

Abstract
Background
To improve responses to tumor microenvironments for achieving a better therapeutic outcome in combination cancer therapy, poly(ε-caprolactone)-SS-poly(methacrylic acid) diblock copolymer (PCL-SS-PMAA) with a disulfide linkage between the hydrophobic and hydrophilic junctions was synthesized.
Materials and Methods
Repeating units of PCL and PMAA in PCL-SS-PMAA were controlled and formulated into polymersomes (PSPps). Truncated octahedral Fe3O4 nanoparticles (IONPs) were synthesized and encapsulated to produce IONPs-PSPps NPs and doxorubicin (DOX) was further loaded to produce IONPs-PSPps@DOX NPs for theranostic applications.
Results
IONPs-PSPps NPs remained a superparamagnetic property with a saturation magnetization value of 85 emu⋅gFe3O4−1 and a relaxivity value of 180 mM−1⋅s−1. Upon exposure to an alternating magnetic field (AMF), IONPs-PSPps NPs increased temperature from 25°C to 54°C within 15 min. Among test groups, the cell apoptosis was greatest in the group exposed to IONPs-PSPps@DOX NPs with AMF and magnet assistance. In vivo T2-weighted magnetic resonance images of A549 tumor-bearing mice also showed highest contrast and greatest tumor suppression in the tumor with AMF and magnet assistance.
Conclusion
IONPs-PSPps@DOX NPs are a potential theranostic agent having multifaceted applications involving magnetic targeting, MRI diagnosis, hyperthermia and chemotherapy.
Keywords: magnetosome, multi-stimuli-responsive copolymer, theranostics, hyperthermia, magnetic resonance imaging
Introduction
In our previous study, a self-assembled poly(methacrylic acid)-poly(ε-caprolactone) copolymer with disulfide linkages in the core of the hydrophobic domain, PMAA-b-PCL-SS-PCL-b-PMAA (S-PCL-PMAA)2, has been synthesized and showed higher cellular uptake than a similar construct with a carbon linkage, PMAA-b-PCL-CC-PCL-b-PMAA (C-PCL-PMAA)2.1 The cleavage of disulfide linkages in the hydrophobic PCL block of the micellar core caused destabilization of self-assembled micelles due to changes in the hydrophobic/hydrophilic balance, leading to enhanced drug release. Since the disulfide linkages were embedded in the center of the hydrophobic PCL segment, the cleavage of the disulfide linkages occurred in a harsh basic condition and with long incubation time, which was not feasible in a physiological condition. In the present study, a simple poly(ε-caprolactone)-SS-poly(methacrylic acid) diblock copolymer PCL-SS-PMAA that had a disulfide linkage between the hydrophobic and hydrophilic junctions was designed and synthesized to improve the reduction response to glutathione (GSH).
GSH is a thiol-containing tripeptide found at different concentrations in intracellular compartments (1–10 mM) and extracellular compartments (2–20 μM) in living cells.2 The cytosolic GSH concentrations in tumor cells are several times higher than those in normal cells.3 Using a GSH concentration gradient could provide a promising cue to the response in tumor microenvironments (TME) and allow the control of anticancer drug release at a tumor site. Amphiphilic block copolymers bearing disulfide linkages have been widely developed for redox-responsive drug delivery systems (DDS). The disulfide linkages can be designed at different positions including within hydrophilic and hydrophobic block junctions,4–6 in the center of hydrophobic block segments7–9 or in pendant chains.10–14 The reduction response of the disulfide linkages embedded within hydrophobic block segments showed slower destabilization than those located within hydrophilic and hydrophobic block junctions. It therefore seems beneficial to design amphiphilic block copolymers with a disulfide linkage between the hydrophobic and hydrophilic junctions. PCL and PMAA polymers approved by the US Food and Drug Administration (FDA) were selected to synthesize PCL-SS-PMAA, an amphiphilic block copolymer containing the disulfide linkages between the hydrophobic and hydrophilic junctions to achieve a good response in TME.
Amphiphilic block copolymers can self-assemble into a variety of nanostructures including spherical micelles, cylindrical micelles and polymersomes by controlling the molecular weight, surfactant, non-polar solvent, surface functionalization and hydrophobic/hydrophilic ratio of a block copolymer.15 Since polymersomes are able to be simultaneously loaded with hydrophobic and hydrophilic agents, repeating units of PCL and PMAA in PCL-SS-PMAA were therefore tuned up to form polymersomes (PSPps). The polymersomes constructed from poly(trimethylene carbonate)-block-poly(L-glutamic acid) (PTMC-b-PGA) have been used to encapsulate superparamagnetic iron oxide nanoparticles (γ-Fe2O3) in the hydrophobic domain and doxorubicin (DOX) in the hydrophilic cavity for hyperthermia and chemotherapy.16
Magnetic nanoparticles (NPs) are widely utilized in biomedical research.17 Applying a magnetic field near a tumor to enhance the confinement of magnetic NPs is a promising strategy for site-specific delivery to the tumor.18 Moreover, magnetic NPs generate heat under an alternating magnetic field (AMF) due to hysteresis losses, which makes them hyperthermia agents that can deliver thermal energy directly to the tumor.19 The advantages of using truncated octahedral Fe3O4 nanoparticles (IONPs) over spherical ones include (1) higher saturation magnetization (Ms), higher Curie temperature, greater crystallization, greater contact area, enhanced magnetic properties, stronger signals and better analytical sensitivity.20
Here, we synthesized a new redox-responsive PCL-SS-PMAA copolymer that could be formulated into PSPps by controlling the repeating units of PCL and PMAA. PSPps was conjugated with rhodamine 123 and tested its endocytotic pathway. The endocytic pathway and subsequent processing strongly depend on the size and morphology of nanostructures.21–23 Three major chemical inhibitors were selected to study the internalization pathways of the construct: (1) chlorpromazine for clathrin-mediated endocytosis, (2) genistein for caveolae-mediated endocytosis and (3) wortmannin for micropinocytosis.24 The PSPps were utilized to encapsulate IONPs in the hydrophobic compartment (IONPs-PSPps NPs) not only as a T2-weighted magnetic resonance imaging (MRI) contrast agent but for inducing hyperthermia in cancer therapy. In addition, DOX was loaded in the center of the hydrophilic compartment to produce IONPs-PSPps@DOX NPs for chemotherapy. The merits of the IONPs-PSPps@DOX NPs include (1) there will be little concern of toxicity for future translational development because PCL and PMAA both are FDA-approved polymers and have been widely used in pharmaceutical applications. (2) The disulfide linkages positioned within the hydrophobic and hydrophilic segments of PSPps accelerate drug release in TME, leading to reduced side effects of the drug. (3) Compared with spherical Fe3O4 NPs, the IONPs showed higher saturated magnetization value, higher T2-weighted image and higher internalization into cells (our unpublished data).
IONPs-PSPps@DOX NPs possess multifaceted functions including negative contrast enhancement for MRI, AMF-induced thermal heat for hyperthermia and DOX release for chemotherapy. The cleavage of disulfide linkages, cellular uptake, internalization pathway and cytotoxicity of PSPps were studied. The Ms, relaxivity (r2) values and the temperature elevation upon exposure to AMF were characterized. The DOX-released profiles were tested in the presence and absence of a reducing agent. The annexin-V-FITC/PI (propidium iodide) dual-staining assay was performed to evaluate cell apoptosis. The in vivo T2-weighted MRI images and tumor suppression of mice dorsal flank tumors were observed in mice injected with the IONPs-PSPps@DOX NPs via the tail vein. The accumulation of the IONPs-PSPps@DOX NPs in the tumor was enhanced by exposing the tumor area to a magnetic field. Preliminary biocompatibility and toxicology were evaluated using hematoxylin and eosin (H&E) staining, and biochemical analysis of the liver function and kidney function in nude mice after intravenous treatments with the IONPs-PSPps@DOX NPs were examined.
