

Abstract
Niemann‐Pick disease type C (NPC) is a neurodegenerative lysosomal storage disorder caused by mutations in either NPC1 (95% of cases) or NPC2. Reduced late endosome/lysosome calcium (Ca2+) levels and the accumulation of unesterified cholesterol and sphingolipids within the late endocytic system characterize this disease. We previously reported impaired lysosome‐related organelle (LRO) function in Npc1 −/− Natural Killer cells; however, the potential contribution of impaired acid compartment Ca2+ flux and LRO function in other cell types has not been determined. Here, we investigated LRO function in NPC1 disease platelets. We found elevated numbers of circulating platelets, impaired platelet aggregation and prolonged bleeding times in a murine model of NPC1 disease. Electron microscopy revealed abnormal ultrastructure in murine platelets, consistent with that seen in a U18666A (pharmacological inhibitor of NPC1) treated megakaryocyte cell line (MEG‐01) exhibiting lipid storage and acidic compartment Ca2+ flux defects. Furthermore, platelets from NPC1 patients across different ages were found to cluster at the lower end of the normal range when platelet numbers were measured and had platelet volumes that were clustered at the top of the normal range. Taken together, these findings highlight the role of acid compartment Ca2+ flux in the function of platelet LROs.
Keywords: calcium (Ca2+), lysosome, lysosome‐related organelle, Niemann‐Pick disease type C
Synopsis.
Defects in lysosome‐related organelles, specifically in platelets, are observed in the mouse model and patients with Niemann‐Pick disease type C1.
1. MATERIALS AND METHODS
1.1. Patient samples
NPC1 patients were enrolled in a longitudinal observational study (NCT00344331) at the National Institutes of Health (Bethesda, Maryland). Participants data was excluded if on an anticoagulant or post splenectomy. The NICHD Institutional Review Board approved this study; normal ranges are from the NIH Clinical Center Department of Laboratory Medicine reference ranges. Informed consent was obtained for all subjects as well as assent, when appropriate. The diagnosis was established by biochemical testing/mutation analysis.
1.2. Animals
Niemann‐Pick disease type C1 mice (BALB/cNctr‐Npc1 m1N/J, Npc1 −/− mice) are null for Npc1 and were from an established colony. All mice were bred under sterile conditions, with food and water available ad lib. Pups were weaned 3 weeks post‐partum. All animal studies were conducted using protocols approved by the UK Home Office for the conduct of regulated procedures under license (Animal scientific Procedures Act, 1986). Npc1 −/− mice have a lifespan of 10 to 12 weeks (average 10.5 weeks) with neurological symptoms presenting from 5 to 6 weeks of age.
1.3. Mice hematological analysis
Blood samples were obtained by cardiac puncture using an EDTA‐rinsed needle/syringe and collected into EDTA‐rinsed tubes. Multiple hematological parameters were determined using an automated blood analyzer (ABX Pentro 60 system, HORIBA‐ABX, UK).
1.4. Isolation of murine platelets
Platelets were harvested at indicated time points of Npc1 −/− and age‐matched control littermates. For preparation of washed platelets, mice were anaesthetized and blood was collected by cardiac puncture. A total of 500 μL of blood per mouse was collected in a tube containing 100 μL acid citrate‐dextrose solution (Sigma, UK) and mixed with 3 mL 100 mM EGTA (pH 6.8) in a modified Tyrode's Ca2+ free buffer (134 mM NaCl, 3 mM KCl, 5 mM HEPES, 5 mM glucose, 2 mM MgCl2, pH 7.4) supplemented with NaHCO3 and BSA. Platelet‐rich plasma (PRP) was obtained by centrifugation at 180g for 10 minutes at room temperature. For preparation of washed platelets, PRP was washed at 1000 g for 8 minutes at room temperature, and the pellet was resuspended in the modified Tyrodes buffer containing prostaglandin E1 (0.25 μmol/L). Isolated mouse platelets were washed twice and either fixed for EM analysis or incubated at 37°C for 30 minutes before use.
