

Summary
Most triacylglycerol-lowering fibrates have been developed in the 1960s–1980s before their molecular target, peroxisome proliferator-activated receptor alpha (PPARα), was identified. Twenty-one ligand-bound PPARα structures have been deposited in the Protein Data Bank since 2001; however, binding modes of fibrates and physiological ligands remain unknown. Here we show thirty-four X-ray crystallographic structures of the PPARα ligand-binding domain, which are composed of a “Center” and four “Arm” regions, in complexes with five endogenous fatty acids, six fibrates in clinical use, and six synthetic PPARα agonists. High-resolution structural analyses, in combination with coactivator recruitment and thermostability assays, demonstrate that stearic and palmitic acids are presumably physiological ligands; coordination to Arm III is important for high PPARα potency/selectivity of pemafibrate and GW7647; and agonistic activities of four fibrates are enhanced by the partial agonist GW9662. These results renew our understanding of PPARα ligand recognition and contribute to the molecular design of next-generation PPAR-targeted drugs.
Subject Areas: Biochemistry, Molecular Physiology, Structural Biology, Protein Structure Aspects
Graphical Abstract
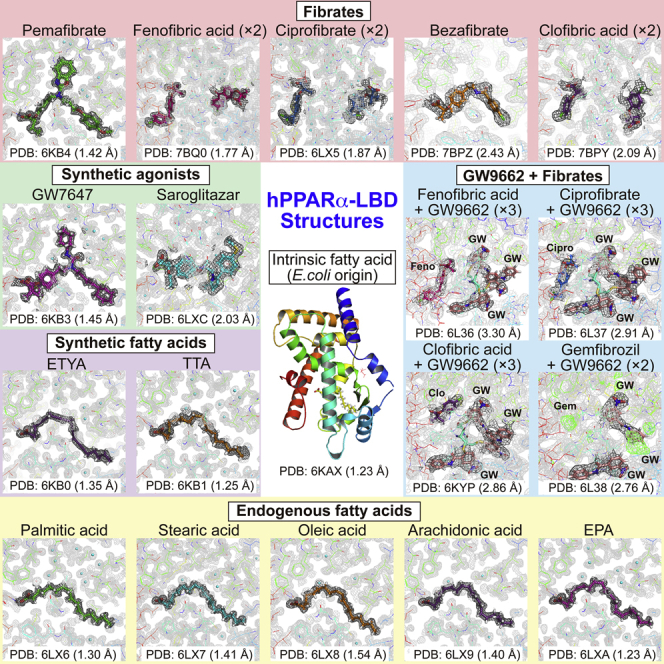
Highlights
-
•
X-ray crystallography reveals 34 high-resolution human PPARα-ligand structures
-
•
Stearic acid and palmitic acid are presumably physiological PPARα ligands
-
•
Coordination to Arm III domain is important for high PPARα potency/selectivity
-
•
Agonistic activities of four fibrates are enhanced by the partial agonist GW9662
Biochemistry; Molecular Physiology; Structural Biology; Protein Structure Aspects
Introduction
Cardiovascular diseases (CVDs) have been identified as the leading cause of deaths worldwide, accounting for 17.9 million deaths annually (WHO, 2017). High blood levels of low-density lipoprotein cholesterol (LDL-C), high triacylglycerol (TG), and low high-density lipoprotein cholesterol (HDL-C) are all considered major risk factors for the development of CVD (Andersson et al., 2014). Although statins, which inhibit β-hydroxy-β-methylglutaryl-CoA reductase, are found to lower blood LDL-C levels, fibrates, which are peroxisome proliferator-activated receptor alpha (PPARα) agonists, lower TG levels and elevate HDL-C levels (Bougarne et al., 2018; Yamashita et al., 2019). Combining statins and fibrates could offer therapeutic advantages for the treatment of refractory combined dyslipidemia (Franssen et al., 2009; Han et al., 2017). However, in general, it has not been recommended because of the increased risks of myopathy and rhabdomyolysis (Franssen et al., 2009; Graham et al., 2004).
Differences in the pharmacokinetic interaction potentials of fibrates with statins have emerged (Franssen et al., 2009), which could act on other cognate PPARs (Yamashita et al., 2019), such as PPAR gamma (PPARγ), which regulates lipid/glucose metabolism, mainly in adipose tissue, and is the target of anti-diabetic thiazolidinedione (glitazone) drugs, and PPAR delta (PPARδ), which is expressed in many tissues to govern a variety of biological processes and whose agonists are expected to treat metabolic/cardiovascular diseases (Da'adoosh et al., 2019; Wu et al., 2017). Differential interactions of fibrates with other PPARs could elicit either beneficial effects or undesirable side effects, although novel effects of fibrates, such as synergistic tumor suppression activity with anti-PD-1 antibody (Chamoto et al., 2017), via unknown mechanisms, are also evident. Two PPARα/γ dual agonists, saroglitazar and lobeglitazone, have already been clinically approved in India and Korea, respectively, as next-generation therapeutics for diabetic dyslipidemia (Han et al., 2017). In 2018, pemafibrate, which is also known as K-877 or SPPARMα (selective PPARα modulator), was approved in Japan as a highly PPARα-selective agonist that is safe for simultaneous use with statins, even in patients with mild kidney diseases (Yamashita et al., 2019, 2020; Yokote et al., 2019). Thus, further development of PPAR dual/pan agonists, including PPARα/γ agonists as anti-diabetic drugs (Artis et al., 2009; Bénardeau et al., 2009) and PPARα/δ agonists as anti-nonalcoholic steatohepatitis (NASH) drugs, and PPARα-specific agonists is anticipated (Han et al., 2017).