Materials and Methods
Materials
Bis(2-hydroxyethyl) disulfide, 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA), copper (I) bromide (CuBr), iron (III) acetylacetonate (Fe(acac)3), oleic acid, tri-octylamine, DOX, (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride) (EDC), N-hydroxysuccinimide (NHS) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Sigma. Amberlite® IR120, 2-bromo-2-methylpropionyl bromide, CL, rhodamine 123 (R123), GSH and pyrene were purchased from Acros. Neutral aluminium oxide (Al2O3) was purchased from Seedchem Company. Tin (Ⅱ) 2-ethylhexanoate (Sn(Oct)2) and o-phenylenediamine were purchased from Alfa Aesar. Dithiothreitol (DTT) was purchased from J.T. Baker. Tert-butyl methacrylate (tBMA) and trimethylamine (TEA) were purchased from TCI. Polyvinyl alcohol (PVA, MW=2000) was purchased from Showa. Minimum essential medium, penicillin, streptomycin and fetal bovine serum (FBS) were purchased from Biological Industries. The annexin-V/PI staining kit was purchased from Strong Biotech Corporation. Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Invitrogen.
Synthesis of HO-SS-Br
2-Hydroxyethyl-2ʹ-(bromoisobutyryl) ethyl disulfide (HO-SS-Br) was synthesized using a procedure slightly modified from the literature.25 Briefly, a mixture consisting of bis (2-hydroxyethyl) disulfide (5.00 g, 32.4 mmol), 2-bromo-2-methylpropionyl bromide (5 mL, 40.4 mmol), TEA (12.0 mL, 86.1 mmol) and tetrahydrofuran (THF, 100 mL) was stirred for 24 h at room temperature. The obtained product was purified using silica-column chromatography with n-hexane and ethyl acetate as eluents. The yield was 4.0 g (40.8%), and Rf=0.45 on silica (3/7 v/v of ethyl acetate/hexane). 1H-NMR findings (CDCl3, ppm): 4.45 (t, 2H, -CH2OC(O)-), 3.90 (t, 2H, HO-CH2-), 3.00 (t, 2H, -SS-CH2-CH2OC(O)-), 2.90 (t, 2H, HOCH2-CH2-SS-) and 1.95 (s, 6H, -C(CH3)2Br). Mass of C8H19BrO3S2: calculated=301.95, measured=301.93.
Synthesis of PCL-SS-Br
Poly(ε-caprolactone)-b-bromide (PCL-SS-Br) was prepared using ring-opening polymerization (ROP) with HO-SS-Br as an initiator and Sn(Oct)2 as a catalyst in toluene.1 In brief, HO-SS-Br (96 mg, 0.31 mmol), CL (2.19 mL, 19.49 mmol), Sn(Oct)2 (15.9 mg, 0.049 mmol) and toluene (2.1 mL) were placed in a two-neck round-bottom flask, and the mixture was degassed by nitrogen gas three times before being put into a glove box. The polymerization was conducted at 110°C for 5.5 h under a nitrogen atmosphere, and then the reaction solution was cooled down to room temperature and precipitated into excess cold methanol. The precipitated white solid was collected and dried under a vacuum.
PCL-SS-PtBMA diblock copolymers were synthesized by atom-transfer radical polymerization (ATRP) in toluene mediated with the CuBr-HMTETA complex using PCL-SS-Br as the initiator. Typically, PCL-SS-Br (200 mg, 0.028 mmol), CuBr (12.3 mg, 0.085 mmol), HMTETA (21 mg, 0.091 mmol), tBMA (617 mg, 4.34 mmol) and toluene (1.5 mL) were added to a two-neck round-bottom flask. The mixture was degassed by nitrogen gas three times before being put into a glove box. The polymerization was allowed to proceed by continuous stirring at 50°C for 6 h under a nitrogen atmosphere. The reaction was stopped by diluting with toluene and the solution was then passed through an alumina column and Amberlite® IR120 to remove the residual copper catalyst. Finally, the product was obtained by precipitation into excess n-hexane and dried under a vacuum.
PCL-SS-PMAA was obtained from the hydrolysis of the tert-butyl esters of PCL-SS-PtBMA using TFA.26 The typical procedure was as follows: PCL-SS-PtBMA copolymer (160 mg) was dissolved in dichloromethane (40 mL) at room temperature, the solution was cooled to 0°C and TFA (4 mL) was slowly added with stirring. Following 24 h of reaction at room temperature, the solution was concentrated and precipitated into cold n-hexane. This procedure was repeated three times using dichloromethane as the solvent and cold n-hexane as the non-solvent. The final product was collected and dried under a vacuum.
Synthesis of R123-Conjugated PCL-SS-PMAA
R123-conjugated PCL-SS-PMAA (PCL-SS-PMAA-R123) was synthesized by dissolving PCL-SS-PMAA (10 mg) in 10 mL of DD water, to which 0.25 mL of 10 mM EDC followed by 10 mM NHS stock solution were added. The solution was stirred at room temperature for 1 h. One milligram of R123 in 0.5 mL of DD water was added to the above solution, which was then stirred at room temperature for 24 h in darkness. The solution was centrifuged at 15,000 rpm for 10 min, and the obtained supernatant was discarded. The residue was washed with 1 mL of DD water followed by centrifugation for a further 10 min. The washing and centrifugation procedure was repeated until no color was observed in the supernatant.
Characterization of Copolymers
A Mercury Plus-200 spectrometer (Varian, Palo Alto, CA) was used for 1H-NMR spectrum analyses to determine the chemical structures of polymers using CDCl3 or DMSO-d6 as a solvent. The molecular weights of copolymers were measured by GPC using an Agilent 1100 series device (Santa Clara, CA) equipped with a Shodex-KF804 column. Samples were dissolved in THF at a concentration of 5 mg⋅mL–1 and filtered through a 0.45-µm filter prior to injection. THF was used as an eluent at a flow rate of 1 mL⋅min–1. Polystyrene standards were used to generate a calibration curve.
Preparation and Characterization of Polymersomes
PSPps were prepared using a water-in-oil-in-water double-emulsion method in accordance with the literature.27 Morphologies were observed using transmission electron microscopy (TEM, JEM-2000 EXII, Jeol). A carbon-coated 200 mesh copper specimen grid (Agar Scientific) was glow discharged for 1.5 min. The obtained powder (2 mg) was dissolved in a vial containing 4 mL of PBS (pH 7.4) with and without treatments with various amounts of GSH. At predetermined time intervals, the solution was taken out from the vial and dropped on the copper grid and allowed to dry at room temperature for 5 days.
Preparation of IONPs- and DOX-Encapsulated PSPps
IONPs were synthesized according to the method reported in the literature.28 IONPs were embedded in the hydrophobic layer between two hydrophilic regions of PSPps. We first prepared a stock concentration of IONPs at 1400 ppm. IONPs (500 μL) and 4 mL of 1 wt% PCL-SS-PMAA in dichloromethane were put in a 50-mL centrifuge tube in an ice bath. PVA solution (500 μL of 1 wt%) was added dropwise into the tube under sonication for 20 min to embed IONPs into the hydrophobic layer. The solution was subsequently added dropwise into another tube containing 12 mL of 1 wt% PVA solution under sonication using a homogenizer in an ice bath for 20 min to form IONPs-PSPps NPs. The solvent was removed using vacuum evaporation for 30 min. The obtained product was washed with DD water and centrifuged at 1000 rpm for 1 min and the supernatant was collected. DOX was simultaneously encapsulated with IONPs-PSPps NPs by using the above-described double-emulsion technique, with the exception that 500 μL of the salt form of DOX at 2 mM was put in 500 μL of 1 wt% PVA solution and added dropwise during the first emulsion process.
DOX Content of IONPs-PSPps@DOX NPs
A DOX calibration curve was generated using a fluorescence spectrometer (Cary Eclipse, Varian) based on the emission intensity at 555 nm. The free DOX remaining in the supernatant during the double-emulsion process was collected and analyzed to estimate the encapsulation efficiency of DOX in IONPs-PSPps@DOX NPs against the DOX standard calibration curve. The drug entrapment efficiency is calculated using the following equations,
Encapsulation efficiency: 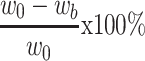 (1)
(1)
Loading efficiency: 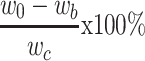 (2)
(2)
where w0 and wb are the amounts of DOX in the feed and the supernatant; wc is the total weight of the NPs, respectively.
