

Abstract
Lung cancer accounts for most cancer-related deaths worldwide and has an overall 5-year survival rate of ~15%. Cell lines have played important roles in the study of cancer biology and potential therapeutic targets, as well as pre-clinical testing of novel drugs. However, most experimental therapies that have cleared preclinical testing using established cell lines have failed phase III clinical trials. This suggests that such models may not adequately recapitulate patient tumor biology and clinical outcome predictions. Here, we discuss and compare different pre-clinical lung cancer models, including established cell lines, patient-derived cell lines, xenografts and organoids, summarize the methodology for generating these models, and review their relative advantages and limitations in different oncologic research applications. We further discuss additional gaps in patient-derived pre-clinical models to better recapitulate tumor biology and improve their clinical predictive power.
Keywords: Lung cancer, preclinical models, organoid, xenograft, cell line, patient-derived models, 3D culture
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide, accounting for 22% or 2.09 million deaths in 2018 (The World Health Organization: https://www.who.int/news-room/fact-sheets/detail/cancer, accessed on Jan 04, 2020). The molecular basis of the highly aggressive biology of lung cancers remains largely unclear, despite efforts to identify complex genomic aberrations occurring in various histological subtypes of this disease (1,2). Treatments of lung cancer patients are determined by multiple factors including tumor histology, stage and biomarker status, the latter include actionable driver mutations and the expression level of immune checkpoint programmed death-ligand 1 (PD-L1) (3). For patients with actionable driver mutations, there are currently many clinically approved targeted therapies, most of which target the protein product of mutated oncogenes. Activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) gene were the first genomic alterations in non-small cell lung cancer (NSCLC) to be successfully targeted (4-8). Since then, many kinase inhibitors have been developed and used as first-line treatment for patients with mutations in other actionable drivers, including ALK, ROS1, NTRK, RET, BRAF and MET, and have shown significant improvement in patient survival (9). Similarly, for patients with high levels of immune checkpoint PD-L1, immunotherapy has also prolonged survival compared to chemotherapies (10,11).
However, the initial responses to therapies almost always result in eventual therapeutic resistance, due to development of additional mutations or yet unidentified factors. Against this backdrop of poor disease prognosis with current approved treatment options, patient-derived lung cancer models continue to be developed for gaining better understanding of the molecular pathobiology of lung cancers, identification of novel therapeutic targets which may also serve as disease or treatment biomarkers, as well as testing of novel agents as new therapies, thus contribute to further improvement of precision medicine. This review will discuss and compare current patient-derived lung cancer models and their limitations, including cell lines, patient-derived xenografts (PDXs) and patient-derived lung cancer organoids (LCOs).
Established cancer cell line models
Cancer cell lines have traditionally been the most commonly used models to study tumor biology and pharmacogenomics. The National Cancer Institute (NCI) and the Hamon Cancer Center (HCC) lines are the two largest series of lung cancer cell lines that have been established; most of these are available to researchers worldwide through the American Type Culture Collection (ATCC, Manassas, VA, USA). Additional cell line repositories have been established in various countries, e.g., CellBank Australia, European Collection of Authenticated Cell Cultures (United Kingdom), Japanese Collection of Research Bioresources Cell Bank, Deutsche Sammlung von Mikroorganismen und Zellkulturen and the Riken BioResource Center Cell Bank (Japan).
Cell line models in lung cancer research
Due to their relative ease of handling and ready availability, cell lines have been widely used in numerous studies in lung cancer. Drug sensitivity observed in cell lines may mimic clinical drug response. For example, cell lines with EGFR aberrations (PC-9 cells with exon 19 deletion and H3255 cells with L858R mutation) showed sensitivity to the EGFR TKIs erlotinib, afatinib, rociletinib and osimertinib. In contrast, H1975 cells, which bear both L858R and T790M mutations, responded only to the new-generation TKIs afatinib, rociletinib and osimertinib (12), although it has been reported that T790M mutation may also confer resistance to afatinib in the clinic (13). Another well-studied EGFR-mutated cell line, HCC827, was also shown to respond to the monoclonal antibody cetuximab (14). Multiple groups have reported that the RAF-inhibitors dabrafenib and vemurafenib, as well as the MEK-inhibitor trametinib, were able to inhibit the growth of cell lines with activating BRAF mutations including V600E (15,16). Similarly, the growth of cell lines bearing ALK fusions could be inhibited by ALK inhibitors (17,18). However, coactivation of EGFR and HER2 could confer resistance to ALK inhibitors in one of these cell lines (17).
The generally simple, low cost and rapid culture of cell lines is practical for large-scale studies, including pharmacogenomics projects such as the Cancer Cell Line Encyclopedia (CCLE) (19), the Genomics of Drug Sensitivity in Cancer (GDSC) (20-22), the Cancer Therapeutics Response Portal (CTRP) (23) and the Genentech Cell Line Screening Initiative (gCSI) (24,25). These projects combine massively parallel-omic profiling including genomics, copy number variation (CNV) analysis and transcriptomics, with screening of drug responses to more than 100 drugs on hundreds of cancer cell lines (including over 100 lung cancer lines), in order to identify associations between molecular markers and drug sensitivity. These valuable databases allow researchers worldwide to identify genes and drug responses in specific cell lines of interest. Machine learning models have also been applied to interrogate these pharmacogenomics datasets based on cancer subtypes or molecular markers (26,27). Another large-scale screening project using cancer cell lines is the Connectivity Map (CMap), which explores gene expression changes after drug treatments (28,29). The CMap study generated over a million gene expression profiles in 3 to 77 cell lines before and after treatments with 42,080 perturbagens, including 19,811 small molecule drugs, 18,493 shRNAs, 3,462 cDNAs and 314 biologics. Their data provide information on drug responses in cancer cell lines on the RNA level, from which researchers have identified drugs that can potentially be repositioned to treat cancer. Using the CMap data, Jahchan et al. (30) reported that the tricyclic antidepressant imipramine, the Histamine H1 receptor antagonist promethazine and the calcium channel blocker bepridil can reverse the small cell lung cancer (SCLC)-associated gene signatures, and showed anti-tumor effects in both in vitro and in vivo SCLC models.
Cell lines are also relatively easy to work with for genetic manipulations, as compared to other models. For instance, a loss-of-function study using the CRISPR-Cas9 system would be technically more challenging if carried out in organoid or PDX models (see next sections). Moreover, cell lines enable clonal selection and expansion to validate and select for positive knock-out cells (Figure 1). CRISPR-Cas9 loss-of-function studies enable us to identify essential genes in different cancer subtypes and test for synthetic lethality relationships between specific genes and driver mutations. For example, CRISPR/Cas9 knockout of focal adhesion kinase (FAK) in NSCLC cells with mutant KRAS showed sensitization to radiotherapy (31). In a further study, mutant EGFR-specific knockout by CRISPR/Cas9 was shown to inhibit the proliferation of the EGFR-mutated lung cancer cell line H1975 and to reduce tumor volumes of xenografts implanted with H1975; this was not observed when using A549 lung cancer cells that carry wild-type EGFR alleles (32). As discussed previously, cell line models facilitate large-scale studies: The Achilles project performed a genome-wide scale CRISPR-Cas9 knockout screen to identify essential genes in 689 lines of different cancer types, including 97 lung cancer cell lines, in the 19Q4 release (33-35). Although still in its infancy, CRISPR-Cas9 cell engineering may hold great promise in future cell therapy.
Figure 1.
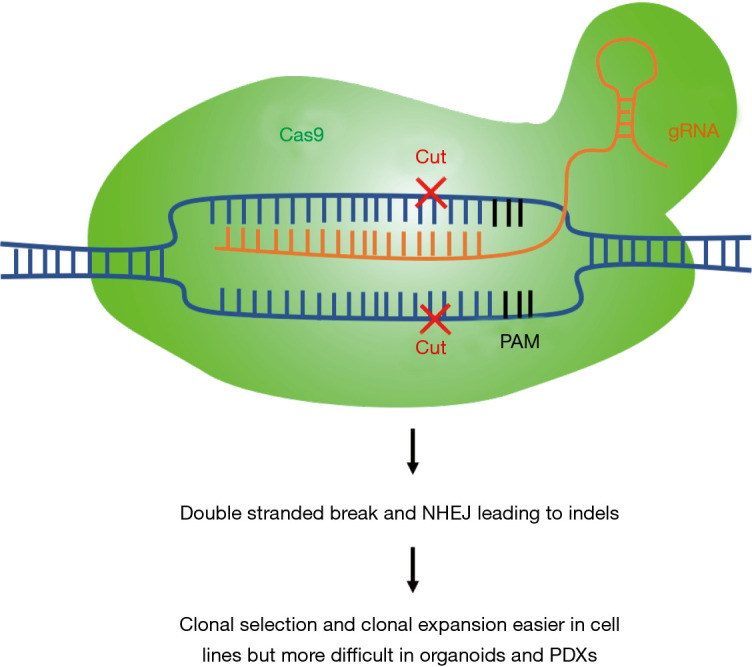
Schematic of CRISPR-Cas9 knockout system. PAM, protospacer adjacent motif; NHEJ, non-homologous end joining; PDX, patient-derived xenograft.