However, compared with PPARγ and PPARδ, structural information for the PPARα-ligand remains limited. Only 21 records of PPARα structures have been deposited in the Protein Data Bank (PDB; http://www.pdb.org/) (Artis et al., 2009; Bernardes et al., 2013; Bénardeau et al., 2009; Kawasaki et al., 2020; Kuwabara et al., 2012; Oyama et al., 2009; Xu et al., 2001, 2002) (Table S1), in contrast to 224 for PPARγ (Artis et al., 2009; Bénardeau et al., 2009; Brust et al., 2018; Fyffe et al., 2006; Itoh et al., 2008; Kuwabara et al., 2012; Nolte et al., 1998; Oyama et al., 2009) and 44 for PPARδ (Artis et al., 2009; Oyama et al., 2009; Xu et al., 1999). In addition, there have been no depositions for endogenous (physiological) ligands or fibrates, except for pemafibrate (Kawasaki et al., 2020). The limited data describe the 1,400 Å3 cavity of the PPARα-ligand-binding domain (LBD) (Xu et al., 2001), which is equivalent to the PPARγ-LBD (1,440 Å3) and PPARδ-LBD (1,300 Å3) but is found much larger than those observed in other nuclear receptors (600–1,100 Å3) (Itoh et al., 2008; Wu et al., 2017; Xu et al., 1999). It is composed of five regions; Arm I, which is surrounded by helices 3, 11, and 12 and loop H11–H12; Arm II, with helices 2′ and 3 and strands 1, 3, 4; Arm III, with helices 3 and 5 and loop H1–H2; Arm X, which is outside of Arm II; and Center, with helices 3, 5, 7, and 12 (which we renamed from Zoete et al., 2007; Figure S1). However, the roles of those regions for fibrate recognition are yet to be determined.
Using X-ray crystallography, this study provided 34 high-resolution PPARα-LBD structures in complexes with 17 ligands (Figure S2), which include endogenous fatty acids, all clinically approved fibrates, and other synthetic PPARα agonists (Han et al., 2017). Although the study was underway, Kawasaki et al. (2020) reported an X-ray crystal structure of the ternary complex of PPARα-LBD/pemafibrate/coactivator peptide at 3.20 Å resolution (1.52 Å in this study), in which quite limited information was available about the location of pemafibrate and its surrounding amino acid side chains.
Results
Structures with Intrinsic Fatty Acids
Unlike PPARγ (Nolte et al., 1998), it was difficult to obtain PPARα apocrystals. First, we reproduced Wy14643-bound crystals (Bernardes et al., 2013) using co-crystallization (Hassell et al., 2007) with highly purified human PPARα-LBD recombinant proteins (Oyama et al., 2009; Capelli et al., 2016) (Figures S3A–S3C). The Wy14643-bound rod-shaped crystals (Figure 1A) that we formed using buffer no. 43 of the Index kit (Table S2, related to Figure 1) provided similar but higher-resolution (1.82 Å) diffraction data, including its two binding sites in Center with Arm X, than the referenced report (Bernardes et al., 2013) (Figures S4A and S4B). Collected crystallography data and refinement statistics (Tables S3 and S4) were deposited in the PDB. Next, we used cross-seeding (Hassell et al., 2007) to obtain crystals containing an intrinsic fatty acid (iFA) of Escherichia coli origin (Figure 1A) from crushes of the Wy14643-bound crystals as crystal nuclei (Benvenuti et al., 2007) (Figure S3C), which gave an atomic resolution (1.23 Å) data (Figures 1B and 1C). The Fo-Fc omit map illustrated the electron density of palmitic acid, stearic acid, oleic acid, or vaccenic acid (18:1, trans-11 (n–7) that is most abundant in E. coli extracts (Fyffe et al., 2006)) (Figure 1C). Mass spectrometry analyses of the lipid contents of PPARα-LBD proteins showed that they are mixtures of palmitic acid, stearic acid, and oleic or vaccenic acid, which is difficult to distinguish when using mass spectrometry (Figure 1D). Among these, palmitic acid and stearic acid displayed PPARγ coactivator 1-alpha (PGC1α) coactivator recruitment activity as a measure of PPARα activation (Figure 1E). When five young male undergraduate student volunteers were fasted for 12 h, their serum-free fatty acid concentrations were 114, 48, 133, and 78 μM for palmitic, stearic, oleic, and linoleic acid, respectively. When they were fed thereafter, these concentrations were dramatically declined to 41, 21, 17, and 7 μM, respectively, after 2 h (Figure 1F). Stearic acid (45 μM) and, more potently, its combination with palmitic acid (110 μM) significantly activated PPARα, but further supplementation of oleic acid (130 μM) plus linoleic acid (75 μM) abolished it (Figure 1G). A thermostability assay also identified higher Tm values in stearic and palmitic acids rather than in oleic acid (Figure 1I). These data suggest that both stearic and palmitic acids are physiological PPARα ligands. Their potency for PPARα activation/stabilization was lower than most synthetic PPARα agonists but equivalent to clofibric acid and higher than gemfibrozil (Figures 1H and 1I). The pharmacological effects of fibrates in clinical use are elicited by their replacement with iFAs in the PPARα-LBD.
Figure 1.

Structures of Intrinsic Fatty Acid-Bound PPARα-LBD and PPARα Activation by Endogenous Fatty Acids
(A) Intrinsic fatty acid (iFA)-bound crystals (right) obtained using crushed Wy14643-bound crystals (left) as crystal nuclei. Scale bars: 100 μm.
(B) High-resolution (1.23 Å) structure of PPARα-LBD and iFA (yellow). α-helices, β-sheets, and glycerol are labeled.
