

Abstract
The hallmark pathologies of the Alzheimer’s disease (AD) brain are Aβ-containing senile plaques and neurofibrillary tangles formed from the microtubule (MT)-binding tau protein. Tau becomes hyperphosphorylated and disengages from MTs in AD, with evidence of resulting MT structure/function defects. Brain-penetrant MT-stabilizing compounds can normalize MTs and axonal transport in mouse models with tau pathology, thereby reducing neuron loss and decreasing tau pathology. MT dysfunction is also observed in dystrophic axons adjacent to Aβ plaques, resulting in accumulation of amyloid precursor protein (APP) and BACE1 with the potential for enhanced localized Aβ generation. We have examined whether the brain-penetrant MT-stabilizing compound, CNDR-51657, might decrease plaque-associated axonal dystrophy and Aβ release in 5XFAD mice that develop an abundance of Aβ plaques. Administration of CNDR-51657 to 1.5-month old male and female 5XFAD mice for 4- or 7-weeks led to decreased soluble brain Aβ that coincided with reduced APP and BACE1 levels, resulting in decreased formation of insoluble Aβ deposits. These data suggest a vicious cycle whereby initial Aβ plaque formation causes MT disruption in nearby axons, resulting in the local accumulation of APP and BACE1 that facilitates additional Aβ generation and plaque deposition. The ability of a MT-stabilizing compound to attenuate this cycle, and also reduce deficits resulting from reduced tau binding to MTs, suggests that molecules of this type hold promise as potential AD therapeutics.
I. Narrative
Contextual Background
Alzheimer’s disease (AD) brain is characterized by the presence of extracellular senile plaques composed of amyloid β (Aβ) peptides, and intracellular inclusions formed from tau protein that are commonly referred to as neurofibrillary tangles (NFTs) [1, 2]. Plaque-forming Aβ peptides are generated after proteolytic cleavage of APP by β-site APP cleaving enzyme (BACE) and γ-secretase [3]. Mutations in APP and in the presenilin (PS) proteins that comprise the enzymatic activity within the γ-secretase complex cause familial forms of AD [4, 5], and the finding of disease-causing mutations in genes linked to Aβ production led to the “amyloid cascade” hypothesis [6, 7], which postulates that Aβ plaques initiate a series of events leading to the neuron loss and cognitive deficits in AD. In this regard, senile plaques may contribute to AD neurodegeneration by facilitating the spread of brain tau pathology from its initial sites in the entorhinal cortex/hippocampus into neocortical regions [8]. Notably, the extent of tau pathology, but not senile plaque burden, correlates with AD cognitive status [9–12], consistent with tau pathology causing neurodegeneration in AD.
The molecular mechanism(s) by which senile plaques might enhance tau pathology is largely unknown. Tau is normally a microtubule (MT)-associated protein that appears to provide stability to the distal labile regions of axonal MTs [13, 14] while also blocking MT severing [15–17]. MTs play a critical role in neurons, as they are the conduits upon which cellular constituents are trafficked along axonal extensions via the process of axonal transport. Tau becomes hyperphosphorylated in AD, which promotes its disengagement from MTs [18–20] and is thought to increase MT dynamicity [21–24] and perhaps MT cleavage [17], with resulting impairment of axonal transport [22, 25]. Reduced tau binding to MTs may also affect the interaction of the molecule motors involved in axonal transport [26, 27]. Impairments of MT structure have been documented in AD brain [23, 24] and have also been demonstrated, along with impaired axonal transport, in transgenic (Tg) mouse models with tau pathology [21, 25, 28]. Increased levels of unbound hyperphosphorylated tau are also likely to promote the formation of the tau inclusions [29, 30] that correlate with AD symptomology. Thus, both loss of the MT-regulating functions of tau, as well as the formation of insoluble tau deposits, may contribute to the neuron loss observed in AD and related tauopathies, such as Pick’s disease, progressive supranuclear palsy and corticobasal degeneration [31].
Interestingly, MT deficits are not only found in models of tau pathology but are also observed in the swollen (dystrophic) neuronal processes that are in proximity to senile plaques in AD and Tg mouse models [32–34]. These plaque-associated MT abnormalities have been suggested to cause local impairment of axonal transport that accounts for the observed peri-plaque accumulation of APP, BACE1, and other proteins [32]. A recent study revealed that tau also accumulates in these dystrophic processes, and this localized tau pool can be converted to insoluble tau inclusions referred to as neuritic plaque tau in Tg mice harboring senile plaques [35]. Thus, plaque-associated neuronal processes are a site where tau pathology can form in proximity to Aβ deposits, representing a confluence of the hallmark AD pathologies that may provide insights into how senile plaques contribute to the cortical spread of tau pathology. Importantly, it appears that MT dysregulation is a key consequence of both Aβ and tau pathologies in AD, providing a potential unifying mechanistic target to slow AD progression.
The evidence of altered axonal MT structure/function in AD has led to the investigation of brain-penetrant MT-stabilizing compounds as potential AD therapeutics. A number of known MT-stabilizing natural products, including paclitaxel [25], epothilone D [21, 22, 28] and dictyostatin [36], proved to be beneficial in Tg mouse models with tau pathology. Importantly, these molecules increased MT density and normalized MT dynamics, with a corresponding improvement of axonal transport and reduction in tau pathology, synapse loss, and neuronal death. More recently, a non-natural product compound referred to as CNDR-51657 (hereafter 51657) [37], which stabilizes MTs by binding to a different site than the aforementioned natural products [38], conferred similar benefits in tau Tg mice [39]. As senile plaque-associated dystrophic axons have also been shown to have MT abnormalities [32, 34], we hypothesized that normalization of MT structure/function in these swollen processes with a previously characterized MT-stabilizing compound might improve regional axonal transport and slow the focal accumulation of cellular proteins such as APP and BACE1, thereby reducing Aβ generation. Here, we have tested this hypothesis by examining the effects of 51657-treatment in the 5XFAD mouse model of robust senile plaque formation [40].
Study Conclusions and Disease Implications
Our results indicate that treatment of young 1.5-month old 5XFAD mice with the MT-stabilizing compound, 51657, for either 4- or 7-weeks leads to a meaningful reduction of Aβ release and senile plaque deposition in 5XFAD mice via a lowering of APP and BACE1 protein levels. This appears to result from an improvement in MT structure/function in plaque-associated dystrophic axons. Thus, the disruption of MTs in these axons appears to initiate a cascade that leads to more fulminant Aβ deposition and further axonal dystrophy, and this cascade can be slowed by treatment with a MT-stabilizing agent such as 51657. Greater reductions of soluble and insoluble Aβ were observed in male than in female 5XFAD mice treated with 51657, likely due to the inherently higher APP expression and greater Aβ production in female than male 5XFAD mice [40, 41].
The study results further emphasize that MT abnormalities appear to be a key feature of AD, as well as other neurodegenerative diseases [31]. Importantly, small molecule MT-stabilizing agents such as 51657 can reduce AD-like pathology and confer neuronal benefits in mouse models with only tau pathology [21, 22, 28, 36, 39] or in mice with only Aβ pathology, as demonstrated here. MT-stabilizing compounds are thus unique in being able to independently affect the two key pathologies found in AD brain. Although the overall reduction of soluble and insoluble Aβ in 5XFAD mice treated with 51657 was relatively modest (20–30%), these mice are among the most aggressive Aβ plaque mouse models, producing an abundance of Aβ42 at early age [40].
As with all studies that employ transgenic mouse models of AD, it is important to recognize the potential shortcomings and limitations of the model when attempting to translate findings to human AD. In this regard, there would be merit in examining a well-characterized brain-penetrant MT-stabilizing molecule in additional Aβ plaque mouse models, particularly in those that show a slower rate of senile plaque accumulation as it is possible that a MT-stabilizing compound might provide even greater benefit in such mice. In fact, a greater reduction of Aβ release and senile plaque deposition might be seen upon treatment of early-stage AD patients with a MT-stabilizing agent than was observed in young 5XFAD mice since plaques accumulate over the course of many years in the human disease. It is also prudent to recognize the limitations of the tau Tg mouse models in which MT-stabilizing compound have proven quite efficacious. These models generally depend on the overexpression of mutated tau, and neither tau mutations or tau overexpression are characteristics of AD. Thus, the observed MT deficits in these models may not fully mimic those that have been reported in AD brain [23, 24]. Similarly, tau knockout mice do not show gross changes in MT structure that would be predicted from loss of tau function, although CNS deficits and compensatory changes in other MT-associated proteins have been noted [42]. Thus, it is possible that that MT abnormalities in tau Tg mouse models or in AD are not simply the result of tau loss-of-function, but perhaps are also due to effects stemming from the formation of tau inclusions. Regardless of mechanism, the observation of MT deficits in AD suggests that MT-stabilizing molecules might provide therapeutic benefit.