1.5. Bleeding time assay
Mice were anaesthetized with isoflurane and placed onto a heated mat (37°C). The tail bleeding time was determined by removing 3 mm of the distal tail tip and immersing the tail into sterile DPBS solution (37°C). The time to cessation of bleeding was measured. 1
1.6. Platelet aggregation
Platelet‐rich plasma (PRP) was prepared by centrifugation at 250g for 10 minutes at room temperature. The platelet count was adjusted with autologous plasma. Aggregation from PRP platelets was monitored by assessing light transmission in a microplate reader (BMG Labtech, UK) with continuous stirring at 1100 rpm at 37°C.
1.7. Cell culture
The human megakaryoblastic leukemia cell line MEG‐01 was obtained from the ATCC and cultured in RPMI‐1640 supplemented with 10% (vol/vol) fetal bovine serum, 1% penicillin/ streptomycin, and 0.3 μg/mL glutamine in a humidified atmosphere at 37°C and 5% CO2. 2
1.8. Electron microscopy
DMSO or U18666A treated MEG‐01 cells were prepared for electron microscopy as previously described. 2 Murine isolated platelets were fixed in 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate for 1 hour. The cells were pelleted at 1000 g for 10 minutes and washed three times in 0.1 M sodium cacodylate prior to incubation with 1% osmium tetroxide/1.5% potassium ferricyanide on ice for 1 hour. Cell pellets were washed three times in 0.1 M sodium cacodylate, stained with 1% tannic acid for 45 minutes, dehydrated in increasing concentration of ethanol and embedded in TAAB‐812 resin. Ultrathin sections stained with lead citrate were viewed on a JOEL 1400+ transmission electron microscope (JOEL, Tokyo, Japan) equipped with a Gatan Orius SC1000B charge‐coupled device camera.
1.9. Lysotracker flow cytometry analysis
Washed murine platelets were diluted to 2.5 × 107 platelets/mL in modified Tyrode's buffer and DMSO or U18666A treated MEG‐01 cells were resuspended in HBSS solution prior to incubation with LysoTracker Green DND‐26 for 15 minutes at room temperature. 3 Cells were then washed with HBSS solution and flow cytometer analysis was immediately carried out.
1.10. Intracellular Ca2+ measurements
MEG‐01 cells were treated either with DMSO or 2 μM U18666A for 72 hours and then allowed to adhere to poly‐l‐lysine‐coated glass coverslips and loaded with 5 μM fura‐2/AM in the presence of 0.03% Pluronic F127 in extracellular midum (ECM, mM: 121 NaCl, 5.4 KCl, 0.8 MgCl2, 1.8 CaCl2, 6 NaHCO3, 25 HEPES, 10 Glucose) for 45 minutes at room temperature, followed by a 15 minutes de‐esterification. Experiments were conducted in Ca2+‐free medium: cells were washed once with Ca2+‐free ECM supplemented with 1 mM EGTA, followed by two washes in Ca2+‐free ECM containing 100 μM EGTA. Cells were imaged using an Olympus IX71 microscope equipped with a 40× UApo/340 objective and excited alternately by 350 nm and 380 nm light using a Cairn monochromator; emission was collected at 480 to 540 nm. Experiments were conducted at room temperature with an image collected every 3 seconds. Autoflourescence was determined at the end of each experiment by addition of 1 μM ionomycin with 4 mM MnCl2 to quench fura‐2. Images were analyzed using custom‐written Magipix software (Ron Jacob, King's College London, London, UK) on a single‐cell basis and the data expressed as the mean ± SEM of the maximum peak fluorescence changes (∆350/380).
1.11. GSL extraction, ceramide glycanase digestion, and 2AA labeling
Glycosphingolipid (GSL) extraction, ceramide glycanase digestion, and 2‐aminobenzoic acid (2‐AA) labeling were performed as described. 4
1.12. Normal‐phase HPLC analysis for glycosphingolipids profile on MEG‐01 cells
Normal‐phase HPLC analysis for glycosphingolipids profiles was performed as described. 4
1.13. Statistics
Data were expressed as the mean ± SEM. For group comparisons, the statistical significance of differences in mean values was determined by a two‐tailed single‐factor ANOVA, Student's t test or multiple t test using GraphPad Prism 5 for Mac OS X (version 5.0d). P ≤ .05 was considered significant.