(C) Magnified view of iFA and surrounding amino acids. The electron density is shown in the mesh by Fo-Fc omit map contoured at 3.0σ. Water molecules are presented as cyan spheres.
(D) Contents of the seven major fatty acids (of E. coli origin) in PPARα-LBD proteins measured by LC-MS/MS analysis.
(E) PPARα activation (PGC1α coactivator recruitment) by endogenous fatty acids. Maximal response by 1 μM GW7647 was set as 100 (%).
(F) Serum fatty acid contents of five healthy male volunteers who were fasted for 12 h and then fed. Serum fatty acid levels were decreased after 2 h.
(G) Effects of fatty acid combinations on PPARα activation.
(H) Concentration-dependent PPARα activation by several ligands; their EC50 values are shown in parentheses.
(I) Effects of several ligands on PPARα-LBD thermostability. Ligands were dissolved in 100% DMSO (left) or 100% ethanol (right), and their final concentrations in assays were 0.1%.
The differences versus control (0 h in (F), no lipids in (G), and iFA-bound proteins in (I)) are significant at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Data are means ± SEM in (D) (n = 3), (E) (n = 3), (G) (n = 4), (H) (n = 3–4), and (I) (n = 4–6), and means ± SD in (F) (n = 5). Myr, myristic acid; Pal, palmitic acid; Ste, stearic acid; Ole, oleic acid; Lin, linoleic acid; αLnn, α-linolenic acid; and Ara, arachidonic acid.
Structures with Various PPARα Agonists
By using the soaking method (Hassell et al., 2007; Kuwabara et al., 2012) (Figure S3C), we were able to obtain crystals with potent agonists; pemafibrate, GW7647, 5,8,11,14-eicosatetraynoic acid (ETYA), saroglitazar, tetradecylthioacetic acid (TTA), and Wy14643 for high-resolution crystallography (Figures 2A and S4C). Crystals with pemafibrate and GW7647 were also acquired through cross-seeding (Figure 2B). Because we failed to obtain crystals with less potent agonists, iFAs were removed using delipidation (Figure S3D) in order to obtain apoproteins. The delipidized PPARα-LBD protein preserved its molecular weight, but ∼80% of the protein lost its iFAs (Figures S3E and S3F). The delipidation induced a slight shift in the circular dichroism (CD) spectrum from its original protein, but this was restored using stearic acid (1 mM) supplementation (Figure S3G). The delipidized protein displayed ligand concentration-dependent PPARα activation (Figure S3H). Therefore, the delipidation was able to maintain PPARα activity. We also obtained crystals of the delipidized proteins bound with fenofibric acid, ciprofibrate (Figure 2C), five endogenous fatty acids (Figure 2D), and other synthetic ligands (Figures S4D and S5). Importantly, PPARα-LBD-ligand structures were conserved in crystals formed by different methods. The coordination of some ligands to PPARα-LBD was independently supported by anomalous dispersion signals from low Z-atoms of sulfur (in ligands or surrounding Met/Cys residues of proteins) and chloride (in ligands). Anomalous difference Fourier maps, which were obtained using 1.8 Å X-ray, detected sulfur signals from Wy14643, GW7647, saroglitazar, and TTA, as well as chloride signals from fenofibric acid and ciprofibrate, in the exact positions that conventional 1.0 Å X-ray crystallography was deployed (Figure S6).
Figure 2.

PPARα-LBD-ligand Structures Obtained Using Soaking, Cross-Seeding, and Delipidation Methods for Crystallization
(A–D) Crystal structures and magnified views of PPARα-LBD and five potent PPARα agonists obtained using soaking (A); two potent PPARα agonists obtained using cross-seeding (see also Figure S5) (B); two intermediately potent PPARα agonists obtained using co-crystallization of the delipidized proteins (C); or five exogenous fatty acids with relatively low potency obtained by cross-seeding of the delipidized proteins (D).
The electron density is shown in the mesh by Feature Enhanced Maps (FEMs) contoured at 1.0σ. PDB identities and resolutions are labeled, and water molecules are presented as cyan spheres.
Ternary Complex with Ligands and Coactivators
Activation Function-2 (AF-2) helix 12 is known to form in the active PPAR conformation (Xu et al., 2002; Zoete et al., 2007). Coexistence with coactivator peptides could enhance PPARα-ligand complex stability through interaction with the AF-2 helix 12 and thereby facilitate crystal formation (Kawasaki et al., 2020; Xu et al., 2001). Indeed, among the 21 structures deposited in PDB (Table S1), 12 structures were obtained with any coactivator: 8 with steroid receptor coactivator-1 (SRC1), 2 with PGC1α, 1 with PPARγ coactivator 1-beta, and 1 with the silencing mediator of retinoic acid and thyroid hormone receptors. Therefore, we produced crystals with a SRC1 peptide. First, we were able to obtain crystals of the ternary complex with an iFA, GW7647, and eicosapentaenoic acid (EPA) using co-crystallization (Figure 3A) and then with pemafibrate by soaking with the PPARα-LBD-iFA-SRC1 cocrystals (Figure 3B). We were also able to obtain structures with clofibric acid and fenofibric acid using co-crystallization with the delipidized protein, in which both fibrates have two binding sites in Arm I and Arm II/X boundary (Figure 3C), and then with bezafibrate by soaking with the PPARα-LBD-clofibric acid-SRC1 cocrystals (Figure 3D). In all cases, SRC1 peptides were associated with helix 12 in a similar manner (Figure 3). We examined molecular interactions between amino acid residues of PPARα-LBD and ligands using LigPlot + analysis (Figure S7). All 15 ligands have a single carboxylic acid surrounded by common residues (S280, Y314, H440, and Y464) located between Arm I and Center regions. Most of the interactions between ligands and amino acid residues, except carboxylic acids, were found to be hydrophobic rather than electrostatic. The location of the first fenofibric acid, ciprofibrate, and clofibric acid molecules in Arm I might flip the neighboring benzyl side chains of Phe273 and the presence of the second those molecules in Arm X could affect the orientation of the helices 2’ (Figures 2C, 3C, and S7), which may affect PPARα activation. Pemafibrate and GW7647 in Arm III might interact with Thr279 via their hydrogen bonds (a water molecule mediated in case of GW7647), thereby stabilizing the helix 3 and the coactivator binding pocket (Figures 2A, 2B, 3A, 3B, and S7).