Characterization of NPs
Electron micrographs were obtained using transmission electron microscopy (TEM, H-7500, Hitachi Koki) or high-resolution TEM (JEM-2100F, Jeol). To measure iron contents of NPs, the as-prepared NPs were digested in aqua regia. The iron concentrations of NPs were measured using inductively coupled plasma (ICP) analysis. XRD signals of NPs were obtained using a X-ray diffractometer (Shimadzu Cu Kα radiation at λ=1.54060 Å). The magnetic properties of NPs were measured using a superconducting quantum interference device (SQUID) vibrating sample magnetometer (Quantum Design MPMS). Cell viabilities were measured using an MTT assay and analyzed using an enzyme-linked immunosorbent assay (ELISA) reader (Multiskan EX, Thermo Scientific).
Temperature Elevation Profiles with AMF
The temperature changes induced by applying AMF at 50–100 kHz were observed. The temperature of an AMF generator was controlled at 25°C using a cooling-water setup. The AMF coil had eight loops with a magnetic field power of 2.5 kA⋅m−1. Seven milliliters of pure DD water or DD water containing NPs were separately put into 15-mL test tubes that were placed at the center of the coil. The iron concentration was maintained at 200 μg⋅mL−1 when measuring the temperature elevations in NPs.
Relaxivity Measurements
MRI analysis was performed using a 9.4-T device (BioSpec 94/20 USR, Bruker) coupled with a high-performance transmitter-receiver RF volume coil. For relaxation time measurements, the IONPs and IONPs-PSPps NPs were dispersed in a 0.5% agarose gel solution at various iron concentrations, and then r2 was calculated by T2-weighted mapping according to a previously reported method.28
In vitro Drug Release
In vitro drug release was investigated in PBS using a dialyzer tube (3.5 kDa DiaEasy, BioVision). In general, 600 μL of 200 μg⋅mL–1 IONPs-PSPps@DOX NPs were placed in a dialyzer tube to measure the amount of DOX released. The dialyzer tube was immersed in 15 mL of PBS at pH 5.5 and pH 7.4 with and without the presence of glutathione (GSH) and then treated with and without exposure to AMF at 10 kW for 15 min. After the AMF treatment, the dialyzer tube was continuously shaking at 100 rpm in an incubator at 37°C. At different time intervals, 2-mL aliquots were taken from the test solutions for DOX-released measurements against the DOX calibration curve, with this replaced by 2 mL of fresh PBS to maintain a constant volume. The mean amount of DOX released was determined in at least triplicate measurements.
Cytotoxicity
HEK-293T, CRL-5802, NCI-520 and A549 cells were all cultured in DMEM (high glucose) supplied with non-essential amino acids and supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C in a humidified environment with 5% CO2. Cells were seeded in 96-well tissue culture plates at a density of 5×103 per well. Cell viabilities were determined after 24 h of incubation at various concentrations (40–250 μg⋅mL−1) using the MTT colorimetric method.1 The number of active cells was estimated by measuring the absorbance at 595 nm. The cell lines were purchased from Bioresource Collection and Research Center (BCRC) of Taiwan. (CRL-5802)
The in vitro effects of hyperthermia and chemotherapy on cell viability were tested in A549 cells. The cells were treated with an equivalent DOX concentration of 4.4 μM and IONPs concentration of 100 μg⋅mL−1 for 24 h. A magnetic field was applied to the cells by placing a MagnetoFACTOR-96 plate underneath the culture plate for 30 min to enhance the internalization of NPs. The medium containing non-internalized NPs was discarded, and the cells were washed with PBS three times before being replaced with fresh medium. The cells were then exposed to AMF at 10 kW for 15 min, and the number of active cells was estimated using the MTT assay as described above.
Detection of Cell Apoptosis by Annexin V/PI Staining
Briefly, A549 cells were seeded in 12-well plates at 1×105 cells per well and incubated for 24 h before being exposed to free DOX, IONPs-PSPps and IONPs-PSPps@DOX NPs (equivalent to 4.4 μM DOX and 100 μg⋅mL−1 IONPs). Similarly, the cells were exposed with and without assistance from a magnet for 30 min, followed by exposure to AMF at 10 kW for 15 min. The cells were then incubated for another 24 h and trypsinized, washed twice with PBS and the pellets were resuspended in 96 μL of binding buffer. Then, 2 μL of annexin V-FITC and 2 μL of PI were added to the cell suspension, mixed and incubated for 30 min at 37°C in an incubator. The cells were then washed with binding buffer and resuspended in 500 μL of binding buffer. Viable cells, early apoptosis cells, late apoptosis cells and necrotic cells were detected using a flow cytometer (Epics XL-MCL, Beckman Coulter).
In vivo MRI of IONPs-PSPps NPs
All of the animals used in in vivo MRI investigations of IONPs-PSPps NPs received human care in compliance with the institutional guidelines for maintaining an AAALAC-certified specific pathogen-free animal facility at Kaohsiung Chang Gung Memorial Hospital (KCGMH) and the use of laboratory animals in research. All of the experimental protocols involving live animals were reviewed and approved by the Institutional Animal Care and Use Committee of KCGMH. The male A549 tumor-bearing nude mice (BALB/cAnN) were subcutaneously injected with 1×107 A549 cells into the right and left subcutaneous regions on the lower back. After 7 days, at which time the tumors had grown to a size of ~25-30 mm3, the IONPs-PSPps NPs were intravenously injected into mice via the tail vein at an iron dose of 10 mg⋅kg−1. To attract IONPs-PSPps NPs to the tumor on the left side, a commercial neodymium magnet (~6300 G) was placed near to the tumor area for 30 min. MRI images were acquired under anesthesia induced using 2% isoflurane (Abbott Laboratories) mixed with 100% O2 delivered via a veterinary anesthesia delivery system (ADS 1000, Engler) in a 9.4-T horizontal-bore animal MRI system with a RARE T2-weighted MRI axial sequence with the following parameters: number of slices, 11; SI, 0.5/0.5 mm; field of view, 30 mm×30 mm; matrix, 256 pixels×256 pixels; RARE factor, 8; TR/TE, 2500/28 ms; and number of excitations, 9; The total scan time was 12 min, in accordance with our previous publication.28
Biodistribution and Toxicology
The tumor-bearing mice were sacrificed at several time points (3, 7, 24, 48, 72 and 168 h) after injecting IONPs-PSPps@DOX NPs at a dose of 24 mg⋅kg−1. The tissues (heart, liver, spleen, lung, kidneys, and tumors with and without assistance from a magnet) were collected and homogenized to analyze iron contents by ICP/atomic emission spectroscopy. In addition, blood was obtained from the heart and a solution containing heparin sodium was added immediately. The clotted blood samples were centrifuged at 1200 rpm for 15 min to collect the serum. The blood biochemistry analysis of aspartate aminotransferase, alanine aminotransferase, blood urea nitrogen (BUN), uric acid (UA) and creatinine (CRE) was performing using a biochemical analyzer (DRI-CHEM 4000i, Fuji). All of the measurements were performed in triplicate.
In vivo Antitumor Efficacy
All of the animal treatments and surgical procedures for determining the in vivo antitumor efficacy were performed in accordance with the guidelines of the Laboratory Animal Center of Kaohsiung Medical University (Kaohsiung, Taiwan). The antitumor efficacy was evaluated using male nude mice (BALB/cAnN). Two tumors were implanted on the mice as described above. IONPs-PSPps@DOX NPs were injected at ~24 mg⋅kg−1 intravenously into the mice via the tail vein. The magnet was then placed over the tumor on the right side to attract the NPs for 30 min. The mice were treated with and without exposure to AMF at 10 kW for 15 min. The tumor volume was measured as the volume of an ellipsoid based on the longest width and the perpendicular length: (length×width2)/2. The tumor size and body weight of each mouse were tracked for up to 30 days. The relevant organs and tumors were dissected from one mouse and sectioned into slices for histopathological examination with H&E staining.