Limitations of cell line models
Important limitations of these models must be considered. Cell lines likely represent a subpopulation of the original tumor and are largely homogenous, due to the selective survival pressures present in culture conditions devoid of the original microenvironment involving interactions with stromal, immune and inflammatory cells (36,37). As a result, there are genetic and epigenetic differences between the cell lines and the original tumors, which make it difficult to evaluate how much of the original tumor biology is retained in established cell line models that have been maintained long-term in vitro (38). The lack of cell-cell interactions within a three-dimensional (3D) environment also limits the translational potential of cell line studies. However, in an effort to elevate the relevance of two-dimensional (2D) cell cultures for modelling patient disease, lung cell lines have been utilized in a variety of studies to develop engineered microenvironments (so-called ‘on-chip’ culture approaches) to allow for growth in 3D, as well as growth in co-cultures with stromal cells (endothelial cells, immune cells and fibroblasts) (39-41). Such 3D on-chip cultures have shown superiority for modeling drug sensitivity over traditional 2D culture (42).
Differences between the 2D and 3D culture conditions may become apparent when examining surface marker expression: for example, expression of TTF1 (a marker for lung adenocarcinoma) and TP63 (a marker for squamous cell carcinoma) was found to be similar between the adenocarcinoma and squamous cell lines included in the CCLE project, with more than half of the squamous cell lines having very low TP63 expression (Figure 2, Raw data obtained from https://portals.broadinstitute.org/ccle, accessed on Sep 3rd, 2019). This contradicts the clinical histopathology finding that 70–80% of lung adenocarcinoma are positive for TTF1, while lung squamous cells are negative for TTF1 and 100% positive for TP63 (43,44). While these discrepancies might be attributed to differences between the output of RNA-seq and immunohistochemistry (IHC) assays, others have reported no significant difference between TTF1 mRNA and IHC protein expression (45). These contradictions highlight the importance of ascertaining whether cell lines of interest in fact retain certain key aspects of tumor biology, prior to their use in specific studies.
Figure 2.
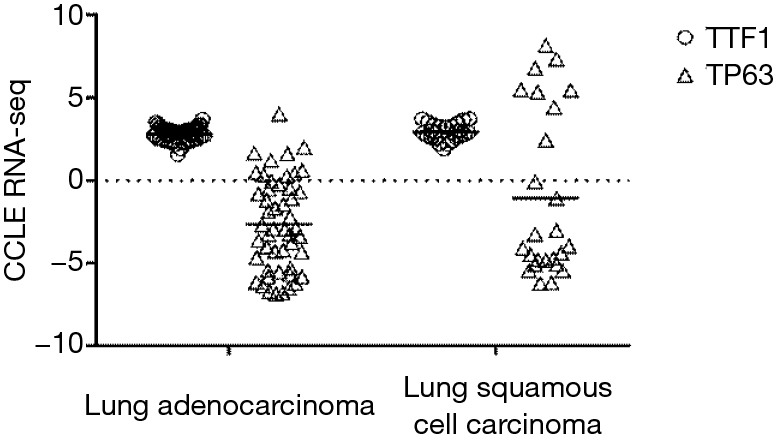
CCLE RNA-seq data on TTF1 and TP63 expression in lung adenocarcinoma and lung squamous cell carcinoma. TTF1, Transcription Termination Factor 1; TP63, Tumor Protein P63. Raw data obtained from https://portals.broadinstitute.org/ccle, assessed on Sep 3rd, 2019.
Theoretically, experimental results generated using the same line in different laboratories should be directly comparable. However, in practice, this may not be the case. Haibe-Kains et al. reported a poor concordance between the CCLE and the GDSC drug response data (46), and although improved consistency was obtained using a different statistical approach, poor correlation was still observed in some cases, especially in cell lines with poor drug sensitivity (47). It should be noted that even when comparing drug response of the same drug and the same cell line, results may be affected by differences in experimental protocols. Standardization of methodology would certainly help to improve the consistency of cell line experimental results across different laboratories.
Primary patient-derived cell culture models
As established cell lines have undergone unspecified long-term passaging in vitro, they may no longer faithfully represent the molecular heterogeneity of primary patient tumors. Comparison of transcriptomic and/or proteomic profiles of patient NSCLC tumors and established cell lines have demonstrated significant divergence (48,49). More recently, a new technique involving Rho-associated kinase inhibitor-induced conditional programming to develop primary lung cancer cell cultures derived from patient biopsy samples has been reported with a ~50% success rate (50,51). These models might better retain the molecular characteristics of patient tumors and be a better alternative to study cancer biology and drug sensitivity. The generation of these primary cell cultures is of particular importance for studies aiming at guiding decision making in the clinic, due to the speed at which they can be established, as well as the concordance of drug responses between the patients and the respective cell lines (52).
Patient-derived tumor xenograft (PDX) models
PDXs are cancer models established by engrafting and growing human tumor tissue/cells in animal host, most commonly immunodeficient or humanized mice (53,54). Unlike cell lines that grow in vitro under non-physiological conditions, PDX models grow in a 3D microenvironment, which includes vasculature that provides in vivo delivery of nutrients and oxygen, and host stromal and immune cells interact and communicate with the tumor cells. PDXs mostly retain the genomic and phenotypic profiles of the original patient tumors, thus may better reproduce the clinically observed drug response (49,55-57). Thus, PDXs hold promise as better pre-clinical models in personalized medicine, for which they may be used to predict drug-responses and potentially guide patient therapies.
Methodology of generating PDX models
PDX tumors have primarily been established from solid tumors and most published lung cancer PDX models were established using surgically resected tumors. However, successful engraftment can also be achieved using smaller tissue samples, such as computed-tomography (CT)- and endobronchial-ultrasound (EBUS)-guided biopsies (58-60). The latter approaches have been particularly useful in generating SCLC PDXs, since these patients seldom undergo surgical resection of the tumor (61,62). Different types of tumor histologies and collection methods may impact on the success rate of engraftment. In NSCLCs, the engraftment rates range from 25–60% (43,49,50,54,57-63) (Table 1), with squamous cell carcinoma having a higher take rate than adenocarcinoma (60,63,67). The ability of resected NSCLC to engraft and form PDX is strongly associated with poorer patient prognosis (49). In SCLCs, EBUS-guided biopsies have a 67–83% success rate in PDX establishment (61,62).
Table 1. Representative large studies of Lung PDX model development.
| Publication | Patients | Tissue collection | Mouse strain | Engraftment rate | Engrafted tumor histology [n] |
|---|---|---|---|---|---|
| Fichtner et al. (55) | NSCLC | Surgical resection | NOD/SCID | 25/102 (24.5%) | Adenocarcinoma [6] |
| Squamous [12] | |||||
| Others [7] | |||||
| Wang et al. (49) | NSCLC | Surgical resection | NOD/SCID | 127/441 (28.8%) | Adenocarcinoma [52] |
| Squamous [62] | |||||
| Others [13] | |||||
| Hao et al. (64) | NSCLC | Surgical resection | NOD/SCID | 23/88 (26.1%) | Adenocarcinoma [11] |
| Squamous [9] | |||||
| Others [3] | |||||
| John et al. (63) | NSCLC | Surgical resection | NOD/SCID | Adenocarcinoma 30/96 (31.1%) Squamous 29/45 (64.4%) |
Adenocarcinoma [30] |
| Squamous [29] | |||||
| Others [4] | |||||
| Nakajima et al. (60) | NSCLC& SCLC | EBUS-guided biopsy | NSG | Adenocarcinoma 3/12 (25%) | Adenocarcinoma [3] |
| Squamous 2/3 (66.7%) | Squamous [2] | ||||
| Small cell carcinoma 2/3 (66.7%) | Small cell carcinoma [2] | ||||
| Large cell carcinoma 1/1 (100%) | Large cell carcinoma [1] | ||||
| Fang et al. (65) | NSCLC& SCLC | Surgical resection | BALB/c nude mice | N/a | Adenocarcinoma [15] |
| Squamous [68] | |||||
| Others [16] | |||||
| Kang et al. (66) | NSCLC | Surgical resection & biopsy | NOD and nude | Adenocarcinoma 30/92 (32.6%) | Adenocarcinoma [30] |
| Squamous 11/30 (36.7%) | Squamous [11] | ||||
| Kita et al. (67) | NSCLC | Surgical resection | NSG & SHO | Adenocarcinoma 13/48 (27.1%) | Adenocarcinoma [13] |
| Squamous 6/10 (60%) | Squamous [6] | ||||
| Large cell carcinoma 1/2 (50%) | Large cell carcinoma [1] | ||||
| Cuenca et al. (68) | NSCLC | Surgical resection | SCID | Adenocarcinoma 9/28 (32.1%) | Adenocarcinoma [9] |
| Squamous 6/19 (31.5%) | Squamous [6] | ||||
| Adenosquamous [1] | |||||
| Drapkin et al. (56) | SCLC | CTC | NSG | 16/42 (38.1%) | Small cell carcinoma [16] |
| Hodgkinson et al. (69) | SCLC | CTC | NSG | 4/6 (66.7%) | Small cell carcinoma [4] |
NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer; CTC, circulating tumor cell; NOD, nonobese diabetic; SCID, severely compromised immune deficient; NSG, NOD-SCID gamma.