Figure 3.

Crystal Structures of Ternary Complexes of PPARα-LBD, Ligand, and SRC1 Coactivator Peptide
(A) Ternary complexes with intrinsic fatty acid (iFA), GW7647, or EPA obtained by co-crystallization.
(B) Ternary complexes with pemafibrate obtained using soaking with iFA/SRC1-bound crystals.
(C) Ternary complexes with clofibric acid or fenofibric acid obtained by co-crystallization of the delipidized proteins. Both fibrates have two binding sites in Arm I and Arm II/X.
(D) Ternary complexes with bezafibrate obtained by soaking with clofibric acid/SRC1-bound crystals.
SRC1 peptides (magenta) and helices 12 (red) are indicated by arrows and arrowheads. The electron density is shown in the mesh by FEMs contoured at 1.0σ. PDB identities and resolutions are labeled, and water molecules are presented as cyan spheres.
Bilateral Effects of GW9662
The PPARγ-selective antagonist GW9662 is considered unique because it covalently cross-links with Cys285 of PPARγ-LBD, which is equivalent to Cys276 in the Center region of PPARα-LBD (Brust et al., 2018), and acts as both an agonist and antagonist against PPARα, whereas it is only antagonistic against PPARγ and PPARδ (Leesnitzer et al., 2002). Indeed, we observed that GW9662 inhibits PPARα activation by potent agonists, such as pemafibrate, GW7647, ETYA, and TTA, whereas it enhances activation by less potent agonists: fenofibric acid, ciprofibrate, clofibric acid, and gemfibrozil (Figure 4A). Furthermore, a thermostability assay has showed that GW9662 lowers the PPARα-LBD stability induced by pemafibrate, GW7647, ETYA, saroglitazar, and TTA, whereas it enhances that induced by fenofibric acid, ciprofibrate, Wy14643, bezafibrate, clofibric acid, and gemfibrozil (Figure 4B). We analyzed the cocrystals with GW9662 and fibrates: fenofibric acid, ciprofibrate, clofibric acid, and gemfibrozil (Figures 4C and S8A). The second molecules of the first three fibrates located at Arm II/X were replaced by three GW9662 molecules via their covalent cross-linking to Cys275/Cys278 in Arm X and Cys276 in Arm II (Figures S8A and S8B). Cross-linking to Cys276 rather than Cys275 was important for both the inhibitory effects and enhancing effects (Figure 4D). We were not able to obtain crystals with gemfibrozil but only those bound with both gemfibrozil and GW9662 (Figure S8A). Although the electron density images of gemfibrozil in Arm I and GW9662, which was bound to Cys275, in Arm X were not clear, two GW9662 molecules were cross-linked to Cys276 in Arm II and Cys278 in Arm X. As GW9662 enhanced gemfibrozil-induced PPARα activation/thermostability (Figures 4A and 4B), gemfibrozil may have the second binding site in Arm II/X, besides the Arm I/Center region, like other fibrates (Figure 4E).
Figure 4.

Structural Basis for Bilateral Effects of GW9662 in PPARα Activation
(A) Concentration-dependent bilateral effects of GW9662 on PGC1α recruitment by agonists with various potencies. Data are means ± SEM (n = 3–4).
(B) Bilateral effects of GW9662 (100 μM) on ligand-induced PPARα-LBD thermostability. Data are means ± SEM (n = 3–4) and effects are found to be significant at ∗∗∗p < 0.001.
(C) Crystal structures of PPARα-LBD bound with single fibrate and two or three GW9662 molecules. See also Figure S8.
(D) Covalent cross-linking to Cys 276, in Arm II, but not to Cys 275, in Arm X, is essential for bilateral effects of GW9662 on PPARα regulation to inhibit GW7647 effects and activate clofibric acid effects. Data are means ± SEM (n = 3).
(E) Summary of bilateral effects of GW9662 and alignment of 16 ligands to LBD pockets in the order of ligand potencies in the coactivator recruitment assay; ligand-induced thermostability data were attached. Structural analyses demonstrated the location (circles), the possible location (triangles), or the absence (crosses) of those ligands in each LBD. One fenofibric acid (F) molecule is located at Arm I (as F1), whereas another fenofibric acid molecule is located across Arms II and X (as F2), and the same applies for GW9662, ciprofibrate, clofibric acid, and gemfibrozil.