The prior findings in tau Tg mice have led to MT-stabilizing compounds being considered as therapeutic candidates for AD. A short 9-week phase 1b trial was conducted in which AD patients received low doses of the natural product MT-stabilizing agent, epothilone D (BMS-241027; clinicaltrials.gov identifier NCT01492374). No adverse events were noted in this small study, although improvements were not observed in cerebrospinal fluid disease biomarkers or in MRI brain volumes [43]. Although this might lead one to surmise that this therapeutic strategy was unsuccessful, the absence of biomarker improvement is frankly unsurprising given that the trial was not powered for these endpoints and because the study duration was a fraction of the typical 18–24 month AD disease modification trial. More recently, AD patients that received the taxane, TPI-287, in a Phase 1b trial were reported to show a significant improvement in MMSE score relative to the placebo group after 12 weeks of dosing (www.corticebiosciences.com; 11/03/17 press release). Although this report is encouraging, the short trial duration and small patient cohorts again limit the conclusions that can be drawn from this study, and thus the clinical potential of MT-stabilizing drugs for AD has not been fully explored. Accordingly, there would seem to be considerable merit in further investigating the potential of brain-penetrant MT-stabilizing drugs in AD.
There are clearly challenges to utilizing drugs of this type for the treatment of AD or other neurodegenerative conditions. One concern is safety, as MT-stabilizing drugs are effective anti-cancer agents due to their ability to promote cell death through alteration of MT-rich mitotic spindles during cancer cell division. However, our prior studies with epothiline D revealed that doses that were ~1/100th those used in cancer trials effectively normalized MT structure and function in the brains of tau Tg mice [28]. Moreover, these low doses of epothilone D did not lead to changes in blood cell counts, including neutrophils [28], which is a leading side-effect when MT-stabilizing drugs are used in oncology settings [44, 45]. Similarly, the doses of 51657 that led to improvements in tau Tg mice, and here in Tg Aβ plaque mice, did not affect neutrophil or other blood cell counts [39]. Finally, the doses of epothilone D used in the short Phase 1b trial, which were similar to those used in Tg mouse studies when scaled for species, did not result in adverse events [43]. The fact that much lower doses of MT-stabilizing compounds are needed to improve brain outcomes in mouse models with AD pathology than are required to affect cell division indicates that the brain MT abnormalities can be “repaired” with lower concentrations of compound than are required to affect cellular mitosis.
Another challenge to the clinical development of MT-stabilizing drugs for AD and related diseases is the absence of a readily assessed marker of target engagement that can be utilized to assist in determining proper drug dosage in patients. Although this situation is not unique to this drug class, it does increase the clinical development risk. The recent development of tau PET ligands that can be used to monitor tau pathological burden [46, 47] provides one potential study endpoint for MT-stabilizing compounds, which have been shown to reduce tau pathology in mouse models. However, improvements based on tau PET imaging are likely to occur over many months and thus limit this methodology as a dosing biomarker. At this time, evaluation of cerebrospinal fluid drug levels to ensure that CNS exposures to MT-stabilizing candidates are consistent with those shown to be efficacious in mouse AD models may have to suffice for human dose estimation, although there would clearly be benefit in identifying a direct or indirect acute biomarker of MT stabilization or axonal transport. Notwithstanding these challenges, the ability of compounds such as 51657 to attenuate the consequences of both Aβ and tau pathology suggest that MT-stabilizing molecules merit further consideration as potential AD therapeutic candidates.
2. Consolidated Results and Study Design
In an initial study, young 1.5-month old 5XFAD mice that have not yet developed Aβ plaque accumulations [40] were treated with 51657 twice-weekly at 3 mg/kg [39] or with vehicle (4 males and 4 females in each treatment group), for a total of 7 weeks, after which fixed brain sections from one hemisphere of each study mouse were stained for detection of Aβ plaques, APP-positive plaque-associated dystrophic axons, and/or β3-tubulin-positive MTs. Tissue from the other hemisphere from each mouse was used for additional biochemical analyses. An evaluation of cortical plaque burden in two different brain regions (bregmas −1.96 and 0.2) revealed a significant reduction in Aβ deposits in male mice treated with 51657 compared to those that received vehicle only. A comparable decrease in cortical plaque staining was not observed in 51657-treated female 5XFAD mice. Notably, there was a highly significant correlation between the cortical Aβ plaque burden and plaque-associated APP-positive dystrophic axon staining, such that mice with less plaques had less axonal dystrophy. A similar trend was observed upon examination of a brain region (bregma −3.08) that contains the hippocampus and subiculum, with the latter being a site of early and robust plaque formation in 5XFAD mice. Male mice that were treated with 51657 again showed decreased Aβ-positive plaques, and interestingly, they also had less MT disruption, as revealed by an attenuation in plaque-associated MT “voids” that could be visualized after staining with β3-tubulin antibody.
The findings obtained upon immunofluorescence evaluation of the 5XFAD mouse brains led to a more quantitative evaluation of Aβ plaques through an assessment of insoluble Aβ peptide levels in homogenate samples prepared from the entire cortex and hippocampus/subiculum from one hemisphere of each study mice. These analyses focused on the longer Aβ42 peptide, which is produced at vastly higher levels than Aβ40 in young 5XFAD mice due to these mice harboring multiple mutations in the APP and PS1 genes that results in Aβ42 over-production [40]. The biochemical measurements revealed an overall significant 28% decrease of insoluble Aβ42 in the combined cortical and hippocampal homogenates in the 51657-treated 5XFAD mice group (males and females combined). As seen by immunofluorescence imaging, male 5XFAD mice had a greater decrease of insoluble Aβ42 upon 51657-treatment than did females, although a trend toward decreased insoluble Aβ was seen in female 5XFAD mice upon sampling of the entire cortical and hippocampal regions. An evaluation of soluble Aβ42 levels from the brain homogenates revealed a significant decrease in the 51657-treated 5XFAD mice (males and females combined), with the magnitude of change being similar to that observed for insoluble Aβ42 with a greater reduction of soluble Aβ42 in male (34%) than in female (10%) mice. There was a highly significant correlation between the relative levels of soluble and insoluble Aβ42 for each study mouse, suggesting that the extent of plaque deposition was directly related to soluble Aβ42 levels. The overall insoluble and soluble Aβ42 levels were higher in the female 5XFAD mice than in their male counterparts, consistent with prior observations [40, 41]. This sex-dependent difference in Aβ generation likely explains why 51657-treatment was less effective in reducing Aβ42 levels in female than male mice. Nonetheless, the >20% reduction of soluble Aβ42 levels after 51657 treatment implies that plaque-associated dystrophic axons account for an appreciable amount of the Aβ42 production in 5XFAD mice, even though these mice produce an abundance of Aβ42 through transgene overexpression of mutated APP and PS1.
To further confirm the finding of reduced Aβ levels in 5XFAD mice treated with 51657, another study was conducted in which 1.5-month old male and female 5XFAD mice were treated with vehicle or 51657 for 4 weeks. This shorter study duration allowed for an examination of changes in Aβ levels during an earlier stage of SP deposition, with plaques in 2.5-month old 5XFAD mice being largely confined to the subiculum. Again, a significant reduction of soluble Aβ42 was observed in combined hippocampal and cortical homogenates in the 5XFAD mice treated with 51657. As in the prior study, the extent of compound-mediated lowering of soluble Aβ was again greater in male (42%) than in female (23%) 5XFAD mice, although both sexes showed a greater decrease of soluble Aβ42 than was observed in the older mice that were dosed for 7 weeks. The measurable reduction of soluble Aβ in the 5XFAD mice treated with the MT-stabilizing agent suggested that a decrease should also be observed in carboxyl-terminal APP fragments generated by BACE1 cleavage of APP. This was confirmed in immunoblots of cortical homogenates from the 5XFAD mice, where there was a significant reduction of the APP fragments generated both by BACE1 and α-secretase cleavage in 51657-treated mice. The lowering of both BACE1 and α-secretase APP cleavage products after 51657-treatment indicated that there was an overall decrease of APP processing, perhaps due to decreased APP accumulation. Immunoblot analyses of APP holoprotein confirmed that there was a significant decrease in the 5XFAD mice receiving the MT-stabilizing compound, with male mice again showing a somewhat greater reduction than females. A similar reduction of BACE1 was observed in the 51657-treated 5XFAD mice, consistent with a reduction of protein accumulation within dystrophic processes due to improved MT-dependent transport.
3. Detailed Methods and Results
Methods
51657 Synthesis
The synthesis of 51657 was conducted at the 0.5 g scale following procedures described previously [37, 39], with the purity and spectroscopic properties being identical to those previously reported.
5XFAD Mouse Studies
5XFAD mice harboring transgenes for human APP and PS1 with a total of five mutations found in familial AD [40] were the kind gift of Dr. Robert Vassar, and are commercially available from Jackson Laboratories (B6SJL-g(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/Mmjax). All methods utilizing these mice were first submitted and approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC). The 5XFAD mice were utilized to evaluate the effect of the MT-stabilizing compound, 51657, on Aβ plaque-associated axonal dystrophy and Aβ biogenesis. Two studies were conducted in which 1.5-month old 5XFAD mice (groups of 4 males and 4 females) were dosed twice-weekly (3 mg/kg, i.p.) with 51657. A 51657 stock solution was prepared in DMSO, and this was diluted to 9% DMSO/91% corn oil and administered at 2 μl/g body weight to achieve the desired dose. An additional group of 5XFAD mice received twice-weekly injections of vehicle only. In one study, the mice were doses for a total of 4 weeks (to 2.5-months of age), whereas in another study dosing continued for 7 weeks (to 3.3-months of age). All mice were monitored for signs of abnormal behavior or distress, and were weighed weekly to monitor body weight. No behavioral abnormalities were observed and body weight gain was similar in the vehicle- and 51657-treated mice. Study mice were perfused with PBS (20 ml) after being deeply anesthetized using a protocol approved by the University of Pennsylvania IACUC. Brains were collected from all study mice, with one hemisphere processed for immunohistochemistry as previously described [22, 28]. The cortex and hippocampus, including subiculum, were manually dissected from the other brain hemisphere for use in biochemical analyses prior to freezing. The midbrain of each mouse was retained for APP mRNA analysis.
