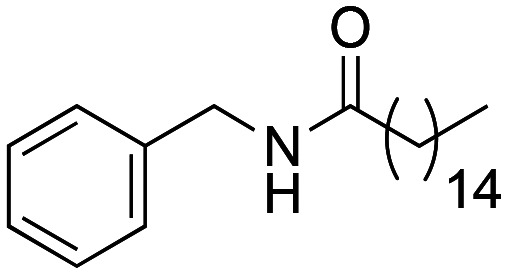
N-Benzylhexadecanamide (1c) was obtained from hexadecanoic acid (788 mg, 1.12 mmol) and benzylamine (119 mg, 1.12 mmol). 580 mg of the synthetic macamide 1c (60% yield) were obtained as a white solid. The product was purified with a Hex–AcOEt mixture (5 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.85 (t, 3H, J = 7.2 Hz), 1.26–1.17 (m, 24H), 1.61 (q, 2H, J = 7.7 Hz), 2.18 (t, 2H, J = 7.4 Hz), 4.41 (d, 2H, J = 5.7 Hz), 5.72 (bs, 1H), 7.32–7.20 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.3, 22.8, 25.9, 29.4, 29.5, 29.5, 29.6, 29.7, 29.8, 29.8, 29.8, 29.9, 29.9, 32.0, 36.9, 43.7, 127.6, 127.9, 128.8, 138.5, 173.1.
N-Benzyloctadecanamide (1d)
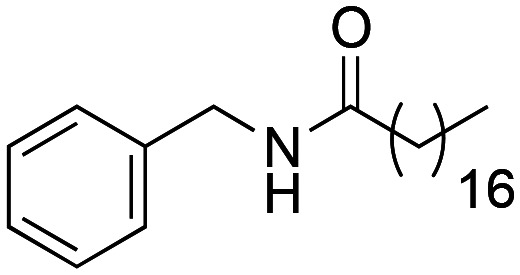
N-Benzyloctadecanamide (1d) was obtained from octadecanoic acid (611 mg, 2.15 mmol) and benzylamine (230 mg, 2.15 mmol). 506 mg of the synthetic macamide 1d (63% yield) were obtained as a white solid. The product was chromatographed with a Hex–AcOEt mixture (5 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.88 (t, 3H, J = 7.2 Hz), 1.28–1.17 (m, 28H), 1.65 (q, 2H, J = 7.7 Hz), 2.21 (t, 2H, J = 7.2 Hz), 4.45 (d, 2H, J = 5.7 Hz), 5.70 (bs, 1H), 7.32–7.20 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 13.8, 22.4, 25.5, 29.0, 29.1, 29.1, 29.2, 29.3, 29.4, 29.4, 29.4, 29.5, 29.5, 29.5, 29.6, 31.7, 36.6, 43.3, 127.2, 127.5, 128.4, 138.2, 172.7.
(9Z)-N-Benzyloctadec-9-enamide (1e)
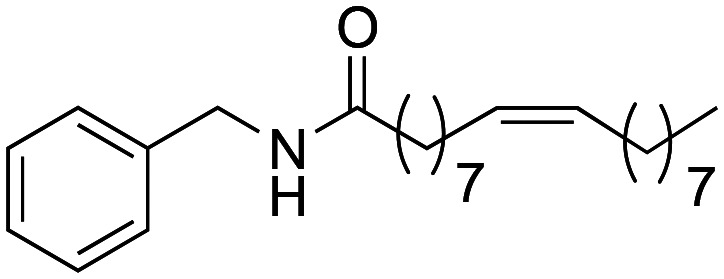
(9Z)-N-Benzyloctadec-9-enamide (1e) was obtained from oleic acid (500 mg, 1.77 mmol) and benzylamine (189 mg, 1.77 mmol). 209 mg of the synthetic macamide 1e (56% yield) were obtained as a white solid. The product was chromatographed with a Hex–AcOEt mixture (5 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.83 (t, 3H, J = 7.0 Hz), 1.32–1.23 (m, 20H), 1.62 (q, 2H, J = 6.9 Hz), 2.02–1.85 (m, 4H), 2.18 (t, 2H, J = 7.2 Hz), 4.40 (d, 2H, J = 5.7 Hz), 5.37–5.26 (m, 2H) 5.70 (bs, 1H), 7.34–7.22 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.3, 22.8, 25.9, 27.3, 27.4, 29.3, 29.4, 29.4, 29.4, 29.5, 29.7, 29.8, 29.9, 32.0, 37.0, 43.7, 127.6, 128.0, 128.8, 129.9, 130.1, 138.6, 173.0.
(9Z,12Z)-N-Benzyloctadeca-9,12-dienamide (1f)
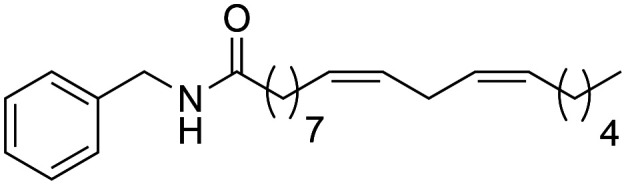
(9Z,12Z)-N-Benzyloctadeca-9,12-dienamide (1f) was obtained from (9Z,12Z)-octadeca-9,12-dienoic acid (400 mg, 1.42 mmol) and benzylamine (151.9 mg, 1.42 mmol). 265 mg of the synthetic macamide 1f (50% yield) were obtained as a colourless liquid. The product was chromatographed with a Hex–AcOEt mixture (5 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 7.0 Hz), 1.31–1.20 (m, 14H), 1.67–1.62 (m, 2H), 2.07–2.01 (m, 4H), 2.21 (t, 2H, J = 7.7 Hz), 2.77 (t, 2H, J = 5.8 Hz), 4.44 (d, 2H, J = 5.7 Hz), 5.47–5.23 (m, 4H) 5.70 (bs, 1H), 7.32 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.2, 22.7, 25.8, 25.9, 27.3, 27.4, 29.3, 29.4, 29.5, 29.5, 29.7, 31.7, 37.0, 43.7, 127.6, 128.0, 128.1, 128.2, 128.9, 130.2, 130.4, 138.6, 173.0. HRESIMS [M + H]+m/z 370.3102 (calculated for C25H40NO 370.3104).
(9Z,12Z)-N-Phenyloctadeca-9,12-dienamide (1g)
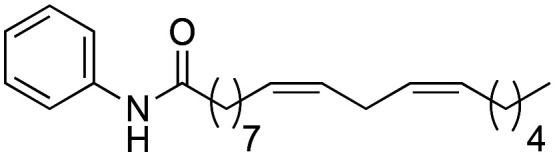
(9Z,12Z)-N-Phenyloctadeca-9,12-dienamide (1g) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (736 mg, 2.47 mmol) and aniline (230 mg, 2.47 mmol). 640 mg of the synthetic macamide 1g (73% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (3 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.8 Hz), 1.34–1.17 (m, 14H), 1.73–1.60 (m, 2H), 2.05 (q, 4H, J = 6.4 Hz), 2.35 (t, 2H, J = 7.7 Hz), 2.77 (t, 2H, J = 6.0 Hz), 5.35–5.21 (m, 4H), 7.10 (t, 1H, J = 7.2 Hz), 7.18 (bs, 1H), 7.31 (t, 2H, J = 8.2 Hz), 7.51 (d, 2H, J = 7.8 Hz). 13C NMR (CDCl3, 75 MHz): δC 14.1, 22.6, 25.6, 25.8, 27.2, 29.2, 29.3, 29.3, 29.6, 31.5, 37.6, 120.3, 124.1, 127.9, 128.0, 128.8, 130.0, 130.2, 138.3, 172.3.