Statistical Analysis
Group comparisons were performed using the two-tailed Student’s t-test and Tukey’s multiple comparison test; both of the methods used the Prism 5 software (GraphPad Software Inc., San Diego, California, USA) for statistical analyses. Experiments were repeated at least four times. Unless otherwise stated, data are expressed as the mean±standard deviation. The MR imaging processing and signal analysis using ImageJ software for imaging processing, and the percentages relative to the in vivo T2-weighted MR images that were compared with pre-injection of each group The results of MTT assay, flow-cytometric analysis, therapeutic outcome of tumor volume, body weight analysis, and MR imaging contrast signals were analyzed using ANOVA. If the result of the ANOVA was significant (p<0.05 versus control), pairwise comparisons between the groups were performed using a post testing. In all cases, p<0.05 was considered statistically significant.
Results and Discussion
Polymer Synthesis and Characterization
The synthesis of PCL-SS-PMAA diblock copolymers was illustrated in Scheme 1. Initially, OH-SS-Br was synthesized and purified according to a previously reported procedure.29 The PCL-SS-Br that had a disulfide linkage was prepared by ROP of CL using OH-SS-Br as an initiator. The degree of polymerization (DP) of PCL was calculated to be 60 using the intensity ratio of peak e and peaks c and d shown in Figure S1A. The PCL-SS-Br was subsequently utilized to perform ATRP reaction using tBMA as the monomer. The repeating units of PtBMA were also controlled at 60 by adjusting the reaction time and the supplied amount of tBMA. Finally, the tert-butyl groups of PCL-SS-PtBMA were hydrolyzed to yield PCL-SS-PMAA in the presence of trifluoroacetic acid (TFA). Their corresponding proton nuclear magnetic resonance (1H-NMR) spectra are shown in Figure S1 Compared with Figure S1B, the peak intensity at ~1.40 ppm of Figure S1C is markedly decreased due to the disappearance of tert-butyl groups.
Scheme 1.

Synthesis of poly(ε-caprolactone)-SS-poly(methacrylic acid) block copolymers (PCL-SS-PMAA).
The PCL-SS-PMAA could be formulated into PSPps with the assistance of PVA as a surfactant. The TEM image of PSPps is shown in Figure 1A, and the hydrophobic layer within two hydrophilic compartments was stained with Nile red and captured by CLSM (Figure 1B). Both of the images clearly demonstrated the successful construction of PSPps with a size of 203.4±12.0 nm, which was close to that of 210.4±20.0 nm as measured using DLS (PDI=0.150, Figure S2). The zeta potential of PSPps was −21.9 mV. PMAA exposed on the surface of PSPps is a polyelectrolyte and can be deprotonated in PBS at pH 7.4 because the pKa value of PMAA is ~4.8.30 The disassembled behaviors of PSPps in solutions at pH 7.4 as determined using DLS for GSH at concentrations of 1, 5, and 10 mM GSH are shown in Figure 1C-E, respectively, at different time points. The DLS profiles of PSPps remained monodispersive since PSPps were left in the solution containing 1 mM GSH for 2 days (Figure 1C). The inserted TEM image showed that the morphology of PSPps was preserved. However, bimodal distribution profiles appeared when PSPps were left in 5 mM GSH solution for 2 h (Figure 1D) and in 10 mM GSH solution for 10 min (Figure 1E). The TEM images clearly illustrated the irregular structure and aggregations of PSPps. It seems that the disulfide bonds of PSPps were stable in the GSH concentration less than 1 mM but became cleavable as the GSH concentration was increased to ≥5 mM.
Figure 1.

Characterization of the (PCL)60-SS-(PMAA)60 polymersomes (PSPps). (A) TEM image of PSPps. (B) Confocal image of PSPps encapsulated with Nile red. Disassembled behaviors of PSPps in PBS at pH 7.4 owing to disulfide bond cleavage triggered with (C) 1 mM GSH, (D) 5 mM GSH, and (E) 10 mM GSH at different time points by DLS. Inserted TEM images are PSPps left in PBS at pH 7.4 with (C) 1 mM GSH for 2 days, (D) 5 mM GSH for 2 h, and (E) 10 mM GSH for 10 min.
The GSH concentration was a key factor determining the disassembly rate of PSPps. Since GSH is a thiol-containing tripeptide found at different concentrations in intracellular and extracellular in living cells, and the cytosolic GSH concentration is several times higher in tumor cells than in normal cells. The response of PSPps to a GSH concentration gradient seems a promising cue for triggering chemotherapeutic agents release at the tumor site. Several self-assembled micelles constructed from polylactide (PLA)-based block copolymers containing disulfide linkages have been reported by Oh and colleagues.6,11,31,32 The response of micelles to reducing agents (GSH or DTT) is strongly affected by the position of disulfide linkages. For example, the PLA-based block copolymer composed of the disulfide linkages in the dual location positioned in the PLA core and PLA/POEOMA (polyoligoethylene oxide methacrylate) interface showed disassembly of micelles in the presence of 10 mM GSH after 20 h.32 Amphiphilic block PLA-SS-POEOMA copolymers functionalized with disulfides at hydrophilic and hydrophobic block junctions degraded in the presence of DTT after 2 h.6 Although SS(PLA-b-POEOMA)2 bearing a single disulfide linkage in the middle of triblock copolymers did not exhibit a marked change in the dynamic particle size after 2 days with DTT, the molecular weight of the extracted residue evaluated by GPC was half that of the original.33 This result suggests that the central single disulfide linkage of SS(PLA-b-POEOMA)2 in the micellar core can be cleaved in response to DTT, to yield amphiphilic HS-PLA-b-POEOMA. Based on the studies of Oh and colleagues as well as our own previous studies,1 polymers with disulfide linkages positioned in the central core of micelles exhibited a prolonged response to reducing agents. We therefore decided to design a disulfide bond between the hydrophobic and hydrophilic junctions in order to improve the response to GSH.
In vitro Cytotoxicity and Cellular Uptake
The relative cell viabilities of PSPps were tested in HEK-293T, CRL-5802, NCI-520 and A549 cells (Figure 2A). No obvious cytotoxicity of PSPps was observed at test concentrations of 40–250 μg⋅mL−1 or any dependence on cell lines used. Even when cells were exposed to the highest concentration of 250 μg⋅mL−1, their viabilities were still ~90%. Flow cytometry was used to study the uptake of PSPps. Figure 2B demonstrates the internalized amount of PSPps into A549 cells increases with an increased incubation time from 1 h to 4 h. To clarify internalization pathways of cells exposed to PSPps, we labelled PSPps with a fluorescent dye. We tested three major internalization pathways using their corresponding chemical inhibitors, the cellular uptake of PSPps into A549 cells clearly decreased when cells were pretreated with chlorpromazine and genistein (Figure 2C), and similar results were found in CRL5802 cells (Figure S3). These leftward shifts of the flow-cytometry diagrams suggest that both A549 cells and CRL5802 cells utilized endocytosis mediated by clathrin and caveolae to internalize PSPps particles. The use of double internalization pathways is beneficial for the development of PSPps in DDS.
Figure 2.

In vitro cytotoxicity and internalization of PSPps. (A) Relative cell viabilities of HEK-293T, CRL-5802, NCI-520 and A549 cells exposed to PSPps at various concentrations. (B) Flow-cytometry diagrams of A549 cells exposed to rhodamine 123 (R123)-linked PSPps at 250 µg⋅mL−1 for 1h and 4h incubation. (C) Flow-cytometry diagrams of A549 cells exposed to R123-linked PSPps at 250 µg⋅mL−1 for 4h incubation after the cells were pretreated with different chemical inhibitors: 10 µg⋅mL−1 chlorpromazine, 200 µM genistein and 50 nM wortmannin for 30 min.
Encapsulation of IONPs with PSPps and Their Characterization
We selected truncated octahedral Fe3O4 nanoparticles (IONPs) instead of the commonly used spherical Fe3O4 nanoparticles because the former offer distinct advantages including higher Ms and higher cellular uptake ability (our unpublished data). Kolhatkar et al also explained that greater crystallization in the multi-domain size regime of truncated octahedral Fe3O4 nanoparticles resulted in stronger binding to substrate surfaces and better magnetic properties.20 The diagonal length of IONPs was found to be 15.6±3.8 nm (Figure S4A), and the X-ray diffraction (XRD) pattern confirmed the crystallization of Fe3O4 according to JCPDS card no. 19–0629 (Figure S4B), while high-resolution TEM images explained the truncated octahedral morphology of IONPs (Figure S4C). The IONPs had lattice spacing (d-spacing) of 2.9 Å, 2.4 Å, and 2.2 Å in the (220), (222) and (004) planes, respectively (Figure S4D).
