

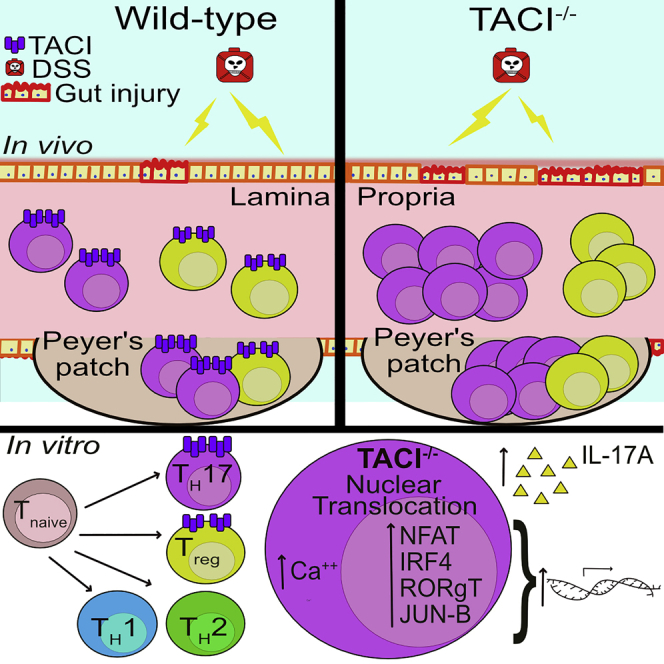
Summary
TACI (transmembrane activator and calcium modulator and cyclophilin ligand interactor) plays critical roles in B cells by promoting immunoglobulin class switching and plasma cell survival. However, its expression and function in T cells remain controversial. We show here that TACI expression can be strongly induced in murine CD4+ T cells in vitro by cytokines responsible for TH17 but not TH1 or TH2 differentiation. Frequencies and numbers of TH17 cells were elevated in TACI−/− compared with wild-type mice as well as among TACI−/− versus wild-type CD4+ T cells in mixed bone marrow chimeras, arguing for a T cell-intrinsic effect in the contribution of TACI deficiency to TH17 cell accumulation. TACI−/− mice were more susceptible to severe colitis induced by dextran sodium sulfate or adoptive T cell transfer, suggesting that TACI negatively regulates TH17 function and limits intestinal inflammation in a cell-autonomous manner. Finally, transcriptomic and biochemical analyses revealed that TACI−/− CD4+ T cells exhibited enhanced activation of TH17-promoting transcription factors NFAT, IRF4, c-MAF, and JUNB. Taken together, these findings reveal an important role of TACI in constraining TH17 pathogenicity and protecting against gut disease.
Subject Areas: Immunology
Graphical Abstract
Highlights
-
•
TACI expression is induced in TH17 but not TH1 or TH2 cells differentiated in vitro
-
•
TACI−/− mice have expanded TH17 and Treg populations in various organs
-
•
TACI−/− mice have enhanced susceptibility to intestinal disease
-
•
Activation of TH17-promoting transcription factors is enhanced in TACI−/− CD4+ T cells
Immunology
Introduction
The transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI encoded by the Tnfrsf13b gene) is a member of the tumor necrosis factor receptor superfamily (TNFRSF) and is closely related to the B cell activating factor receptor (BAFF-R encoded by the Tnfrsf13c gene) and B cell maturation antigen (BCMA encoded by the Tnfrsf17 gene) (Mackay and Schneider, 2008). TACI shares one of its binding ligands, BAFF, with BAFF-R and BCMA and the other ligand, a proliferation-inducing ligand (APRIL), with BCMA. Previous studies have shown TACI to be expressed in B cells (Rickert et al., 2011; Salzer et al., 2007), macrophages (Allman et al., 2015), and possibly also in activated T cells (Von Bülow and Bram, 1997). As expression of TACI is most prominent in B cells, most studies have focused on defining the functions of TACI in B cells, including its negative regulation of B cell expansion (Yan et al., 2001) and germinal center (GC) B cell formation (Ou et al., 2012) and promotion of plasma cell survival (Bossen et al., 2008; Ou et al., 2012) and immunoglobulin (Ig) isotype switching, diversification, and production (Figgett et al., 2015; He et al., 2010; Salzer et al., 2007; Seshasayee et al., 2003). TACI has also been shown to mediate BAFF- and APRIL-induced signals in innate immune cells that favor M1 macrophage polarization (Allman et al., 2015).
The expression of TACI in T cells is controversial, and the exact role of TACI in T cell function remains obscure, with studies reporting conflicting results (Mackay and Leung, 2006). TACI was first found to be expressed in activated mouse and human T cells using flow cytometry (Von Bülow and Bram, 1997; Wang et al., 2001). TACI expression was also observed on the surface of a small subset of CD3+ T cells in synovial tissues of human patients with rheumatoid arthritis (Seyler et al., 2005). In contrast, an independent study failed to detect TACI+ T cells derived from the blood and secondary lymphoid organs of mouse and human origin (Ng et al., 2004). Moreover, another study found TACI mRNA levels to be at least an order of magnitude weaker in human T cells compared with B cells, which was further decreased upon T cell activation (Wu et al., 2000). Characterization of a possible role of TACI in T cell signaling has been largely confined to transiently over-expressing TACI in Jurkat T cells and cross-linking the receptor with TACI-specific antibodies (Abs). Such an inquiry showed that TACI activates the transcription factors nuclear factor of activated T cells (NFAT), nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) and activator protein (AP)-1 (Von Bülow and Bram, 1997). However, the physiological role of TACI in T cells remains unaddressed.
In this study, we sought to clarify the role of TACI, if any, in T cell function and differentiation. We found TACI expression to be strongly induced in CD3+CD4+ T cells by TGF-β and further enhanced by IL-6 but not IL-2. TACI-deficient (TACI−/−) T cells were shown to exhibit enhanced activation of the transcription factors NFAT, interferon regulatory factor 4 (IRF4), Musculo Aponeurotic Fibrosarcoma oncogene (c-MAF), and JunB proto-oncogene (JUNB), which are known to be important for the differentiation of IL-17-producing T helper (TH17) cells. Accordingly, we found the frequencies and numbers of TH17 cells in the various lymphoid organs to be higher in TACI−/− compared with wild-type (WT) mice. Similar expansion of TH17 cells observed among TACI−/− versus WT CD4+ T cells in competitive chimeras that were reconstituted with equal ratio of WT and TACI−/− bone marrow cells suggested a cell-intrinsic effect of TACI in constraining TH17 accumulation. Furthermore, the induction of experimental colitis with dextran sodium sulfate (DSS) led to more severe colitis in TACI−/− compared with WT mice, as did the transfer of naive TACI−/− versus WT T cells into immune-deficient RAG1−/− mice. Collectively, our findings reveal for the first time a unique physiological role of TACI in limiting TH17 cell activation and pathogenicity, possibly to maintain gut homeostasis.