Circulating tumor cells (CTCs) can be non-invasively collected from blood, and these cells generally have acquired greater metastatic potential (70). Given the low number of CTCs in NSCLC patients, generating NSCLC PDXs from CTCs has been technically challenging (71), with only one case study found in the literature (72). In contrast, most SCLC patients with advanced stage disease release high numbers of CTCs (73,74), and since surgical resection is rarely performed in SCLC patients, CTCs have become an important material source for SCLC PDXs establishment. Reported engraftment rate for CTC-derived SCLC PDXs ranged between 38% (16/42) to 67% (4/6), depending on the CTC counts in patient blood samples (56,69).
In most studies, immunodeficient mice are used as the animal host to generate PDX models. One of the most commonly used immunodeficient mice is the non-obese diabetic (NOD)-severely compromised immune deficient (SCID) mouse, which lacks mature T and B cells and has deficient natural killer cell function (57,75). To enhance engraftment rates, NOD-SCID gamma (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice, also known as NSG mice, have been developed from a NOD-SCID background with additional interleukin 2 receptor gamma chain impairment. This causes deficient cytokine signaling and consequently enhanced immunodeficiency. NSG mice also have longer lifespans compared to NOD-SCID mice, which appear to have higher rates of developing thymic lymphomas (76,77). However, PDXs generated in immunodeficient mice cannot efficiently be used to study immunotherapies, since an intact immune system, and preferably one of human origin, is required. For this purpose, humanized mice have been developed by engrafting human leukocytes and purified CD34+ hematopoietic stem cells into immunodeficient mice. Hematopoietic stem cells can be obtained from different tissue sources, including umbilical cord blood, bone marrow, fetal liver and neonatal thymus (78-81). Rosato et al. demonstrated increased human cytokine levels in not only the plasma, but also the tumors of humanized NSG mice compared to non-humanized NSG mice (82). Further, it has also been shown that the growth of human stromal and immune cells can be maintained in humanized mice, therefore better mimicking the original tumor microenvironment (83).
Few technical considerations may help to enhance the success rate of PDX tumors establishment with a higher degree of patient fidelity. For example, solid tumors can be first dissociated and injected into host animals as single-cell suspension; this may potentially avoid selection of clonal subpopulation of the tumor for engraftment (77). This approach also allows genetic manipulations of the tumor cells prior to engrafting. Dissociated tumor cells can be mixed with matrix protein cocktail Matrigel to increase the rate of successful engraftment and growth of tumor cells (84). Patient tumors can be implanted heterotopically or orthotopically into the host animals. Most studies on lung cancer PDX models use heterotopic implantation, which provides the advantages of an easy procedure and accurate tumor size measurements. On the other hand, orthotopic implantation should better recapitulate the native tumor environment of lung cancer and enable the study of metastasis, but this is technically challenging (85). Only few labs have used this approach to make lung cancer PDX models, where a tumor fragment is sewn into the left lung or placed into the left thoracic cavity in a thoracotomy procedure (68,86). Cuenca et al. reported a 30.8% (16/52) engraftment rate in NSCLC models using orthotopic implantation (68).
PDX models in lung cancer research
Our lab has demonstrated that NSCLC PDXs with EGFR exon 19 deletion and L858R mutations responded to first/second generation EGFR TKIs (gefitinib, erlotinib, dacomitinib and afatinib), consistent with clinical observations (87). In contrast, a PDX model with both EGFR exon 19 deletion and T790M mutations is resistant to gefitinib/erlotinib, but responded to cetuximab (87,88). Zhang et al. have reported that a lung PDX model with KRAS mutation was resistant to gefitinib (89), agreeing with clinical data (90,91). We have also shown that lung squamous carcinoma models bearing PIK3CA E542K mutations are sensitive to PI3K inhibitors, and CDK4/6 inhibitors conferred a synergistic anti-tumor activity when used in combination with PI3K inhibitors (92). Several groups have demonstrated sensitivity of PDXs established from lung cancer patients with ALK gene rearrangement to ALK inhibitors (65,66). In contrast, a PDX model established from a patient who developed resistance to multiple ALK inhibitors demonstrated a similar response profile as observed clinically (93). Ambrogio et al. demonstrated that dual inhibition of DDR1 and the Notch signaling pathway induced tumor regression in KRAS-mutated PDXs, opening up the possibility of using the combination of dasatinib and Notch inhibitor (e.g., demcizumab) to treat patients with KRAS mutations (94,95), for whom there currently is no targeted therapy available. Both NSCLC (55) and SCLC (96) PDX models show responses to chemotherapies similar to those observed in clinical settings. In SCLC, EZH2 inhibition provides synergistic tumor suppressive effect when combined with chemotherapies, even in chemoresistant PDX models (97). Nevertheless, PDXs do not always accurately predict patient response. For example, although it has been reported that the ERK1/2 inhibitor CC-90003 (98) and the XPO-1 inhibitor KPT-330 (99) were able to repress tumor growth in PDXs, neither showed encouraging results in clinical trials.
Immunotherapies have revolutionized cancer treatment and have led to promising clinical outcomes in various types of advanced cancers, including lung cancer (3). There is increasing interest in testing immunotherapy responses using PDX models established in humanized mice. Hu et al. observed an anti-tumor response upon adoptively transferring human T cells that express melanoma antigen MART-1-specific T cell receptors to their humanized melanoma PDXs (100). Anti-PD-1 antibodies have also shown anti-tumor effects in humanized triple-negative breast cancer (82), osteosarcoma (101) and lung cancer (102) models. In lung cancer specifically, anti-PD-1 checkpoint inhibitors were shown to inhibit tumor growth in humanized mice engrafted with A549 lung tumor cells (102).
In contrast to in vitro cell line assays, large-scale experiments are more difficult to perform in PDXs. Nevertheless, efforts in this direction are underway and Gao et al. (103), for example, recently published a high-throughput drug screening project using PDXs of various cancer types with different driver mutations, allowing discoveries of associations between biomarkers, pathway activation and drug sensitivity in different cancer subtypes.
Limitations of PDX models
Although PDXs retain tumor heterogeneity and potentially better mimic tumor biology and microenvironment compared to cell lines, several limitations and challenges apply to this type of models. Compared to cell lines and 3D organoids (see next section), the establishment of PDX models is more time-consuming and costly, especially when using humanized mice. Concerns have been raised regarding potential genetic drift of tumor cells in xenografts of late passages, though several studies have shown that no major genetic difference could be found through at least 10 passages. Nevertheless, it is common practice to limit PDX experiments to less than 10 passages (104-106). When non-humanized mice are used, the original human stromal and immune cells are replaced by mouse stromal cells after serial passages, thereby losing the contribution of human stromal cells to the original tumor biology (55,107). Overall, the methodology of PDX establishment remains suboptimal and current efforts in this field are devoted to better recapitulate the patient tumor and microenvironment, and to increase engraftment rate in order to lower the cost.
Patient-derived LCOs
Because 2D cell cultures retain only limited analogy to their parent tissue (108), scientific focus has shifted to 3D cell culture strategies (109). Specifically, patient-derived cell populations can be grown in a 3D extracellular matrix, giving rise to structures termed organoids, which are characterized by their capacity to self-organize into structures that are reflective of the tissue they are derived from (110). Tumor organoids have been generated from a large number of tissues, most recently also from lung.
Because of the genotype-phenotype similarity to their parent tumors, as well as the possibility of rapid expansion for drug sensitivity assays, tumor organoids represent preclinical tools of great relevance to patient disease. For example, a recent study comparing DNA copy numbers in breast tumor organoids and 2D cell lines to patient tumors found greater concordance between tumor samples and organoids over 2D cell lines (111), indicating that organoids may be a more apt culture system to model patient tissue heterogeneity (112,113).
Methodology of generating LCO models
Patient-derived LCOs require growth within a supporting 3D matrix, most commonly Matrigel (114,115), as well as a defined cocktail of growth factors and inhibitors. The formulation of this growth media varies between laboratories (113,116-119) (Table 2), but all formulations contain factors for lung stem cell maintenance. While the only factor common to all reported LCO-specific media formulations is the B27 supplement, an optimized serum substitute initially utilized for neuronal cell cultures (120), growth factors included are either epidermal growth factor (EGF) or members of the fibroblast growth factor (FGF) family, and inhibitors/activators of specific pathways, namely TGF-β and Rho-associated protein kinase (inhibited) and Wnt (activated). LCOs may be generated from tumor biopsies or resections and can be maintained in both short- and long-term (> passage 10) cultures, with reported high overall success rates of culture initiation (approx. 90%) (113,118). Specifically, tumor tissue is minced and dissociated enzymatically to obtain single cells, which are then typically mixed with Matrigel, deposited in so-called domes (i.e., large droplets) in 24- or 48-well plates, and finally topped with the appropriate growth medium. Whether or not this procedure yields viable and proliferating LCOs depends on a number of factors, including cell viability after tissue digestion and cellularity of the tumor tissue processed. Alternatively, LCOs can be established form CTCs isolated from blood (73,121).