(9Z,12Z)-N-Phenethyloctadeca-9,12-dienamide (1h)
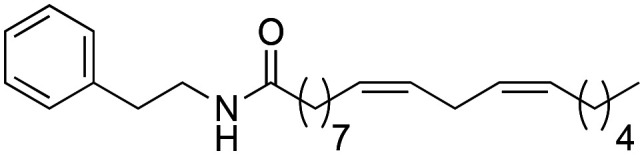
(9Z,12Z)-N-Phenethyloctadeca-9,12-dienamide (1h) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (736 mg, 2.47 mmol) and phenethylamine (300 mg, 2.47 mmol). 663 mg of the synthetic macamide 1h (70% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (4 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.9 Hz), 1.35–1.27 (m, 14H), 1.58–1.53 (m, 3H), 2.15–2.03 (m, 5H), 2.85–2.73 (m, 3H), 3.52 (q, 2H, J = 6.8 Hz), 5.46–5.32 (m, 4H), 7.35–7.18 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.0, 22.5, 25.6, 25.7, 27.1, 29.1, 29.2, 29.3, 29.6, 31.5, 35.6, 36.6, 40.6, 126.4, 127.9, 128.5, 128.7, 130.0, 130.1, 139.0, 173.4.
(9Z,12Z)-N-(3-Phenylpropyl)-octadeca-9,12-dienamide (1i)
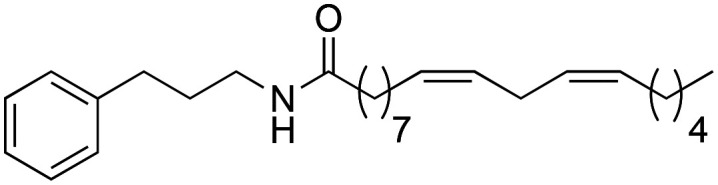
(9Z,12Z)-N-(3-Phenylpropyl)-octadeca-9,12-dienamide (1i), 250 mg of the amine were introduced and 573 mg of the synthetic macamide 1i were obtained (78% yield). Column with Hex–AcOEt (6 : 1) pale yellow liquid. 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 7.0 Hz), 1.40–1.24 (m, 14H), 1.69–1.56 (m, 3H), 1.84 (quint, 2H, J = 7.0 Hz), 2.15–2.02 (m, 5H), 2.65 (t, 2H, J = 7.8 Hz), 2.77 (t, 2H, J = 5.9 Hz), 3.29 (q, 2H, J = 6.9 Hz), 5.42–5.28 (m, 4H), 7.22–7.15 (m, 3H), 7.30–7.26 (m, 2H). 13C NMR (CDCl3, 75 MHz): δC 14.0, 22.5, 25.6, 25.8, 27.2, 29.1, 29.3, 29.3, 29.6, 31.2, 31.5, 33.3, 36.7, 39.2, 125.9, 127.9, 128.0, 128.3, 128.4, 130.0, 130.2, 141.5, 173.4.
(9Z,12Z)-N′-Phenyloctadeca-9,12-dienehydrazide (2a)
(9Z,12Z)-N′-Phenyloctadeca-9,12-dienehydrazide (2a) was obtained from phenylhydrazine (215 mg, 2 mmol), CH2Cl2 (3 mL mmol–1), Et3N (201 mg, 2 mmol) and (9Z,12Z)-octadeca-9,12-dienoyl chloride (500 mg, 1.66 mmol). 350 mg of synthetic macamide 2a (70% yield) were obtained as a colourless liquid with a yield. The product was chromatographed with a Hex–AcOEt mixture (2 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.81 (t, 3H, J = 7.1 Hz), 1.40–1.22 (m, 14H), 1.72–1.59 (m, 2H), 2.08–1.97 (m, 4H), 2.18 (t, 1H, J = 7.8 Hz), 2.69 (q, 2H, J = 5.2 Hz), 5.41–5.27 (m, 4H), 6.94–6.75 (m, 2H), 7.38–7.19 (m, 3H). 13C NMR (CDCl3, 75 MHz): δC 14.1, 22.6, 25.5, 25.6, 27.2, 29.2, 29.3, 29.3, 29.6, 31.5, 34.2, 113.5, 120.9, 127.9, 128.1, 129.0, 130.0, 130.2, 130.2, 148.1, 173.8.
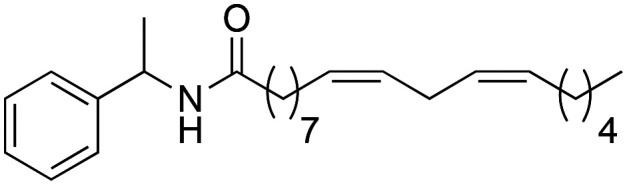
(9Z,12Z)-N-(1-Phenylethyl)-octadeca-9,12-dienamide (2b)
(9Z,12Z)-N-(1-Phenylethyl)-octadeca-9,12-dienamide (2b) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (593 mg, 1.99 mmol) and 1-methyl benzylamine (241 mg, 1.99 mmol). 470 mg of the synthetic macamide 2b (73% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (5 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 7.0 Hz), 1.36–1.25 (m, 14H), 1.49 (d, 3H, J = 6.9 Hz), 1.65–1.56 (m, 4H), 2.04–2.00 (m, 4H), 2.16 (t, 2H, J = 7.8 Hz) 2.77 (t, 2H, J = 5.9 Hz), 5.15 (quint, 1H, J = 7.1 Hz), 5.43–5.29 (m, 4H), 5.61 (bd, 1H), 7.36–7.24 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.1, 21.8, 22.6, 25.7, 25.8, 27.2, 29.2, 29.3, 29.3, 29.4, 29.6, 31.5, 36.8, 48.5, 126.2, 127.2, 127.9, 128.1, 128.6, 130.1, 130.2, 143.5, 172.3.
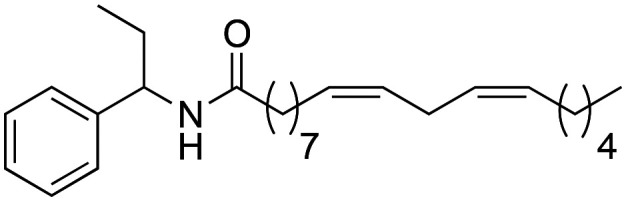
(9Z,12Z)-N-(1-Phenylpropyl)-octadeca-9,12-dienamide (2c)
(9Z,12Z)-N-(1-Phenylpropyl)-octadeca-9,12-dienamide (2c) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (658 mg, 2.21 mmol) and 1-ethyl benzylamine (300 mg, 2.21 mmol). 608 mg of the synthetic macamide 2c (69% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (5 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.87 (t, 6H, J = 7.4 Hz), 1.34–1.23 (m, 14H), 1.62–1.57 (m, 4H), 1.85–1.74 (m, 2H), 2.06–1.98 (m, 3H), 2.15 (t, J = 7.8 Hz), 2.75 (t, 1H, J = 5.7 Hz), 4.87 (q, 1H, J = 7.3 Hz), 5.41–5.26 (m, 3H), 5.65 (d, 1H, J = 6.7 Hz), 7.34–7.22 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 10.8, 14.1, 22.6, 25.7, 25.9, 27.3, 29.2, 29.2, 29.3, 29.3, 29.4, 29.7, 31.6, 36.9, 54.8, 126.7, 127.3, 128.0, 128.1, 128.6, 130.1, 130.3, 142.4, 172.6.