2. INTRODUCTION
The lysosome is a regulated Ca2+ store and releases Ca2+ in response to the potent second‐messenger nicotinic acid adenine dinucleotide phosphate (NAADP). 5 , 6 The discovery of acidic Ca2+ store was made in specialized lysosome‐related organelles (LROs) found in sea urchin oocytes (yolk platelets/reserve granules). 6 The finding that sea urchin oocyte LROs were regulated Ca2+ stores was subsequently replicated in conventional lysosomes, giving rise to the field of acidic store Ca2+ signaling. 7 The biological roles of acidic store Ca2+ remain incompletely understood, but there is growing evidence that they are important in the homeostasis of the late endocytic system and in signaling cross‐talk with the endoplasmic reticulum (ER) at IP3 receptors. 8 , 9 LROs and secretary lysosomes are tissue/cell type specific organelles that share some common features with conventional lysosomes, including acidic pH. 10 However, they exhibit unique morphological and functional properties, enabling them to mediate specialized functions that are distinct from conventional lysosomes. They play a particularly important role within multiple cell types of the hematopoietic system.
One approach to better understand the physiological functions of acidic store Ca2+ is to study inborn errors of metabolism in which this store is compromised. It has previously been identified that there is a lysosomal Ca2+ defect in the rare neurodegenerative lysosomal storage disease, Niemann‐Pick disease type C (NPC). 11 NPC is caused by inherited defects in either NPC1 or NPC2. 12 We found reduced levels of Ca2+ in LE/Lys of NPC disease cells, likely the result of a Ca2+ store‐filling defect or enhanced Ca2+ leak. 6 This results in insufficient Ca2+ release from late endosomes/lysosomes in response to the second messenger NAADP, blocking fusion, and leading to the secondary storage of multiple lipids in late endosomes (LE)/Lys, including cholesterol, sphingosine, sphingomyelin and multiple glycosphingolipids. 11 However, the role of Ca2+ in LRO function and in NPC remains incompletely understood.
We previously reported that LRO function in Natural Killer (NK) cells was compromised in a mouse model of NPC disease, reducing the ability of these cells to kill target cells. 13 Whether this is unique to NK cell biology or indicative of a broader defect in LRO function in NPC remains unknown. In this study, we have therefore investigated whether the acidic store Ca2+ defect found in NPC disease also affects LRO function more generally focusing on platelets, as they have both LROs and conventional lysosomes. Defects in platelets, including reduced aggregation responses to collagen, absent secondary response to epinephrine, and mean platelet volumes at the lower normal range limits, were previously observed in three NPC patients. 14 These platelet abnormalities were further characterized in a zebrafish model of NPC disease, 14 however it remains unclear if cholesterol storage or the calcium imbalance played a key role in contributing to these phenotypes. In this study, we detail functional deficits in platelets from NPC1 null mice and human data, supporting the hypothesis that platelet regulation is affected in NPC patients as a result of the acidic store Ca2+ defect.