Results
TACI Expression in CD4+ T Cells Is Induced by TGF-β and Further Enhanced by IL-6 but Not IL-2
Previous data have shown that TACI expression in T cells can be induced by phorbol-12-myristate-13-acetate (PMA) and ionomycin treatment (Von Bülow and Bram, 1997). We sought to ascertain if TCR-mediated activation signals, in combination with those triggered by cytokines, could more readily induce TACI expression in CD3+CD4+ T cells. We stimulated CD4+ T cells isolated from the spleens (Spl) of WT mice with anti-CD3 and anti-CD28 Abs and various combinations of cytokines known to promote T helper 1 (TH1), T helper 2 (TH2), T helper 17 (TH17), or T regulatory (Treg) cell differentiation and assessed the level of TACI expression in these polarized cells, with TACI−/− CD4+ T cells cultured under corresponding conditions used as negative controls. Activation of CD4+ T cells by anti-CD3 and anti-CD28 Abs in non-polarizing (TH0; anti-IFN-γ + anti-IL-4 Abs), TH1-polarizing (IL-12 + anti-IL-4 Ab), or TH2-polarizing (IL-4 + anti-IFN-γ Ab) conditions failed to induce TACI cell surface protein (Figures 1A and 1B) and mRNA (Figure 1C) expression. Strikingly, cytokines promoting TH17 (TGF-β + IL-6 + IL-23) or Treg (TGF-β + IL-2) differentiation strongly upregulated TACI protein and mRNA expression, with TH17-promoting cytokines eliciting the most significant increase. In contrast, expression of the closely related TNFRSF members BAFF-R and BCMA were similar in WT and TACI−/− CD4+ T cells stimulated under various polarizing conditions, with that of BAFF-R being modestly increased versus isotype control in both WT and TACI−/− TH17 and Treg cells (Figure S1). These data suggest that cytokine combinations driving TH17 and Treg lineage commitment specifically induced TACI but not BAFF-R or BCMA expression in these lineages.
Figure 1.

Induction by TGF-β and Enhancement by IL-6 of TACI Expression in CD3+CD4+ T Cells
(A and B)Cell surface expression of TACI in CD3+CD4+ T cells isolated from Spl of WT and TACI−/− mice (A), or WT mice only (B), stimulated under TH0-, TH1-, TH2-, TH17-, or Treg-polarizing conditions for 3 days and assessed by flow cytometry.
(C) Transcript levels of TACI in WT CD3+CD4+ T cells stimulated as in (A) and assessed by quantitative real-time PCR. Transcript levels in TH1, TH2, TH17, or Treg cells were normalized to that in TH0 cells.
(D and E) Cell surface expression of TACI in WT CD3+CD4+ T cells activated with anti-CD3 and anti-CD28 Abs in the presence of indicated cytokines for 3 days.
Data in (A), (D), and (E) are representative of three independent experiments. Data in (B) and (C) are the mean ± SEM of three independent experiments.
To determine the relative contributions of the cytokines involved in TH17 and Treg differentiation in eliciting TACI expression, we incubated anti-CD3-and anti-CD28-activated CD4+ T cells with individual or specific combinations of cytokines and examined their cell surface expression of TACI (Figures 1D and 1E). We found TGF-β, but not IL-6, IL-23, or combination of the latter two, to robustly upregulate TACI expression (27.7% versus < 1% TACI+ cells induced by TGF-β versus IL-6, IL-23 or IL-6 + IL-23). Interestingly, IL-6, but not IL-23, when administered together with TGF-β, further enhanced TACI expression (47.0% versus 27.0% TACI+ cells induced by TGF-β + IL-6 versus TGF-β + IL-23) on activated CD4+ T cells (Figure 1D). We also examined if IL-2 supplementation could further enhance TGF-β-induced TACI expression on activated CD4+ T cells and found no such effect (Figure 1E). Taken together, our findings define a previously unappreciated role for cytokines, specifically TGF-β that promotes TH17 and Treg development, in inducing TACI expression in CD4+ T cells.
TACI Limits IL-17A Production by In Vitro Differentiated TH17 Cells
We next asked whether specific upregulation of TACI expression in TH17 and Treg but not TH1 and TH2 cells correlated with an important role for TACI in modulating the in vitro differentiation of TH17 and Treg but not TH1 and TH2 cells. To this end, we activated WT and TACI−/− CD4+ T cells under various polarizing conditions for 3 days as shown in Figure 1 and assessed their production of signature cytokines or expression of Foxp3. We confirmed that in vitro differentiated WT and TACI−/− TH1 cells produced high but comparable concentrations of IFN-γ (Figure S2A). IL-4 production by WT and TACI−/− TH1 cells was undetectable as expected (Figure S2B). WT and TACI−/− TH2 cells, on the other hand, produced expectedly high but indistinguishable concentrations of IL-4 (Figure S2B). Both produced low levels of IFN-γ (Figure S2A). In contrast, TACI−/− TH17 cells produced significantly higher concentration of IL-17A compared with their WT counterparts, although WT and TACI−/− CD4+ T cells differentiated under Treg-polarizing conditions yielded similar frequencies of Foxp3+ cells (Figure S2D).
To ascertain if the modulation of TH17 differentiation by TACI is dependent on its binding of ligands, we added increasing concentrations (1, 10, or 100 ng/mL) of recombinant BAFF or APRIL to WT CD4+ T cells differentiated under TH17- or Treg-polarizing conditions (Figure S3B) and found neither BAFF nor APRIL reduced the frequencies of TH17 (IL-17A+; top panel) or Treg (Foxp3+; bottom panel) cells after 3 days in culture compared with vehicle control regardless of the concentration of ligand employed. This was not due to lack of functional potency of the ligands since, consistent with the well-known role of these ligands to co-stimulate B cell responses, each ligand enhanced proliferation of LPS-stimulated B cells in a fashion dependent on the same dose escalation administered to TH17 or Treg cultures, compared with B cells activated by LPS alone, as assessed by CFSE dilution over 4 days of culture (Figure S3A). Using Fc-tagged recombinant BAFF or APRIL in combination with fluorochrome-conjugated secondary anti-Fc Ab, we also found specific binding of BAFF and APRIL ligands to splenic B cells which exceeded background binding by secondary Ab alone as anticipated (B cells; Figure S3C) but not to CD4+ T cells (CD4+ T cells; Figure S3C). This suggests that soluble BAFF or APRIL unlikely bound CD4+ T cells when they were added at the beginning of TH17 and Treg polarization cultures. Taken together, the preceding data allude to TACI being specifically upregulated in TH17 cells as a negative feedback mechanism to limit their IL-17A production in vitro in a manner that is likely to be ligand independent.