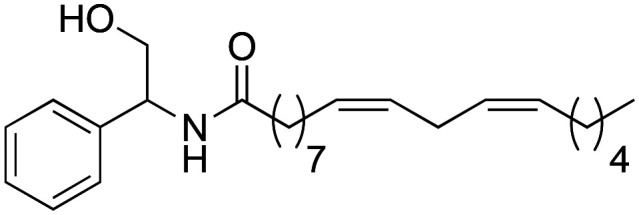
(9Z,12Z)-N-(2-Hydroxy-1-phenylethyl)-octadeca-9,12-dienamide (2d)
(9Z,12Z)-N-(2-Hydroxy-1-phenylethyl)-octadeca-9,12-dienamide (2d) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (593 mg, 2.18 mmol) and dl-2-phenylglycinol (300 mg, 2.18 mmol). 535 mg of the synthetic macamide 2d (77% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (5 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.9 Hz), 1.39–1.26 (m, 14H), 1.69–1.59 (m, 4H), 2.07–2.01 (m, 3H), 2.25 (t, 2H, J = 7.4 Hz), 2.77 (t, 2H, J = 6.9 Hz), 3.91–3.87 (m, 2H), 5.07 (q, 1H, J = 6.2 Hz), 5.41–5.29 (m, 3H), 6.1 (m, d, J = 6.6 Hz), 7.41–7.27 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.2, 22.7, 25.7, 25.8, 27.3, 29.3, 29.4, 29.4, 29.4, 29.7, 31.6, 36.8, 55.9, 66.5, 126.8, 127.9, 128.0, 128.2, 128.9, 130.1, 130.3, 139.3, 174.1.
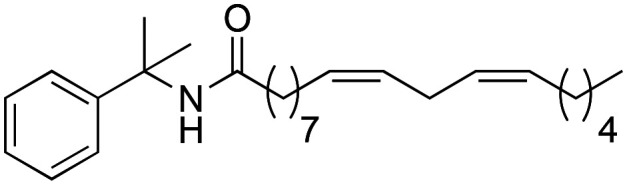
(9Z,12Z)-N-(2-Phenylpropan-2-yl)-octadeca-9,12-dienamide (2e)
(9Z,12Z)-N-(2-Phenylpropan-2-yl)-octadeca-9,12-dienamide (2e) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (658 mg, 2.21 mmol) and 1-methyl-1-phenyl ethylamine (300 mg, 2.21 mmol). 704 mg of the synthetic macamide 2e (80% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (7 : 1). 300 mg of the amine were introduced and 704 mg of product were obtained (80% yield). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 7.9 Hz), 1.39–1.25 (m, 14H), 1.66–1.56 (m, 4H), 1.70 (s, 6H), 2.08–2.02 (m, 3H), 2.14 (t, 2H, J = 7.4 Hz), 2.77 (t, 1H, J = 5.9 Hz), 5.39–5.29 (m, 4H), 5.66 (bs, 1H), 7.41–7.19 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.1, 22.6, 25.7, 25.8, 27.2, 29.2, 29.2, 29.3, 29.3, 29.4, 29.7, 31.5, 37.3, 55.7, 124.8, 126.5, 128.0, 128.1, 128.3, 130.1, 130.2, 147.1, 172.5.
(9Z,12Z)-N-Benzyl-N-methyloctadeca-9,12-dienamide (2f)
(9Z,12Z)-N-Benzyl-N-methyloctadeca-9,12-dienamide (2f) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (572 mg, 1.92 mmol) and N-benzyl methylamine (233 mg, 1.92 mmol). 477 mg of the synthetic macamide 2f (65% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (4 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 7.0 Hz), 1.36–1.25 (m, 14H), 1.49 (d, 3H, J = 6.9 Hz), 1.65–1.56 (m, 4H), 2.04–2.00 (m, 4H), 2.16 (t, 2H, J = 7.8 Hz) 2.77 (t, 2H, J = 5.9 Hz), 2.88 (s, 3H), 4.56 (s, 2H), 5.43–5.29 (m, 4H), 5.61 (bd, 1H), 7.36–7.15 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.1, 21.8, 22.6, 25.7, 25.8, 27.2, 29.2, 29.3, 29.3, 29.4, 29.6, 31.5, 36.8, 50.6, 126.2, 127.8, 127.9, 128.4, 128.6, 129.9, 130.2, 137.4, 173.3.
(9Z,12Z)-1-(Benzyloxy)-octadeca-9,12-dien-1-olate (2g)
(9Z,12Z)-1-(Benzyloxy)-octadeca-9,12-dien-1-olate (2g) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (581 mg, 1.94 mmol) and benzyl alcohol (200 mg, 1.94 mmol). 640 mg of the synthetic macamide 2g (80% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (4 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.90 (t, 3H, J = 6.9 Hz), 1.38–1.27 (m, 14H), 1.70–1.60 (m, 2H), 2.11–2.01 (m, 4H), 2.36 (t, 2H, J = 7.6 Hz), 2.78 (t, 2H, J = 5.9 Hz), 5.12 (s, 2H), 5.40–5.30 (m, 4H), 7.38–7.35 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.1, 22.6, 25.0, 25.7, 29.1, 29.2, 29.4, 29.6, 31.6, 34.3, 66.0, 128.0, 128.1, 128.1, 128.2, 128.5, 130.0, 130.2, 136.2, 173.5 HRESIMS [M + Na]+m/z 425.2633 (calculated for C25H32O2Na 393.2764).
(9Z,12Z)-1-(Benzylthio)-octadeca-9,12-dien-1-olate (2h)
(9Z,12Z)-1-(Benzylthio)-octadeca-9,12-dien-1-olate (2h) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (357 mg, 1.20 mmol) and benzyl mercaptan (150 mg, 1.20 mmol). 355 mg of the synthetic macamide 2h (76% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (4 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.88 (t, 3H, J = 6.9 Hz), 1.29 (m, 14H), 1.64 (m, 2H), 2.02 (m, 4H), 2.54 (t, 2H, J = 7.7 Hz), 2.76 (t, 2H, J = 5.9 Hz), 4.10 (s, 2H), 5.38–5.25 (m, 4H), 7.33–7.23 (m, 5H) 13C NMR (CDCl3, 75 MHz): δC 14.1, 22.6, 25.6, 25.7, 27.2, 27.3, 28.9, 29.1, 29.2, 29.4, 29.6, 31.6, 33.1, 43.8, 127.2, 128.0, 128.1, 128.6, 128.8, 130.0, 130.2, 137.8, 198.6.
(9Z,12Z)-N-(1-Phenylethyl)-octadeca-9,12-dien-1-amine (2i)
(9Z,12Z)-N-(1-Phenylethyl)-octadeca-9,12-dien-1-amine (2i) was obtained from derivative 1f (150 mg, 0.40 mmol) and LiAlH4 (76 mg, 2 mmol). 119 mg of the synthetic macamide 2i (83% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (1 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.85 (t, 3H, J = 6.9 Hz), 1.33–1.20 (m, 14H), 1.32 (d, 3H, J = 10.4 Hz), 2.06–1.96 (m, 4H), 2.49–2.34 (m, 2H), 2.73 (t, 2H, J = 5.8 Hz), 3.71 (q, 1H, J = 6.6 Hz), 5.41–5.26 (m, 4H), 7.16–7.28 (m, 5H) 13C NMR (CDCl3, 75 MHz): δC 14.2, 22.7, 24.4, 25.8, 27.3, 27.3, 27.5, 29.4, 29.5, 29.6, 29.8, 30.4, 31.7, 48.0, 58.5, 126.7, 126.9, 128.1, 128.1, 128.5, 130.2, 130.3, 146.0. HRESIMS [M + H]+m/z 370.3468 (calculated for C26H44N 370.3468).