Immunofluorescence Analyses of 5XFAD Brain Sections
Paraffin-embedded sections of brain hemispheres (6 μm) from bregma −3.08, −1.96 and 0.2 were prepared from each study mouse essentially as previously described [28, 39] and stained to visualize Aβ plaques, APP-positive dystrophic processes and/or areas of MT voids, utilizing the following antibodies and dyes:
Aβ: NAB228 monoclonal antibody (mouse anti-Aβ1–11; in-house [48]) or X34 dye [49].
APP: 22C11 mouse monoclonal antibody (APP N-terminus; Millipore, Inc.).
MT: TUJ1 (βIII-tubulin mouse monoclonal antibody; Biolegend, Inc.).
Bound primary antibodies were detected with one of the following secondary antibodies:
Goat anti-mouse IgG2a conjugated with Alexa Fluor 647 (ThermoFisher).
Goat anti-mouse IgG1 conjugated with Alexa Fluor 488 (ThermoFisher)
Goat anti-mouse IgG1 conjugated with Alexa Fluor 568 (Millipore)
Immunostained sections from all study mice were masked to treatment identification and were imaged using a 10x microscopic objective. Cortical APP-positive dystrophic processes were manually annotated to avoid confounding signal found in neuronal soma. The curated area for APP was then transferred onto the NAB228 channel to quantify NAB228-positive SP signal, with exclusion of NASB228-positive neuronal soma. All images were quantified using Image J software, with the area and average optical density (OD) obtained and results reported as NAB228 or APP area x OD divided by the total area of the measured image. A similar process of manual APP annotation followed by overly onto the X34 channel was used to identify X34-positive SPs, with quantification of X34 area x OD divided by the total area of the measured image.
ELISA Quantification of Aβ42
Frozen whole cortical and hippocampal brain samples from a hemisphere of each study mouse were each homogenized in RAB high-salt buffer (0.1 M MES, pH 7.0, 1 mM EGTA, 0.5 M MgSO4, 0.75 M NaCl and 0.02 M NaF), with 0.5 ml used for cortical samples and 0.15 ml for hippocampal samples, followed by repeated sonication on ice. A portion of the cortical lysate (0.3 ml) was frozen for later immunoblot analyses, and the remainder of the cortical homogenate and hippocampal homogenates were centrifuged at 100,000 × g for 30 min at 40C. The supernatant was collected as the high-salt fraction, and the pellet was resuspended in 0.1 ml of RAB high-salt buffer and re-centrifuged as above, with the resulting supernatant added to the first high-salt sample. This process was repeated again, to yield the complete high-salt fraction. The pellet remaining after the three RAB high-salt homogenizations were treated with 0.2 ml of RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% Na deoxycholate, 1% NP-40 and 0.1% SDS). The pellet was sonicated, and then centrifuged as above. The supernatant was removed, and the pellet was treated with 0.1 ml of 70% formic acid followed by sonication to solubilize plaque Aβ. These samples were centrifuged as above, and the supernatant retained as the formic acid fraction. Total proteins levels were determined in all of the high-salt samples by BCA analysis. Aβ42 levels were assessed in the high-salt and formic acid fractions using a specific ELISA as previously described [50, 51]. Dilutions of the high-salt and formic acid samples were made to ensure that resulting Aβ42 measurements were within the linear range of the Aβ42 standard curve. Aβ42 levels were normalized to the amount of total protein within the high-salt fraction. For the formic acid samples, the samples were normalized to the corresponding protein amount in the high-salt fraction based on the fraction of formic acid sample used in the Aβ42 ELISA. To determine the combined cortical and hippocampal/subiculum Aβ42 values, the total cortical and hippocampal protein amounts were first determined from the high-salt fractions (total homogenate volume multiplied by protein concentration). The total protein fraction attributable to the cortex and hippocampus if the samples were to be combined was determined (typically ~60–65% cortical and 35–40% hippocampal/subiculum). These fractional values were then multiplied by the cortical and hippocampal/subiculum Aβ values to yield the weighted values, which were summed to yield the combined cortical and hippocampal/subiculum Aβ values. Typically, these values were normalized to the mean of the vehicle group for each sex to eliminate known differences in Aβ levels between males and females.
Immunoblot Assessments of APP, BACE1 and APP C-terminal Fragments
Cortical RAB high-salt homogenates that had been retained prior to centrifugation as described above were utilized for immunoblot analyses of APP holoprotein, BACE1 and APP carboxy-terminal C99/C89 and C83 fragments. 0.1 ml of cortical homogenates were treated with 25 μl of 10% SDS to solubilize membrane proteins, followed by centrifugation at 100,000 × g for 20 min at 40C. The supernatant was collected, and the pellet was re-extracted with 0.1 ml of 10% SDS in water. After centrifugation, this supernatant was combined with the first supernatant sample, and the total protein determined by BCA. A total of 30 μg of protein from each study mouse underwent SDS-PAGE (4–12% gradient gels), followed by blotting to nitrocellulose membranes as previously described [50]. Intact APP holoprotein and carboxy-terminal APP fragments were detected using the carboxy-terminal APP monoclonal antibody 5685 [48], as previously described [50]. BACE1 was also detected (Abcam), and GAPDH (Advanced Immunochemical) was used as a loading control. Secondary antibodies and imaging were essentially as described [50], using the Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE). All APP, APP fragments and BACE1 integrated signals were normalized to GAPDH integrated signal for each sample. The quantification was conducted by an individual masked to the blot lane assignments.
APP mRNA Analysis
Frozen midbrain samples (~30 mg) from male 5XFAD mice treated with vehicle or 51657 were homogenized in 0.6 ml of lysis buffer (RLT buffer containing 1% β-mercaptoethanol; Qiagen RNeasy mini-kit). Total RNA was subsequently isolated using the protocol specified by the manufacturer (Qiagene RNeasy mini-kit). Human APP and mouse GAPDH mRNA were analyzed by qPCR using previously described primers and quantification methods [50].
Statistical Analyses
The statistical methods utilized for each graph, including replicate numbers and statistical details, are noted in the figure legends. All statistical analyses were conducted with GraphPad Prism software. Briefly, vehicle- and 51657-treated 5XFAD mice were compared by unpaired 2-tailed t-test, with error bars on all graphs representing SEM. Correlation plots underwent linear fit analyses to obtain r2 and p-values, with dashed lines representing the 95% confidence intervals.
Results
5XFAD mice develop an abundance of senile plaques in the cortex, hippocampus/subiculum and some midbrain regions as a result of overexpression of human APP and PS1 that harbor a total of 5 mutations linked to early-onset AD [40]. Neuronal processes, particularly axons, in proximity to plaques in 5XFAD mice are swollen and show accumulations of several neuronal proteins [32, 34], replicating what is observed in human AD brain [32, 52, 53]. Notably, there is focal MT disruption in these axons that may be the cause of the accumulated cellular proteins [32], including APP and BACE1, and it has been suggested that these dystrophic axons are a site of Aβ production [32, 33]. This led us to investigate whether 51657 [37, 39] might reduce Aβ plaque-associated MT deficits in 5XFAD mice, with a resulting decrease in plaque biogenesis and associated axonal dystrophy.
Young 1.5-month old 5XFAD mice that have not yet developed appreciable Aβ plaque accumulations [40] were treated with 51657 twice-weekly at 3 mg/kg [39] or with vehicle for a total of 7 weeks, after which formalin-fixed coronal brain sections from one hemisphere of each study mouse were stained for detection of Aβ plaques and APP-positive plaque-associated processes. Tissue from the other hemisphere from each mouse was saved for biochemical analyses. Masked quantification was conducted on stained sections encompassing cortical regions with appreciable Aβ plaques (bregmas −1.96 and 0.2). Interestingly, a significant reduction in plaques was observed in male mice treated with 51657 when the results from both bregma levels were combined (Fig. 1C), with the compound-induced decrease being particularly noticeable at bregma −1.96 (Fig. 1A). A reduction in cortical plaques was not seen in 51657-treated female 5XFAD mice, where a higher Aβ plaque burden and a greater degree of variability was observed (Fig. 1C). The finding of greater plaque deposition in female 5XFAD mice is consistent with prior observations [40]. A reduction of plaque-associated APP staining, indicative of decreased axonal dystrophy, was also observed in the male mice and there was a strong correlation between Aβ plaque burden and plaque-associated APP staining for both male and female 5XFAD mice (Fig. 1B and 1D). To further examine Aβ plaque load in the 5XFAD mice, triple-stained sections from the plaque-rich subiculum (bregma −3.08) were examined for amyloid plaques (X34 dye; [49]), APP and MTs (βIII-tubulin antibody) (Fig. 2A). As observed in the cortex, masked quantification revealed a greater reduction of X34-positive plaques in the subiculum of male than female mice receiving 51657, although this did not reach statistical significance (Fig. 2B). At this bregma level, the female and male mice had comparable amounts of X34-positive plaques. Notably, and in agreement with prior findings [32], there were plaque-associated βIII-tubulin “voids” indicative of MT disruptions that coincided with X34-positive plaques and APP-positive dystrophic processes, and these were visibly reduced in the male mice that showed reduction of plaques (Fig. 2A).