The TEM image in Figure 3A shows that the IONPs-PSPps NPs had a size of 222.0±20.1 nm. Changes in the size of PSPps and IONPs-PSPps NPs in PBS at pH 7.4 within 60 days were examined by DLS (Figure S5). No obvious change in size was followed. The hydrodynamic sizes of PSPps and IONPs-PSPps NPs were ~200 nm and ~250 nm, respectively. The Ms values of IONPs and IONPs-PSPps NPs were 93 and 85 emu⋅gFe3O4−1 (Figure 3B). The IONPs embedded in the hydrophobic domain of PSPps slightly reduced Ms, while maintaining strong superparamagnetism. To evaluate the potential of IONPs-PSPps NPs as MRI contrast agents, test samples were prepared in 0.5 wt% agarose gel at various iron concentrations. The r2 values of IONPs and IONPs-PSPps NPs were 217 and 180 mM−1⋅s−1, respectively (Figure 3C and D). Comparisons with the conventional superparamagnetic products Resovist (151.0 mM−1⋅s−1),34 AMI-25 (100 mM−1⋅s−1)35 and Combidex (65 mM−1⋅s−1)36 reveal that the IONPs-PSPps NPs are potentially useful as an MRI contrast agent.
Figure 3.

Characterization of PSPps encapsulated with truncated octahedral Fe3O4 nanoparticles (IONPs) in the hydrophobic compartment (IONPs-PSPps NPs). (A) TEM image of IONPs-PSPps NPs. (B) Hysteresis loops of IONPs (blue curve) and IONPs-PSPps NPs (orange curve) at 300 K. The insets are enlarged hysteresis curves, and BR and HC correspond to their remnant magnetization and coercivity, respectively. Saturation magnetizations (MS) were calculated based on the weight of Fe3O4. Calculated relaxivity (r2) values of (C) IONPs and (D) IONPs-PSPps NPs based on the T2 relaxation rate versus iron ion concentration.
Encapsulation of DOX in IONPs-PSPps NPs and Their Thermal Response
DOX and IONPs were simultaneously encapsulated into PSPps during the double emulsion. The optimal amount of DOX encapsulated with IONPs-PSPps@DOX NPs was found by using different amounts of DOX in feed. A DOX calibration curve was established to calculate the drug loading efficiency and encapsulation efficiency, which were ~1.4% and 65%, respectively (Figure S6A and B). Temperature elevations were examined in pure double-distilled (DD) water and in solutions containing IONPs, IONP-PSPps NPs and IONP-PSPps@DOX NPs upon AMF (Figure 4A). The power of AMF was maintained at 10 kW and the iron concentration was set at 200 μg⋅mL−1. The thermal image of DD water recorded before applying AMF was used as control group (Figure 4B). Figure 4C shows that exposure to AMF for 15 min increased the temperatures of test samples from 25°C to 28°C for DD water, to 60°C for IONPs, to 54°C for IONPs-PSPps NPs and to 52°C for IONP-PSPps@DOX NPs; their corresponding thermal images are shown in Figure 4D-G, respectively. The temperature clearly increased in all samples containing IONPs, indicating the rapid and efficient conversion of the energy in AMF into thermal energy.
Figure 4.

Thermal behaviors and doxorubicin (DOX) release profiles of test samples. (A) A photograph of the setup for obtaining thermal images of test samples after exposure to an alternating magnetic field (AMF). Temperatures were recorded using a digital near-infrared thermal imaging camera. (B) Recorded thermal image before applying AMF, as the control group. (C) Increases in temperatures of test samples exposed to AMF versus time. Representative thermal images of test samples exposed to AMF: (D) double-distilled (DD) water, (E) IONPs, (F) IONPs-PSPps NPs, and (G) IONPs-PSPps@DOX NPs. The iron concentration of IONPs, IONPs-PSPps NPs, and IONPs-PSPps@DOX NPs was controlled at 200 μg⋅mL−1. The AMF power was set at 10 kW for 15 min. (H) Drug release profiles of IONPs-PSPps@DOX NPs dispersed in PBS at pH 5.5 or 7.4 in the presence or absence of 5 mM glutathione (GSH) at 37 °C, and with or without exposure to AMF at 10 kW for 15 min, respectively. Experiments were performed in triplicate (n=3, **p<0.01).
The in vitro DOX release from IONPs-PSPps@DOX NPs was performed in PBS at pH 5.5 and pH 7.4 in the presence and absence of 5 mM GSH, and treated with and without exposure to AMF at 10 kW for 15 min, respectively. After the AMF treatment, test samples were continuously shaking at 37°C. Figure 4H shows that ~27% and ~48% of DOX were released from IONPs-PSPps@DOX NPs at pH 7.4 and pH 5.5. The DOX-released percentages for samples treated with AMF were ~33% at pH 7.4 and ~60% at pH 5.5, while those in the presence of GSH without exposure to AMF increased to ~78% at pH 7.4 and ~95% at pH 5.5, respectively. The DOX-released percentages were slightly increased upon AMF and they were ~80% at pH 7.4 and ~96% at 5.5.
The DOX-released behavior depends not only on the presence of the reducing agent but on the pH value. The degree of deprotonation of the PMAA segment in PCL-SS-PMAA was larger in pH 7.4 than in pH 5.5 because the pKa value of PMAA is ~4.8.30 The higher deprotonated carboxylic groups resulted in stronger interaction of PMAA with the salt form of DOX at pH 7.4, leading to the slow release of DOX. The pH-responsive drug release behavior of IONPs-PSPps@DOX NPs is advantageous in cancer therapy because the pH value of the tumor microenvironment is typically acidic. The extracellular pH is typically between 6.0 and 7.0 for a tumor, whereas it is ~7.4 in normal tissues and blood.37,38 NPs are generally taken up by endocytosis into cells and this endocytic pathway begins near the physiological pH of 7.4, then dropping to pH 5.5–6.0 in endosomes.39
To check whether AMF-induced hyperthermia changed the morphology of IONPs-PSPps@DOX NPs, TEM images were examined after test samples were exposed to AMF at 10 kW for 15 min and left in PBS either at pH 7.4 or pH 5.5 in the presence and absence of 5 mM GSH for different time points. As shown in Figure S7A, the IONPs-PSPps@DOX NPs exposed to AMF at pH 7.4 could preserve the spherical shape till 24 h, aggregate and disassemble after 96 h. Figure S7B is the size evolution profiles of IONPs-PSPps@DOX NPs estimated from TEM images shown in Figure S7A. Those exposed to AMF at pH 5.5 formed aggregates but preserved the spherical shape at 2 h and became denser and denser aggregates with prolonged incubation time. Nevertheless, those exposed to AMF in pH 5.5 PBS containing 5 mM GSH showed that the polymersome nanostructure disappeared at 2 h and completely disassembled at 8 h. The IONPs-PSPps@DOX NPs endowed a triple-responsive character, including pH-sensitivity, redox-response, and AMF-induced hyperthermia; all of them could accelerate the drug release in the tumor site.
Cell Viability and Cell Apoptosis
We checked the cytotoxicity of IONPs-PSPps NP against HEK-293T, CRL-5802, NCI-520 and A549 cells (Figure 5A). No obvious cytotoxicity of IONPs-PSPps NPs was observed for test concentrations used or any dependence on cell lines. After ensuring non-toxicity of IONPs-PSPps NPs, we optimized the exposure power and duration of AMF applied to IONPs-PSPps@DOX NPs and the annexin-V/PI dual-staining assay was carried out to evaluate an apoptosis percentage in A549 cells. The effect of exposure to AMF at 10 kW on cell apoptosis for different time points is shown in Figure S8A and B. The summation of Q1 (necrotic cells), Q2 (late apoptotic cells) and Q4 (apoptotic cells) indicates percentages of the cell death for applied AMF. They were ~80, 35, and 10% corresponding to the exposure time of 15, 10 and 5 min, respectively. In parallel, the time point of 15 min was fixed to test cell apoptosis as the cells were exposed for different AMF powers. As shown in Figure S9A and B, the cell death reached almost 100%, implying that the powers of 13.5 and 18 kW were too high to see the cell-killing effect of the released DOX from IONPs-PSPps@DOX NPs in later study. On the other hand, ~50% cell death was observed for AMF at 4.5 kW, which might be too low. Thus, the power of 10 kW and exposure time of 15 min were then selected for the subsequent AMF testing.