(9Z,12Z)-N-(1-Phenylethyl)-octadeca-9,12-dienothiamide (2j)
(9Z,12Z)-N-(1-Phenylethyl)-octadeca-9,12-dienothiamide (2j) was obtained from derivative 1f (130 mg, 0.34 mmol) and Lawesson's reagent (206 mg, 0.51 mmol). 135 mg of the synthetic macamide 2j (99% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (15 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.83 (t, 3H, J = 6.9 Hz), 1.32–1.18 (m, 14H), 1.50 (d, 3H, J = 6.9 Hz), 1.72–1.62 (m, 2H), 2.03–1.93 (m, 2H), 2.54 (t, 2H, J = 7.8 Hz), 2.71 (t, 2H, J = 5.9 Hz), 5.36–5.25 (m, 4H), 5.70 (quint, 1H, J = 7.1 Hz), 7.27–7.17 (m, 5H), 7.78 (bd, 1H, J = 8.0 Hz). 13C NMR (CDCl3, 75 MHz): δC 14.0, 19.9, 22.4, 25.5, 27.1, 28.7, 29.0, 29.1, 29.2, 29.4, 29.5, 31.4, 47.0, 54.2, 126.4, 127.5, 127.8, 127.9, 128.6, 129.8, 130.0, 141.3, 204.2.
(9Z,12Z)-N-(1-Cyclohexylethyl)-octadeca-9,12-dienamide (3a)
(9Z,12Z)-N-(1-Cyclohexylethyl)-octadeca-9,12-dienamide (3a) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (187 mg, 0.63 mmol) and cyclohexyl ethylamine (80 mg, 0.63 mmol). 178 mg of the synthetic macamide 3a (73% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (5 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.88 (t, 3H, J = 6.9 Hz), 1.29 (m, 14H), 1.64 (m, 2H), 2.02 (m, 4H), 2.54 (t, 2H, J = 7.7 Hz), 2.76 (t, 2H, J = 5.9 Hz), 4.10 (s, 2H), 5.38–5.25 (m, 4H), 7.33–7.23 (m, 5H). 13C NMR (CDCl3, 75 MHz): δC 14.2, 18.2, 22.7, 25.8, 26.0, 26.3, 26.3, 26.6, 27.3, 29.2, 29.3, 29.4, 29.4, 29.5, 29.7, 31.7, 37.3, 43.2, 49.3, 128.1, 128.2, 130.2, 130.3, 172.4.
(9Z,12Z)-N-(3,3-Dimethylbutan-2-yl)-octadeca-9,12-dienamide (3b)
(9Z,12Z)-N-(3,3-Dimethylbutan-2-yl)-octadeca-9,12-dienamide (3b) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (294 mg, 0.98 mmol) and 2-amino-3,3-dimethylbutane (100 mg, 0.98 mmol). 310 mg of the synthetic macamide 3b (77% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (7 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.83 (s, 3H), 0.83 (t, 9H, J = 8.0 Hz), 0.98 (d, 3H, J = 7.8 Hz), 1.24 (m, 14H), 1.57 (m, 2H), 1.98 (q, 4H, J = 6.4 Hz), 2.11 (t, 2H, J = 7.9 Hz), 2.70 (t, 2H, J = 5.9 Hz), 3.83 (m, 1H), 5.30 (m, 4H), 5.52 (bd, 1H, J = 9.5 Hz). 13C NMR (CDCl3, 75 MHz): δC 14.0, 16.2, 22.6, 25.6, 26.0, 26.2, 27.2, 29.3, 29.3, 29.3, 29.6, 31.5, 34.1, 37.1, 52.4, 127.9, 128.0, 130.0, 130.1, 172.3.
(9Z,12Z)-N-(1-((3r,5r,7r)-Adamantan-1-yl)-ethyl)-octadeca-9,12-dienamide (3c)
(9Z,12Z)-N-(1-((3r,5r,7r)-Adamantan-1-yl)-ethyl)-octadeca-9,12-dienamide (3c) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (250 mg, 0.84 mmol) and 1-adamantylamine (150 mg, 0.84 mmol). 240 mg of the synthetic macamide 3c (65% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (7 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.88 (t, 3H, J = 6.8 Hz), 1.00 (d, 3H, J = 6.9 Hz), 1.42–1.25 (m, 14H), 1.60–1.40 (m, 6H), 1.75–1.57 (m, 8H), 2.10–1.93 (m, 6H), 2.16 (t, 2H, J = 7.8 Hz), 2.77 (t, 2H, J = 5.9 Hz), 3.77–3.67 (m, 1H), 5.42–5.22 (m, 4H). 13C NMR (CDCl3, 75 MHz): δC 14.2, 14.8, 22.7, 25.8, 26.1, 27.3, 28.5, 29.3, 29.4, 29.5, 29.5, 29.8, 31.7, 35.9, 37.2, 37.4, 38.6, 52.8, 128.1, 128.2, 130.2, 130.4, 172.5.
(9Z,12Z)-N-(1-(4-Hydroxyphenyl)-ethyl)-octadeca-9,12-dienamide (3d)
(9Z,12Z)-N-(1-(4-Hydroxyphenyl)-ethyl)-octadeca-9,12-dienamide (3d) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (271 mg, 0.91 mmol) and 1-(4-hydroxyphenyl)-ethylamine (125 mg, 0.91 mmol). 251 mg of the synthetic macamide 3d (69% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (4 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.8 Hz), 1.44–4.17 (m, 14H), 1.47 (d, 3H, J = 6.9 Hz), 1.66–1.58 (m, 2H), 1.79–1.70 (m, 2H), 2.11–2.02 (m, 4H), 2.15 (t, 2H, J = 7.9 Hz), 2.54 (t, 2H, J = 7.6 Hz), 2.82–2.74 (m, 4H), 5.16 (quint, 1H, J = 7.3 Hz), 5.44–5.28 (m, 4H), 5.61 (bd, 1H, J = 7.9 Hz), 7.03 (d, 2H, J = 8.6 Hz), 7.32 (d, 2H, J = 8.6 Hz). 13C NMR (CDCl3, 75 MHz): δC 14.2, 22.7, 25.8, 27.3, 29.2, 29.2, 29.3, 29.3, 29.4, 29.5, 29.7, 31.7, 121.9, 127.5, 128.1, 128.2, 130.2, 130.4, 140.8, 150.0, 172.2.