Figure 1.
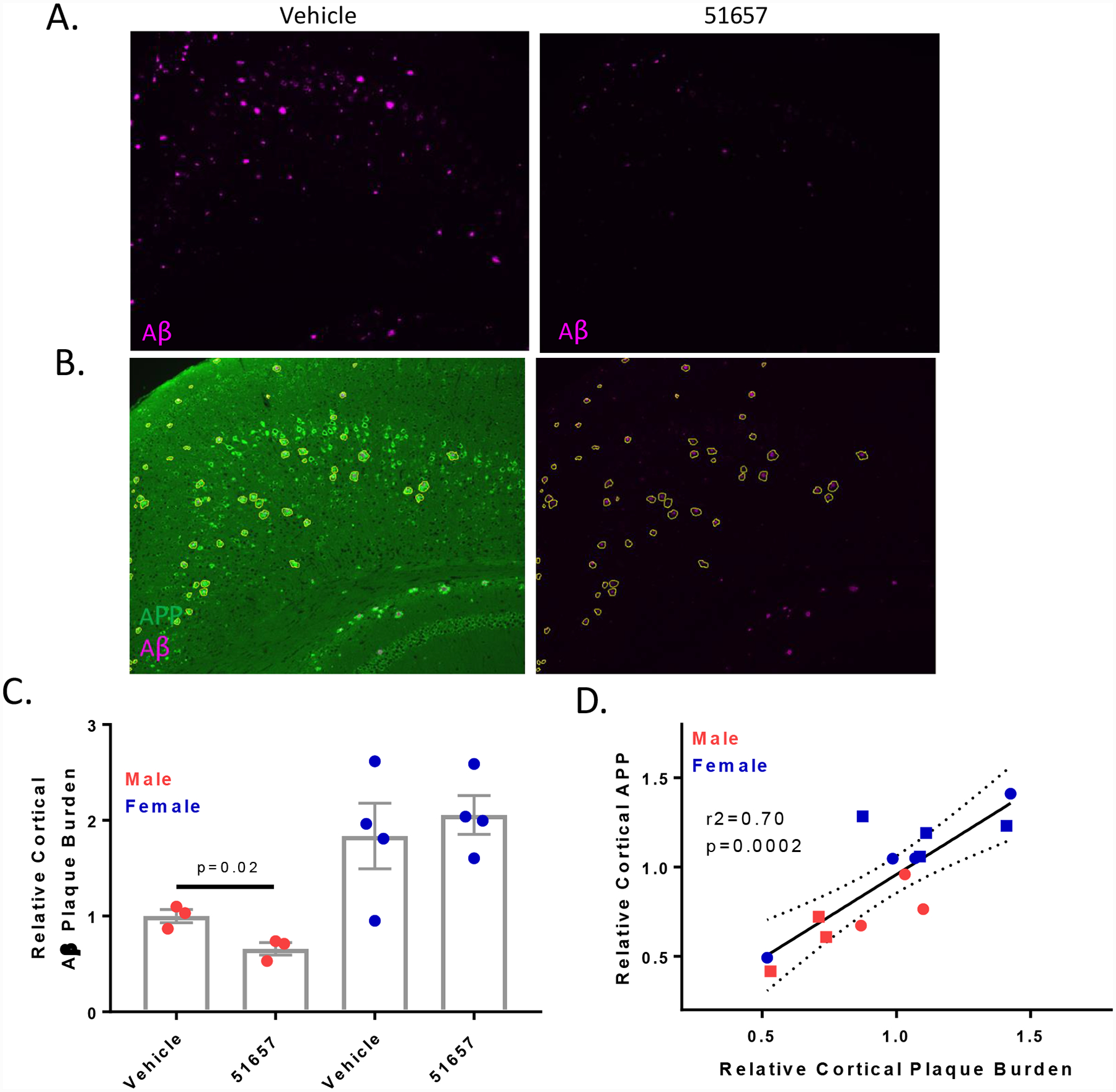
1.5-month old 5XFAD mice treated with 51657 for 7 weeks have reduced SPs and axonal dystrophy. A. Male 5XFAD mice treated with 51657 show reduced cortical SPs compared to those treated with vehicle that is evident in sections from bregma −1.96. B. Representative image of a cortical section (bregma −1.96) stained for APP (22C11; green) and SPs (NAB228; cyan), with the adjacent image showing annotation of SP-associated APP-positive processes, with exclusion of confounding APP-positive neuronal soma. C. Quantification of cortical Aβ (NAB228) integrated signal normalized to total measured area for combined bregma −1.96 and 0.2 analyses. A single high outlier mouse (p<0.05 by Grubb’s test) from both the male vehicle and 51657 group was omitted. Data were normalized to the mean of the male vehicle group, with n=3 vehicle- and 51657-treated male 5XFAD mice, and n=4 vehicle- and 51657-treated female 5XFAD mice. Statistical comparison of the male 5XFAD mice was by unpaired 2-tailed t-test (t=3.60, df=4). Error bars represent SEM. D. Correlation plot of the relative Aβ and plaque-associated APP signals from combined bregma −1.96 and 0.2 data for each 5XFAD study mouse after normalization to the vehicle mean for each sex. Data underwent linear fit analysis, with r2 and p-value shown in the graph and dashed lines representing the 95% confidence interval. Circles=Vehicle; Squares=51657-treated.
Figure 2.
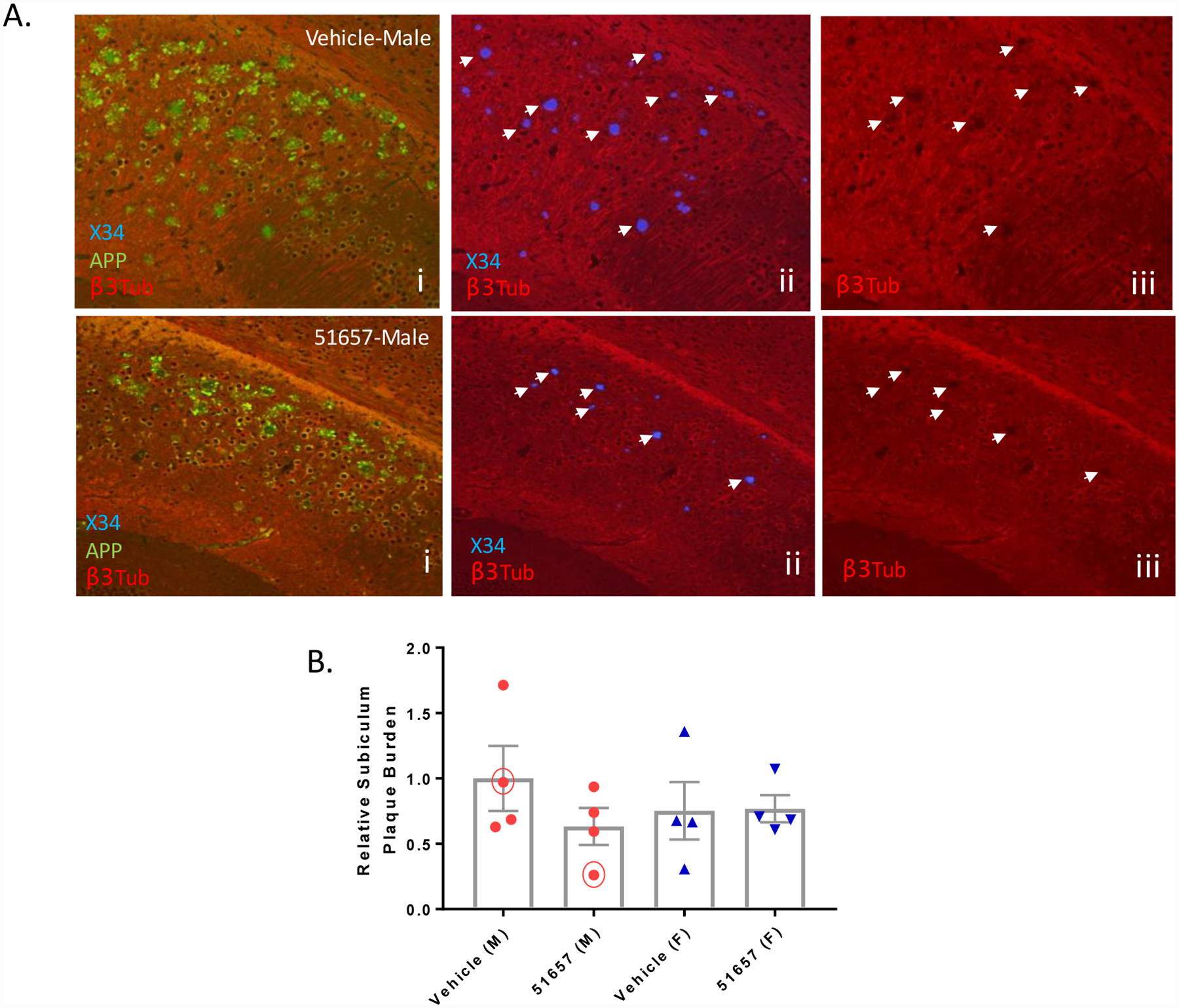
Male 5XFAD mice with decreased X34-positive SPs have decreased focal MT disruptions. A. Sections from the subiculum (bregma −3.08) of 5XFAD study mice i. stained for Aβ plaques (X34), APP-positive dystrophic processes and MTs (βIII-tubulin) show evidence of SP-associated voids of βIII-tubulin staining, indicative of MT disruption (plaques highlighted with arrowheads in panels ii with associated βIII-tubulin voids at identical sites shown in iii). The number of MT voids were reduced in male mice with reduced X34-positive plaques and APP-positive processes (compare upper and lower panels). B. Quantification of X34-positive plaques in the subiculum of the 5XFAD study mice after normalization to the mean of the male vehicle group, with n=4 vehicle- and 51657-treated male and female 5XFAD mice. The trend toward decreased plaque burden in the male 51657 treatment group did not reach statistical significance by unpaired 2-tailed t-test (t=1.28, df=6). Circled data points represent the mice shown in A. Error bars represent SEM.