Discussion
The transcription factor/nuclear receptor PPARα is a master regulator of lipid metabolism (or metabolism in general) that is activated upon fasting in liver and other tissues and regulates the expression of hundreds of genes involved in lipid (/glucose/amino acid) metabolism as well as biotransformation and inflammation (Rakhshandehroo et al., 2010; Kamata et al., 2018). This study has demonstrated thirty-four structures of human PPARα-LBD with seventeen ligands (and a coactivator) (Table 1), highlighting its versatile recognition of those ligands (Figure 4E). Among several endogenous PPARα ligand fatty acid candidates (Han et al., 2017), stearic acid and palmitic acid, the prevalent fatty acids released to bloodstream from cellular TG stores upon fasting (Figure 1F), have been found to bind to and activate PPARα (Figures 2D and 1G) for the integral control of lipid metabolism. Meanwhile, oleic acid, arachidonic acid, and EPA could bind to PPARα-LBD and form helix 12 (a hallmark of PPAR activation) (Figure 2D) but not activate it (Figure 1H). Although the helix 12 structure-function model has been popular in the nuclear receptor field, it is challenged by a new concept that agonists with graded activity display gradations in their ability to stabilize the LBD (Kojetin and Burris, 2013); as for PPARγ, in particular on helix 3, 11, and 12, all in a manner that correlates with the graded response of the ligand (Bruning et al., 2007). Therefore, the further experiments utilizing amide H/D exchange or paramagnetic relaxation enhancement NMR may reveal differences in the PPARα-LBD stability induced by those fatty acids. Although in vitro binding experiments suggest that multiple endogenous fatty acids could bind to PPARα/γ/δ-LBD (Xu et al., 1999), only oleic acid and arachidonic acid were found to be bound to PPARγ-LBD (Shang et al., 2018) and EPA to PPARδ-LBD (Xu et al., 1999) in X-ray crystallography, in addition to our PPARα-LBD analyses in this study. It is notable that oleic acid and arachidonic acid were found to bind to almost identical loci of PPARα-LBD (Figure 2D) but to different loci of PPARγ-LBD (Shang et al., 2018). EPA was also bound to different loci of PPARα-LBD (Figure 2D) and PPARδ-LBD (Xu et al., 1999).
Table 1.
Thirty-Four PPARα-LBD Crystal Structures Newly Deposited in the Protein Data Bank (PDB) from This Study
| No. | PDB ID | Resolution (Å) | Ligand(s) (±SRC1) | Methods | Deposited Date |
|---|---|---|---|---|---|
| 1 | 6KAX | 1.23 | Intrinsic fatty acid | Cross-seeding | 2019/6/24 |
| 23 | 6LXA | 1.23 | EPA | Delipidation/cross-seeding | 2020/2/10 |
| 10 | 6KB1 | 1.25 | TTA | Soaking | 2019/6/24 |
| 19 | 6LX6 | 1.30 | Palmitic acid | Delipidation/cross-seeding | 2020/2/10 |
| 8 | 6KB0 | 1.35 | ETYA | Soaking | 2019/6/24 |
| 22 | 6LX9 | 1.40 | Arachidonic acid | Delipidation/cross-seeding | 2020/2/10 |
| 20 | 6LX7 | 1.41 | Stearic acid | Delipidation/cross-seeding | 2020/2/10 |
| 4 | 6KB4 | 1.42 | Pemafibrate | Delipidation/cross-seeding | 2019/6/24 |
| 11 | 6KB6 | 1.43 | TTA | Delipidation/cross-seeding | 2019/6/24 |
| 7 | 6KB3 | 1.45 | GW7647 | Delipidation/cross-seeding | 2019/6/24 |
| 6 | 6KB8 | 1.47 | GW7647 | Cross-seeding | 2019/6/24 |
| 2 | 6KAZ | 1.48 | Pemafibrate | Soaking | 2019/6/24 |
| 32 | 7BQ2 | 1.52 | Pemafibrate + SRC1 | Soaking (in no.31 crystals) | 2020/3/23 |
| 31 | 7BQ1 | 1.52 | iFA + SRC1 | Co-crystallization | 2020/3/23 |
| 21 | 6LX8 | 1.54 | Oleic acid | Delipidation/cross-seeding | 2020/2/10 |
| 3 | 6KB9 | 1.55 | Pemafibrate | Cross-seeding | 2019/6/24 |
| 34 | 7BQ4 | 1.62 | EPA + SRC1 | Delipidation/co-crystallization | 2020/3/23 |
| 5 | 6KAY | 1.73 | GW7647 | Soaking | 2019/6/24 |
| 30 | 7BQ0 | 1.77 | Fenofibric acid + SRC1 | Delipidation/co-crystallization | 2020/3/23 |
| 15 | 6KBA | 1.82 | Wy14643 | Co-crystallization | 2019/6/24 |
| 18 | 6LX5 | 1.87 | Ciprofibrate | Delipidation/co-crystallization | 2020/2/10 |
| 14 | 6KB2 | 1.95 | Wy14643 | Soaking | 2019/6/24 |
| 9 | 6KB5 | 1.95 | ETYA | Delipidation/cross-seeding | 2019/6/24 |
| 33 | 7BQ3 | 1.98 | GW7647 + SRC1 | Delipidation/co-crystallization | 2020/3/23 |
| 13 | 6LXC | 2.03 | Saroglitazar | Delipidation/cross-seeding | 2020/2/10 |
| 28 | 7BPY | 2.09 | Clofibric acid + SRC1 | Delipidation/co-crystallization | 2020/3/23 |
| 17 | 6LX4 | 2.13 | Fenofibric acid | Delipidation/co-crystallization | 2020/2/10 |
| 16 | 6KB7 | 2.14 | Wy14643 | Delipidation/cross-seeding | 2019/6/24 |
| 12 | 6LXB | 2.36 | Saroglitazar | Soaking | 2020/2/10 |
| 29 | 7BPZ | 2.43 | Bezafibrate + SRC1 | Soaking (in no.28 crystals) | 2020/3/23 |
| 27 | 6L38 | 2.76 | GW9662 + Gemfibrozil | Delipidation/co-crystallization | 2019/10/9 |
| 24 | 6KYP | 2.86 | GW9662 + Clofibric acid | Delipidation/co-crystallization | 2019/9/19 |
| 26 | 6L37 | 2.91 | GW9662 + Ciprofibrate | Delipidation/co-crystallization | 2019/10/9 |
| 25 | 6L36 | 3.30 | GW9662 + Fenofibric acid | Delipidation/co-crystallization | 2019/10/9 |
Data are ordered by resolution, and crystal numbers (no.) are identical to those in Tables S3 and S4. No.1 crystals were used as a common seed for cross-seeding and as an original crystal in soaking except for nos. 29 and 32. Nos. 28 and 31 crystals were used in soaking for nos. 29 and 32, respectively. Delipidized proteins were used for co-crystallization in delipidation. SRC1 is a PPARα coactivator peptide.