TACI Deficiency in Mice Results in T Cell-Intrinsic Accumulation of TH17 and Treg Cells
As TACI is specifically and highly expressed in in vitro differentiated TH17 and Treg cells (Figure 1) and it in turn modulates TH17 differentiation (Figure S2), we next asked what role TACI plays in TH17 and Treg development in vivo. To address this question, we examined the proportions of TH17 and Treg with respect to total CD3+CD4+ T cells in TACI−/− and WT mice. Using gating strategies applied to representative dot plots as depicted in Figures S4A and S4B for cells residing in lamina propria and Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 65 for cells in each of the organs examined, we found the frequencies and numbers of IL-17A-expressing (TH17) and IL-10-expressing (predominantly Treg) CD3+CD4+ T cells in the spleens (Spl), mesenteric lymph nodes (mLN), Peyer's patches (PP), and lamina propria (LP) to be higher in TACI−/− compared with WT mice and with most differences reaching statistical significance (Figures 2A and 2B). This is consistent with higher IL-17A production by in vitro generated TACI−/− compared with WT TH17 cells (Figure S2C). Concordantly, the frequencies and numbers of CD3+CD4+RORγt+ T (TH17) cells were higher in all organs examined in TACI−/− compared with WT mice with almost all differences reaching significance (Figure 2C). The numbers of CD3+CD4+Foxp3+ T (Treg) cells were also significantly elevated in most organs of TACI−/− mice, although the proportions of Foxp3+ cells in TACI−/− mice were not significantly different from those in WT mice (Figure 2D). In contrast, the frequencies and numbers of IFN-γ-expressing TH1 cells (Figures S7A and S7B) and IL-4-expressing TH2 cells (Figures S7C and S7D) in the Spl and mLN were comparable between TACI−/− and WT mice, consistent with similar concentrations of IFN-γ produced by TACI−/− and WT TH1 and of IL-4 produced by TACI−/− and WT TH2 cells differentiated in vitro (Figures S2A and S2B).
Figure 2.

Accumulation of TH17 and Treg Cells in TACI−/− Compared with WT Mice
Cells isolated from the Spl, mLN, PP, and LP of TACI−/− (filled circles) or WT (open circles) mice were stimulated with PMA and ionomycin for 4 h and assessed for intracellular IL-17A (A) and IL-10 (B) expression in CD3+CD4+ T cells by flow cytometry. Cells from various organs of TACI−/− or WT mice as in (A and B) were assessed for RORγt (C) and Foxp3 (D) expression in CD3+CD4+ T cells. Data are based on five or more mice analyzed with each symbol representing one mouse and horizontal bars indicating the mean; Mann-Whitney U test, ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ns, not significant. Actual p values are indicated in parentheses below the asterisks or “ns.”
Figure 3.

Expansion of TH17 and Treg Cells among TACI−/− versus WT CD4+ T Cells in BM Chimeras
Cells isolated from the Spl, mLN, PP, and LP of BM chimeric mice were stimulated with PMA and ionomycin for 4 h and assessed for intracellular IL-17A (A) and IL-10 (B) expression in TACI−/− CD45.2+ (filled circles) or WT CD45.1+ (open circles) CD3+CD4+ T cells by flow cytometry. Cells from various organs of BM chimeric mice as in (A) and (B) were assessed for RORγt (C) and Foxp3 (D) expression in TACI−/− or WT CD3+CD4+ T cells. Data are based on 11 or 12 mice analyzed with each symbol representing one mouse and horizontal bars indicating the mean; Mann-Whitney U test, ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ns, not significant. Actual p values are indicated in parentheses below the asterisks or “ns.”
Figure 4.

TACI Deficiency Led to More Severe Colitis in Mice
(A and B) The progression of wasting disease was monitored in terms of (A) change in body weights of TACI−/− (filled circles) and WT (open circles) mice expressed as percentage of their original weights and (B) Kaplan-Meier analysis of the survival of mice following feeding with 2% DSS in drinking water for 15 days.
(C–I) (C) Histological evaluation and scoring of DSS-induced colitis according to extent of ulceration, hyperplasia, and dysplasia, and infiltration by inflammatory cells in colons of TACI−/− compared with WT mice after feeding with DSS for 6 days. Degree of epithelial damage and leukocyte infiltration (D) as well as crypt cell proliferation (E) as assessed, respectively, by H&E and Ki-67 staining of colons of unchallenged (day 0) or 6-day DSS-fed mice. LP CD3+CD4+ T cells of unchallenged or DSS-fed mice at 3 or 6 days were assessed for RORγt (F and G) and Foxp3 (H and I) expression by flow cytometry.
(J) The ratio of the frequencies of RORγt+CD3+CD4+ to Foxp3+CD3+CD4+ T cells in the LP of TACI−/− (filled bars) and WT (open bars) mice as in (G) and (I).
Data in (A) and (B) are based on five male mice of each genotype; repeated-measures ANOVA, ∗∗∗∗, p < 0.0001 (A); log rank (Mantel-Cox) test, ∗∗∗, p = 0.0008 (B). Data in (C) are the mean ± SEM of histological scores of colons from eight TACI−/− and WT mice. Features shown in (D) and (E) are representative colon sections from one mouse each of 3 TACI−/− and WT mice examined. Scale bars represent 300 μm. Data in (F) to (I) are based on five or more mice analyzed with each symbol representing one mouse and horizontal bars indicating the mean; Mann-Whitney U test, ∗∗, p < 0.01; ns, not significant. Data in (J) are the mean ± SEM of ratios computed from five or more mice as in (G) and (I); unpaired parametric t-test, ∗∗∗∗, p < 0.0001. Actual p values are indicated in parentheses below the asterisks or “ns.”
Figure 5.

CD4+ T Cell-Intrinsic Role of TACI in Colitis
Change in body weights as percentage of original weights (A) and Kaplan-Meier survival analysis (D) of TACI−/− μMT (filled circles) compared with μMT (open circles) control mice after feeding with DSS in drinking water for 15 days. Histological scoring (B) and H&E assessment (C) of colitis in colons of unchallenged (day 0) or 6-day DSS-fed TACI−/− μMT and μMT mice. Percentage change in body weight curves of RAG1−/− mice that received TACI−/− (filled circles) or WT (open circles) CD4+CD62LhiCD44loCD25− Tnaive cells (E), WT Tnaive cells only (gray circles) or concurrently with TACI−/− (black circles) or WT (open circles) CD4+CD62LhiCD44loCD25+ Treg cells (G). Histological scoring (F and H) and H&E assessment (I) of colitis in colons of RAG1−/− mice at day 0 (before cell transfer) or 30 days after receiving Tnaive or Tnaive in combination with Treg cells from mice of genotypes described in (E) and (G). Scale bars in (C) and (I) represent 200 μm. Data in (A) and (D) are based on five female mice of each genotype; repeated-measures ANOVA, ∗∗, p = 0.0032 (A); log rank (Mantel-Cox) test, ns, p = 0.1573 (D). Data in (B) are the mean ± SEM of histological scores of colons from five TACI−/− μMT and μMT mice. Features shown in (C) are representative colon sections from one mouse each of three TACI−/− μMT and μMT mice examined. Data in (E) and (G) are based on five female RAG1−/− recipient mice in each cohort receiving TACI−/− or WT Tnaive cells; repeated-measures ANOVA, ∗∗, p = 0.0270 (E) or WT Tnaive in combination with TACI−/− or WT Treg cells; ∗∗∗∗, p < 0.0001; ns, p = 0.4651 (G). Data in (F) and (H) are the mean ± SEM of histological scores of colons from five RAG1−/− mice. Features shown in (I) are representative colon sections from one mouse of five RAG1−/− mice in each cohort examined. ns, not significant. Actual p values are indicated in parentheses below the asterisks or “ns.”