Given the decreased Aβ plaque staining in the male 5XFAD mice treated with 51657 upon an immunofluorescence survey of brain sections, we conducted a quantitative assessment of insoluble Aβ from hemisphere homogenates comprising the entire cortex and hippocampus/subiculum of the study mice. As young 5XFAD mice synthesize ~25-fold greater amounts of Aβ42 than Aβ40, and because plaques in 5XFAD mice harbor predominantly Aβ42 at this age [40], only Aβ42 levels were reliably quantified by ELISA. As revealed in Fig. 3A, a significant 28% reduction in insoluble Aβ42 was observed in combined cortex and hippocampus/subiculum samples from the 5XFAD mice treated with 51657 after normalization by sex to account for the difference in Aβ plaque burden between males and females. As was seen by imaging, male 5XFAD mice showed a greater decrease of insoluble Aβ42 upon 51657-treatment than did females, although sampling of the entire cortical and hippocampal regions revealed a trend toward decreased insoluble Aβ in female 5XFAD mice. There was greater insoluble Aβ42 in the hippocampus/subiculum samples than in cortical samples for each sex (Fig 3B), consistent with the subiculum having abundant plaques in 5XFAD mice (Fig. 2A and [40]), with female mice showing greater insoluble Aβ burden in both cortex and hippocampus/subiculum than male mice [40]. Treatment with 51657 resulted in trends toward reductions of both cortical and hippocampal insoluble Aβ42 accumulation in both sexes of 5XFAD mice, although statistical significance was not achieved in these subgroup analyses where the group sizes were smaller (Fig. 3B).
Figure 3.
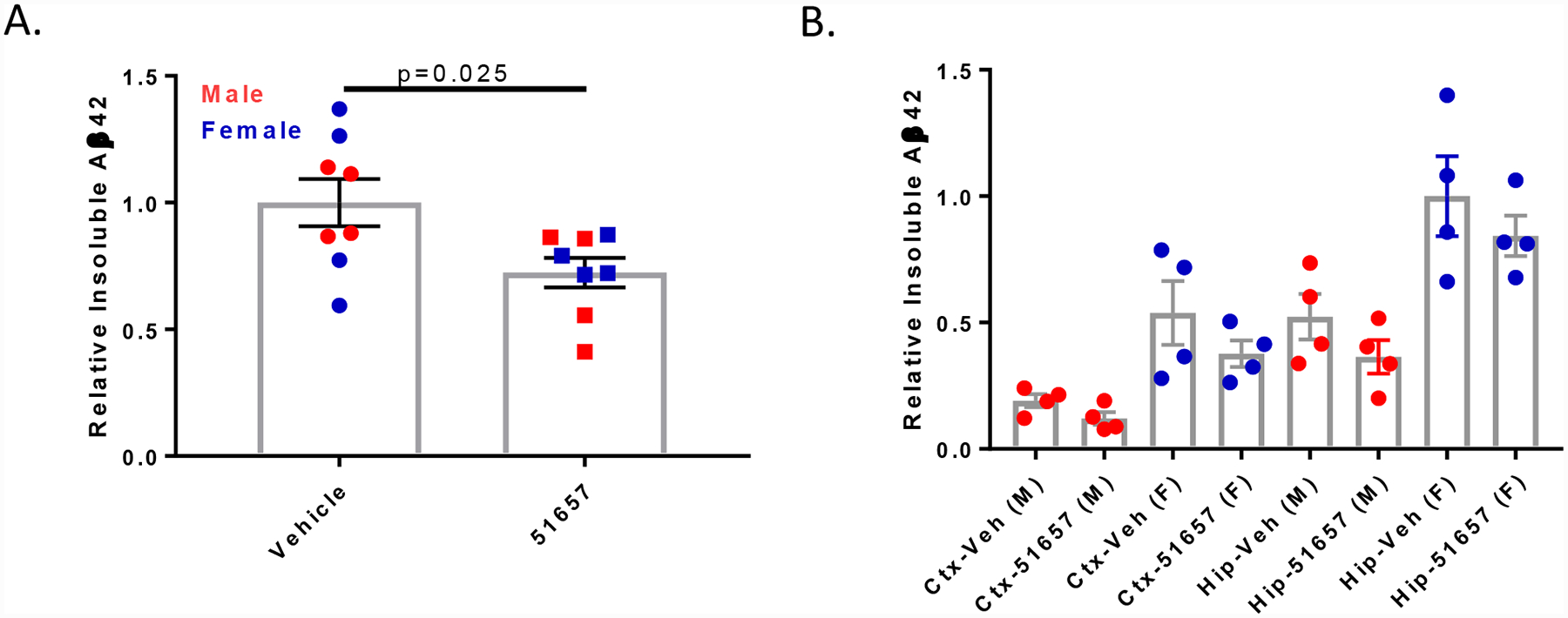
1.5-month old 5XFAD mice treated with 51657 for 7 weeks have reduced insoluble Aβ. A. Insoluble Aβ42 from ELISA analyses of combined cortex (Ctx) and hippocampus/subiculum (Hip) homogenates from 5XFAD study mice. Data were normalized to the vehicle mean for each sex, with n=4 vehicle- and 51657-treated male and female 5XFAD mice. Statistical comparison was by unpaired 2-tailed t-test (t=2.51, df=14). Error bars represent SEM. B. Insoluble Aβ42 in the cortex and hippocampus/subiculum of the 5XFAD study mice plotted by sex, with normalization to the vehicle mean from the female hippocampus/subiculum group. Error bars represent SEM.
As plaque-associated dystrophic processes may be a local source of Aβ production [32], the reduction of insoluble Aβ observed in the 51657-treated mice may have resulted from a lowering of Aβ42 release. Indeed, ELISA measurements revealed a significant reduction in combined cortical and hippocampal soluble Aβ42 in the 5XFAD mice receiving 51657 (Fig. 4A), with the magnitude of change being similar to that observed for insoluble Aβ42 (Fig. 3A) and a greater reduction of soluble Aβ42 in male (34%) than in female (10%) 5XFAD mice. Greater amounts of soluble Aβ42 were found in the hippocampus/subiculum than in the cortex of 5XFAD mice of both sexes (Fig. 4B), as was observed for insoluble Aβ42. Moreover, the reductions of soluble Aβ42 in the 51657-treatment group generally mirrored those observed with insoluble Aβ42 when examined by brain region and sex (compare Figs. 3B and 4B), and there was a significant correlation between the relative levels of soluble and insoluble Aβ42 for each study mouse (Fig. 4C), suggesting that the extent of plaque deposition was directly related to soluble Aβ42 levels. We note that whereas soluble Aβ42 in the hippocampus of vehicle-treated male and female 5XFAD mice were comparable, insoluble Aβ42 levels were higher in the female than male mice (Figs. 3B and 4B), suggesting that female mice had high levels of hippocampal soluble Aβ42 at a younger age than the male mice that resulted in greater plaque deposition by the time of the study completion. The greater overall Aβ release and more pronounced plaque deposition in female 5XFAD mice likely explains why 51657-treatment was less effective in reducing soluble and insoluble Aβ42 in female than male mice. Nonetheless, the >20% reduction of soluble Aβ42 levels on average after 51657 treatment implies that plaque-associated dystrophic axons account for an appreciable amount of the Aβ42 production in 5XFAD mice, even though these mice already produce abundant Aβ42 through overexpression of mutated APP and BACE1.
Figure 4.