Figure 5.

Cell apoptosis of test samples with different treatments. In vitro cytotoxicity analysis evaluated by the MTT assay for (A) IONPs-PSPps NPs against four cell lines and (B) test samples against A549 cells with different treatments. Statistical analysis was performed using the two-tailed Student’s t-test (n=8, *p<0.05, **p<0.01). (C) Annexin-V/PI (propidium iodide) dual-staining assay by flow cytometry analysis. (D) The sum of Q1, Q2 and Q4 accounted for the percentage of the death cells. A549 cells were used as a model for examining the antitumor efficacy of IONPs-PSPps and IONPs-PSPps@DOX NPs with or without exposure to AMF at 10 kW for 15 min. The four quadrants from Q1 to Q4 corresponded to necrotic cells, late apoptotic cells, viable cells, and apoptotic cells, respectively. The experiments were performed in triplicate, and statistical analysis was performed using the two-tailed Student’s t-test (n=3, *p<0.05, **p<0.01).
For antitumor activity testing, A549 cells were exposed to the relevant IONPs at a concentration of 100 μg⋅mL−1 and a free DOX concentration of 4.4 μM. To improve internalization of test samples containing IONPs, a commercially available MagnetoFACTOR-96 plate was placed underneath the cell culture plate for 30 min before cells were incubated at 37°C for 24 h with and without exposure to AMF at 10 kW for 15 min. Figure 5B shows the cell viability percentages of test samples relative to the control for different treatments, ie, 35% for free DOX, 45% for IONPs-PSPps@DOX (-AMF), 35% for IONPs-PSPps (+AMF), and 12% for IONPs-PSPps@DOX (+AMF), respectively. These results clearly indicate that cell viability decreased significantly as cells were exposed to AMF. In the presence of AMF, the IONPs-PSPps@DOX NPs showed significantly higher cytotoxicity than the IONPs-PSPps NPs against A549 cells (p<0.01). Consistent results were obtained using the annexin-V/PI dual-staining assay, as shown in Figure 5C, and the sum of Q1, Q2 and Q4 quantitative percentage of the death cells as plotted in Figure 5D, ie, ~60% for free DOX, ~80% for IONPs-PSPps@DOX (-AMF), ~80% for IONPs-PSPps (+AMF), and 90% for IONPs-PSPps@DOX (+AMF), respectively. Compared with free DOX, a significant effect on killing cells was found for IONPs-PSPps@DOX NPs with a combination of hyperthermia and chemotherapy.
In vivo MRI Contrast
Following 7 days of A549-tumor xenografting in nude mice (BALB/cAnN), tumors grew to 25–30 mm3. Mice were injected with PBS or PBS containing IONPs-PSPps@DOX NPs intravenously, and MRI was performed at pre-injection and post-injections at 1, 3, 5 h with assistance from a magnet or without assistance from a magnet for 30 min using a 9.4-T animal micro-MRI system (Figure 6A). The tumor area was indicated by white arrows and the blood vessel was indicated by red arrows. Negative contrast enhancement in mouse tumors were clearly observed in the group treated with IONPs-PSPps@DOX (+M) with assistance from a magnet. In addition, relative contrast intensities in in vivo T2-weighted images of mice dorsal flank tumors shown in Figure 6A were calculated in each group (Figure S10). The negative contrast enhancement with non-specific binding was observed in IONPs-PSPps@DOX (-M) without assistance from a magnet at post 1 h. Compared with pre-injection, the signal intensity decreased at this time point. After that the contrast enhancement of the tumor region recovered at post 3 h and 5 h with the circulation. The signal intensity was similar to sham control (PBS group) in T2-weighted MR imaging. The blood vessels in the tumor regions were also traced with MR imaging in each group (indicated with red arrows in Figure 6A).
Figure 6.

In vivo magnetic resonance imaging and biodistribution. (A) In vivo T2-weighted images of PBS (control group) and IONPs-PSPps@DOX NPs at a dose of 10 mg.kg−1 using a 9.4-T animal magnetic resonance imaging (MRI) system. Mice were injected with IONPs-PSPps@DOX NPs intravenously, and MRI images were captured at preinjection and 1, 3, and 5h post-injection with magnet assistance or without magnet assistance for 30 min (tumor was indicated by white arrows; blood vessel was indicated by red arrows). (B) The residual iron contents in different tissues of A549 tumor-bearing mice. Mice were injected with IONPs-PSPps@DOX NPs in PBS via the tail vein at a dose of 24 mg⋅kg−1. In some groups, a magnet was placed near one of the tumors for 30 min (+M). (-M) indicates the tumor without the magnetic assistance. Three mice were sacrificed at the indicated time points, and the tissues were dissected for iron content measurements by inductively coupled plasma/atomic emission spectroscopy. The mice administered with PBS were used as a control group, and statistical analysis was performed using the two-tailed Student’s t-test (n=3, **p<0.01).
The quantitatively contrast intensities of the mouse tumors were also included in Figure S10 (the percentage of the in vivo T2-weighted MR images relative to the pre-injection of each group was processed using ImageJ software). Based on the imaging processing and signal analysis results, the significant difference in the negative MRI contrast enhancement with and without assistance from a magnet was found at 3 h and 5 h post-injection (p<0.05). The MR signal intensity decreased to 91.6±2.2% (post 3 h) and 88.6±4.2% (post 5 h) for IONPs-PSPps@DOX (+M). The MR signal intensity did not show any significant change in the group of sham control and IONPs-PSPps@DOX (-M). This fact indicates that the IONPs-PSPps@DOX NPs could be guided by magnetic attraction and accumulated near the tumor region. Besides, the immature angiogenesis and the enhanced permeability and retention (EPR) effect of the tumor also increased accumulation of NPs at the tumor site; leading to enhancement of the negative signal in the tumor region at post 3 h and 5 h for the IONPs-PSPps@DOX (+M) group. When we compared the relative signal change in T2-weighted MR images at post 24 h, the signal intensity recovered, respectively, to be 102±4% for sham control, 91±5% for IONPs-PSPps@DOX (+M), and 102±1% for IONPs-PSPps@DOX (-M) (data were not shown), indicating that the IONPs might be eliminated by circulation after 24 h circulation. Similar results were found in our previous study, where the IONPs coated with a double layer of Au/Ag hybrid metals showed the highest contrast enhancement at 1h post-injection, and then became vague thereafter.28
Biodistribution, Biocompatibility and Therapeutic Effect of IONPs-PSPps@DOX NPs
The biodistribution of IONPs-PSPps@DOX NPs was investigated by measuring the residual iron contents in different tissues of A549 tumor-bearing mice. Mice were injected with PBS (control group) or PBS containing IONPs-PSPps@DOX NPs via the tail vein at a dose of 24 mg⋅kg−1 and sacrificed at different time points after injecting the IONPs-PSPps@DOX NPs for 3, 7, 24, 48, 72 and 168 h. Organ tissues of the heart, liver, spleen, lung, kidney, tumor (+M) with assistance from a magnet and tumor (-M) without assistance from a magnet were resected from mice for biodistribution analysis. The results show that the IONPs-PSPps@DOX NPs were the most abundant in the tumor with assistance from a magnet after 3 h, and then subsequently excreted (Figure 6B). A significant difference in the amount of iron between the groups with and without assistance from a magnet was found at 3 and 7 h post-injection (p<0.01). Even in mice in which a magnet was used, the iron content in the tumor decreased dramatically at 7 h post-injection. The similar iron content in the tumor without assistance from a magnet explains that the optimized circulation time of the IONPs-PSPps@DOX NPs in mice was within 3–7 h and that most of the IONPs-PSPps@DOX NPs could be attracted to the tumor when the magnet was placed nearby. This finding was consistent with the MRI contrast enhancement result. In addition, a large amount of iron ions accumulated in the spleen and liver at 3 and 7 h post-injection. Both of these organs are involved in metabolism.