3. RESULTS
3.1. Increased numbers of platelets (PLT), plateletcrit (PCT), and impaired thrombin‐stimulated platelet aggregation in Npc1−/− mice
To measure hematological parameters, whole blood was collected from presymptomatic (5 weeks), early symptomatic (7 week), and late stage (9 week) Npc1 −/− mice. There was a significant increase in the circulating platelet count (PLT) (Figure 1A) and plateletcrit (percentage volume of platelets in the blood, PCT) in Npc1 −/− mice (Figure 1B). The number of platelets in the Npc1 −/− mice was significantly elevated from 5 weeks of age (Figure 1A, PLT: 5 weeks, P = .0098; 7 week, P = .0026; 9 weeks, P = .0086), as was the plateletcrit compared to wild type controls (Figure 1B PCT: 5 weeks, P = .0127; 7 weeks, P = .0009; 9 weeks, P = .004). However, there was no significant change in mean platelet volume (MPV) in Npc1 −/− platelets compared with control (Npc1 +/+) platelets at any age (data not shown). There was a significant decrease in Npc1 −/− platelet aggregation by 40.7% in response to thrombin stimulation (1 U/mL) compared with control (Npc1 +/+) mouse platelets (Figure 1C,D; P = .0029). However, there were no significant differences in Npc1 −/− platelet aggregation in response to the Ca2+ ionophore A23187 compared with controls (Figure 1E,F; P = .2714).
FIGURE 1.
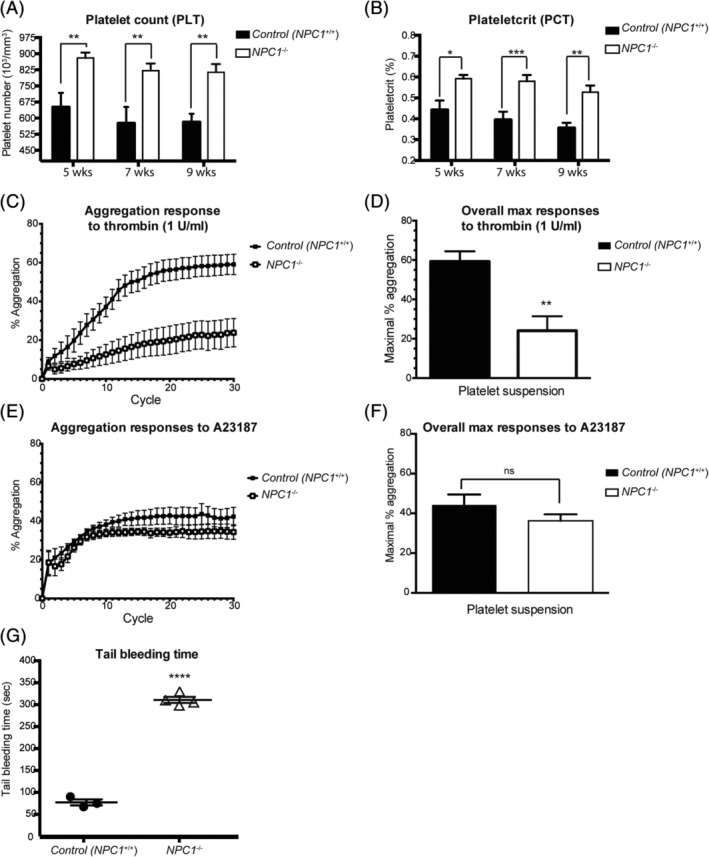
Increased platelet number (PLT), plateletcrit (PCT), impaired thrombin‐stimulated platelet aggregation and increased tail bleeding time in Npc1 −/− mice. A,B, Platelet‐related parameters, including platelet number (PLT) and plateletcrit (PCT), were determined using an automatic blood analyzer. Data shown are mean ± SEM, n = 5‐7, per group. *P < .05, **P < .005, ***P < .0005, calculated by an unpaired t test using GraphPad Prism v5. C,D, Isolated platelets from 10‐week‐old control (Npc1 +/+) and Npc1 −/− mice were stimulated with thrombin (1 U/mL). C, The time‐course of the aggregation response. D, Average maximal Npc1 −/− mouse platelet aggregation was at 40.7% of the average maximal response relative to control mouse platelets (n = 6, per group). Data presented as mean ± SEM, P < .05. E,F, Isolated platelets from 10‐week‐old control (Npc1 +/+) and Npc1 −/− mice were stimulated with the Ca2+ ionophore A23187 (60 μM) in the presence of CaCl2 (1 mM). E, The time‐course of the aggregation response. F, The maximal aggregation responses compared to the average platelet aggregation in response to A23187 (60 μM) was 44.2% for control mouse platelets and 36.2% for Npc1 −/− mouse platelets, respectively. This difference was not statistically significant, n = 4‐5, per group. Data presented as mean ± SEM. G, Tail bleeding assays were performed as described on 10.5‐week‐old control (Npc1 +/+) and Npc1 −/− mice. The time from incision to cessation of bleeding was recorded. Data represent mean ± SEM, n = 3‐4, per group
3.2. Increased bleeding time in Npc1−/− mice
As Npc1 −/− mice have an abnormal circulating platelet count and impaired thrombin induced‐platelet aggregation, we next investigated whether this would lead to bleeding abnormalities. A tail‐bleeding assay was performed on anaesthetized 10.5‐week‐old Npc1 −/− mice (late‐stage disease) and we found a marked hemostatic defect in Npc1 −/− mice (Figure 1G). Whereas bleeding spontaneously stopped after approximately 2 minutes in control age‐matched (Npc1 +/+) mice, tail‐bleeding time was markedly increased in 10.5‐week‐old Npc1 −/− mice (Figure 1G, P < .0001).