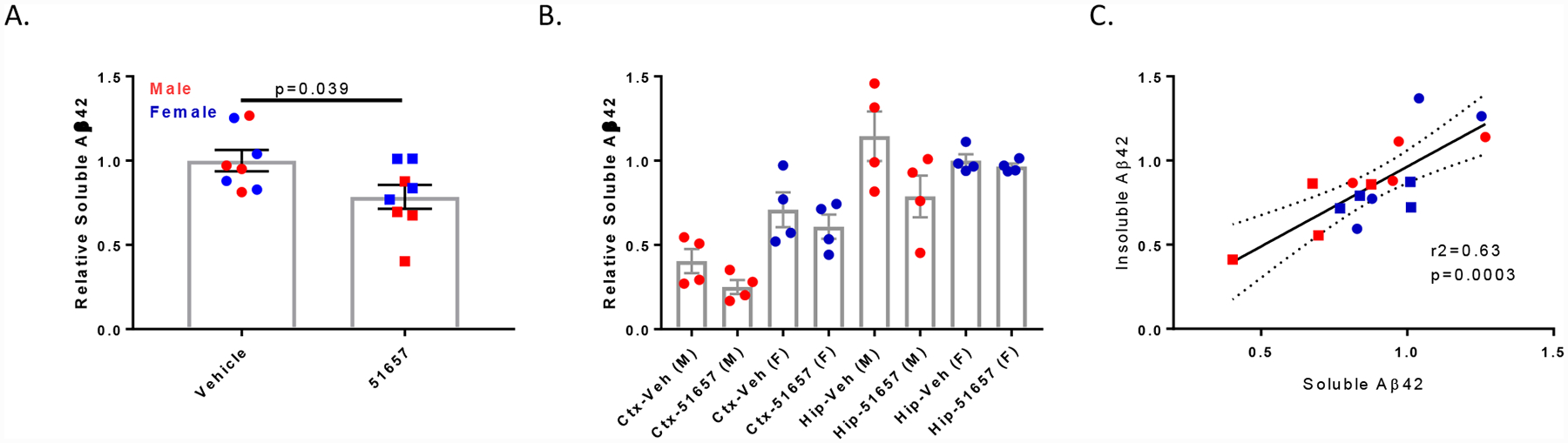
1.5-month old 5XFAD mice treated with 51657 for 7 weeks have reduced cortical and hippocampal/subiculum soluble Aβ. A. Soluble Aβ42 in the combined cortex and hippocampus/subiculum of 5XFAD study mice after normalization to the vehicle mean for each sex, with n=4 vehicle- and 51657-treated male and female 5XFAD mice. Statistical comparison was by unpaired 2-tailed t-test (t=2.28, df=14). Error bars represent SEM. B. Soluble Aβ42 in the cortex (Ctx) and hippocampus/subiculum (Hip) of the 5XFAD study mice graphed by sex, with normalization to the vehicle mean from the female hippocampus/subiculum group. Error bars represent SEM. C. Correlation plot of the relative soluble and insoluble Aβ42 in combined cortical and hippocampal/subiculum samples from the 5XFAD study mice. Data underwent linear fit analysis, with r2 and p-value shown in the graph and dashed lines representing the 95% confidence interval. Circles=Vehicle; Squares=51657-treated.
To further confirm the finding of reduced Aβ levels in 5XFAD mice treated with 51657, another study was conducted in which 1.5-month old male and female 5XFAD mice were treated with vehicle or 51657 for 4 weeks. This shorter study duration allowed for an examination of changes in Aβ levels during an earlier stage of Aβ plaque deposition, with plaques in 2.5-month old 5XFAD mice being largely confined to the subiculum. Whole cortex and hippocampus/subiculum were processed to obtain soluble and insoluble Aβ, and a significant reduction of soluble Aβ42 was again observed in combined hippocampal and cortical homogenates in the 5XFAD mice treated with 51657 (Fig. 5A). As in the prior study, the extent of compound-mediated lowering of soluble Aβ was again greater in male (42%) than in female (23%) 5XFAD mice, although both sexes showed a greater decrease of soluble Aβ42 than was observed in the mice dosed for 7 weeks. Hippocampus/subiculum levels of soluble Aβ42 were higher than cortical levels (Fig. 5B), and there was a trend toward reduced soluble Aβ in the cortex and hippocampus of both sexes of 51657-treated 5XFAD mice. As anticipated, the levels of insoluble Aβ42 levels were much lower in the cortex than in the hippocampus/subiculum of the 2.5-month old 5XFAD mice (Fig. 5C), although the overall trends toward reductions of insoluble Aβ42 in the cortex and hippocampus of 51657-treated male and female mice were similar to what was observed in the mice treated with compound for 7 weeks (Fig. 3B).
Figure 5.
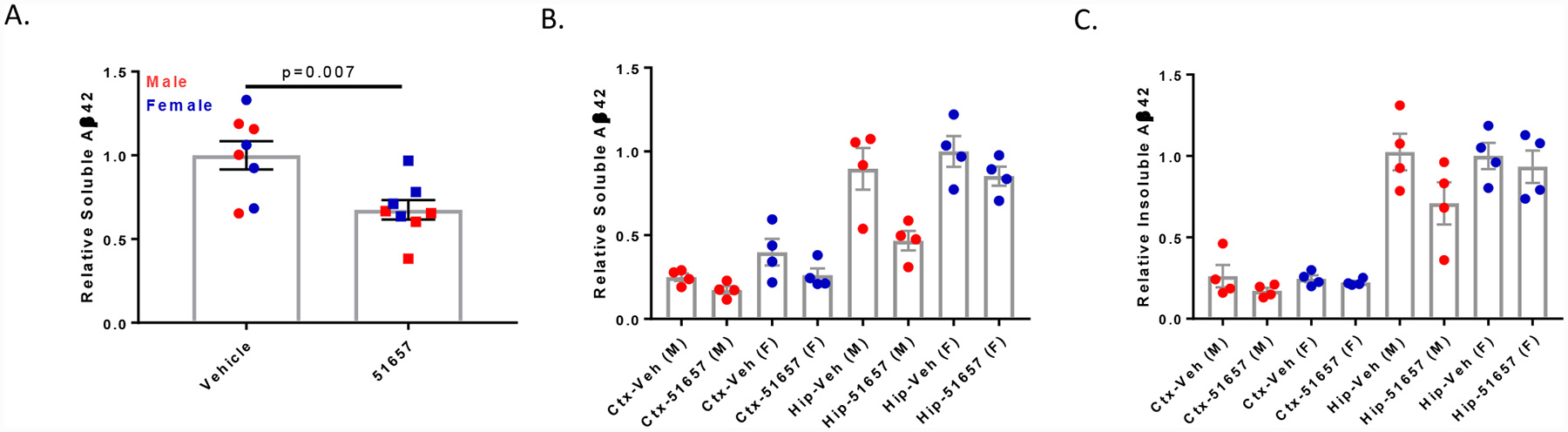
1.5-month old 5XFAD mice treated with 51657 for 4 weeks have reduced soluble Aβ. A. Soluble Aβ42 in the combined cortex and hippocampus/subiculum of the 5XFAD study mice. Data were normalized to the vehicle mean for each sex, with n=4 vehicle- and 51657-treated male and female 5XFAD mice. Statistical comparison was by unpaired 2-tailed t-test (t=3.17, df=14). Error bars represent SEM. B. Soluble Aβ42 in the cortex (Ctx) and hippocampus/subiculum (Hip) of the 5XFAD study mice graphed by sex, with normalization to the vehicle mean from the female hippocampus/subiculum group. Error bars represent SEM, with n=4 vehicle- and 51657-treated male and female 5XFAD mice. C. Insoluble Aβ42 in the cortex and hippocampus/subiculum of the 5XFAD study mice graphed by sex, with normalization to the vehicle mean from the female hippocampus/subiculum group. Error bars represent SEM, with n=4 vehicle- and 51657-treated male and female 5XFAD mice.
The measurable reduction of soluble Aβ in the 5XFAD mice treated with the MT-stabilizing agent for 4 weeks suggested that a reduction should also be observed in carboxyl-terminal APP fragments generated by BACE1 cleavage of APP. The levels of these APP fragments were examined in immunoblots of cortical homogenates from the 5XFAD mice (Fig. 6A), and quantification revealed a significant reduction of the C99/C89 APP fragments generated by BACE1 cleavage in 51657-treated mice, as well as the C83 fragment generated by α-secretase cleavage (Fig. 6B and 6C). The lowering of both BACE1 and α-secretase APP cleavage products after 51657-treatment suggested that the observed reduction in soluble Aβ42 resulted from an overall decrease of APP processing, perhaps due to decreased APP accumulation in dystrophic axons. Immunoblot analyses of APP holoprotein in the vehicle- and 51657-treated 5XFAD mice (Fig. 6A) confirmed that there was a modest but significant decrease in the 5XFAD mice receiving the MT-stabilizing compound (Fig. 7A), with male mice again showing a somewhat greater reduction than females. A similar reduction of BACE1 was also observed in the 51657-treated 5XFAD mice (Fig. 6A and Fig. 7B), consistent with a reduction of protein accumulation within dystrophic processes. To further confirm that there was a coordinate reduction of APP and BACE1, the relative APP and BACE1 levels were plotted for each study mouse, revealing a highly significant correlation (Fig. 7C). An analysis of APP mRNA levels from midbrain samples of the male 5XFAD mice confirmed that the decrease in APP protein was not the result of reduced APP mRNA expression (Fig. 7D). Given the high levels of transgene-driven APP and BACE1 expression in the 5XFAD mice, it is notable that significant reductions of APP and BACE1 levels can be achieved with a MT-stabilizing compound. In summary, these studies indicate that 51657 treatment results in a meaningful reduction of Aβ release and plaque deposition in 5XFAD mice via a lowering of APP and BACE1 proteins that appears to result from improved MT structure/function in SP-associated dystrophic axons.
Figure 6.