Table 2. Summary of LCO culture strategies.
| Publication | Media composition | ECM composition | Culture setup | Reported successful culture time |
|---|---|---|---|---|
| Neal et al. (116) | ADMEM/F12 + HEPES, Glutamax, Pen-Strep, NALC, B-27 (- vit. A), R-spondin 1, Noggin, Nicotinamide, A 83-01, SB-202190, Wnt3a, Gastrin, EGF | Rat tail type 1 collagen | Tumor pieces in ALI culture | Up to 100 days in culture |
| Tamura et al. (117) | FBIM002 medium | NA | Tumor pieces in suspension culture | Over 6 months |
| Sachs et al. (118) | ADMEM/F12 + NALC, B-27, R-spondin, Noggin, Nicotinamide, A 83-01, SB202190, Y-27632, FGF7, FGF10 | BME type 2 | Dome culture | NA |
| Kim et al. (119) | DMEM/F12 + Pen-Strep, B-27, Y27, bFGF, EGF, N2 | GFR MG | Dome culture | Over 6 months |
| Shi et al. (113) | ADMEM/F12 + HEPES, Glutamax, Pen-Strep, NALC, B-27, Noggin, A 83-01, Y-27632, FGF10, FGF4, EGF, CHIR 99021, SAG | GFR MG | Dome culture | Over 10 passages, over 3 months in culture |
ADMEM, advanced DMEM; Pen-Strep, penicillin/streptomycin; EGF, epidermal growth factor; FGF, fibroblast growth factor; SAG, smoothened agonist; NALC, N-Acetyl-L-cysteine; FBIM002 - media pertaining the Fukushima Translational Research Project; NA, not applicable; ALI, air-liquid interface; BME, basement membrane extract; GFR MG, growth factor reduced Matrigel.
Contamination of cultures by normal lung epithelium was identified in a number of studies attempting to grow LCOs (118,119,122), whereby normal cell clusters tend to outgrow LCOs in these cultures (113). This is possibly due to the genetic instability and consequently increased cell death in LCOs (123) or because all published LCO media are based on stem cell media formulations supportive of normal epithelial growth. Besides manually separating LCOs from normal lung organoids based on their morphological appearance (the feasibility of this approach remains unclear), Sachs et al. achieved pure LCO cultures through the addition of Nutlin-3A, which specifically inhibits the growth of TP53 wild type cells and thus, when added to TP53 mutant LCOs, will lead to drop-out of normal epithelium from cultures (118). Alternatively, the use of a suboptimal medium for LCO culture was most recently described by Kim et al., which did not support normal cell growth (119).
To this date, only a handful of studies have documented the establishment of patient-derived LCOs (113,116-119,122,124,125), whereby the success rates of long-term culture establishment vary substantially (Table 2), and detailed descriptions of the number of passages achieved and split ratios used were typically not reported. An exception to this is the distinction between long and short-term models described by our lab (113), where long-term LCO cultures were defined as achieving passage 10 (and a corresponding culture time of over 3 months), while the rest were deemed short-term cultures. The efficiencies of culture establishment for these models were 15% and 72%, for long- and short-term, respectively. The following sections largely focus on insights from the few available LCO studies to date, as well as from pertinent papers investigating tumor organoids in other tissues.
LCO resources and characterization
Large collections of patient-derived LCOs, as well as normal lung organoids, are being established and biobanked (123,126). At the basis of such efforts lies the observation that LCOs closely resemble the genotypic and histological heterogeneity of their parental tumors. Immunohistochemical analyses of LCOs saw major lung tumor subtypes reflected in LCO cultures, using markers such as TTF-1, cytokeratin 5 and synaptophysin for adenocarcinoma, squamous cell carcinoma and small cell carcinoma, respectively (113,116,119). Implantation of patient-derived LCOs into mice resulted in growth of tumor xenografts at variable success rates (30–100%) and with preserved parental tumor histology, proving LCO tumorgenicity (113,118,119).
LCOs further maintained defining genetic characteristics from their parental tumors. Next generation sequencing of LCOs and tumors revealed the presence of matching somatic mutations, such as TP53, KRAS, EGFR and others, which are commonly associated with NSCLC (1,127); no discordant driver mutations were observed. Analyses including SNP genotyping, variant allele frequency distribution analysis and CNV profiling similarly showed that concordance could generally be maintained between LCOs and their parental tissues during long-term culture, highlighting that clonal stability in LCOs is, at least for a certain number of passages, preserved (113,118,119).
Gene-drug correlation studies
Cancer organoids can be used to model human diseases and as a platform for drug screening (128). Across tumor types, studies correlating drug sensitivity and genotype data, have demonstrated that drug responses generated in patient-derived organoids are in line with the molecular profiles of their parental tumors (126). For example, erlotinib was effective against the growth of LCOs with EGFR exon 19 deletions (113). Kim et al. (119) observed resistance to erlotinib in one LCO line, despite the presence of an EGFR mutation, but genomic analysis revealed the presence of MET amplification, which is associated with EGFR TKI resistance. Our lab further demonstrated the use of LCOs to identify potential combination treatments (113). Using an FGFR1-mutated LCO line insensitive to FGFR1 inhibitor BGJ398, we examined the efficacy of BGJ398 in combination with either a MEK inhibitor (trametinib) or a PI3K inhibitor (BKM120), both of which have been reported to decrease the growth of LUSC in cell line studies (129,130), and found a synergistic effect of the former combination in LCO cultures. This finding was then validated in the parental PDX model.
LCOs can also be generated from tumors further along the metastatic cascade. For example, SCLC CTCs spontaneously formed ‘tumorospheres’ in culture, even without the need for lung stem cell media (74). These CTC-derived tumorospheres expressed all typical SCLC markers and showed reduced chemosensitivity, compared to CTC single cells. This may possibly suggest that the growth of CTCs as clusters, rather than as single cells, could explain the commonly observed chemoresistance in relapsed SCLC patients.
Exploring different treatment strategies in patient-derived LCOs during the patients’ lifetime would represent a powerful approach for informing personalized care. Such efforts are underway: for NSCLC specifically, patient-derived LCOs are being screened with 8 different therapeutic agents at The Netherlands Cancer Institute, with the aim of identifying effective drugs, in order to offer patients the option to adjust their treatment accordingly (126).
Screening for drug target discovery
One of the most cited reasons for the low success rate in clinical trials is the utilization of 2D cell cultures as pre-clinical disease model systems (131). Tumor organoids may represent an improved tool for drug screens aiming to identify novel actionable molecular targets. While no large-scale drug screen has been attempted using LCOs yet, the overall approach shows promise in other tumor types, including colorectal cancer (132) and ovarian cancer (133). Pauli et al. (134) described high-throughput dose response screening on four tumor organoid lines against 160 compounds (FDA approved and under development) in mono- and combination treatment. Subsequent to hit validation in PDXs, this study was able to identify effective treatment strategies for two endometrial carcinoma lines, for which there are no currently approved targeted therapies, and two colorectal carcinoma lines.
Studies have found that different media formulations (see Table 2) are supportive of growth of tumor cells that carry genetic markers (e.g., EGFR), as well as protein markers (e.g., TTF-1, p63, cytokeratin 5) of interest, confirming that the in vitro cultures generated are, as far as these markers are concerned, representative of patient material. Beyond this, it remains unclear how the presence of different growth factors in media formulations affects marker expression at the protein level and the response to different drugs. To our knowledge, to date there has been no comparative study of organoid marker expression or drug responses in the different published media formulations.
Modeling of the tumor microenvironment
As the presence and heterogeneity of stromal cells within a tumor mass may alter treatment response, LCO cultures will need to include stromal cell types, in order to model disease in a more comprehensive way (135). Given the very encouraging successes in immunotherapy, the first studies investigating tumor-stroma relationships using LCOs have focused on incorporating immune cells into LCO cultures; to this date, studies specifically exploring fibroblast- LCO or endothelial cell-LCO relationships are still lacking.
While defined media are successful for growing LCOs, the same media also lead to the loss of stromal cell fractions during LCO establishment. This is because certain factors are specifically inhibitory to fibroblast proliferation, [e.g., Noggin, B27 supplement and various TGBβ inhibitors (136)], and immune and endothelial cells drop out from cultures over time (116). To include stromal populations into LCO cultures, two approaches have been reported. Neal et al. have generated 20 en-bloc LCO cultures that maintain their original stromal cell fraction for several passages by culturing minced tumor tissues in air-liquid interphase (ALI) culture (116), which recapitulated the tumor T- and B-cell repertoire. Anti-PD-1 antibody induced TIL-mediated check-point inhibition in one out of three NSCLC LCO lines examined and tumor cell killing was further assessed in tumor organoids from various tissues. Overall, this study demonstrated the possibility of culturing en-bloc LCOs for short term studies, which preserve a functional immune component.
A second approach for establishing tumor-stroma LCOs is to recombine stromal and parenchymal tumor fractions after culturing them separately. For example, the interaction between patient-derived LCOs and infiltrating T cells was investigated by co-culturing tumor epithelial cells and autologous blood lymphocytes. In this system, tumor cell killing by reactive T-cells occurred, indicating the utility of this culture method for assessing patient sensitivity to T-cell based therapies (122).
This re-combination approach has also been utilized for co-culturing tumor organoids and activated fibroblasts in pancreatic cancer studies. To model the functions of carcinoma associated fibroblasts (CAFs) present in desmoplastic PDAC stroma, PDAC organoids were established and co-cultured with murine fibroblast progenitors (136). Upon activation by tumor organoids, CAFs adopted one of two identified phenotypes: CAFs in direct contact with organoids upregulated α-smooth muscle actin (α-sma) expression and TGFb response genes, while CAFs located further away from organoids secreted IL-6, a potent pro-survival signal for PDAC organoids. Separately, in a library of 39 PDAC organoids, CAFs were found to provide a Wnt niche required for maintenance of tumor stem cells, thereby supporting early tumor development (137). Tsai et al. also established co-culture pancreatic tumor organoids to study pancreatic cancer progression and fibroblasts activation, as well as tumor-dependent lymphocyte infiltration (138). Such studies particularly apply to LCOs, as desmoplasia commonly occurs in lung cancers and CAF-mediated effects on tumor growth and drug sensitivity have been amply reported (139,140). For example, one study using lung tumor spheroids formed by the PC-9 cells in 3D co-culture with CAFs identified a pro-proliferative role of specifically podoplanin-positive CAFs on lung tumor cells (141), further demonstrating the heterogeneous effects of CAF subpopulations on disease progression.