We also revealed PPARα-LBD structures with all six fibrates in clinical use. Although fibrates are less popular than statins as anti-dyslipidemia drugs in most countries including Japan, the occurrence of fibrates and their metabolites in source and drinking water was reported in China and other countries (Ido et al., 2017) and the world recipient population is estimated to be huge. Therefore, our findings will attract over millions of patients who take fibrates and doctors who prescribe them. As Bernardes et al. (2013) reported with Wy14643-bound crystals, fenofibric acid, ciprofibrate, clofibric acid, and, perhaps, gemfibrozil, have been found to possess two binding sites. Moreover, single fibrates, fenofibric acid, ciprofibrate, and clofibric acid and three GW9662 could occupy PPARα-LBD at the same time, and the PPARα-LBD cavity was determined to be large enough to accept multiple ligands with various structures (Figure S8C). Among them, the coordination to Arm III and carboxylic acid-binding site is a key for the highest PPARα activity/selectivity. Molecular extension into the greater depth of Arm III deserves consideration for PPARα-selective agonists, and effective utilization of the other spaces, even by multiple same or different ligands, may be important for the development of more potent PPARα agonists. Furthermore, the precise structure comparison analyses of human PPARα/γ/δ-LBD contributes to the development of new PPAR dual/pan agonists as next-generation metabolic disease drugs. Finally, all previously deposited PPARα-LBD X-ray crystal structures except PDB 1KKQ are the ligand-bound active forms (Table S1) and all 34 structures obtained in this study (Table 1) are also the ligand-bound active forms with helix 12. The high-resolution PPARα-LBD structures that we provided here may help the design of high-affinity PPARα antagonists that stabilize the inactive form (with a corepressor).
Limitations of the Study
The coactivator recruitment and thermostability assays demonstrated graded responses (in efficacy and potency) between several PPARα ligands; however, the thirty-four PPARα-LBD-ligand structures we obtained were all similar activated forms. The amide H/D exchange or paramagnetic relaxation enhancement NMR could detect differences in the PPARα-LBD-ligand stability, and the cryogenic electron microscopy might reveal altered PPARα-LBD structures depending on the ligands. The acquisition of structures of PPARα-LBD inactivated forms is awaited.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Isao Ishii (isao-ishii@umin.ac.jp).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The data supporting the findings of this study is available within this paper and its Supplemental Information. PDB IDs for the 34 PPARα-ligand structures reported in this paper are: 6KAX (intrinsic fatty acid); 6KAZ, 6KB9, 6KB4 (pemafibrate); 6KAY, 6KB8, 6KB3 (GW7647); 6KB0, 6KB5 (ETYA); 6KB1, 6KB6 (TTA); 6KXB, 6LXC (saroglotazar); 6KB2, 6KBA, 6KB7 (Wy14643); 6LX4 (fenofibric acid); 6LX5 (ciprofibrate); 6LX6 (palmitic acid); 6LX7 (stearic acid); 6LXB8 (oleic acid); 6LX9 (arachidonic acid); 6LXA (EPA); 6KYP (GW9662 + clofibric acid); 6L36 (GW9662 + fenofibric acid); 6L37 (GW9662 + ciprofibrate); 6L38 (GW9662 + gemfibrozil); 7BPY (clofibric acid + SRC1); 7BPZ (bezafibrate + SRC1); 7BQ0 (fenofibric acid + SRC1); 7BQ1 (intrinsic fatty acid + SRC1); 7BQ2 (pemafibrate + SRC1); 7BQ3 (GW7647 + SRC1); and 7BQ4 (EPA + SRC1).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Prof. Jerold Chun (Sanford Burnham Prebys Medical Discovery Institute) for helpful comments. S.K. and I.I. acknowledge fundings from Grants-in-Aid for Scientific Research from Japan Society for the Promotion of Sciences (JSPS), Japan (grant numbers: 19K16359 and 16H05107), Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research [BINDS]) from AMED, Japan (grant number: JP19am0101071; support number: 1407), and Showa Pharmaceutical University, Japan. T.O. acknowledges fundings from a Grant-in-Aid for Scientific Research from JSPS (grant number: 18K06081) and Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) from Japan Science and Technology Agency, Japan (grant number: JPMJTM19AT). This work was performed under the approval of the Photon Factory Program Advisory Committee, Japan (proposal number: 2018G658).
Author Contributions
S.K., T.O., and I.I. conceived the study. S.K., T.O., K. Saito, A.H., K. Suda, and T.I. preformed crystal preparation and X-ray crystallography. S.K. and I.I. deposited all PDB data. S.K., A.H., and Y.Y. performed TR-FRET assay. S.K. and A.H. performed CD spectrometry. S.K., T.O., K. Saito, R.I., and T.I. performed crystal structure analyses. Y.W. performed blood drawing. T.S. and K.U. performed lipid analyses. S.K., T.O., K.S., A.H., T.I., Y.W., M.S., and I.I. interpreted data. T.O. and I.I. supervised the study. I.I. wrote the paper, with input from all authors.
Declaration of Interests
The authors declare no competing interest.
Published: November 20, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101727.