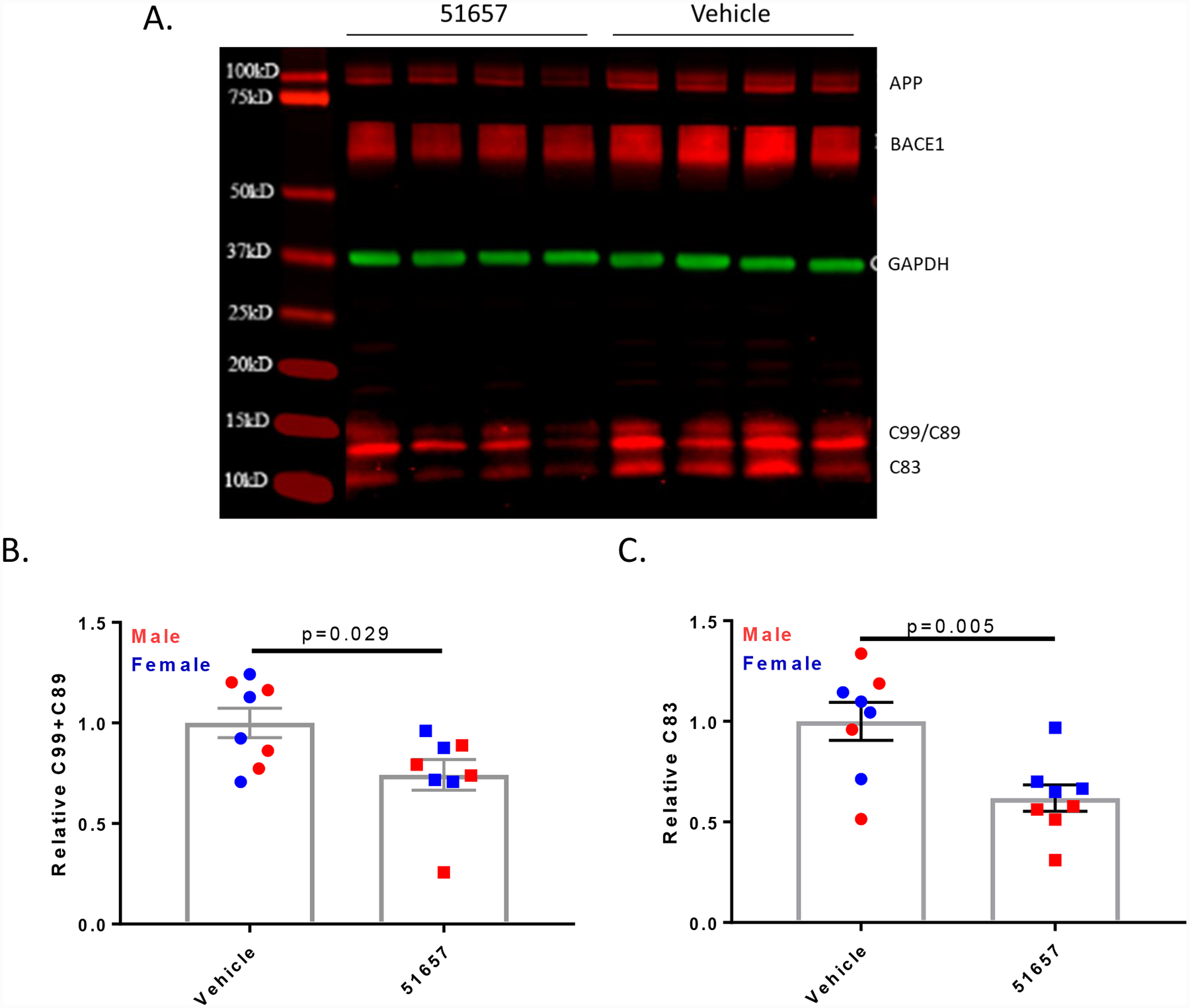
1.5-month old 5XFAD mice treated with 51657 for 4 weeks have reduced levels of APP fragments generated by β- and α-secretase. A. A representative immunoblot of cortical homogenates from 5XFAD mice reveals a reduction of C99/C89 and C83 APP fragments. Each lane represents samples from each male study mouse, with MW markers also shown. GAPDH was used as a loading control. Quantification of immunoblots revealed B. reduced β-secretase APP fragments C99/C89 and C. decreased α-secretase APP fragment C83 in cortical homogenates of 5XFAD mice treated with 51657. Statistical comparisons for B. and C. were by unpaired 2-tailed t-test, with n=4 vehicle- and 51657-treated male and female 5XFAD mice. Error bars represent SEM with statistical values of (t=2.42, df=14) and (t=3.31, df=14), respectively.
Figure 7.
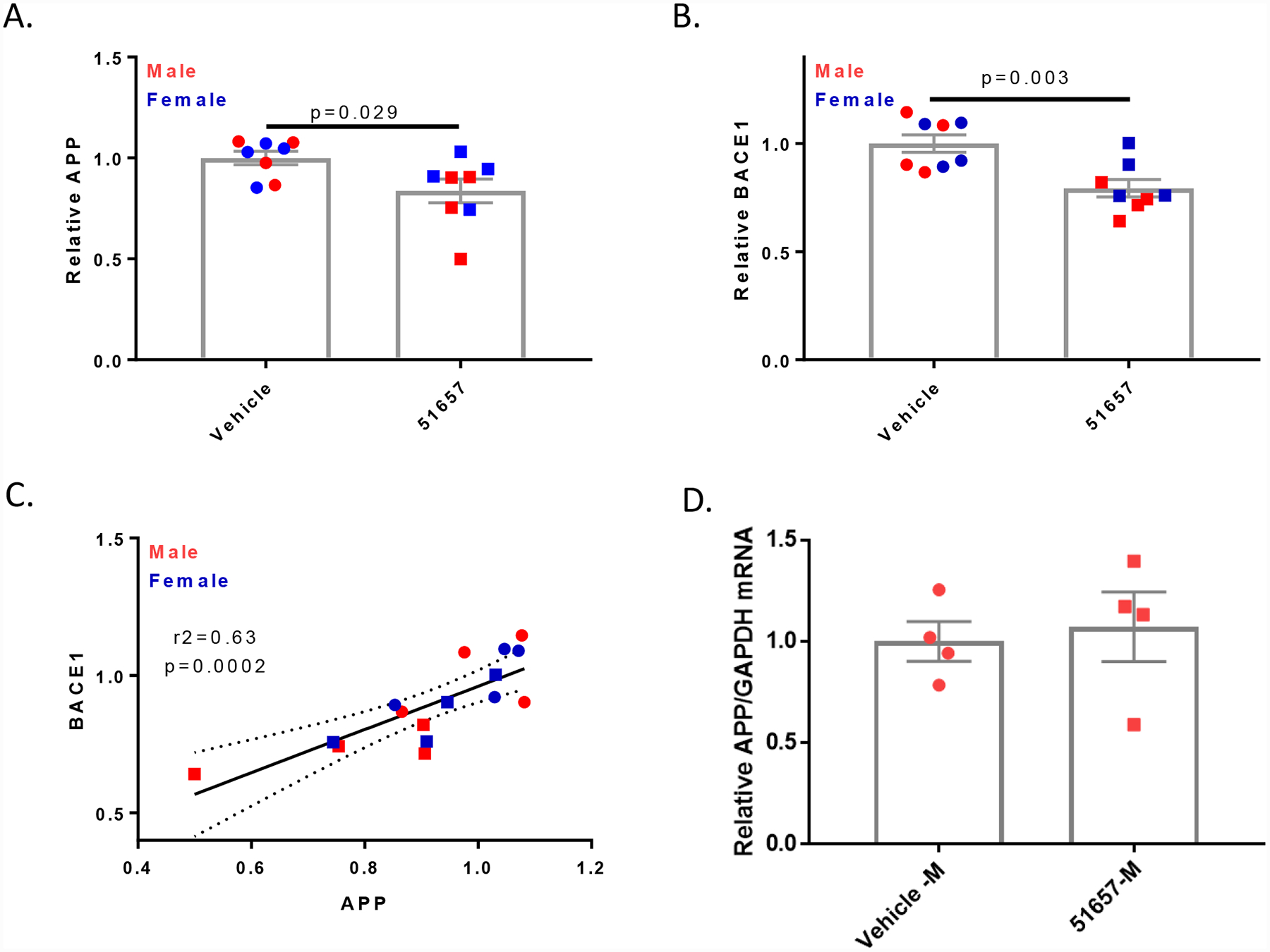
Quantification of immunoblots (Fig. 6A) reveals that 51657-treated 5XFAD mice have reduced cortical A. APP and B. BACE1, with normalization to the vehicle mean for each sex. Statistical comparisons for A. and B. were by unpaired 2-tailed t-test, with n=4 vehicle- and 51657-treated male and female 5XFAD mice. Error bars represent SEM with statistical values of (t=2.43, df=14) and (t=3.64, df=14), respectively. C. A correlation plot of normalized APP and BACE1 levels for each study mouse. Data underwent linear fit analysis, with r2 and p-value shown in the graph and dashed lines representing the 95% confidence interval. Circles=Vehicle; Squares=51657-treated. D. Treatment of 1.5-month old 5XFAD mice with 51657 for 4 weeks does not change brain human APP mRNA levels. Total RNA was isolated from the midbrain of male 5XFAD mice treated with 51657 or vehicle, and APP and GAPDH mRNA were quantified by qPCR. The APP/GAPDH amounts were normalized to the mean of the vehicle group, with n=4 vehicle- and 51657-treated male 5XFAD mice. No difference was observed between the two treatment groups by unpaired 2-tailed t-test (p=0.73, t=0.37, df=6).
Acknowledgements:
The authors thank Pyry Koivula and Thalia Delizannis for their technical assistance, and Alex Crowe for review of the manuscript. These studies were supported by NIH grants R01AG044332 and U01AG061173.