Mice were treated with IONPs-PSPps NPs or IONPs-PSPps@DOX NPs, and one of the tumors was either exposed to or not exposed to a magnet for 30 min, followed by either AMF exposure or no AMF exposure. As shown in Figure 7A and B, the control group (PBS group) shows a continuous growth in tumor size over 30 days, approaching up to 60 times in size compared with its initial size (Figure 7C). Mice injected with IONPs-PSPps@DOX NPs, which endowed a combination effect of hyperthermia when AMF was applied and chemotherapy when DOX was released, exhibited the highest antitumor activity, with a ~100% reduction in tumor volume without significant changes in body weight (Figure S11). Tumor growth sizes in A549 tumor-bearing mice at Day 30 were recorded, and statistical analysis was performed using the Tukey’s multiple comparison test. Figure 7D shows the comparison result among mice with different treatments. Compared therapeutic outcome with free DOX, no significant difference was found for IONPs-PSPps@DOX (+M) (-AMF) with assistance from a magnet and without exposure to AMF but a significant difference for IONPs-PSPps@DOX (+M) (+AMF) with dual stimuli (p<0.05). This result explains that the applied AMF indeed enhanced the DOX release for better chemotherapy. Tumors collected from different groups of mice at the endpoint of treatments were photographed to demonstrate the superior antitumor efficacy of mice treated with IONPs-PSPps@DOX NPs, a magnet and AMF-induced hyperthermia (Figure 7B). The toxicity of IONPs-PSPps@DOX NPs was evaluated by examining H&E-stained histological images of the dissected tissues from the mice that underwent various treatments. No obvious organ damage or inflammation was observed (Figure S12). It was worth mentioning that the morphology of the spleen, which is an important organ in blood purification, exhibited no appreciable abnormality. This finding will be useful when developing the IONPs-PSPps@DOX NPs for biomedical applications.
Figure 7.

In vivo therapeutic outcome of test samples. (A) Photographs of the mice before (0th day) and after (30th day) treatments (the red arrow indicated the position of magnetic attraction). (B) Tumors collected from different groups of the mice at the endpoint of treatments (30th day). (C) Tumor growth curves in A549 tumor-bearing mice after different treatments. Statistical analysis was performed using the two-tailed Student’s t-test (n=4, *p<0.05, **p<0.01) (D) Tumor growth sizes in A549 tumor-bearing mice after different treatments at Day 30. Statistical analysis was performed using the Tukey’s multiple comparison test (n=4, ***p<0.05). The Mice were treated with IONPs-PSPps NPs or IONPs-PSPps@DOX NPs, and one of the tumors received or did not receive assistance from a magnet for 30 min as well as with (+AMF) or without (-AMF) exposure to AMF at 10 kW for 15 min.
Blood samples were also collected for biochemical analysis. To ensure that the metabolized IONPs-PSPps@DOX residues would not damage the liver or urinary tract, the activities of liver (GOT and GPT) and kidney (BUN, CRE, and UA) of the mice treated with IONPs-PSPps@DOX NPs were evaluated (Figure S13). All levels of the IONPs-PSPps@DOX-treated mice were similar to those of PBS-treated mice (control group). These preliminary toxicological results imply that the IONPs-PSPps@DOX NPs are a promising theranostic agent for use in future translational applications.
Conclusion
A pH and redox dual-responsive PCL-SS-PMAA copolymer was successfully synthesized via ROP, ATRP and hydrolysis. By tuning the numbers of PCL and PMAA repeating units, the PCL-SS-PMAA copolymer could be formulated into PSPps, which had a rapid response to physiological GSH concentrations. IONPs and DOX were simultaneously encapsulated into PSPps during double emulsion to yield IONPs-PSPps@DOX NPs. The high values of Ms and r2 implied that the IONPs-PSPps@DOX NPs are a potential MRI contrast agent. DOX released from IONPs-PSPps@DOX NPs was accelerated in a low-pH condition and in the presence of a reducing agent, and similarly responsive in TME. In vitro evaluations demonstrated that the PSPps were non-toxic against three lung cancer cell lines, but the IONPs-PSPps@DOX NPs showed an enhanced cell-killing effect as AMF was applied with assistance from a magnet. The biodistribution results implied that the accumulated amount of IONPs-PSPps@DOX NPs peaked within 3–7 h, followed by metabolization. In vivo studies in A549 tumor-bearing mice showed a significant reduction in the tumor size of the group administered with IONPs-PSPps@DOX NPs and combined with assistance from a magnet and AMF when compared with the other groups. The preliminary toxicological results demonstrated that the IONPs-PSPps@DOX NPs did not produce any major damage to the heart, liver, spleen, lung, and kidney.
In conclusion, the IONPs-PSPps@DOX NPs possess multifaceted characteristics including negative contrast enhancement for MRI, AMF-induced thermal heat for hyperthermia and DOX release for chemotherapy. The IONPs-PSPps@DOX NPs therefore have potential as a theranostic agent in cancer therapy.
Supporting Information
Supporting information can be found as the attached supplementary materials (Supplementary Figures S1-S13) including 1H-NMR, dynamic light scattering, confocal microscopy images, TEM images, flow cytometry, MR imaging processing, and blood biochemistry analysis.
Acknowledgments
We are grateful for financial support from the Ministry of Science and Technology of Taiwan (grant nos. MOST106-2320-B-037-004-MY3 and MOST108-2221-E-037-002-MY3 for L.-F Wang, and MOST106-2314-B-182A-008-MY3 for C.-H. Su). This study was also supported by Kaohsiung Medical University under grant no. KMU-DK(個)109001, by the NSYSU-KMU Joint Research Project under grant no. NSYSUKMU 109-P009, and by the Chang Gung Medical Foundation under grant no. CMRPG8I0141-3 and CMRPG8J0451-3. We appreciate the experimental support of a confocal laser scanning microscope, IVIS and a transmission electron microscope provided by the Center for Research Resources and Development of Kaohsiung Medical University.
Disclosure
The authors declare that they have no conflicts of interest.