Figure 6.

Augmented Expression of NFAT-Dependent Transcriptional Pathway Promoting TH17 Development in TACI−/− Tnaive Cells
(A) GSEA-based enrichment plot of the calcineurin-regulated NFAT-dependent transcriptional pathway (normalized enrichment score = 2.42; FDR q-value < 0.001) based on microarray analysis of Tnaive cells FACS-sorted as in Figures 5C and 5D from pooled Spl and LN of TACI−/− and WT mice (n = 2 each). Vertical black lines indicate genes for which expression was detected by microarray as listed in (B).
(B) Expression heatmap of genes involved in regulation of the pathway as described in (A). Columns represent WT or TACI−/− Tnaive cells derived from two mice of each genotype. Red and blue denote high and low transcript expression levels, respectively.
(C) Transcript levels of TH17-associated genes in WT (open bars) and TACI−/− (filled bars) Tnaive cells as assessed by quantitative real-time PCR.
(D and E) Expression of TH17-associated proteins in the nuclei of WT and TACI−/− Tnaive cells at 0, 4, and 24 h stimulation with anti-CD3 and anti-CD28 Abs (D) or 24 h culture in TH17-polarizing conditions in the presence or absence of CsA as assessed by western blotting (E). Levels of HDAC1 serve as loading control.
(F) Real-time intracellular calcium flux in WT or TACI−/− CD4+ T cells stimulated with biotinylated anti-CD3 Ab and streptavidin as assessed by flow cytometry.
Data in (C) are the mean ± SEM of three technical replicates and representative of two independent experiments; unpaired parametric t-test; ∗, p < 0.05; ∗∗, p < 0.01; ns, not significant. Actual p values are indicated adjacent to the asterisks or “ns.” Data in (D)–(F) are representative of two independent experiments showing similar results.
The accumulation of TH17 and Treg cells in TACI−/− mice could be T cell-intrinsic or due to T cell-extrinsic factors contributed by other immune cell types lacking TACI such as B cells, dendritic cells, or macrophages to promote the preferential differentiation of CD4+ T to TH17 and Treg cells. To distinguish between these possibilities, we generated chimeric mice reconstituted with 1:1 ratio of WT (CD45.1+) and TACI−/− (CD45.2+) bone marrow (BM) cells. Similar analyses using gating strategies as depicted in Figures S4C and S4D and Figure S6 of TH17 and Treg populations in various organs of these mice revealed that the frequencies and numbers of IL-17A+ (Figure 3A), IL-10+ (Figure 3B), RORγt+ (Figure 3C), and Foxp3+ (Figure 3D) cells among TACI−/− CD45.2+CD3+CD4+ T cells were significantly higher compared with those of corresponding cells among WT counterparts existing within the same microenvironment. We observed more consistent expansion of TH17 (IL-17A+) and Treg (Foxp3+) cells among TACI−/− versus WT T cells in chimeric mice compared with TH17 and Treg cells in separate TACI−/− versus WT mice (Figures 3A and 3D versus Figures 2A and 2D). These data suggested that TACI−/− TH17 and Treg cells harbored competitive advantage over their WT counterparts in mixed chimeras, especially in gut-associated lymphoid tissues such as PP and LP, in which the elevation of TACI−/− and the concomitant depression of WT TH17 and Treg frequencies and numbers were most pronounced. Collectively, these results underscore the importance of TACI in attenuating TH17 and Treg but not TH1 or TH2 development in a T cell-intrinsic manner in vivo, which is correlated with the preferential expression of TACI in TH17 and Treg cells (Figure 1).
Loss of TACI Results in Greater Severity of Acute Colitis
CD3+CD4+ T cells, particularly TH1 and TH17 cells, have been shown to promote the pathogenesis of inflammatory bowel disease (IBD) and experimental colitis (Feng et al., 2011), whereas Treg cells dampen IBD (Shale et al., 2013). Since TACI deficiency expanded TH17 cells in vivo (Figure 2), we examined the susceptibility of TACI−/− and WT mice to experimental colitis. TACI−/− and WT mice were fed DSS ad libitum to induce acute colitis and monitored up to 15 days. Mice of both genotypes started losing weight 4 days post induction with TACI−/− mice displaying significantly more rapid weight loss compared with WT counterparts (Figure 4A). This was accompanied by greater mortality of TACI−/− mice, all of which succumbed to disease by day 10, whereas all WT mice survived (Figure 4B). Histological examination showed that the crypt architecture of the colons of TACI−/− mice was drastically perturbed with clear evidence of massive leukocyte infiltration compared with that of WT mice, which was considerably less disrupted after 6 days of DSS treatment (Figure 4D; day 6), corroborated by twice the mean histopathological score attributed to TACI−/− versus WT colons (Figure 4C). The presence of Ki-67+ proliferating crypt cells, important for orchestrating the regeneration of intestinal epithelium following pathological damage, was also dramatically reduced in TACI−/− compared with WT colons (Figure 4E).
TH17 and Treg cells have been demonstrated to play reciprocal roles in promoting and suppressing the development and course of colitis (Duchmann and Zeitz, 2006; Feng et al., 2011). We examined the proportions of LP TH17 and Treg cells in TACI−/− and WT mice before and after 3 or 6 days of DSS administration. The frequencies of RORγt+CD4+ T (TH17) cells were significantly higher in TACI−/− compared with WT LP at all time points examined (Figures 4F and 4G). On the other hand, the frequencies of LP Foxp3+CD4+ T (Treg) cells were only higher in unchallenged TACI−/− compared with WT mice, but there was no difference in their Treg frequencies during the progression of colitis (Figures 4H and 4I). Importantly, the ratios of TH17/Treg cells in TACI−/− mice consistently exceeded those of WT mice over 6 days of DSS challenge (Figure 4J). This imbalance in favor of TH17 cells provides a likely explanation for the development of more severe colitis in TACI−/− mice.
TACI Functions in CD4+ T Cells to Limit Acute and Chronic Colitis
TACI−/− mice are more susceptible to acute colitis (Figure 4), but it remains unclear if there exists a CD4+ T cell-intrinsic role for TACI in disease susceptibility. To address this question, we generated TACI−/− μMT mice lacking B cells to exclude the contribution of TACI−/− B cells in pathogenesis. After induction of colitis in mice via DSS administration, we again observed that TACI−/− μMT mice lost weight more rapidly than control μMT mice (Figure 5A), albeit the difference in weight loss was not as pronounced as between B cell-sufficient TACI−/− and WT mice (Figure 4A). TACI−/− μMT colons scored higher on average versus μMT colons in histopathological features (Figure 5B) as the extent of epithelial cell loss and leukocyte infiltration was greater in colons of TACI−/− μMT compared with μMT mice following 6 days of DSS treatment (Figure 5C; day 6). DSS-treated TACI−/− μMT mice also exhibited greater mortality compared with similarly treated μMT mice (Figure 5D). These experiments eliminated the contribution of TACI in B cells and suggested that TACI acts in other leukocytes to modulate the severity of colitis.