Overall, the field of tumor organoid co-cultures is in its infancy, with disease modelling being the primary goal of current studies. Particularly for LCOs, no methods for stable, long-term cultures of stromal-parenchymal organoids have been identified, nor have media formulations across laboratories been standardized. However, even initial results, particularly with respect to immune cell-tumor cell interactions, indicate that the use of stroma-parenchymal LCOs for biomarker identification, drug screening and modeling of therapy resistance lies in the near future.
Drawing inspiration from the established on-chip models initially generated for use with cell lines, efforts are underway for using LCOs in microfluidic or otherwise engineered microenvironments. On-chip culture platforms have been used to model various tumors using organoids and other cell types (including endothelial cells for neovascularization), and the reader is referred to expert reviews describing the different on-chip setups, their uses and limitations (142-145). Because culture media for LCO growth have only recently emerged, to the best of our knowledge only one study has described the use of LCOs in an on-chip application (125). Jung et al. used a microfluidic setup with continuous media flow for generating small cell LCOs for sensitivity testing of the drugs cisplatin and etoposide; LCO death was identified in a dose-dependent manner. Microfluidic devices have further been used to isolate and expand lung CTCs and patient immune cells from liquid biopsies (146), by using laser ablation to generate an array of microwells, and seeding CTCs and white blood cells into these to allow cluster formation. This platform can then be used to assess CTC drug response.
Limitations of LCOs
Patient-derived LCOs have to date effectively been used for disease modeling, genotype-phenotype studies and drug sensitivity testing, and have already proven to be exceptionally useful models for evaluating targeted therapeutics in a patient-specific fashion. However, certain aspects of current LCO culture methods represent hurdles that hamper the use of this model for different applications.
Firstly, robust strategies for parenchymal-stromal co-cultures are lacking, thereby impeding long-term studies of stromal involvement in tumor growth, drug sensitivity and resistance (147). Secondly, factors present in LCO medium, such as small molecule inhibitors, may alter drug responses. A second issue with regard to media formulations is that it remains unclear to what extent tissue ‘maturation’ is inhibited in LCO cultures, i.e., to what extent media factors inhibit cellular differentiation (148). Similarly, it is known that Matrigel, the most common matrix environment for LCO growth, is an abundant source of growth factors and plays a role in suppressing differentiation. In fact, a number of alternative, natural and synthetic, polymers are being explored for organoid growth (148). Further, culture scalability represents a major bottleneck, especially for high-throughput drug screening applications, as LCOs vary wildly in growth dynamics (149). Finally, due to the lack of viable blood supply and waste removal, LCOs suffer from size-dependent cell death; incorporation of endothelial cells or appropriate use of engineered culture technologies may eliminate this phenomenon (150).
Conclusions
Patient-derived models help researchers better understand cancer biology and genetics, allowing identification of biomarkers for clinical diagnosis and prediction of drug response. Some models, i.e., cell lines, have been used extensively in lung cancer research, while others have recently emerged, such as organoids, and their exploitation remains in infancy. This review has discussed the methodology and applications, as well as the advantages and limitations specific to each preclinical model and provides an overall summary of these observations (Figure 3 and Table 3).
Figure 3.

Methodology and applications of preclinical patient-derived lung cancer models. Solid lines represent greater compatibility between models and applications. Dashed lines represent lesser compatibility between models and applications. EBUS, endobronchial ultrasound.
Table 3. Summary of the readouts, advantages and limitations of the available lung cancer preclinical models.
| Lung cancer preclinical model | Readouts | Advantage | Disadvantage |
|---|---|---|---|
| Cell lines | Growth/metabolic assays | Ease and low cost of culture and scale-up | Lack of clonal heterogeneity |
| 2D Migration | Compatible with screening applications | Poor representation of cell-cell and cell-substrate interactions in patient tumor | |
| Marker expression | Unlimited passaging | Possible changes in marker expression after long-term culture | |
| Multi-omics profiling | Clonal homogeneity | ||
| Tumor cell killing | Well characterized | ||
| 3D invasion | Easy transferability among laboratories | ||
| Ease of genetic manipulation (e.g., CRISPR) | |||
| Co-cultures of specific cell types of interest possible | |||
| PDXs | Tumor volume | In vivo drug response assessment mimicking clinical situation | Long-term lines |
| Metastases | Mouse or humanized tumor microenvironment | Labor intensive and costly to use or maintain | |
| Histology | Humanized mice difficult to establish | ||
| Omics | Difficulty to scale up (low throughput) | ||
| Possible genetic drift after numerous passages | |||
| LCOs | Growth/metabolic assays | Maintenance of clonal heterogeneity | Difficulties with culture initiation and long-term maintenance |
| Histology | Model closest to patient tumor | Scale-up more cumbersome than cell lines | |
| Multi-omics profiling | Genetic manipulations are possible | Lack of robust co-culture strategies for tumor epithelial and stromal cells | |
| Tumor cell killing | Large number of passages (case dependent) | Requirement for costly and non-standardized culture ingredients | |
| 3D invasion | Co-cultures between specific cell types of interest possible | ||
| Immune cell recruitment |
Cell lines are easy and fast to culture, making them the first choice for large-scale studies, but the lack of heterogeneity and cell-cell interactions within a 3D environment limits the translational potential of findings from cell line studies. This limitation has been in part offset by culturing cells as 3D spheroids or in 3D on-chip environments (40). Compared to cell lines, PDX models retain tumor heterogeneity and the in-situ tumor microenvironment, but are more time-consuming and expensive, therefore hampering large-scale experiments. Organoids on the other hand, are established and grown in a 3D environment within a comparatively short time frame and have the potential to be co-cultured with other cell types of interest. While it takes 3–6 months to establish a PDX model, LCO generation form the same tumor specimen can be accomplished in as little as 4 weeks (118,119,123), and may therefore have the potential to inform clinical decisions during the patient’s lifetime. Moreover, cell lines and organoids allow gain- and loss-of-function genetic manipulations to study genes of interest, which are technically much more challenging to perform in PDXs. However, among all the patient-derived cancer models, PDXs may best recapitulate tumor biology and microenvironment, especially when using orthotopic implantation or humanized mice for engraftment. PDXs also have the advantage over other models in drug response studies, as they allow tumor size measurements and toxicity examinations in animals. Patient-matched PDXs with co-engrafted stromal and immune cells from the tumor donors may provide a better option for clinical decision making in precision medicine.
While there is no ideal model for lung cancer research, study design must consider all the advantages and disadvantages of different models, as well as the compatibility with different experiments, before selecting one to answer relevant biological questions.
Supplementary
The article’s supplementary files as
Acknowledgments
We would like to thank Drs. Nadeem Moghal and Sebastiao N. Martins-Filho for helpful scientific discussion.
Funding: This work is supported by the Canadian Institute of Health Research (CIHR) Foundation grant FDN-148395 (MST). D’Arcangelo is supported by the Princess Margaret Post-doctoral Fellowship.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editor (Helmut H. Popper) for the series “New Developments in Lung Cancer Diagnosis and Pathological Patient Management Strategies” published in Translational Lung Cancer Research. The article was sent for external peer review organized by the Guest Editor and the editorial office.
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tlcr-20-154). The series “New Developments in Lung Cancer Diagnosis and Pathological Patient Management Strategies” was commissioned by the editorial office without any funding or sponsorship. MST reports grants and personal fees from AstraZeneca, personal fees from BMS, grants and personal fees from Bayer, personal fees from Hoffmann La Roche, personal fees from Amgen, personal fees from Pfizer, personal fees from Takeda, outside the submitted work. MST serves as an unpaid editorial board member of Translational Lung Cancer Research from Jul 2014 to Jul 2021. The other authors have no conflicts of interest to declare.