(9Z,12Z)-N-(1-(4-Methoxyphenyl)-ethyl)-octadeca-9,12-dienamide (3e)
(9Z,12Z)-N-(1-(4-Methoxyphenyl)-ethyl)-octadeca-9,12-dienamide (3e) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (197 mg, 0.66 mmol) and 1-(4-methoxyphenyl)-ethylamine (100 mg, 0.66 mmol). 226 mg of the synthetic macamide 3e (83% yield) were obtained as a yellowish liquid. 1H NMR (CDCl3, 300 MHz): δH 0.87 (t, 3H, J = 7.0 Hz), 1.40–1.27 (m, 14H), 1.42 (d, 3H, J = 7.0 Hz), 1.54–1.61 (m, 2H), 2.02–2.10 (m, 4H), 2.15 (t, 2H, J = 7.8 Hz), 2.75 (t, 2H, J = 5.9 Hz), 3.74 (s, 3H), 5.05 (quint, 1H, J = 10.7 Hz), 5.44–5.32 (m, 4H), 6.12 (bd, 1H, J = 7.9 Hz), 6.82 (d, 2H, J = 8.7 Hz), 7.20 (d, 2H, J = 8.7 Hz). 13C NMR (CDCl3, 75 MHz): δC 14.0, 21.7, 22.5, 25.6, 25.8, 27.2, 29.1, 29.3, 29.3, 29.3, 29.6, 31.5, 36.8, 47.9, 55.2, 113.9, 127.3, 130.0, 130.2, 135.6, 158.7, 172.2.
(9Z,12Z)-N-(1-(p-Tolyl)-ethyl)-octadeca-9,12-dienamide (3f)
(9Z,12Z)-N-(1-(p-Tolyl)-ethyl)-octadeca-9,12-dienamide (3f) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (289 mg, 0.97 mmol) and 1-(4-methylphenyl)-ethylamine (130 mg, 0.97 mmol). 331 mg of the synthetic macamide 3f (80% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (6 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.90 (t, 3H, J = 6.9 Hz), 1.25–1.40 (m, 14H), 1.45 (d, 3H, J = 6.9 Hz), 1.66–1.58 (m, 2H), 2.01–2.08 (m, 4H), 2.15 (t, 2H, J = 8.0 Hz), 2.32 (s, 3H), 2.78 (t, 2H, J = 5.9 Hz), 5.10 (quint, 1H, J = 7.0 Hz), 5.27–5.43 (m, 4H), 6.02 (bd, J = 7.8 Hz), 7.12 (d, 2H, J = 8.0 Hz), 7.20 (d, 2H, J = 8.1 Hz) 13C NMR (CDCl3, 75 MHz): δC 14.1, 21.04, 21.8, 22.6, 25.7, 25.8, 27.2, 29.2, 29.3, 29.31, 29.4, 31.6, 36.8, 48.3, 126.1, 128.0, 128.1, 129.3, 130.1, 130.2, 136.8, 140.5, 172.2.
(9Z,12Z)-N-(1-(4-(Trifluoromethyl)-phenyl)-ethyl)-octadeca-9,12-dienamide (3g)
(9Z,12Z)-N-(1-(4-(Trifluoromethyl)-phenyl)-ethyl)-octadeca-9,12-dienamide (3g) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (141 mg, 0.47 mmol) and 1-(4-trifluoromethylphenyl)-ethylamine (90 mg, 0.47 mmol). 156 mg of the synthetic macamide 3g (73% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (6 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.88 (t, 3H, J = 6.8 Hz), 1.25–1.40 (m, 14H), 1.48 (d, 3H, J = 6.9 Hz), 1.66–1.58 (m, 2H), 2.01–2.08 (m, 4H), 2.18 (t, 3H, J = 8.1 Hz), 2.77 (t, 2H, J = 6.2 Hz), 5.17 (quint, 1H, J = 7.2 Hz), 5.27–5.43 (m, 4H), 5.74 (bd, 1H, J = 7.1 Hz), 7.41 (d, 2H, J = 8.1 Hz), 7.58 (d, 2H, J = 8.1 Hz). 13C NMR (CDCl3, 75 MHz): δC 14.2, 22.0, 22.7, 25.8, 25.8, 27.3, 27.3, 25.8, 25.8, 29.3, 29.4, 29.5, 29.7, 31.7, 36.9, 48.5, 175.7 (q, J = 3.7 Hz), 126.6, 128.0, 128.2, 130.1, 130.1, 130.4.
(9Z,12Z)-N-(1-(4-Fluorophenyl)-ethyl)-octadeca-9,12-dienamide (3h)
(9Z,12Z)-N-(1-(4-Fluorophenyl)-ethyl)-octadeca-9,12-dienamide (3h) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (246 mg, 0.82 mmol) and 4-fluoro-α-methyl benzylamine (115 mg, 0.82 mmol). 255 mg of the synthetic macamide 3h (79% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (3 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.7 Hz), 1.42–1.25 (m, 14H), 1.45 (d, 3H, J = 6.9 Hz), 1.54–1.61 (m, 2H), 2.04–2.10 (m, 4H), 2.16 (t, 2H, J = 8.1 Hz), 2.16 (t, 2H, J = 8.1 Hz), 2.77 (t, 2H, J = 6.1 Hz), 5.11 (quint, 1H, J = 7.2 Hz), 5.42–5.34 (m, 4H), 5.92 (bd, 1H), 6.9–7.1 (m, 2H), 7.26–7.36 (m, 2H). 13C NMR (CDCl3, 75 MHz): δC 14.1, 21.9, 22.7, 25.7, 25.8, 27.3, 29.2, 29.3, 29.3, 29.4, 29.7, 31.6, 36.9, 48.0, 115.5 (d, J = 79.9 Hz), 127.9 (d, J = 8.0 Hz), 128.1 (d, J = 12.5 Hz), 130.2 (d, J = 16.0 Hz), 139.3 (d, J = 3.1 Hz), 162.1 (d, J = 245.4 Hz), 172.3. 19F NMR (CDCl3, 75 MHz): δC –115.5.
(9Z,12Z)-N-(1-(4-Bromophenyl)-ethyl)-octadeca-9,12-dienamide (3i)
(9Z,12Z)-N-(1-(4-Bromophenyl)-ethyl)-octadeca-9,12-dienamide (3i) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (201 mg, 0.67 mmol) and 4-bromo-α-methyl benzylamine (135 mg, 0.67 mmol). 255 mg of the synthetic macamide 3i (82% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (4 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.87 (t, 3H, J = 4.9 Hz), 1.42–1.25 (m, 14H), 1.45 (d, 3H, J = 7.0 Hz), 1.56–1.61 (m, 2H), 2.05 (q, 4H, J = 6.4 Hz), 2.12 (t, 2H, J = 8.1 Hz), 2.75 (t, 2H, J = 5.9 Hz), 5.00 (quint, 1H, J = 7.3 Hz), 5.42–5.32 (m, 4H), 6.45 (bd, 1H, J = 7.8 Hz), 7.12 (d, 2H, J = 8.4 Hz), 7.38 (d, 2H, J = 8.4 Hz). 13C NMR (CDCl3, 75 MHz): δC 14.1, 21.7, 22.6, 25.6, 25.8, 27.2, 29.2, 29.3, 29.3, 29.3, 29.6, 31.5, 36.6, 48.0, 120.9, 128.0, 127.9, 128.1, 130.0, 130.2, 131.5, 142.8, 172.4.