To directly assess the function of TACI in CD4+ T cells, we adoptively transferred CD4+CD62LhiCD44loCD25- Tnaive cells purified from pooled Spl and LN of TACI−/− or WT mice into RAG1−/− recipients to induce chronic colitis and monitored the recipient mice for wasting disease in terms of weight loss over a duration of 40 days. WT Tnaive cells predictably induced progressive weight loss in RAG1−/− mice over 20–30 days after cell transfer. RAG1−/− cohorts that received TACI−/− Tnaive cells, however, developed more aggressive inflammatory disease relative to RAG1−/− recipients of WT Tnaive cells, as evidenced by their accelerated weight loss (Figure 5E), with divergence in percent body weight curves occurring as early as 10 days after adoptive cell transfer. TACI−/− Tnaive cells inflicted substantially greater colonic damage compared with their WT counterparts in Rag1−/− mice as histologically scored (Figure 5F) and visualized by H&E staining (Figure 5I; TACI−/− Tnaive versus WT Tnaive) 30 days post cell transfer. Because Treg cells, in addition to TH17 cells, also numerically expanded in most organs of resting TACI−/− compared with WT mice (Figures 2D and 4I), we asked if TACI−/- Treg cells were functionally competent to suppress colitis. To address this, WT Tnaive cells were transferred alone or concurrently with TACI−/− or WT CD4+CD62LhiCD44loCD25+ Treg cells into RAG1−/− mice. As expected, mice receiving WT Tnaive cells alone progressively shed weight, which was completely prevented by the co-transfer of WT Treg cells (Figure 5G; WT Tnaive + WT Treg versus WT Tnaive), a finding corroborated by the lower mean histological score (Figure 5H) and degree of pathology (Figure 5I) of colons from Rag1−/− mice that received WT Tnaive together with WT Treg versus those that received WT Tnaive cells alone. Importantly, co-transferred TACI−/− Treg suppressed disease evoked by WT Tnaive cells as efficaciously as WT Treg cells (Figure 5G; WT Tnaive + TACI−/− Treg versus WT Tnaive + WT Treg), which was confirmed by histological scoring and examination (Figures 5H and 5I; WT Tnaive + TACI−/− Treg versus WT Tnaive + WT Treg), suggesting no defect in TACI−/− Treg function. Taken together, the above data argue for a CD4+ T cell-autonomous requirement of TACI in limiting TH17-mediated colitis. These conclusions are consistent with a CD4+ T cell-intrinsic role for TACI in constraining TH17 differentiation in vivo (Figure 3).
CD4+ T Cells Lacking TACI Exhibited Enhanced Activation of TH17-Promoting Transcription Factors
To understand the mechanisms underlying increased frequency of TH17 cells associated with aggravated colitis in TACI−/− mice, we performed microarray-based transcriptome analysis to identify genes that were differentially expressed (DEGs) in TACI−/− versus WT Tnaive cells that were FACS-sorted from pooled Spl and LN of mice. We identified 97 genes that were significantly upregulated and 66 genes that were significantly downregulated in TACI−/− compared with WT Tnaive cells (absolute fold change >1.5, p < 0.05). Gene Set Enrichment Analysis (GSEA, http://software.broadinstitute.org/gsea/index.jsp; Subramanian et al., 2005) comprising gene sets from Pathway Interaction Database, BioCarta, KEGG, Reactome, and Gene Ontology revealed that genes of the calcineurin-regulated NFAT-dependent transcriptional pathway were the most significantly upregulated in TACI−/− versus WT Tnaive cells compared with other signaling pathways (Figure 6A; normalized enrichment score = 2.42, FDR q-value < 0.001). Indeed, expression of genes involved in the NFAT pathway were found to be significantly changed (restricted to those with fold change >1.5; unpaired t-test, p < 0.05; Figure 6B). Notably, the list of genes, all of which have been shown to regulate TH17 differentiation, included NFAT1 and 2 (Dietz et al., 2015; Reppert et al., 2015), IRF4 (Brüstle et al., 2007; Gaffen et al., 2014; Huber et al., 2008), c-MAF (Bauquet et al., 2009; Gabryšová et al., 2018), solute carrier 3A2 (SLC3A2; Kurihara et al., 2015), Fos proto-oncogene (FOS) and JUNB (Carr et al., 2017; Hasan et al., 2017; Moon et al., 2017; Schraml et al., 2009), and early growth response 2 (EGR2; Du et al., 2014). Increased expression of some of these TH17-associated genes in TACI−/− compared with WT Tnaive cells was subsequently verified by quantitative real-time PCR (Figure 6C), although the increase in levels of JUNB and AP-1 transcripts did not reach statistical significance. Notably, when WT and TACI−/− Tnaive cells were activated with anti-CD3 and anti-CD28 Abs for 4 and 24 h, we observed increased localization of NFAT1, NFAT2, NFAT4, IRF4, c-MAF, and JUNB proteins in the nuclei of TACI−/− compared with WT Tnaive cells, suggesting increased activation of the mutant T cells, particularly after 24 h of stimulation (Figure 6D). Similarly, exaggerated levels of nuclear NFATs, IRF4, JUNB, and RORγt were also observed in TACI−/− Tnaive cells activated under TH17-polarizing conditions for 24 h (Figures 6E and S8), indicating hyper-activation of these transcription factors in TACI−/− cells undergoing TH17 differentiation. Treatment for 24 h with cyclosporin A (CsA), a well-known calcineurin inhibitor that dampens NFAT activation, abolished the nuclear translocation of IRF4, JUNB, and RORγt proteins in both WT and TACI−/− cells (Figures 6E and S8). Nuclear expression of NFAT 1, 2, and 4, however, was extinguished in WT but only reduced in TACI−/− cells at the CsA concentration applied, suggesting that TACI−/− Tnaive cells harbored higher levels of activated NFAT than WT counterparts. Since NFATs have been implicated in transcriptional activation of IRF4 (Biswas et al., 2010) and RORγt (Zhou and Littman, 2009), our data are consistent with a model in which loss of TACI promotes the activation of IRF4 and RORγt via increased NFAT induction.
NFAT activation is highly dependent on calcium influx (Gabriel et al., 2016; Macian, 2005). Hence, we next measured intracellular calcium flux in WT and TACI−/− CD4+ T cells. Upon stimulation with biotinylated anti-CD3 Ab and streptavidin, TACI−/− cells accumulated calcium to a greater degree than WT cells as visualized by increased levels of Indo-1-bound intracellular calcium (Figure 6F), in agreement with heightened resistance of TACI−/− cells to CsA-mediated inhibition of NFAT activation. This greater amplitude of intracellular calcium was sustained till the addition of ionomycin, which triggered the same expected calcium spike in both WT and TACI−/− cells (Figure 6F). Moreover, TCRβ expression in WT and TACI−/− Tnaive cells, whether left unstimulated or stimulated with anti-CD3 and anti-CD28 Abs for 24 h, was comparable (Figure S9), clarifying that the higher calcium influx in TACI−/− cells was not due to increased TCRβ expression.