References
- 1.Liu Y-S, Huang S-J, Huang X-S, et al. The synthesis and comparison of poly(methacrylic acid)–poly(ε-caprolactone) block copolymers with and without symmetrical disulfide linkages in the center for enhanced cellular uptake. RSC Adv. 2016;6(79):75092–75103. doi: 10.1039/C6RA15307K [DOI] [Google Scholar]
- 2.Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. [DOI] [PubMed] [Google Scholar]
- 3.Huang L, Chaurasiya B, Wu D, et al. Versatile redox-sensitive pullulan nanoparticles for enhanced liver targeting and efficient cancer therapy. Nanomedicine. 2018;14(3):1005–1017. [DOI] [PubMed] [Google Scholar]
- 4.Wei H, Volpatti LR, Sellers DL, et al. Dual responsive, stabilized nanoparticles for efficient in vivo plasmid delivery. Angew Chem Int Ed Engl. 2013;52(20):5377–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song N, Liu W, Tu Q, Liu R, Zhang Y, Wang J. Preparation and in vitro properties of redox-responsive polymeric nanoparticles for paclitaxel delivery. Colloids Surf B Biointerfaces. 2011;87(2):454–463. [DOI] [PubMed] [Google Scholar]
- 6.Khorsand Sourkohi B, Cunningham A, Zhang Q, Oh JK. Biodegradable block copolymer micelles with thiol-responsive sheddable coronas. Biomacromolecules. 2011;12(10):3819–3825. [DOI] [PubMed] [Google Scholar]
- 7.Li C, Madsen J, Armes SP, Lewis AL. A new class of biochemically degradable, stimulus-responsive triblock copolymer gelators. Angew Chem Int Ed Engl. 2006;45(21):3510–3513. [DOI] [PubMed] [Google Scholar]
- 8.Ding M, Li J, He X, et al. Molecular engineered super-nanodevices: smart and safe delivery of potent drugs into tumors. Adv Mater. 2012;24(27):3639–3645. [DOI] [PubMed] [Google Scholar]
- 9.Cunningham A, Ko NR, Oh JK. Synthesis and reduction-responsive disassembly of PLA-based mono-cleavable micelles. Colloids Surf B Biointerfaces. 2014;122:693–700. [DOI] [PubMed] [Google Scholar]
- 10.Khorsand B, Lapointe G, Brett C, Oh JK. Intracellular drug delivery nanocarriers of glutathione-responsive degradable block copolymers having pendant disulfide linkages. Biomacromolecules. 2013;14(6):2103–2111. doi: 10.1021/bm4004805 [DOI] [PubMed] [Google Scholar]
- 11.Zhang Q, Aleksanian S, Nohbc SM, Oh JK. Thiol-responsive block copolymer nanocarriers exhibiting tunable release with morphology changes. Polym. Chem. 2013;4(2):351–359. doi: 10.1039/C2PY20582C [DOI] [Google Scholar]
- 12.Chen K, Liao SS, Guo SW, et al. Multistimuli-responsive PEGylated polymeric bioconjugate-based nano-aggregate for cancer therapy. Chem Eng J. 2020;391. [Google Scholar]
- 13.Pan DY, Zheng XL, Zhang QF, et al. Dendronized-Polymer Disturbing Cells’ Stress Protection by Targeting Metabolism Leads to Tumor Vulnerability. Adv Mater. 2020;32:14. [DOI] [PubMed] [Google Scholar]
- 14.Zhang XQ, Wu YH, Li ZQ, et al. Glycodendron/pyropheophorbide-a (Ppa)-functionalized hyaluronic acid as a nanosystem for tumor photodynamic therapy. Carbohyd Polym. 2020;247. [DOI] [PubMed] [Google Scholar]
- 15.Tritschler U, Pearce S, Gwyther J, Whittell GR, Manners I. 50th Anniversary Perspective: functional Nanoparticles from the Solution Self-Assembly of Block Copolymers. Macromolecules. 2017;50(9):3439–3463. [Google Scholar]
- 16.Oliveira H, Perez-Andres E, Thevenot J, Sandre O, Berra E, Lecommandoux S. Magnetic field triggered drug release from polymersomes for cancer therapeutics. J Control Release. 2013;169(3):165–170. [DOI] [PubMed] [Google Scholar]
- 17.Lu AH, Salabas EL, Schuth F. Magnetic nanoparticles: synthesis, protection, functionalization, and application. Angew Chem Int Ed Engl. 2007;46(8):1222–1244. [DOI] [PubMed] [Google Scholar]
- 18.Wang M, Li J, Li X, et al. Magnetically and pH dual responsive dendrosomes for tumor accumulation enhanced folate-targeted hybrid drug delivery. J Controlled Release. 2016;232:161–174. [DOI] [PubMed] [Google Scholar]
- 19.Zolata H, Afarideh H, Davani FA. Triple Therapy of HER2+ Cancer Using Radiolabeled Multifunctional Iron Oxide Nanoparticles and Alternating Magnetic Field. Cancer Biother Radiopharm. 2016;31(9):324–329. [DOI] [PubMed] [Google Scholar]
- 20.Kolhatkar AG, Chen YT, Chinwangso P, et al. Magnetic Sensing Potential of Fe3O4 Nanocubes Exceeds That of Fe3O4 Nanospheres. ACS Omega. 2017;2(11):8010–8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harush-Frenkel O, Debotton N, Benita S, Altschuler Y. Targeting of nanoparticles to the clathrin-mediated endocytic pathway. Biochem Biophys Res Commun. 2007;353(1):26–32. [DOI] [PubMed] [Google Scholar]
- 22.Harush-Frenkel O, Rozentur E, Benita S, Altschuler Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules. 2008;9(2):435–443. [DOI] [PubMed] [Google Scholar]
- 23.Chen GJ, Hsu C, Ke JH, Wang LF. Imaging and Chemotherapeutic Comparisons of Iron Oxide Nanoparticles Chemically and Physically Coated with Poly(ethylene glycol)-b-Poly(ε-caprolactone)-g-Poly(acrylic acid). J Biomed Nanotech. 2015;11(6):951–963. [DOI] [PubMed] [Google Scholar]
- 24.Liu YS, Cheng RY, Lo YL, et al. Distinct CPT-induced deaths in lung cancer cells caused by clathrin-mediated internalization of CP micelles. Nanoscale. 2016;8(6):3510–3522. [DOI] [PubMed] [Google Scholar]
- 25.Chan N, An SY, Oh JK. Dual location disulfide degradable interlayer-crosslinked micelles with extended sheddable coronas exhibiting enhanced colloidal stability and rapid release. Polym Chem-Uk. 2014;5(5):1637–1649. [Google Scholar]
- 26.Yang YQ, Zheng LS, Guo XD, Qian Y, Zhang LJ. pH-Sensitive micelles self-assembled from amphiphilic copolymer brush for delivery of poorly water-soluble drugs. Biomacromolecules. 2011;12(1):116–122. [DOI] [PubMed] [Google Scholar]
- 27.Li WP, Su CH, Chang YC, Lin YJ, Yeh CS. Ultrasound-Induced Reactive Oxygen Species Mediated Therapy and Imaging Using a Fenton Reaction Activable Polymersome. ACS Nano. 2016;10(2):2017–2027. [DOI] [PubMed] [Google Scholar]
- 28.Tsai MF, Hsu C, Yeh CS, Hsiao YJ, Su CH, Wang LF. Tuning the Distance of Rattle-Shaped IONP@Shell-in-Shell Nanoparticles for Magnetically-Targeted Photothermal Therapy in the Second Near-Infrared Window. ACS Appl Mater Interfaces. 2018;10(2):1508–1519. [DOI] [PubMed] [Google Scholar]
- 29.Xiong D, Zhang R, Luo W, Gu H, Peng S, Zhang L. Hydrazone cross-linked micelles based on redox degradable block copolymer for enhanced stability and controlled drug release. React Funct Polym. 2017;119:64–74. [Google Scholar]
- 30.Schüwer N, Klok H-A. Tuning the pH Sensitivity of Poly(methacrylic acid) Brushes. Langmuir. 2011;27(8):4789–4796. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Qiu X, Ouyang J, Kong J, Zhong W, Xing MM. pH and reduction dual-sensitive copolymeric micelles for intracellular doxorubicin delivery. Biomacromolecules. 2011;12(10):3601–3611. [DOI] [PubMed] [Google Scholar]
- 32.Ko NR, Oh JK. Glutathione-Triggered Disassembly of Dual Disulfide Located Degradable Nanocarriers of Polylactide-Based Block Copolymers for Rapid Drug Release. Biomacromolecules. 2014;15(8):3180–3189. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham A, Oh JK. New Design of Thiol-Responsive Degradable Polylactide-Based Block Copolymer Micelles. Macromol Rapid Commun. 2013;34(2):163–168. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y-XJ. Superparamagnetic iron oxide based MRI contrast agents: current status of clinical application. Quant Imaging Med Surg. 2011;1(1):35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jun YW, Lee JH, Cheon J. Chemical design of nanoparticle probes for high-performance magnetic resonance imaging. Angew Chem Int Ed Engl. 2008;47(28):5122–5135. [DOI] [PubMed] [Google Scholar]
- 36.Estelrich J, Sanchez-Martin MJ, Busquets MA. Nanoparticles in magnetic resonance imaging: from simple to dual contrast agents. Int J Nanomedicine. 2015;10:1727–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danhier F, Feron O, Préat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Controlled Release. 2010;148(2):135–146. [DOI] [PubMed] [Google Scholar]
- 38.Breunig M, Bauer S, Goepferich A. Polymers and nanoparticles: intelligent tools for intracellular targeting? Eur J Pharm Biopharm. 2008;68(1):112–128. [DOI] [PubMed] [Google Scholar]
- 39.Park JS, Han TH, Lee KY, et al. N-acetyl histidine-conjugated glycol chitosan self-assembled nanoparticles for intracytoplasmic delivery of drugs: endocytosis, exocytosis and drug release. J Controlled Release. 2006;115(1):37–45. [DOI] [PubMed] [Google Scholar]