(9Z,12Z)-N-(1-(4-Ethylphenyl)-ethyl)-octadeca-9,12-dienamide (3j)
(9Z,12Z)-N-(1-(4-Ethylphenyl)-ethyl)-octadeca-9,12-dienamide (3j) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (159 mg, 0.53 mmol) and 1-(4-ethylphenyl)-ethylamine (80 mg, 0.53 mmol). 157 mg of the synthetic macamide 3j (73% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (4 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.8 Hz), 1.23 (t, 3H, J = 7.6 Hz), 1.40–1.26 (m, 14H), 1.47 (d, 3H, J = 6.9 Hz), 1.68–1.59 (m, 2H), 2.06 (m, 4H), 2.15 (t, 2H, J = 7.9 Hz), 2.63 (q, 2H, J = 7.6 Hz), 2.77 (t, 2H, J = 5.7 Hz), 2.12 (quint, 1H, J = 7.0 Hz), 5.47–5.28 (m, 4H), 5.63 (bd, 1H, J = 7.9 Hz), 7.17 (d, 2H, J = 8.1 Hz), 7.25 (d, 2H, J = 8.1 Hz). 13C NMR (CDCl3, 75 MHz): δC 13.8, 15.2, 21.3, 22.3, 25.4, 25.4, 26.9, 28.2, 28.9, 29.0, 29.1, 29.3, 31.2, 36.7, 48.0, 126.0, 127.6, 127.8, 127.8, 129.8, 130.0, 140.2, 143.1, 171.8.
(9Z,12Z)-N-(1-(o-Tolyl)-ethyl)-octadeca-9,12-dienamide (3k)
(9Z,12Z)-N-(1-(o-Tolyl)-ethyl)-octadeca-9,12-dienamide (3k) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (176 mg, 0.59 mmol) and o-tolyl ethylamine (80 mg, 0.59 mmol). 162 mg of the synthetic macamide 3k were obtained (77% yield) as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (7 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.97 (t, 3H, J = 6.7 Hz), 1.46–1.34 (m, 14H), 1.55 (d, 3H, J = 6.8 Hz), 1.73–1.66 (m, 2H), 2.16–2.10 (m, 2H), 2.22 (t, 2H, J = 8.1 Hz), 2.46 (s, 3H), 2.85 (t, 2H, J = 6.0 Hz), 5.52–5.36 (m, 4H), 5.69 (bd, 1H, J = 8.0 Hz), 7.40–7.22 (m, 4H). 13C NMR (CDCl3, 75 MHz): δC 14.2, 19.3, 21.2, 22.7, 25.8, 25.9, 29.3, 29.4, 29.5, 29.7, 31.7, 36.9, 45.3, 124.8, 126.4, 127.4, 128.0, 128.2, 130.2, 130.4, 130.9, 136.2, 141.3, 172.1.
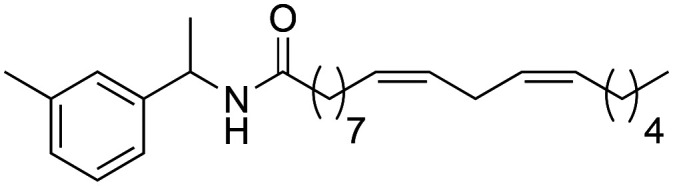
(9Z,12Z)-N-(1-(m-Tolyl)-ethyl)-octadeca-9,12-dienamide (3l)
(9Z,12Z)-N-(1-(m-Tolyl)-ethyl)-octadeca-9,12-dienamide (3l) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (176 mg, 0.59 mmol) and m-tolyl ethylamine (80 mg, 0.59 mmol). 162 mg of the synthetic macamide 3l (77% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (7 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.9 Hz), 1.41–1.25 (m, 14H), 1.45 (d, 2H, J = 6.9 Hz), 1.63 (m, 3H), 2.08–2.01 (m, 4H), 2.16 (t, 2H, J = 7.9 Hz), 2.35 (s, 3H), 2.77 (t, 2H, J = 5.9 Hz), 5.10 (quint, 1H, J = 7.2 Hz), 5.43–5.28 (m, 4H), 5.60 (d, 1H, J = 7.8 Hz), 7.26–7.06 (m, 4H) 13C NMR (CDCl3, 75 MHz): δC 14.2, 21.6, 21.9, 22.7, 25.8, 25.9, 27.4, 29.3, 29.4, 29.5, 29.8, 31.7, 37.1, 48.7, 123.3, 127.2, 128.1, 128.2, 128.2, 128.7, 130.2, 130.4, 138.5, 143.4, 172. HRESIMS [M + H]+m/z 398.3354 (calculated for C27H44NO 398.3359).
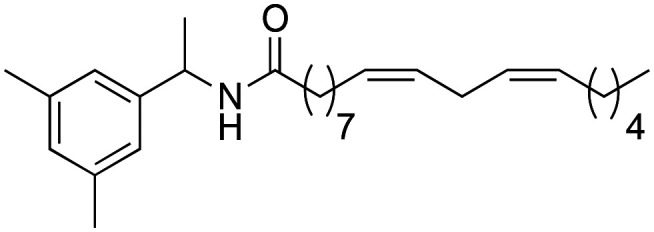
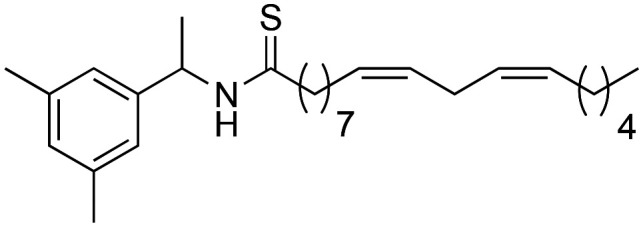
(9Z,12Z)-N-(1-(3,5-Dimethylphenyl)-ethyl)-octadeca-9,12-dienamide (3m)
(9Z,12Z)-N-(1-(3,5-Dimethylphenyl)-ethyl)-octadeca-9,12-dienamide (3m) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (239 mg, 0.80 mmol) and 1-(3,5-dimethylphenyl)-ethylamine (120 mg, 0.80 mmol). 162 mg of the synthetic macamide 3m (75% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (7 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.7 Hz), 1.41–1.25 (m, 14H), 1.45 (d, 2H, J = 6.9 Hz), 1.63 (m, 2H), 2.08–2.01 (m, 4H), 2.15 (t, 2H, J = 7.9 Hz), 2.24 (s, 3H), 2.26 (s, 3H), 2.77 (t, 2H, J = 5.9 Hz), 5.10 (quint, 1H, J = 7.2 Hz), 5.43–5.28 (m, 4H), 5.60 (d, 1H, J = 7.8 Hz), 7.12–7.03 (m, 3H) 13C NMR (CDCl3, 75 MHz): δC 14.2, 19.5, 20.0, 21.9, 22.7, 25.8, 25.9, 27.4, 29.3, 29.4, 29.5, 29.8, 31.7, 37.1, 48.7, 123.6, 127.8, 128.0, 128.2, 130.0, 130.2, 130.4, 137.0, 140.9, 172.1. HRESIMS [M + H]+m/z 412.3566 (calculated for C28H46NO 412.3574).