Discussion
Extensive studies have delineated enigmatic but yet important functions for TACI in B cells, which highly express the receptor at later stages of their differentiation. These include inhibition of B cell proliferation and GC B cell formation (Ou et al., 2012; Yan et al., 2001) but promotion of plasma cell survival and Ig class switch recombination (Figgett et al., 2015; He et al., 2010; Ou et al., 2012; Salzer et al., 2007; Seshasayee et al., 2003). TACI is weakly expressed in dendritic cells (Wu et al., 2000) and macrophages (Allman et al., 2015), whereas its expression and function in T cells remain controversial (Mackay and Leung, 2006; Ng et al., 2004; Seyler et al., 2005; Von Bülow and Bram, 1997; Wu et al., 2000).
Although TACI was shown to be upregulated in mouse T cells activated with PMA and ionomycin (Von Bülow and Bram, 1997), it is unclear whether TCR- and/or cytokine-mediated signals induce TACI expression and whether TACI plays a physiological role in T cell function. We showed that activation of Tnaive cells with anti-CD3 and anti-CD28 Abs alone was insufficient to induce TACI expression in CD4+ Tnaive cells. However, TACI expression could be induced by administering the anti-inflammatory TH17 and Treg-promoting cytokine TGF-β to activated T cells and this effect was enhanced by IL-6 but not IL-2. In contrast, cytokines directing TH1 and TH2 commitment failed to induce TACI expression in T cells. Although extensive studies demonstrated that BAFF can co-stimulate proliferation of TCR-activated T cells, only a very small subset of 2%–3% of murine splenic T cells expresses BAFF-R at steady state and this expression increased 2-fold upon activation (Huard et al, 2001, 2004; Ng et al., 2004; Scapini et al., 2010; Ye et al., 2004). Unlike TACI expression, which was substantially increased, BAFF-R expression was slightly increased, whereas BMCA was not expressed in TH17 and Treg cells. Further studies are needed to more carefully assess if expression of TACI, BAFF-R, and BCMA in CD4+ T cells under various polarizing conditions changes at culture time points not examined in the current study or when strong mitogens such as PMA are utilized to activate T cells. Nevertheless, our current data suggest BAFF-R and BCMA, compared with TACI, may not play a major role in regulating TH17 and Treg differentiation.
Given the selective expression of TACI in TH17 and Treg cells, we investigated a possible role of TACI in TH17 and Treg development. TACI−/− TH17 cells differentiated in vitro produced significantly higher concentration of IL-17A compared with their WT counterparts. Consistent with this, the homeostatic proportions of TH17 and Treg cells in Spl, mLN, PP, and LP were consistently elevated in TACI−/− compared with WT mice and among TACI−/− versus WT CD4+ T cells in BM chimeras, with the most pronounced increases observed in the PP and LP. This may be due to the much higher concentration of TGF-β in the gut (including PP and LP) compared with Spl and mLN (Neurath, 2014). These data suggest that TH17 and Treg accumulation in TACI−/− mice is T cell-intrinsic and that TACI in WT CD4+ T cells constrains TH17 and Treg development. To determine if the latter requires binding of BAFF or APRIL ligand to TACI, we added BAFF or APRIL to CD4+ T cells at the beginning of TH17 or Treg polarization and found neither cytokine reduced TH17 or Treg frequencies at the end of culture. This was likely because neither BAFF nor APRIL bound CD4+ T cells when added at the start of TH17 and Treg culture. It is plausible that low levels of TACI expressed in initially polarizing TH17 and Treg cells are saturated by endogenous BAFF and APRIL and thus unable to bind exogenous cytokines. Alternatively, TACI expressed in TH17 and Treg cells may only bind membrane-bound but not soluble BAFF or APRIL. It also remains possible that BAFF or APRIL binds TH17 and Treg cells to modulate their cytokine expression and survival beyond the differentiation period of 3 days. These hypotheses warrant future investigation to determine whether the functions of TACI in CD4+ T cells depend on interaction with its ligands BAFF and/or APRIL.
Since TH17 cells have been implicated in the pathogenesis of IBD (Feng et al., 2011; Shale et al., 2013), we chemically induced colitis in mice, which revealed that TACI deficiency led to worsened wasting disease, which was accompanied by increased mortality. There was more severe epithelial tissue destruction attributable to markedly reduced presence of proliferating crypt cells that facilitate tissue repair. Because of the prominent function of TACI in B cells and its role in promoting IgA class switching, we eliminated the contribution of B cells by generating TACI−/− mice devoid of B cells (TACI−/−- μMT mice) and found that TACI deficiency in the absence of B cells again led to manifestation of more severe colitis. Furthermore, adoptive transfer of TACI−/− Tnaive cells into RAG1−/− mice evoked more severe disease, which, together with findings from TACI−/− μMT mice, argued for a CD4+ T cell-autonomous requirement for TACI in alleviating colitis. However, we cannot rule out the possibility that TACI may nonetheless function in B cells to modulate colitis since disease severity in TACI−/− μMT versus control μMT mice was less than B cell-sufficient TACI−/− versus WT mice. Moreover, dendritic cells (Wu et al., 2000) and macrophages (Allman et al., 2015), which weakly express TACI, could also regulate colitis. The relative contributions of TACI function in T cells, B cells, dendritic cells, and macrophages to colitis await the generation of mice with the TACI gene ablated conditionally in these cell types.
To understand why TACI−/− CD4+ T cells have propensity to adopt TH17 phenotype, we conducted microarray analysis of TACI−/− versus WT Tnaive cells, which revealed that the former over-expressed a constellation of TH17-associated genes constituting the NFAT-dependent transcriptional pathway, including transcription factors NFAT, IRF4, c-MAF, and JUNB known to promote TH17 differentiation and the production of signature TH17 cytokines (Hermann-Kleiter and Baier, 2010). Tnaive rather than total CD4+ T cells were employed to avoid analysis of DEGs being confounded by increased proportions of activated T cells in resting TACI−/− compared with WT mice. TACI−/− CD4+ T cells fluxed aberrantly higher levels of calcium that promote enhanced dephosphorylation of cytoplasmic NFATs by calcineurin and accelerated nuclear import of NFATs (Macian, 2005), thereby increasing NFAT, IRF4, and RORγt activation. These data suggest that the suppression of the calcineurin-NFAT signaling axis by TACI in CD4+ T cells inhibits TH17 development, similar to what was earlier observed in B cells (Ou et al., 2012). Elucidation of how TACI specifically restricts signaling by components of the NFAT pathway in CD4+ T cells will be the subject of future investigation. This is crucial to devising interventions that cripple TH17-mediated pathology in IBD without excessively compromising TACI-mediated functions in other immune cell types that are important to preserve intestinal homeostasis, e.g., production of gut IgA by B and plasma cells (Cerutti and Rescigno, 2008).
TACI has been shown to play opposing, context-dependent roles in the pathogenesis of autoimmune disease that include promoting inflammation and joint destruction in collagen-induced arthritis (Wang et al., 2001), autoantibody production in systemic lupus erythematosus (Figgett et al., 2015), and Ig transcription in GC+ synovitis while limiting IFN-γ production in aggregate and diffuse synovitis (Seyler et al., 2005). Our current data support a working model in which TACI is induced selectively in CD4+ T cells differentiating into the TH17 lineage and in turn constrains their activation and pathogenicity by restricting their levels of intracellular calcium flux to limit TH17 responses. Besides extending the diverse roles TACI plays in various immune cell types to CD4+ T cells, our findings advance the understanding of the context-specific mechanisms governing TACI function that will inform the development of new therapeutics to target TACI for the amelioration of autoimmune diseases, infection, and cancer.