References
- 1.Cancer Genome Atlas Research Network . Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519-25. 10.1038/nature11404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543-50. 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ettinger DS, Wood DE, Aggarwal C, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 1.2020. J Natl Compr Canc Netw 2019;17:1464-72. 10.6004/jnccn.2019.0059 [DOI] [PubMed] [Google Scholar]
- 4.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350:2129-39. 10.1056/NEJMoa040938 [DOI] [PubMed] [Google Scholar]
- 5.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004;304:1497-500. 10.1126/science.1099314 [DOI] [PubMed] [Google Scholar]
- 6.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101:13306-11. 10.1073/pnas.0405220101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vansteenkiste J, Wauters E. Tyrosine kinase inhibition of EGFR: a successful history of targeted therapy for NSCLC since 20 years. Ann Oncol 2018;29:i1-2. 10.1093/annonc/mdx724 [DOI] [PubMed] [Google Scholar]
- 8.Sequist LV, Lynch TJ. EGFR tyrosine kinase inhibitors in lung cancer: an evolving story. Annu Rev Med 2008;59:429-42. 10.1146/annurev.med.59.090506.202405 [DOI] [PubMed] [Google Scholar]
- 9.Chakravarty D, Gao J, Phillips SM, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017. doi: . 10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016;375:1823-33. 10.1056/NEJMoa1606774 [DOI] [PubMed] [Google Scholar]
- 11.Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 2016;387:1540-50. 10.1016/S0140-6736(15)01281-7 [DOI] [PubMed] [Google Scholar]
- 12.Hirano T, Yasuda H, Tani T, et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget 2015;6:38789-803. 10.18632/oncotarget.5887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang JC, Ahn MJ, Kim DW, et al. Osimertinib in Pretreated T790M-Positive Advanced Non-Small-Cell Lung Cancer: AURA Study Phase II Extension Component. J Clin Oncol 2017;35:1288-96. 10.1200/JCO.2016.70.3223 [DOI] [PubMed] [Google Scholar]
- 14.Amann J, Kalyankrishna S, Massion PP, et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res 2005;65:226-35. [PubMed] [Google Scholar]
- 15.Yao Z, Torres NM, Tao A, et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015;28:370-83. 10.1016/j.ccell.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noeparast A, Teugels E, Giron P, et al. Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of Trametinib and Dabrafenib. Oncotarget 2016;8:60094-108. 10.18632/oncotarget.11635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res 2008;14:4275-83. 10.1158/1078-0432.CCR-08-0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furugaki K, Mochizuki M, Kohno M, et al. Expression of C-terminal ALK, RET, or ROS1 in lung cancer cells with or without fusion. BMC Cancer 2019;19:301. 10.1186/s12885-019-5527-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603-7. 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012;483:570-5. 10.1038/nature11005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang W, Soares J, Greninger P, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955-61. 10.1093/nar/gks1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iorio F, Knijnenburg TA, Vis DJ, et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016;166:740-54. 10.1016/j.cell.2016.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basu A, Bodycombe NE, Cheah JH, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013;154:1151-61. 10.1016/j.cell.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klijn C, Durinck S, Stawiski EW, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 2015;33:306-12. 10.1038/nbt.3080 [DOI] [PubMed] [Google Scholar]
- 25.Haverty PM, Lin E, Tan J, et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 2016;533:333-7. 10.1038/nature17987 [DOI] [PubMed] [Google Scholar]
- 26.Chiu YC, Chen HH, Zhang T, et al. Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC Med Genomics 2019;12:18. 10.1186/s12920-018-0460-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suphavilai C, Bertrand D, Nagarajan N. Predicting Cancer Drug Response using a Recommender System. Bioinformatics 2018;34:3907-14. 10.1093/bioinformatics/bty452 [DOI] [PubMed] [Google Scholar]
- 28.Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006;313:1929-35. 10.1126/science.1132939 [DOI] [PubMed] [Google Scholar]
- 29.Subramanian A, Narayan R, Corsello SM, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017;171:1437-52.e17. 10.1016/j.cell.2017.10.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jahchan NS, Dudley JT, Mazur PK, et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov 2013;3:1364-77. 10.1158/2159-8290.CD-13-0183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang KJ, Constanzo JD, Venkateswaran N, et al. Focal Adhesion Kinase Regulates the DNA Damage Response and Its Inhibition Radiosensitizes Mutant KRAS Lung Cancer. Clin Cancer Res 2016;22:5851-63. 10.1158/1078-0432.CCR-15-2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koo T, Yoon AR, Cho HY, et al. Selective disruption of an oncogenic mutant allele by CRISPR/Cas9 induces efficient tumor regression. Nucleic Acids Res 2017;45:7897-908. 10.1093/nar/gkx490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hart T, Chandrashekhar M, Aregger M, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015;163:1515-26. 10.1016/j.cell.2015.11.015 [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Birsoy K, Hughes NW, et al. Identification and characterization of essential genes in the human genome. Science 2015;350:1096-101. 10.1126/science.aac7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyers RM, Bryan JG, McFarland JM, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 2017;49:1779-84. 10.1038/ng.3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilding JL, Bodmer WF. Cancer Cell Lines for Drug Discovery and Development. Cancer Research 2014;74:2377-84. 10.1158/0008-5472.CAN-13-2971 [DOI] [PubMed] [Google Scholar]
- 37.Gazdar AF, Gao B, Minna JD. Lung cancer cell lines: Useless artifacts or invaluable tools for medical science? Lung Cancer (Amsterdam, Netherlands) 2010;68:309-18. 10.1016/j.lungcan.2009.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gazdar AF, Girard L, Lockwood WW, et al. Lung cancer cell lines as tools for biomedical discovery and research. J Natl Cancer Inst 2010;102:1310-21. 10.1093/jnci/djq279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sontheimer-Phelps A, Hassell BA, Ingber DE. Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer 2019;19:65-81. 10.1038/s41568-018-0104-6 [DOI] [PubMed] [Google Scholar]
- 40.Rodenhizer D, Dean T, D'Arcangelo E, et al. The Current Landscape of 3D In Vitro Tumor Models: What Cancer Hallmarks Are Accessible for Drug Discovery? Adv Healthc Mater 2018;7:e1701174. 10.1002/adhm.201701174 [DOI] [PubMed] [Google Scholar]
- 41.Infanger DW, Lynch ME, Fischbach C. Engineered culture models for studies of tumor-microenvironment interactions. Annu Rev Biomed Eng 2013;15:29-53. 10.1146/annurev-bioeng-071811-150028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aref AR, Huang RY, Yu W, et al. Screening therapeutic EMT blocking agents in a three-dimensional microenvironment. Integr Biol (Camb) 2013;5:381-9. 10.1039/C2IB20209C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rekhtman N, Ang DC, Sima CS, et al. Immunohistochemical algorithm for differentiation of lung adenocarcinoma and squamous cell carcinoma based on large series of whole-tissue sections with validation in small specimens. Mod Pathol 2011;24:1348-59. 10.1038/modpathol.2011.92 [DOI] [PubMed] [Google Scholar]
- 44.Inamura K. Update on Immunohistochemistry for the Diagnosis of Lung Cancer. Cancers (Basel) 2018. doi: . 10.3390/cancers10030072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang H, Bai X, Meng F, et al. Comparison of immunohistochemistry and mRNA in situ hybridization in detecting thyroid transcription factor-1 expression in non-small cell lung carcinomas tissue. Oncol Lett 2015;10:3581-4. 10.3892/ol.2015.3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haibe-Kains B, El-Hachem N, Birkbak NJ, et al. Inconsistency in large pharmacogenomic studies. Nature 2013;504:389-93. 10.1038/nature12831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cancer Cell Line Encyclopedia C, Genomics of Drug Sensitivity in Cancer C. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015;528:84-7. 10.1038/nature15736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007;131:1190-203. 10.1016/j.cell.2007.11.025 [DOI] [PubMed] [Google Scholar]
- 49.Wang D, Pham NA, Tong J, et al. Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int J Cancer 2017;140:662-73. 10.1002/ijc.30472 [DOI] [PubMed] [Google Scholar]
- 50.Crystal AS, Shaw AT, Sequist LV, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014;346:1480-6. 10.1126/science.1254721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Ory V, Chapman S, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 2012;180:599-607. 10.1016/j.ajpath.2011.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kodack DP, Farago AF, Dastur A, et al. Primary Patient-Derived Cancer Cells and Their Potential for Personalized Cancer Patient Care. Cell Rep 2017;21:3298-309. 10.1016/j.celrep.2017.11.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tentler JJ, Tan AC, Weekes CD, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012;9:338-50. 10.1038/nrclinonc.2012.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai Y, Wei X, Lin S, et al. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol 2017;10:106. 10.1186/s13045-017-0470-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fichtner I, Rolff J, Soong R, et al. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res 2008;14:6456-68. 10.1158/1078-0432.CCR-08-0138 [DOI] [PubMed] [Google Scholar]