3.3. Abnormal ultrastructure in Npc1−/− platelets
To determine whether there were any ultrastructural changes in Npc1 −/− platelets we analyzed them by transmission electron microscopy. Compared to control wild type (Npc1 +/+) platelets (Figure 2A), numerous abnormal and heterogeneous electron‐dense structures resembling lysosomal storage bodies as well as increased cytosolic vacuoles were observed in the Npc1 −/− platelets (Figure 2B, alpha granules (A) and cytosolic vacuoles (C) indicated in inset). In general, the electron density of the Npc1 −/− platelets was also higher than in control platelets consistent with lipid storage. In line with the EM images, the volume of the isolated platelet acidic organelles was measured using LysoTracker. There was a significant increase in acidic organelle volume in the Npc1 −/− platelets as compared to the wild type Npc1 +/+ platelets (P = .0025, Figure 2C).
FIGURE 2.
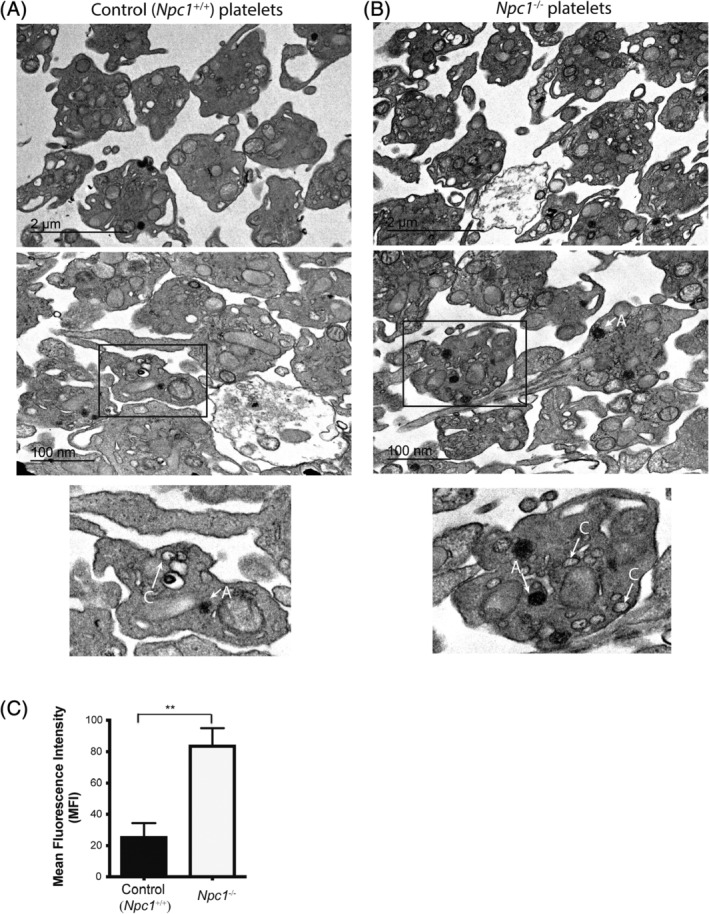
Abnormal platelet lysosomal morphology in the Npc1 −/− mice. Representative images of transmission electron microscopy analysis from, A, control (Npc1 +/+) and B, Npc1 −/− murine platelets. Arrows indicate alpha granules (A) and cytoplasmic vacuoles (C). C, Significant enlargement of the lysosomal/acidic compartment volume using lysotracker staining in Npc1 −/− murine platelets was observed compared to control Npc1 +/+ platelets. Data were quantified by flow cytometry from three independent experiments. *P < .05. Data are presented as mean ± SEM, n = 3, calculated by student t test and plotted using GraphPad Prism v5
3.4. Increased LysoTracker fluorescence, presence of storage bodies, and elevated GSLs in U18666A‐treated human megakaryoblastic MEG‐01 cell line
To further investigate the underlying mechanisms of Npc1 −/− platelet dysfunction, we used the pharmacological agent U18666A (2 μM) to induce NPC1 disease‐associated cellular phenotypes in the human megakaryocyte line MEG‐01, as these cells are responsible for the production of platelets. U18666A is an inhibitor of NPC1 and has recently been shown to directly bind to the NPC1 protein. 