Limitations of the Study
We initially observed accumulation of TH17 and Treg cells in TACI−/− mice that could be attributed to T cell-intrinsic effects or extrinsic factors produced by other immune cell types lacking TACI. We then generated chimeric mice reconstituted with equal numbers of congenically distinguished WT and TACI−/− bone marrow (BM) cells to clarify that TACI functions intrinsically in CD4+ T cells to constrain TH17 and Treg differentiation. Limitations of the chimeric system include variability in BM reconstitution of irradiated mice as well as perturbations of the gut microbiome induced by antibiotics administered to mice shortly following irradiation to minimize opportunistic infections. To overcome these, future studies could consider engineering Tnfrsf13b (encoding TACI) floxed (Tnfrsf13bf/f) mice and crossing them to Cd4cre mice to generate mice harboring T cell-specific deletion of TACI. These Tnfrsf13bf/fCd4Cre mice will provide an independent model to substantiate findings in the current study using TACI−/− and BM chimeric mice. Furthermore, crossing Tnfrsf13bf/f mice to commercially available Il17cre or Foxp3cre mice will enable more specific dissection of the roles of TACI in TH17 and Treg populations. Whether and how TACI deficiency affects the gut microbiome and in turn contributes to colitis pathogenesis remains to be examined.
Resource Availability
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Microarray data were deposited in NCBI with accession number GSE152077 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152077).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank staff of the Biological Resource Centre (BRC) for care and maintenance of mice and members of the laboratory for insightful discussions. The Advanced Molecular Pathology Laboratory (AMPL) of the Institute of Molecular and Cell Biology assisted in the preparation and histological staining of colon samples. This research was supported by Bioprocessing Technology Institute, Agency for Science, Technology and Research, Singapore (A∗STAR).
Author Contributions
A.H.-M.T., G.H.W.T., P.-Y.T., X.O., and K.-P.L. conceived and designed the study. G.H.W.T., B.Z., P.-Y.T., X.O., S.-W.N., A.X.F.W., S.J.X.T., A.S., and S.S.-Y.K. performed experiments and generated data. A.H.-M.T., B.Z., G.H.W.T., P.-Y.T., X.O., S.-W.N., A.P.L., S.X., and K.-P.L. analyzed data. A.H.-M.T. and K.-P.L. supervised the study, interpreted data, and wrote the manuscript. All authors reviewed and approved the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 20, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101707.
Contributor Information
Andy Hee-Meng Tan, Email: andy_tan@bti.a-star.edu.sg.
Kong-Peng Lam, Email: lam_kong_peng@bti.a-star.edu.sg.
Supplemental Information
References
- Allman W.R., Dey R., Liu L., Siddiqui S., Coleman A.S., Bhattacharya P., Yano M., Uslu K., Takeda K., Nakhasi H.L., Akkoyunlu M. TACI deficiency leads to alternatively activated macrophage phenotype and susceptibility to Leishmania infection. Proc. Natl. Acad. Sci. U S A. 2015;112:E4094–E4103. doi: 10.1073/pnas.1421580112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauquet A.T., Jin H., Paterson A.M., Mitsdoerffer M., Ho I.C., Sharpe A.H., Kuchroo V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH -17 cells. Nat. Immunol. 2009;10:167–175. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas P.S., Bhagat G., Pernis A.B. IRF4 and its regulators: evolving insights into the pathogenesis of inflammatory arthritis? Immunol. Rev. 2010;233:79–96. doi: 10.1111/j.0105-2896.2009.00864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossen C., Cachero T.G., Tardivel A., Ingold K., Willen L., Dobles M., Scott M.L., Maquelin A., Belnoue E., Siegrist C.A. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood. 2008;111:1004–1012. doi: 10.1182/blood-2007-09-110874. [DOI] [PubMed] [Google Scholar]
- Brüstle A., Heink S., Huber M., Rosenplänter C., Stadelmann C., Yu P., Arpaia E., Mak T.W., Kamradt T., Lohoff M. The development of inflammatory TH-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 2007;8:958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- Carr T.M., Wheaton J.D., Houtz G.M., Ciofani M. JunB promotes Th17 cell identity and restrains alternative CD4+ T-cell programs during inflammation. Nat. Commun. 2017;8:301. doi: 10.1038/s41467-017-00380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti A., Rescigno M. The biology of intestinal immunoglobulin A responses. Immunity. 2008;28:740–750. doi: 10.1016/j.immuni.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz L., Frommer F., Vogel A.L., Vaeth M., Serfling E., Waisman A., Buttmann M., Berberich-Siebelt F. NFAT1 deficit and NFAT2 deficit attenuate EAE via different mechanisms. Eur. J. Immunol. 2015;45:1377–1389. doi: 10.1002/eji.201444638. [DOI] [PubMed] [Google Scholar]
- Du N., Kwon H., Li P., West E.E., Oh J., Liao W., Yu Z., Ren M., Leonard W.J. EGR2 is critical for peripheral naíve T-cell differentiation and the T-cell response to influenza. Proc. Natl. Acad. Sci. U S A. 2014;111:16484–16489. doi: 10.1073/pnas.1417215111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchmann R., Zeitz M. T regulatory cell suppression of colitis: the role of TGF-β. Gut. 2006;55:604–606. doi: 10.1136/gut.2005.083592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T., Qin H., Wang L., Benveniste E.N., Elson C.O., Cong Y. Th17 cells induce colitis and promote Th1 cell responses through IL-17 induction of innate IL-12 and IL-23 production. J. Immunol. 2011;186:6313–6318. doi: 10.4049/jimmunol.1001454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figgett W.A., Deliyanti D., Fairfax K.A., Quah P.S., Wilkinson-Berka J.L., Mackay F. Deleting the BAFF receptor TACI protects against systemic lupus erythematosus without extensive reduction of B cell numbers. J. Autoimmun. 2015;61:9–16. doi: 10.1016/j.jaut.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Gabriel C.H., Gross F., Karl M., Stephanowitz H., Hennig A.F., Weber M., Gryzik S., Bachmann I., Hecklau K., Wienands J. Identification of novel nuclear factor of activated T cell (NFAT)-associated proteins in T cells. J. Biol. Chem. 2016;291:24172–24187. doi: 10.1074/jbc.M116.739326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabryšová L., Alvarez-Martinez M., Luisier R., Cox L.S., Sodenkamp J., Hosking C., Pérez-Mazliah D., Whicher C., Kannan Y., Potempa K. C-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells article. Nat. Immunol. 2018;19:497–507. doi: 10.1038/s41590-018-0083-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen S.L., Jain R., Garg A.V., Cua D.J. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan Z., Koizumi S.I., Sasaki D., Yamada H., Arakaki N., Fujihara Y., Okitsu S., Shirahata H., Ishikawa H. JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat. Commun. 2017;8:15628. doi: 10.1038/ncomms15628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Santamaria R., Xu W., Cols M., Chen K., Puga I., Shan M., Xiong H., Bussel J.B., Chiu A. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat. Immunol. 2010;11:836–845. doi: 10.1038/ni.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann-Kleiter N., Baier G. NFAT pulls the strings during CD4+ T helper cell effector functions. Blood. 2010;115:2989–2997. doi: 10.1182/blood-2009-10-233585. [DOI] [PubMed] [Google Scholar]