Supplemental Information
References
- Andersson C., Lyass A., Vasan R.S., Massaro J.M., D'Agostino R.B. Sr, Robins S.J. Long-term risk of cardiovascular events across a spectrum of adverse major plasma lipid combinations in the Framingham Heart Study. Am. Heart J. 2014;168:878–883.e1. doi: 10.1016/j.ahj.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Artis D.R., Lin J.J., Zhang C., Wang W., Mehra U., Perreault M., Erbe D., Krupka H.I., England B.P., Arnold J. Scaffold-based discovery of indeglitazar, a PPAR pan-active anti-diabetic agent. Proc. Natl. Acad. Sci. U S A. 2009;106:262–267. doi: 10.1073/pnas.0811325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénardeau A., Benz J., Binggeli A., Blum D., Boehringer M., Grether U., Hilpert H., Kuhn B., Märki H.P., Meyer M. Aleglitazar, a new, potent, and balanced dual PPARalpha/gamma agonist for the treatment of type II diabetes. Bioorg. Med. Chem. Lett. 2009;19:2468–2473. doi: 10.1016/j.bmcl.2009.03.036. [DOI] [PubMed] [Google Scholar]
- Benvenuti M., Mangani S. Crystallization of soluble proteins in vapor diffusion for x-ray crystallography. Nat. Protoc. 2007;2:1633–1651. doi: 10.1038/nprot.2007.198. [DOI] [PubMed] [Google Scholar]
- Bernardes A., Souza P.C., Muniz J.R., Ricci C.G., Ayers S.D., Parekh N.M., Godoy A.S., Trivella D.B., Reinach P., Webb P. Molecular mechanism of peroxisome proliferator-activated receptor alpha activation by WY14643: a new mode of ligand recognition and receptor stabilization. J. Mol. Biol. 2013;425:2878–2893. doi: 10.1016/j.jmb.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Bougarne N., Weyers B., Desmet S.J., Deckers J., Ray D.W., Staels B., De Bosscher K. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr. Rev. 2018;39:760–802. doi: 10.1210/er.2018-00064. [DOI] [PubMed] [Google Scholar]
- Bruning J.B., Chalmers M.J., Prasad S., Busby S.A., Kamenecka T.M., He Y., Nettles K.W., Griffin P.R. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Brust R., Shang J., Fuhrmann J., Mosure S.A., Bass J., Cano A., Heidari Z., Chrisman I.M., Nemetchek M.D., Blayo A.L. A structural mechanism for directing corepressor-selective inverse agonism of PPARgamma. Nat. Commun. 2018;9:4687. doi: 10.1038/s41467-018-07133-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capelli D., Cerchia C., Montanari R., Loiodice F., Tortorella P., Laghezza A., Cervoni L., Pochetti G., Lavecchia A. Structural basis for PPAR partial or full activation revealed by a novel ligand binding mode. Sci. Rep. 2016;6:34792. doi: 10.1038/srep34792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoto K., Chowdhury P.S., Kumar A., Sonomura K., Matsuda F., Fagarasan S., Honjo T. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. U S A. 2017;114:E761–E770. doi: 10.1073/pnas.1620433114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da'adoosh B., Marcus D., Rayan A., King F., Che J., Goldblum A. Discovering highly selective and diverse PPAR-delta agonists by ligand based machine learning and structural modeling. Sci. Rep. 2019;9:1106. doi: 10.1038/s41598-019-38508-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen R., Vergeer M., Stroes E.S., Kastelein J.J. Combination statin-fibrate therapy: safety aspects. Diabetes Obes. Metab. 2009;11:89–94. doi: 10.1111/j.1463-1326.2008.00917.x. [DOI] [PubMed] [Google Scholar]
- Fyffe S.A., Alphey M.S., Buetow L., Smith T.K., Ferguson M.A., Sørensen M.D., Björkling F., Hunter W.N. Recombinant human PPAR-beta/delta ligand-binding domain is locked in an activated conformation by endogenous fatty acids. J. Mol. Biol. 2006;356:1005–1013. doi: 10.1016/j.jmb.2005.12.047. [DOI] [PubMed] [Google Scholar]
- Graham D.J., Staffa J.A., Shatin D., Andrade S.E., Schech S.D., La Grenade L., Gurwitz J.H., Chan K.A., Goodman M.J., Platt R. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585–2590. doi: 10.1001/jama.292.21.2585. [DOI] [PubMed] [Google Scholar]
- Han L., Shen W.J., Bittner S., Kraemer F.B., Azhar S. PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-alpha. Future Cardiol. 2017;13:259–278. doi: 10.2217/fca-2016-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassell A.M., An G., Bledsoe R.K., Bynum J.M., Carter H.L., 3rd, Deng S.J., Gampe R.T., Grisard T.E., Madauss K.P., Nolte R.T. Crystallization of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2007;63:72–79. doi: 10.1107/S0907444906047020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ido A., Hiromori Y., Meng L., Usuda H., Nagase H., Yang M., Hu J., Nakanishi T. Occurrence of fibrates and their metabolites in source and drinking water in Shanghai and Zhejiang, China. Sci. Rep. 2017;7:45931. doi: 10.1038/srep45931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T., Fairall L., Amin K., Inaba Y., Szanto A., Balint B.L., Nagy L., Yamamoto K., Schwabe J.W. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat. Struct. Mol. Biol. 2008;15:924–931. doi: 10.1038/nsmb.