15 The volume of acidic organelles in MEG‐01 cells were measured and there was a significant increase in LysoTracker‐Green signal intensity in U18666A‐treated MEG‐01 cells compared with vehicle‐treated (DMSO) cells (P < .0001) (Figure 3A). Transmission electron microscopy analysis revealed the presence of numerous enlarged electron dense storage bodies in U18666A‐treated MEG‐01 cells (Figure 3B) suggesting that U18666A‐treated MEG‐01 cells could act as a model for studying phenotypes relevant to mouse platelets. Furthermore, U18666A or vehicle‐treated MEG‐01 cellular glycosphingolipid (GSL) levels were measured by HPLC analysis and we found significantly elevated levels of Gb3 and Gb4 in U18666A‐treated MEG‐01 cells, compared with vehicle treated cells (Figure 3C,D, Gb3 P = .0074, Gb4 P = .0004).
FIGURE 3.
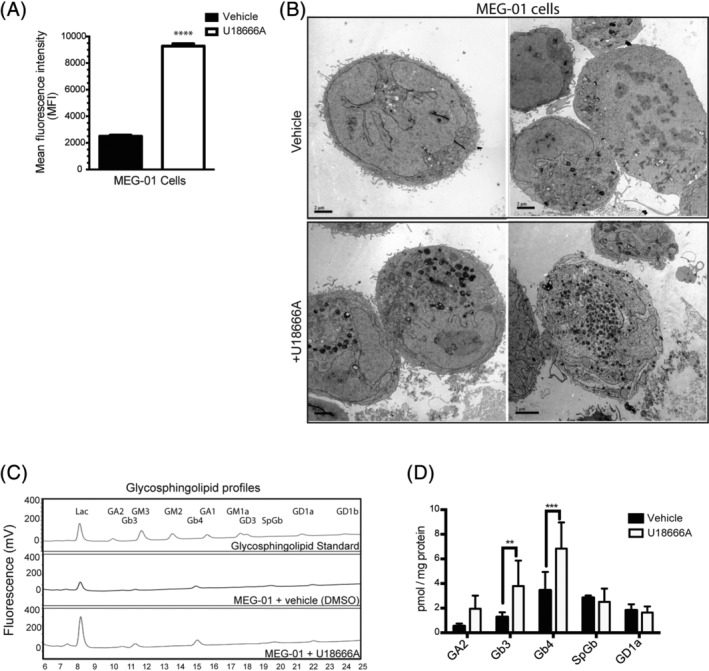
Enlarged acidic compartments are present and accumulates GSLs in U18666A‐treated MEG‐01 cell line. A, Quantitative data of flow cytometric analysis of the numbers/volume changes of acidic compartments in vehicle (DMSO) or U18666A‐treated MEG‐01 cells using lysotracker staining. Data were quantified from three independent experiments. ****P < .00005. Data are presented as mean ± SEM, n = 3, calculated by student t test and plotted using GraphPad Prism v5. B, Representative images of transmission electron microscopy analysis from vehicle (DMSO) and U18666A‐treated human megakaryoblastic MEG‐01 cell lines. Scale bar = 2 μm. C, HPLC analysis of GSLs compositions of DMSO, U18666A treated MEG‐01 cells. Profiles are representative of three independent analyses. D, Bar graph comparing GSLs contents in DMSO, U18666A treated MEG‐01 cells. MEG‐01 cells were treated either with DMSO or U18666A for 72 hours as described in Section 1. Data shown are mean ± SEM, n = 3, per group. *P ≤ .05, **P ≤ .01, ***P ≤ .001, calculated by one‐way ANOVA with multiple comparisons using GraphPad Prism v5
3.5. Defects in acidic compartment Ca2+ flux in U18666A‐treated MEG‐01 cell line