- 56.Drapkin BJ, George J, Christensen CL, et al. Genomic and Functional Fidelity of Small Cell Lung Cancer Patient-Derived Xenografts. Cancer Discov 2018;8:600-15. 10.1158/2159-8290.CD-17-0935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res 2013;73:5315-9. 10.1158/0008-5472.CAN-13-1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moghal N, Pham N, Shi R, et al. MTE01.02 Lung Patient Derived Xenograft and Organoid. J Thorac Oncol 2018;13:S206-7. 10.1016/j.jtho.2018.08.048 [DOI] [Google Scholar]
- 59.Pham NA, Wang D, Tong J, et al. Abstract B32: Fidelity of genomic and proteomic features of patient-derived xenografts of lung cancers. Cancer Clin Res 2016;22:B32. 10.1158/1557-3265.PDX16-B32 [DOI] [Google Scholar]
- 60.Nakajima T, Geddie W, Anayama T, et al. Patient-derived tumor xenograft models established from samples obtained by endobronchial ultrasound-guided transbronchial needle aspiration. Lung Cancer 2015;89:110-4. 10.1016/j.lungcan.2015.05.018 [DOI] [PubMed] [Google Scholar]
- 61.Anderson WC, Boyd MB, Aguilar J, et al. Initiation and characterization of small cell lung cancer patient-derived xenografts from ultrasound-guided transbronchial needle aspirates. PLoS One 2015;10:e0125255. 10.1371/journal.pone.0125255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leong TL, Marini KD, Rossello FJ, et al. Genomic characterisation of small cell lung cancer patient-derived xenografts generated from endobronchial ultrasound-guided transbronchial needle aspiration specimens. PLoS One 2014;9:e106862. 10.1371/journal.pone.0106862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.John T, Kohler D, Pintilie M, et al. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin Cancer Res 2011;17:134-41. 10.1158/1078-0432.CCR-10-2224 [DOI] [PubMed] [Google Scholar]
- 64.Hao C, Wang L, Peng S, et al. Gene mutations in primary tumors and corresponding patient-derived xenografts derived from non-small cell lung cancer. Cancer Lett 2015;357:179-85. 10.1016/j.canlet.2014.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fang DD, Zhang B, Gu Q, et al. HIP1-ALK, a novel ALK fusion variant that responds to crizotinib. J Thorac Oncol 2014;9:285-94. 10.1097/JTO.0000000000000087 [DOI] [PubMed] [Google Scholar]
- 66.Kang HN, Choi JW, Shim HS, et al. Establishment of a platform of non-small-cell lung cancer patient-derived xenografts with clinical and genomic annotation. Lung Cancer 2018;124:168-78. 10.1016/j.lungcan.2018.08.008 [DOI] [PubMed] [Google Scholar]
- 67.Kita K, Fukuda K, Takahashi H, et al. Patient-derived xenograft models of non-small cell lung cancer for evaluating targeted drug sensitivity and resistance. Cancer Sci 2019;110:3215-24. 10.1111/cas.14171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cuenca RE, Takita H, Bankert R. Orthotopic engraftment of human lung tumours in SCID mice for the study of metastasis. Surg Oncol 1996;5:85-91. 10.1016/S0960-7404(96)80005-3 [DOI] [PubMed] [Google Scholar]
- 69.Hodgkinson CL, Morrow CJ, Li Y, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med 2014;20:897-903. 10.1038/nm.3600 [DOI] [PubMed] [Google Scholar]
- 70.Plaks V, Koopman CD, Werb Z. Cancer. Circulating tumor cells. Science 2013;341:1186-8. 10.1126/science.1235226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kapeleris J, Kulasinghe A, Warkiani ME, et al. The Prognostic Role of Circulating Tumor Cells (CTCs) in Lung Cancer. Front Oncol 2018;8:311. 10.3389/fonc.2018.00311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morrow CJ, Trapani F, Metcalf RL, et al. Tumourigenic non-small-cell lung cancer mesenchymal circulating tumour cells: a clinical case study. Ann Oncol 2016;27:1155-60. 10.1093/annonc/mdw122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tellez-Gabriel M, Cochonneau D, Cadé M, et al. Circulating Tumor Cell-Derived Pre-Clinical Models for Personalized Medicine. Cancers 2018;11:19. 10.3390/cancers11010019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Klameth L, Rath B, Hochmaier M, et al. Small cell lung cancer: model of circulating tumor cell tumorospheres in chemoresistance. Sci Rep 2017;7:5337. 10.1038/s41598-017-05562-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morton CL, Houghton PJ. Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc 2007;2:247-50. 10.1038/nprot.2007.25 [DOI] [PubMed] [Google Scholar]
- 76.Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 2005;174:6477-89. 10.4049/jimmunol.174.10.6477 [DOI] [PubMed] [Google Scholar]
- 77.Jung J, Seol HS, Chang S. The Generation and Application of Patient-Derived Xenograft Model for Cancer Research. Cancer Res Treat 2018;50:1-10. 10.4143/crt.2017.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Douglas DN, Kneteman NM. Generation of improved mouse models for the study of hepatitis C virus. Eur J Pharmacol 2015;759:313-25. 10.1016/j.ejphar.2015.03.022 [DOI] [PubMed] [Google Scholar]
- 79.Zhao Y, Shuen TWH, Toh TB, et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018;67:1845-54. 10.1136/gutjnl-2017-315201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pearson T, Greiner DL, Shultz LD. Creation of "humanized" mice to study human immunity. Curr Protoc Immunol 2008;Chapter 15:Unit 15.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brown ME, Zhou Y, McIntosh BE, et al. A Humanized Mouse Model Generated Using Surplus Neonatal Tissue. Stem Cell Reports 2018;10:1175-83. 10.1016/j.stemcr.2018.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rosato RR, Davila-Gonzalez D, Choi DS, et al. Evaluation of anti-PD-1-based therapy against triple-negative breast cancer patient-derived xenograft tumors engrafted in humanized mouse models. Breast Cancer Res 2018;20:108. 10.1186/s13058-018-1037-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morton JJ, Bird G, Keysar SB, et al. XactMice: humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene 2016;35:290-300. 10.1038/onc.2015.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gock M, Kuhn F, Mullins CS, et al. Tumor Take Rate Optimization for Colorectal Carcinoma Patient-Derived Xenograft Models. Biomed Res Int 2016;2016:1715053. 10.1155/2016/1715053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morgan KM, Riedlinger GM, Rosenfeld J, et al. Patient-Derived Xenograft Models of Non-Small Cell Lung Cancer and Their Potential Utility in Personalized Medicine. Front Oncol 2017;7:2. 10.3389/fonc.2017.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X, Fu X, Hoffman RM. A new patient-like metastatic model of human lung cancer constructed orthotopically with intact tissue via thoracotomy in immunodeficient mice. Int J Cancer 1992;51:992-5. 10.1002/ijc.2910510626 [DOI] [PubMed] [Google Scholar]
- 87.Stewart EL, Mascaux C, Pham NA, et al. Clinical Utility of Patient-Derived Xenografts to Determine Biomarkers of Prognosis and Map Resistance Pathways in EGFR-Mutant Lung Adenocarcinoma. J Clin Oncol 2015;33:2472-80. 10.1200/JCO.2014.60.1492 [DOI] [PubMed] [Google Scholar]
- 88.Martin P, Stewart E, Pham NA, et al. Cetuximab Inhibits T790M-Mediated Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor in a Lung Adenocarcinoma Patient-Derived Xenograft Mouse Model. Clin Lung Cancer 2016;17:375-83.e2. 10.1016/j.cllc.2016.01.002 [DOI] [PubMed] [Google Scholar]
- 89.Zhang XC, Zhang J, Li M, et al. Establishment of patient-derived non-small cell lung cancer xenograft models with genetic aberrations within EGFR, KRAS and FGFR1: useful tools for preclinical studies of targeted therapies. J Transl Med 2013;11:168. 10.1186/1479-5876-11-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005;23:5900-9. 10.1200/JCO.2005.02.857 [DOI] [PubMed] [Google Scholar]
- 91.Massarelli E, Varella-Garcia M, Tang X, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res 2007;13:2890-6. 10.1158/1078-0432.CCR-06-3043 [DOI] [PubMed] [Google Scholar]
- 92.Shi R, Li M, Raghavan V, et al. Targeting the CDK4/6-Rb Pathway Enhances Response to PI3K Inhibition in PIK3CA-Mutant Lung Squamous Cell Carcinoma. Clin Cancer Res 2018;24:5990-6000. 10.1158/1078-0432.CCR-18-0717 [DOI] [PubMed] [Google Scholar]
- 93.Makimoto G, Ohashi K, Tomida S, et al. Rapid Acquisition of Alectinib Resistance in ALK-Positive Lung Cancer With High Tumor Mutation Burden. J Thorac Oncol 2019;14:2009-18. 10.1016/j.jtho.2019.07.017 [DOI] [PubMed] [Google Scholar]
- 94.Ambrogio C, Gomez-Lopez G, Falcone M, et al. Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med 2016;22:270-7. 10.1038/nm.4041 [DOI] [PubMed] [Google Scholar]
- 95.Dual Therapy with DDR1 and Notch Inhibitors Kills KRAS-Mutant Lung Tumors . Available online: https://cancerdiscovery.aacrjournals.org/content/6/4/OF10
- 96.Owonikoko TK, Zhang G, Kim HS, et al. Patient-derived xenografts faithfully replicated clinical outcome in a phase II co-clinical trial of arsenic trioxide in relapsed small cell lung cancer. J Transl Med 2016;14:111. 10.1186/s12967-016-0861-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gardner EE, Lok BH, Schneeberger VE, et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017;31:286-99. 10.1016/j.ccell.2017.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aronchik I, Dai Y, Labenski M, et al. Efficacy of a Covalent ERK1/2 Inhibitor, CC-90003, in KRAS-Mutant Cancer Models Reveals Novel Mechanisms of Response and Resistance. Mol Cancer Res 2019;17:642-54. 10.1158/1541-7786.MCR-17-0554 [DOI] [PubMed] [Google Scholar]