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata S., Yamamoto J., Ohtani H., Tosaka Y., Yoshikawa S., Akahoshi N., Ishii I. 2D DIGE proteomic analysis reveals fasting-induced protein remodeling through organ-specific transcriptional factor(s) in mice. FEBS Open Bio. 2018;8:1524–1543. doi: 10.1002/2211-5463.12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki M., Kambe A., Yamamoto Y., Arulmozhiraja S., Ito S., Nakagawa Y., Tokiwa H., Nakano S., Shimano H. Elucidation of molecular mechanism of a selective PPARalpha modulator, pemafibrate, through combinational approaches of X-ray crystallography, Thermodynamic analysis, and first-principle calculations. Int. J. Mol. Sci. 2020;21:361. doi: 10.3390/ijms21010361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojetin D.J., Burris T.P. Small molecule modulation of nuclear receptor conformation dynamics: implication for functin and drug discovery. Mol. Pharm. 2013;83:1–8. doi: 10.1124/mol.112.079285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara N., Oyama T., Tomioka D., Ohashi M., Yanagisawa J., Shimizu T., Miyachi H. Peroxisome proliferator-activated receptors (PPARs) have multiple binding points that accommodate ligands in various conformations: phenylpropanoic acid-type PPAR ligands bind to PPAR in different conformations, depending on the subtype. J. Med. Chem. 2012;55:893–902. doi: 10.1021/jm2014293. [DOI] [PubMed] [Google Scholar]
- Leesnitzer L.M., Parks D.J., Bledsoe R.K., Cobb J.E., Collins J.L., Consler T.G., Davis R.G., Hull-Ryde E.A., Lenhard J.M., Patel L. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41:6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- Nolte R.T., Wisely G.B., Westin S., Cobb J.E., Lambert M.H., Kurokawa R., Rosenfeld M.G., Willson T.M., Glass C.K., Milburn M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- Oyama T., Toyota K., Waku T., Hirakawa Y., Nagasawa N., Kasuga J.I., Hashimoto Y., Miyachi H., Morikawa K. Adaptability and selectivity of human peroxisome proliferator-activated receptor (PPAR) pan agonists revealed from crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2009;65:786–795. doi: 10.1107/S0907444909015935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhshandehroo M., Knoch B., Muller M., Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010;2010:612089. doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Brust R., Mosure S.A., Bass J., Munoz-Tello P., Lin H., Hughes T.S., Tang M., Ge Q., Kamenekca T.M. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARgamma. Elife. 2018;7:e43320. doi: 10.7554/eLife.43320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Cardiovascular Dieseases. 2017. https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)
- Wu C.C., Baiga T.J., Downes M., La Clair J.J., Atkins A.R., Richard S.B., Fan W., Stockley-Noel T.A., Bowman M.E., Noel J.P. Structural basis for specific ligation of the peroxisome proliferator-activated receptor delta. Proc. Natl. Acad. Sci. U S A. 2017;114:E2563–E2570. doi: 10.1073/pnas.1621513114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.E., Lambert M.H., Montana V.G., Parks D.J., Blanchard S.G., Brown P.J., Sternbach D.D., Lehmann J.M., Wisely G.B., Willson T.M. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- Xu H.E., Lambert M.H., Montana V.G., Plunket K.D., Moore L.B., Collins J.L., Oplinger J.A., Kliewer S.A., Gampe R.T., Jr., McKee D.D. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. U S A. 2001;98:13919–13924. doi: 10.1073/pnas.241410198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.E., Stanley T.B., Montana V.G., Lambert M.H., Shearer B.G., Cobb J.E., McKee D.D., Galardi C.M., Plunket K.D., Nolte R.T. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–817. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- Yamashita S., Masuda D., Matsuzawa Y. Clinical applications of a novel selective PPARalpha modulator, pemafibrate, in dyslipidemia and metabolic diseases. J. Atheroscler. Thromb. 2019;26:389–402. doi: 10.5551/jat.48918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S., Masuda D., Matsuzawa Y. Pemafibrate, a new selective PPARα modulator: drug concept and its clinical applications for dyslipidemia and metabolic diseases. Curr. Atheroscler. Rep. 2020;22:5. doi: 10.1007/s11883-020-0823-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokote K., Yamashita S., Arai H., Araki E., Suganami H., Ishibashi S., Of The K-Study Group OB. Long-term efficacy and safety of pemafibrate, a novel selective peroxisome proliferator-activated receptor-alpha modulator (SPPARMalpha), in dyslipidemic patients with renal impairment. Int. J. Mol. Sci. 2019;20:706. doi: 10.3390/ijms20030706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V., Grosdidier A., Michielin O. Peroxisome proliferator-activated receptor structures: ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta. 2007;1771:915–925. doi: 10.1016/j.bbalip.2007.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study is available within this paper and its Supplemental Information. PDB IDs for the 34 PPARα-ligand structures reported in this paper are: 6KAX (intrinsic fatty acid); 6KAZ, 6KB9, 6KB4 (pemafibrate); 6KAY, 6KB8, 6KB3 (GW7647); 6KB0, 6KB5 (ETYA); 6KB1, 6KB6 (TTA); 6KXB, 6LXC (saroglotazar); 6KB2, 6KBA, 6KB7 (Wy14643); 6LX4 (fenofibric acid); 6LX5 (ciprofibrate); 6LX6 (palmitic acid); 6LX7 (stearic acid); 6LXB8 (oleic acid); 6LX9 (arachidonic acid); 6LXA (EPA); 6KYP (GW9662 + clofibric acid); 6L36 (GW9662 + fenofibric acid); 6L37 (GW9662 + ciprofibrate); 6L38 (GW9662 + gemfibrozil); 7BPY (clofibric acid + SRC1); 7BPZ (bezafibrate + SRC1); 7BQ0 (fenofibric acid + SRC1); 7BQ1 (intrinsic fatty acid + SRC1); 7BQ2 (pemafibrate + SRC1); 7BQ3 (GW7647 + SRC1); and 7BQ4 (EPA + SRC1).