- Huard B., Arlettaz L., Ambrose C., Kindler V., Mauri D., Roosnek E., Tschopp J., Schneider P., French L.E. BAFF production by antigen-presenting cells provides T cell co-stimulation. Int. Immunol. 2004;16:467–475. doi: 10.1093/intimm/dxh043. [DOI] [PubMed] [Google Scholar]
- Huard B., Schneider P., Mauri D., Tschopp J., French L.E. T cell costimulation by the TNF ligand BAFF. J. Immunol. 2001;167:6225–6231. doi: 10.4049/jimmunol.167.11.6225. [DOI] [PubMed] [Google Scholar]
- Huber M., Brüstle A., Reinhard K., Guralnik A., Walter G., Mahiny A., Von Löw E., Lohoff M. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc. Natl. Acad. Sci. U S A. 2008;105:20846–20851. doi: 10.1073/pnas.0809077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T., Arimochi H., Bhuyan Z.A., Ishifune C., Tsumura H., Ito M., Ito Y., Kitamura A., Maekawa Y., Yasutomo K. CD98 heavy chain is a potent positive regulator of CD4+ T cell proliferation and interferon-γ production in vivo. PLoS One. 2015;10:e0139692. doi: 10.1371/journal.pone.0139692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Mackay F., Leung H. The role of the BAFF/APRIL system on T cell function. Semin. Immunol. 2006;18:284–289. doi: 10.1016/j.smim.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Mackay F., Schneider P. TACI, an enigmatic BAFF/APRIL receptor, with new unappreciated biochemical and biological properties. Cytokine Growth Factor Rev. 2008;19:263–276. doi: 10.1016/j.cytogfr.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Moon Y.M., Lee S.Y., Kwok S.K., Lee S.H., Kim D., Kim W.K., Her Y.M., Son H.J., Kim E.K., Ryu J.G. The fos-related antigen 1-JUNB/activator protein 1 transcription complex, a downstream target of signal transducer and activator of transcription 3, induces T helper 17 differentiation and promotes experimental autoimmune arthritis. Front. Immunol. 2017;8:1793. doi: 10.3389/fimmu.2017.01793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014;14:329–342. doi: 10.1038/nri3661. [DOI] [PubMed] [Google Scholar]
- Ng L.G., Sutherland A.P.R., Newton R., Qian F., Cachero T.G., Scott M.L., Thompson J.S., Wheway J., Chtanova T., Groom J. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J. Immunol. 2004;173:807–817. doi: 10.4049/jimmunol.173.2.807. [DOI] [PubMed] [Google Scholar]
- Ou X., Xu S., Lam K.P. Deficiency in TNFRSF13B (TACI) expands T-follicular helper and germinal center B cells via increased ICOS-ligand expression but impairs plasma cell survival. Proc. Natl. Acad. Sci. U S A. 2012;109:15401–15406. doi: 10.1073/pnas.1200386109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert S., Zinser E., Holzinger C., Sandrock L., Koch S., Finotto S. NFATc1 deficiency in T cells protects mice from experimental autoimmune encephalomyelitis. Eur. J. Immunol. 2015;45:1426–1440. doi: 10.1002/eji.201445150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert R.C., Jellusova J., Miletic A.V. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol. Rev. 2011;244:115–133. doi: 10.1111/j.1600-065X.2011.01067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer U., Jennings S., Grimbacher B. To switch or not to switch - the opposing roles of TACI in terminal B cell differentiation. Eur. J. Immunol. 2007;37:17–20. doi: 10.1002/eji.200636914. [DOI] [PubMed] [Google Scholar]
- Scapini P., Hu Y., Chu C.L., Migone T.S., DeFranco A.L., Cassatella M.A., Lowell C.A. Myeloid cells, BAFF, and IFN-γ establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J. Exp. Med. 2010;207:1757–1773. doi: 10.1084/jem.20100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraml B.U., Hildner K., Ise W., Lee W.L., Smith W.A.E., Solomon B., Sahota G., Sim J., Mukasa R., Cemerski S. The AP-1 transcription factor Batf controls T H 17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshasayee D., Valdez P., Yan M., Dixit V.M., Tumas D., Grewal I.S. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18:279–288. doi: 10.1016/s1074-7613(03)00025-6. [DOI] [PubMed] [Google Scholar]
- Seyler T.M., Park Y.W., Takemura S., Bram R.J., Kurtin P.J., Goronzy J.J., Weyand C.M. BLyS and APRIL in rheumatoid arthritis. J. Clin. Invest. 2005;115:3083–3092. doi: 10.1172/JCI25265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shale M., Schiering C., Powrie F. CD4+ T-cell subsets in intestinal inflammation. Immunol. Rev. 2013;252:164–182. doi: 10.1111/imr.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Bülow G.U., Bram R.J. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science. 1997;278:138–141. doi: 10.1126/science.278.5335.138. [DOI] [PubMed] [Google Scholar]
- Wang H., Marsters S.A., Baker T., Chan B., Lee W.P., Fu L., Tumas D., Yan M., Dixit V.M., Ashkenazi A., Grewal I.S. TACI-ligand interactions are required for T cell activation and collagen-induced arthritis in mice. Nat. Immunol. 2001;2:632–637. doi: 10.1038/89782. [DOI] [PubMed] [Google Scholar]
- Wu Y., Bressette D., Carrell J.A., Kaufman T., Feng P., Taylor K., Gan Y., Cho Y.H., Garcia A.D., Gollatz E. Tumor necrosis factor (TNF) receptor superfamily member TACI is a high affinity receptor for TNF family members APRIL and BLyS. J. Biol. Chem. 2000;275:35478–35485. doi: 10.1074/jbc.M005224200. [DOI] [PubMed] [Google Scholar]
- Yan M., Wang H., Chan B., Roose-Girma M., Erickson S., Baker T., Tumas D., Grewal I.S., Dixit V.M. Activation and accumulation of B cells in TACI-deficient mice. Nat. Immunol. 2001;2:638–643. doi: 10.1038/89790. [DOI] [PubMed] [Google Scholar]
- Ye Q., Wang L., Wells A.D., Tao R., Han R., Davidson A., Scott M.L., Hancock W.W. BAFF binding to T cell-expressed BAFF-R costimulates T cell proliferation and alloresponses. Eur. J. Immunol. 2004;34:2750–2759. doi: 10.1002/eji.200425198. [DOI] [PubMed] [Google Scholar]
- Zhou L., Littman D.R. Transcriptional regulatory networks in Th17 cell differentiation. Curr. Opin. Immunol. 2009;21:146–152. doi: 10.1016/j.coi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data were deposited in NCBI with accession number GSE152077 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE152077).