- 99.Kim J, McMillan E, Kim HS, et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016;538:114-7. 10.1038/nature19771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu Z, Xia J, Fan W, et al. Human melanoma immunotherapy using tumor antigen-specific T cells generated in humanized mice. Oncotarget 2016;7:6448-59. 10.18632/oncotarget.7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zheng B, Ren T, Huang Y, et al. PD-1 axis expression in musculoskeletal tumors and antitumor effect of nivolumab in osteosarcoma model of humanized mouse. J Hematol Oncol 2018;11:16. 10.1186/s13045-018-0560-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Meraz IM, Majidi M, Meng F, et al. An improved patient-derived xenograft humanized mouse model for evaluation of lung cancer immune responses. Cancer Immunol Res 2019;7:1267-79. 10.1158/2326-6066.CIR-18-0874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gao H, Korn JM, Ferretti S, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 2015;21:1318-25. 10.1038/nm.3954 [DOI] [PubMed] [Google Scholar]
- 104.DeRose YS, Wang G, Lin YC, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 2011;17:1514-20. 10.1038/nm.2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Julien S, Merino-Trigo A, Lacroix L, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res 2012;18:5314-28. 10.1158/1078-0432.CCR-12-0372 [DOI] [PubMed] [Google Scholar]
- 106.Mattie M, Christensen A, Chang MS, et al. Molecular characterization of patient-derived human pancreatic tumor xenograft models for preclinical and translational development of cancer therapeutics. Neoplasia 2013;15:1138-50. 10.1593/neo.13922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cassidy JW, Caldas C, Bruna A. Maintaining Tumor Heterogeneity in Patient-Derived Tumor Xenografts. Cancer Res 2015;75:2963-8. 10.1158/0008-5472.CAN-15-0727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fang Y, Eglen RM. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov 2017;22:456-72. 10.1177/1087057117696795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang C, Tang Z, Zhao Y, et al. Three-dimensional in vitro cancer models: a short review. Biofabrication 2014;6:022001. 10.1088/1758-5082/6/2/022001 [DOI] [PubMed] [Google Scholar]
- 110.Drost J, Clevers H. Organoids in cancer research. Nature Reviews Cancer 2018;18:407-18. 10.1038/s41568-018-0007-6 [DOI] [PubMed] [Google Scholar]
- 111.Duarte AA, Gogola E, Sachs N, et al. BRCA-deficient mouse mammary tumor organoids to study cancer-drug resistance. Nat Methods 2018;15:134-40. 10.1038/nmeth.4535 [DOI] [PubMed] [Google Scholar]
- 112.Yang H, Sun L, Liu M, et al. Patient-derived organoids: a promising model for personalized cancer treatment. Gastroenterol Rep (Oxf) 2018;6:243-5. 10.1093/gastro/goy040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shi R, Radulovich N, Ng C, et al. Organoid cultures as preclinical models of non-small cell lung cancer. Clin Cancer Res 2020;26:1162-74. 10.1158/1078-0432.CCR-19-1376 [DOI] [PubMed] [Google Scholar]
- 114.Barkauskas CE, Chung MI, Fioret B, et al. Lung organoids: current uses and future promise. Development 2017;144:986-97. 10.1242/dev.140103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dutta D, Heo I, Clevers H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol Med 2017;23:393-410. 10.1016/j.molmed.2017.02.007 [DOI] [PubMed] [Google Scholar]
- 116.Neal JT, Li X, Zhu J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018;175:1972-88.e16. 10.1016/j.cell.2018.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tamura H, Higa A, Hoshi H, et al. Evaluation of anticancer agents using patient-derived tumor organoids characteristically similar to source tissues. Oncol Rep 2018;40:635-46. 10.3892/or.2018.6501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, et al. Long-term expanding human airway organoids for disease modeling. EMBO J 2019. doi: . 10.15252/embj.2018100300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim M, Mun H, Sung CO, et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat Commun 2019;10:3991. 10.1038/s41467-019-11867-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brewer GJ, Torricelli JR, Evege EK, et al. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 1993;35:567-76. 10.1002/jnr.490350513 [DOI] [PubMed] [Google Scholar]
- 121.Praharaj PP, Bhutia SK, Nagrath S, et al. Circulating tumor cell-derived organoids: Current challenges and promises in medical research and precision medicine. Biochim Biophys Acta Rev Cancer 2018;1869:117-27. 10.1016/j.bbcan.2017.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dijkstra KK, Cattaneo CM, Weeber F, et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018;174:1586-98.e12. 10.1016/j.cell.2018.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weeber F, Ooft SN, Dijkstra KK, et al. Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem Biol 2017;24:1092-100. 10.1016/j.chembiol.2017.06.012 [DOI] [PubMed] [Google Scholar]
- 124.Takahashi N, Hoshi H, Higa A, et al. An In Vitro System for Evaluating Molecular Targeted Drugs Using Lung Patient-Derived Tumor Organoids. Cells 2019. doi: . 10.3390/cells8050481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jung DJ, Shin TH, Kim M, et al. A one-stop microfluidic-based lung cancer organoid culture platform for testing drug sensitivity. Lab Chip 2019;19:2854-65. 10.1039/C9LC00496C [DOI] [PubMed] [Google Scholar]
- 126.Aboulkheyr Es H, Montazeri L, Aref AR, et al. Personalized Cancer Medicine: An Organoid Approach. Trends Biotechnol 2018;36:358-71. 10.1016/j.tibtech.2017.12.005 [DOI] [PubMed] [Google Scholar]
- 127.Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543-50. 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lancaster MA, Huch M. Disease modelling in human organoids. Dis Model Mech 2019. doi: . 10.1242/dmm.039347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chae YK, Ranganath K, Hammerman PS, et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget 2017;8:16052-74. 10.18632/oncotarget.14109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bockorny B, Rusan M, Chen W, et al. RAS-MAPK Reactivation Facilitates Acquired Resistance in FGFR1-Amplified Lung Cancer and Underlies a Rationale for Upfront FGFR-MEK Blockade. Mol Cancer Ther 2018;17:1526-39. 10.1158/1535-7163.MCT-17-0464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Langhans SA. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front Pharmacol 2018;9:6. 10.3389/fphar.2018.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.van de Wetering M, Francies HE, Francis JM, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015;161:933-45. 10.1016/j.cell.2015.03.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Phan N, Hong JJ, Tofig B, et al. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun Biol 2019;2:78. 10.1038/s42003-019-0305-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pauli C, Moch H, Rubin MA. Establishment of a living biobank: Improved guidance of precision cancer care with in vitro and in vivo cancer models. Pathologe 2017;38:160-8. 10.1007/s00292-017-0346-1 [DOI] [PubMed] [Google Scholar]
- 135.Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell 2016;164:1233-47. 10.1016/j.cell.2016.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Öhlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579-96. 10.1084/jem.20162024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Seino T, Kawasaki S, Shimokawa M, et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018;22:454-67.e6. 10.1016/j.stem.2017.12.009 [DOI] [PubMed] [Google Scholar]
- 138.Tsai S, McOlash L, Palen K, et al. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018;18:335. 10.1186/s12885-018-4238-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kunicki TJ, Nurden AT, Pidard D, et al. Characterization of human platelet glycoprotein antigens giving rise to individual immunoprecipitates in crossed-immunoelectrophoresis. Blood 1981;58:1190-7. 10.1182/blood.V58.6.1190.1190 [DOI] [PubMed] [Google Scholar]
- 140.Bremnes RM, Donnem T, Al-Saad S, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol 2011;6:209-17. 10.1097/JTO.0b013e3181f8a1bd [DOI] [PubMed] [Google Scholar]
- 141.Nakamura H, Sugano M, Miyashita T, et al. Organoid culture containing cancer cells and stromal cells reveals that podoplanin-positive cancer-associated fibroblasts enhance proliferation of lung cancer cells. Lung Cancer 2019;134:100-7. 10.1016/j.lungcan.2019.04.007 [DOI] [PubMed] [Google Scholar]
- 142.Saglam-Metiner P, Gulce-Iz S, Biray-Avci C. Bioengineering-inspired three-dimensional culture systems: Organoids to create tumor microenvironment. Gene 2019;686:203-12. 10.1016/j.gene.2018.11.058 [DOI] [PubMed] [Google Scholar]
- 143.Takebe T, Zhang B, Radisic M. Synergistic Engineering: Organoids Meet Organs-on-a-Chip. Cell Stem Cell 2017;21:297-300. 10.1016/j.stem.2017.08.016 [DOI] [PubMed] [Google Scholar]
- 144.Skardal A, Shupe T, Atala A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov Today 2016;21:1399-411. 10.1016/j.drudis.2016.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Grebenyuk S, Ranga A. Engineering Organoid Vascularization. Front Bioeng Biotechnol 2019;7:39. 10.3389/fbioe.2019.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Khoo BL, Grenci G, Lim YB, et al. Expansion of patient-derived circulating tumor cells from liquid biopsies using a CTC microfluidic culture device. Nat Protoc 2018;13:34-58. 10.1038/nprot.2017.125 [DOI] [PubMed] [Google Scholar]
- 147.Xu H, Lyu X, Yi M, et al. Organoid technology and applications in cancer research. J Hematol Oncol 2018;11:116. 10.1186/s13045-018-0662-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Akkerman N, Defize LH. Dawn of the organoid era: 3D tissue and organ cultures revolutionize the study of development, disease, and regeneration. Bioessays 2017. doi: . 10.1002/bies.201600244 [DOI] [PubMed] [Google Scholar]
- 149.Huch M, Knoblich JA, Lutolf MP, et al. The hope and the hype of organoid research. Development 2017;144:938-41. 10.1242/dev.150201 [DOI] [PubMed] [Google Scholar]
- 150.Kaushik G, Ponnusamy MP, Batra SK. Concise Review: Current Status of Three-Dimensional Organoids as Preclinical Models. Stem Cells 2018;36:1329-40. 10.1002/stem.2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as