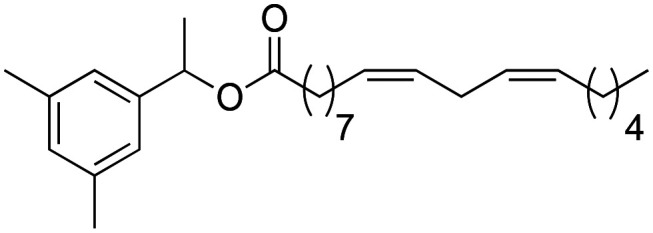
(9Z,12Z)-1-(1-(3,5-Dimethylphenyl)-ethoxy)-octadeca-9,12-dien-1-olate (4a)
(9Z,12Z)-1-(1-(3,5-Dimethylphenyl)-ethoxy)-octadeca-9,12-dien-1-olate (4a) was obtained from (9Z,12Z)-octadeca-9,12-dienoyl chloride (139 mg, 0.46 mmol) and 1-(3,5-dimethylphenyl)-ethan-1-ol (70 mg, 0.46 mmol). 128 mg of the synthetic macamide 4a (80% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (6 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.89 (t, 3H, J = 6.7 Hz), 1.41–1.25 (m, 14H), 1.45 (d, 2H, J = 6.9 Hz), 1.63 (m, 2H), 2.08–2.01 (m, 4H), 2.15 (t, 2H, J = 7.9 Hz), 2.26 (s, 6H), 2.77 (t, 2H, J = 5.9 Hz), 4.08 (q, 1H, J = 6.9 Hz) 5.10 (quint, 1H, J = 7.2 Hz), 5.43–5.28 (m, 4H), 5.60 (d, 1H, J = 7.8 Hz), 6.92 (s, 2H), 6.84 (s, 1H). 13C NMR (CDCl3, 75 MHz): δC 14.2, 19.5, 20.0, 21.9, 22.7, 25.8, 25.9, 27.4, 29.3, 29.4, 29.5, 29.8, 31.7, 37.1, 72.1, 123.8, 127.8, 128.0, 129.5, 130.0, 130.2, 130.4, 138.2, 141.8, 173.1.
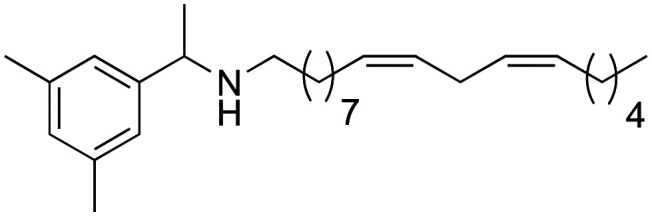
(9Z,12Z)-N-(1-(3,5-Dimethylphenyl)-ethyl)-octadeca-9,12-dien-1-amine (4b)
(9Z,12Z)-N-(1-(3,5-Dimethylphenyl)-ethyl)-octadeca-9,12-dien-1-amine (4b) was obtained from derivative 7c (80 mg, 0.20 mmol) and LiAlH4 (38 mg, 1 mmol). 59 mg of the synthetic macamide 4b (77% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (1 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.85 (t, 3H, J = 6.9 Hz), 1.33–1.20 (m, 14H), 1.32 (d, 3H, J = 10.4 Hz), 2.06–1.96 (m, 4H), 2.49–2.34 (m, 2H), 2.73 (t, 2H, J = 5.8 Hz), 3.71 (q, 1H, J = 6.6 Hz), 5.41–5.26 (m, 4H), 6.91 (s, 1H), 7.00 (s, 2H). 13C NMR (CDCl3, 75 MHz): δC 14.2, 21.5, 22.7, 25.8, 27.3, 27.4, 29.4, 29.5, 29.6, 29.8, 30.4, 31.7, 48.0, 58.7, 124.8, 126.9, 128.1, 128.1, 130.1, 130.3, 130.4, 138.3.
(9Z,12Z)-N-(1-(3,5-Dimethylphenyl)-ethyl)-octadeca-9,12-dienethioamide (4c)
(9Z,12Z)-N-(1-(3,5-Dimethylphenyl)-ethyl)-octadeca-9,12-dienethioamide (4c) was obtained from derivative 3m (70 mg, 0.17 mmol) and Lawesson's reagent (103 mg, 0.25 mmol). 62 mg of the synthetic macamide 4c (93% yield) were obtained as a yellowish liquid. The product was chromatographed with a Hex–AcOEt mixture (15 : 1). 1H NMR (CDCl3, 300 MHz): δH 0.83 (t, 3H, J = 6.9 Hz), 1.32–1.18 (m, 14H), 1.50 (d, 3H, J = 6.9 Hz), 1.72–1.62 (m, 2H), 2.03–1.93 (m, 2H), 2.54 (t, 2H, J = 7.8 Hz), 2.71 (t, 2H, J = 5.9 Hz), 5.36–5.25 (m, 4H), 5.70 (quint, 1H, J = 7.1 Hz), 6.95 (s, 2H), 7.11 (s, 1H). 13C NMR (CDCl3, 75 MHz): δC 14.0, 19.9, 22.4, 25.5, 27.1, 28.7, 29.0, 29.1, 29.2, 29.4, 29.5, 31.4, 47.0, 54.2, 124.5, 128.0, 128.2, 129.6, 130.1, 130.4, 138.6, 138.9, 141.4, 204.2.
3.8. Cell culture
The THP-1 cell line (human peripheral blood monocyte, TIB-202) was used in this study. Cells were grown in specific media according to ATCC recommendations. The incubation condition for the cells was in an atmosphere of 95% air and 5% CO2 at 37 °C. DMEM (Sigma-Aldrich, St. Louis, MO, USA), FBS (Summit Biotechnology; Ft. Collins, CO) and PBS (SAFC Biosciences, Inc. Andover-Hampshire, UK) were used as culture mediums. l-Glutamine was obtained from Applichem. Penicillin and streptomycin were purchased from Fisher Scientific (Pittsburgh, PA). For cytotoxicity and activity assays, the synthetic macamides were dissolved in DMSO (Merck) at a concentration of 10 mM.
3.9. MTT-cytotoxicity assay
THP-1 cells were seeded in 96-well plates at a density of 5 × 104 cells per well and incubated overnight at 37 °C in a humidified atmosphere of 5% CO2. Subsequently, cells were treated with test synthetic macamides at the indicated concentrations and DMSO as control for 72 h. An MTT solution (50 μL, 7 mg mL–1 in PBS) was added to each well, and plates were incubated for an additional 2 h. Next, the medium was removed and 100 μL of DMSO was added to each well to dissolve the formazan crystals that were formed. The absorbance was measured at 590 nm using a spectrophotometric ELISA plate reader (SpectraMax® i3, Molecular Devices, CA, USA). Samples were considered non-toxic for the THP-1 cell line when cell viability was ≥90%. Actinomycin D (≥95% Sigma-Aldrich, CAS Number 50-76-0) was used as a positive control at a concentration of 0.008 μM, showing cell death.
3.10. TNF-α inhibition assay
THP-1 cell (5 × 104 cells per well) were seeded on a 96-well culture plate and incubated for 12 h. The cells were then pre-treated with various concentrations of the synthetic macamides for 2 h, before stimulation with LPS (0.1 μg mL–1) with or without synthetic macamides samples for 12 h. Supernatants were then collected and the protein expression levels of TNF-α were measured by using an enzyme-linked immunosorbent assay kit (ELISA), according to the manufacturer's instructions (Diaclone Company, Besancon, France). Absorbance was read at 450 nm on a spectrophotometric ELISA plate reader (Anthos 2020, Version 2.0.5, Biochrom Ltd., UK). The percentage of TNF-α inhibition was calculated from the ratio between the observed TNF-α amount secreted by treated cells (μM) and the baseline secretion of TNF-α (pg mL–1). C87 (Sigma-Aldrich; 332420-90-3) was used as positive control at a concentration of 0.11 μM. Results were normalised to the DMSO solvent control (1%).
3.11. Statistical analysis
CC50 and IC50 values were determined by non-linear regression. All the experiments were performed in triplicate. One-way ANOVA statistical analysis (Tukey's multiple comparisons test, *p < 0.05; ***p < 0.001) was performed to evaluate the significant differences among values. All the analysis was performed using GraphPad Prism, version 8.4.0.