Footnotes
Competing Interests: The authors have no competing interests
Citations
- [1].Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annual Review of Neuroscience. 2001;24:1121–59. [DOI] [PubMed] [Google Scholar]
- [2].Ballatore C, Lee VMY, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nature Reviews Neuroscience. 2007;8:663–72. [DOI] [PubMed] [Google Scholar]
- [3].Selkoe DJ, Schenk D. Alzheimer’s disease: Molecular understanding predicts amyloid-based therapeutics. Annual Review of Pharmacology and Toxicology. 2003;43:545–84. [DOI] [PubMed] [Google Scholar]
- [4].George-Hyslop PH, Petit A. Molecular biology and genetics of Alzheimer’s disease. C RBiol. 2005;328:119–30. [DOI] [PubMed] [Google Scholar]
- [5].Tanzi RE, Bertram L. New frontiers in Alzheimer’s disease genetics. Neuron. 2001;32:181–4. [DOI] [PubMed] [Google Scholar]
- [6].Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (New York, NY). 2002;297:353–6. [DOI] [PubMed] [Google Scholar]
- [7].Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. [DOI] [PubMed] [Google Scholar]
- [8].van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21:21–35. [DOI] [PubMed] [Google Scholar]
- [9].Arriagada PV, Growdon JH, Hedleywhyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimers disease. Neurology. 1992;42:631–9. [DOI] [PubMed] [Google Scholar]
- [10].Wilcock GK, Esiri MM. Plaques, tangles and dementia - a quantitative study. Journal of the Neurological Sciences. 1982;56:343–56. [DOI] [PubMed] [Google Scholar]
- [11].Cho H, Choi JY, Hwang MS, Lee JH, Kim YJ, Lee HM, et al. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology. 2016;87:375–83. [DOI] [PubMed] [Google Scholar]
- [12].Teng E, Ward M, Manser PT, Sanabria-Bohorquez S, Ray RD, Wildsmith KR, et al. Cross-sectional associations between [(18)F]GTP1 tau PET and cognition in Alzheimer’s disease. Neurobiol Aging. 2019;81:138–45. [DOI] [PubMed] [Google Scholar]
- [13].Black MM, Slaughter T, Moshiach S, Obrocka M, Fischer I. Tau is enriched on dynamic microtubules in the distal region of growing axons. J Neurosci. 1996;16:3601–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kempf M, Clement A, Faissner A, Lee G, Brandt R. Tau binds to the distal axon early in development of polarity in a microtubule- and microfilament-dependent manner. J Neurosci. 1996;16:5583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Qiang L, Yu W, Andreadis A, Luo M, Baas PW. Tau protects microtubules in the axon from severing by katanin. J Neurosci. 2006;26:3120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sudo H, Baas PW. Strategies for diminishing katanin-based loss of microtubules in tauopathic neurodegenerative diseases. Hum Mol Genet. 2011;20:763–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tan R, Lam AJ, Tan T, Han J, Nowakowski DW, Vershinin M, et al. Microtubules gate tau condensation to spatially regulate microtubule functions. Nat Cell Biol. 2019;21:1078–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Alonso AD, GrundkeIqbal I, Iqbal K. Abnormally phosphorylated-tau from Alzheimer-disease brain depolymerizes microtubules. Neurobiology of Aging. 1994;15:S37–S.7700457 [Google Scholar]
- [19].Dayanandan R, Van Slegtenhorst M, Mack TGA, Ko L, Yen SH, Leroy K, et al. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. Febs Letters. 1999;446:228–32. [DOI] [PubMed] [Google Scholar]
- [20].Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct Tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–7. [DOI] [PubMed] [Google Scholar]
- [21].Barten DM, Fanara P, Andorfer C, Hoque N, Wong PYA, Husted KH, et al. Hyperdynamic Microtubules, Cognitive Deficits, and Pathology Are Improved in Tau Transgenic Mice with Low Doses of the Microtubule-Stabilizing Agent BMS-241027. Journal of Neuroscience. 2012;32:7137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30:13861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cash AD, Aliev G, Siedlak SL, Nunomura A, Fujioka H, Zhu XW, et al. Microtubule reduction in Alzheimer’s disease and aging is independent of tau filament formation. American Journal of Pathology. 2003;162:1623–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hempen B, Brion JP. Reduction of acetylated alpha-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer’s disease. Journal of Neuropathology and Experimental Neurology. 1996;55:964–72. [DOI] [PubMed] [Google Scholar]
- [25].Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dixit R, Ross JL, Goldman YE, Holzbaur ELF. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vershinin M, Carter BC, Razafsky DS, King SJ, Gross SP. Multiple-motor based transport and its regulation by Tau. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, cognitive deficits, neurotoxicity and Alzheimer-like pathology in an interventional study with aged tau transgenic mice Journal of Neuroscience. 2012;32:3601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–21. [DOI] [PubMed] [Google Scholar]
- [30].Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, et al. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. Journal of Neuropathology and Experimental Neurology. 1999;58:545-. [DOI] [PubMed] [Google Scholar]
- [31].Brunden KR, Lee VM, Smith AB 3rd, Trojanowski JQ, Ballatore C. Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol Dis. 2017;105:328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sadleir KR, Kandalepas PC, Buggia-Prevot V, Nicholson DA, Thinakaran G, Vassar R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol. 2016;132:235–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kandalepas PC, Sadleir KR, Eimer WA, Zhao J, Nicholson DA, Vassar R. The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013;126:329–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O’Connor T, et al. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci. 2007;27:3639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].He Z, Guo JL, McBride JD, Narasimhan S, Kim H, Changolkar L, et al. Amyloid-beta plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med. 2018;24:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Makani VZ B; Han H; Yao Y; Lassalas P; Lou K; Paterson I; Lee VM-Y; Trojanowski JQ; Ballatore C; Smith AB III; Brunden KR Evaluation of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in the PS19 tau transgenic mouse model of tauopathy. Acta Neuropathol Commun. 2016;4:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kovalevich J, Cornec AS, Yao Y, James M, Crowe A, Lee VM, et al. Characterization of Brain-Penetrant Pyrimidine-Containing Molecules with Differential Microtubule-Stabilizing Activities Developed as Potential Therapeutic Agents for Alzheimer’s Disease and Related Tauopathies. J Pharmacol Exp Ther. 2016;357:432–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Saez-Calvo G, Sharma A, Balaguer FA, Barasoain I, Rodriguez-Salarichs J, Olieric N, et al. Triazolopyrimidines Are Microtubule-Stabilizing Agents that Bind the Vinca Inhibitor Site of Tubulin. Cell Chem Biol. 2017;24:737–50 e6. [DOI] [PubMed] [Google Scholar]
- [39].Zhang B, Yao Y, Cornec A-S, Oukoloff K, James M, Koivula P, Trojanowski JQ, Smith AB III, Lee VM-Y, Ballatore C, and Brunden KR A brain-penetrant triazolopyrimidine enhances microtubule-stability, reduces axonal dysfunction and decreases tau pathology in a mouse tauopathy model Mol Neurodegen. 2018;13:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sadleir KR, Eimer WA, Cole SL, Vassar R. Abeta reduction in BACE1 heterozygous null 5XFAD mice is associated with transgenic APP level. Mol Neurodegener. 2015;10:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ke YD, Suchowerska AK, van der Hoven J, De Silva DM, Wu CW, van Eersel J, et al. Lessons from tau-deficient mice. Int J Alzheimers Dis. 2012;2012:873270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Malamut RW J-S; Savant I; Xiao H; Sverdlov O; Tendolkar AV; Keswani SC A randomized, double-blind, placebo-controlled, multiple ascending dose study to evaluate the safety, tolerability and pharmacokinetics of a microtubule stabilizer (BMS-241027) in healthy females. Alzheimers and Dementia. 2013;9:P668–9. [Google Scholar]
- [44].Bedard PL, Di Leo A, Piccart-Gebhart MJ. Taxanes: optimizing adjuvant chemotherapy for early-stage breast cancer. Nature Reviews Clinical Oncology. 2010;7:22–36. [DOI] [PubMed] [Google Scholar]
- [45].Cortes J, Baselga J. Targeting the microtubules in breast cancer beyond taxanes: The epothilones. The Oncologist. 2007;12:271–80. [DOI] [PubMed] [Google Scholar]
- [46].Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain. 2017;140:764–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019;76:915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, et al. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005;168:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Styren SD, Hamilton RL, Styren GC, Klunk WE. X-34, a fluorescent derivative of Congo red: a novel histochemical stain for Alzheimer’s disease pathology. J Histochem Cytochem. 2000;48:1223–32. [DOI] [PubMed] [Google Scholar]
- [50].Herbst-Robinson KJ, Liu L, James M, Yao Y, Xie SX, Brunden KR. Inflammatory Eicosanoids Increase Amyloid Precursor Protein Expression via Activation of Multiple Neuronal Receptors. Sci Rep. 2015;5:18286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Soper JH, Sugiyama S, Herbst-Robinson K, James MJ, Wang X, Trojanowski JQ, et al. Brain-penetrant tetrahydronaphthalene thromboxane A2-prostanoid (TP) receptor antagonists as prototype therapeutics for Alzheimer’s disease. ACS chemical neuroscience. 2012;3:928–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proc Natl Acad Sci U S A. 1991;88:7552–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Su JH, Cummings BJ, Cotman CW. Plaque biogenesis in brain aging and Alzheimer’s disease. II. Progressive transformation and developmental sequence of dystrophic neurites. Acta Neuropathol. 1998;96:463–71. [DOI] [PubMed] [Google Scholar]