Author contribution
VTP and JAG experimental design of the chemistry study; ARS and LAT contributed to the analysis of the spectral data; LAT and AMS contributed to the conception and experimental design of the pharmacological study and analysis of the measurements of the clog P values; LAT contributed to the writing and review of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the Fundación de la Universidad Autónoma de Madrid (FUAM).
Footnotes
†Electronic supplementary information (ESI) available: 1H- and 13C- NMR spectra of the synthetic macamides analysed in this study are provided as supporting information (Fig. 1S–70S). See DOI: 10.1039/d0md00208a
References
- Leulier F., MacNeil L. T., Lee W. J., Rawls J. F., Cani P. D., Schwarzer M., Zhao L., Simpson S. J. Cell Metab. 2017;25:522–534. doi: 10.1016/j.cmet.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulkhaleq L. A., Assi M. A., Abdullah R., Zamri-Saad M., Taufiq-Yap Y. H., Hezmee M. N. M. Vet. World. 2018;11:627–635. doi: 10.14202/vetworld.2018.627-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. M., An J. Int. Anesthesiol. Clin. 2007;45:27–37. doi: 10.1097/AIA.0b013e318034194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelová H., Hošek J. Inflammation Res. 2013;62:641–651. doi: 10.1007/s00011-013-0633-0. [DOI] [PubMed] [Google Scholar]
- Bradley J. R. J. Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- Belgaumi A. F., Al-Bakrah M., Al-Mahr M., Al-Jefri A., Al-Musa A., Saleh M., Salim M. F., Osman M., Osman L., El-Solh H. Cancer. 2003;97:2898–2903. doi: 10.1002/cncr.11390. [DOI] [PubMed] [Google Scholar]
- Apaza L. T., Tena V. P., Serban A. M., Alonso M. J. N., Rumbero A. S. J. Ethnopharmacol. 2019;235:199–205. doi: 10.1016/j.jep.2019.02.015. [DOI] [PubMed] [Google Scholar]
- Xia C., Deng J., Chen J., Zhu Y., Song Y., Zhang Y., Li H., Lin C. Rev. Bras. Farmacogn. 2019;29:702–709. doi: 10.1016/j.bjp.2019.05.009. [DOI] [Google Scholar]
- Apaza L. T., Tena V. P., Bermejo P. B. J. Ethnopharmacol. 2020;247:112152. doi: 10.1016/j.jep.2019.112152. [DOI] [PubMed] [Google Scholar]
- Chen J. J., Gong P. F., Liu Y. L., Liu B. Y., Eggert D., Guo Y. H., Zhao M. X., Zhao Q. S., Zhao B. J. Food Sci. 2018;83:966–974. doi: 10.1111/1750-3841.14083. [DOI] [PubMed] [Google Scholar]
- Wu H., Kelley C. J., Pino-Figueroa A., Vu H. D., Maher T. J. Bioorg. Med. Chem. 2013;21:5188–5197. doi: 10.1016/j.bmc.2013.06.034. [DOI] [PubMed] [Google Scholar]
- D'Oca C. D. A. R., Coelho T., Marinho T. G., Hack C. R., Duarte R. D. A. C., da Silva P. A., D'Oca M. G. Bioorg. Med. Chem. Lett. 2010;20:5255–5257. doi: 10.1016/j.bmcl.2010.06.149. [DOI] [PubMed] [Google Scholar]
- Suzuki Y. Yakugaku Zasshi. 1977;97:5–13. doi: 10.1248/yakushi1947.97.1_5. [DOI] [PubMed] [Google Scholar]
- de Castellarnau C., Pich I., Chanquia C., Vila L., Lagunas C., Fontcuberta J., Rutllant M. Toxicology. 1993;81:181–194. doi: 10.1016/0300-483x(93)90011-g. [DOI] [PubMed] [Google Scholar]
- King A. M., De Ryck M., Kaminski R., Valade A., Stables J. P., Kohn H. J. Med. Chem. 2011;54:6432–6442. doi: 10.1021/jm200760a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Wang H., Guo F., Si N., Brantner A., Yang J., Han L., Wei X., Zhao H., Bian B. Molecules. 2018;23:E2929. doi: 10.3390/molecules23112929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X. X., Xiong C., He M., He C., Yin Z. Lett. Drug Des. Discovery. 2016:13. doi: 10.2174/1570180813888160201100814. [DOI] [Google Scholar]
- Neises B., Steglich W. Angew. Chem., Int. Ed. Engl. 1978;17(7):522–524. doi: 10.1002/anie.197805221. [DOI] [Google Scholar]
- Gilles V., Vieira M. A., Lacerda V., Castro E. V. R., Santos R. B., Orestes E., Carneiro J. W. M., Greco S. J. J. Braz. Chem. Soc. 2015;26(1):74–83. doi: 10.5935/0103-5053.20140216. [DOI] [Google Scholar]
- Sahariah P., Benediktssdóttir B. E., Hjálmarsdóttir M. Á., Sigurjonsson O. E., Sørensen K. K., Thygesen M. B., Jensen K. J., Másson M. Biomacromolecules. 2015;16(5):1449–1460. doi: 10.1021/acs.biomac.5b00163. [DOI] [PubMed] [Google Scholar]
- Waring M. J., Arrowsmith A., Leach A. R., Leeson P. D., Mandrell S., Owen R. M., Pairaudeau G., Pennie W. D., Pickett S. D., Wang J., Wallace O., Weir A. Nat. Rev. Drug Discovery. 2015;14:475–486. doi: 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- Arnott J. A., Planey S. L. Expert Opin. Drug Discovery. 2012;10:863–875. doi: 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- Alasmari M., Böhlke M., Kelley C., Maher T., Pino-Figueroa A. Mol. Neurobiol. 2019;56:1770–1781. doi: 10.1007/s12035-018-1115-8. [DOI] [PubMed] [Google Scholar]
- Rengstl D., Diat O., Klein R., Kunz W. Langmuir. 2013;29(8):2506–2519. doi: 10.1021/la304431c. [DOI] [PubMed] [Google Scholar]
- Apaza L. T., Rodríguez M. C., Potente G., Rumbero A. S. Planta Med. Int. Open. 2020;7:e88–e99. doi: 10.1055/a-1159-4242. [DOI] [Google Scholar]
- Gung B. W., Wekesa F., Barnes C. L. J. Org. Chem. 2008;73(5):1803–1808. doi: 10.1021/jo702354x. [DOI] [PubMed] [Google Scholar]
- Sander T., Freyss J., von Korff M., Rufener C. J. Chem. Inf. Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- Stasyuk O. A., Szatylowicz H., Krygowski T. M., Fonseca Guerra C. Phys. Chem. Chem. Phys. 2016;18(17):11624–11633. doi: 10.1039/c5cp07483e. [DOI] [PubMed] [Google Scholar]
- Ferreira J. E. V., da Costa C. H. S., de Miranda R. M., Figueiredo A. F., Ginarte Y. M. A. Educ. Quim. 2014;25(4):418–424. doi: 10.1016/s0187-893x(14)70061-8. [DOI] [Google Scholar]
- Moncada J. L., Morán G. S. Quim. Nova. 2008;31(5):1255–1258. doi: 10.1590/s0100-40422008000500057. [DOI] [Google Scholar]
- Habtemariam S. Planta Med. 2002;66:303–313. doi: 10.1055/s-2000-8660. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.