To assess the effect of U18666A treatment on lysosomal Ca2+, we monitored Ca2+ content in MEG‐01 cells indirectly by releasing Ca2+ from the lysosome lumen to the cytosol with the lysomotropic agent glycyl‐l‐phenylalamine 2‐napthylamide (GPN). 16 All experiments were performed in Ca2+‐free medium to eliminate Ca2+ influx. We have previously shown that GPN responses faithfully reflect lysosomal Ca2+ levels in NPC1 patients‐derived human fibroblasts and U18666A‐treated RAW cells. 11 In agreement with known NPC cellular phenotypes, 11 , 13 U18666A‐treated MEG‐01 cells exhibited a significant decrease in GPN‐stimulated Ca2+ release compared to vehicle (DMSO)‐treated cells (Figure 4A,B, P < .0001), consistent with less Ca2+ within the lysosomes of U18666A‐treated MEG‐01 cells. The major effect of U18666A on the GPN response is on the maximum amplitude of the Ca2+ response (48% reduction), whilst there is only a small (13%) difference in the kinetics (time from GPN addition to the maximum amplitude) (Figure 4C, P = .0014).
FIGURE 4.
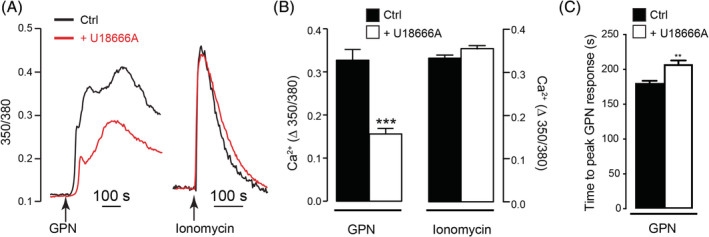
Defects in acidic compartment Ca2+ flux in U18666A‐treated MEG‐01 cell line. A, Representative single‐cell Ca2+ traces showing 350/380 ratios of fura‐2 fluorescence and B, maximum Ca2+ changes upon addition of 200 μM GPN or 1 μM ionomycin in MEG‐01 cells with or without 72 hours pretreatment with 2 μM U18666A. DMSO was used as a vehicle control and no Ca2+ release was observed (not shown). C, Time‐to‐maximum response upon GPN addition following DMSO (Ctrl) or U18666A pretreatment in MEG‐01 cells. n = 383 (GPN Ctrl), 450 (GPN U18666A), 285 (Iono Ctrl), 279 (Iono U18666A) cells. Data are represented as mean ± SEM and statistical significance was determined using an unpaired 2‐tailed t test. **P < .01. ****P < .0001, ns P > .05
Because GPN responses can be a summation of Ca2+ release from both the lysosome and the endoplasmic reticulum (ER), 16 the reduced GPN response could also reflect a lower ER Ca2+ content. To discount this explanation, the ER content was assessed with the Ca2+ ionophore, ionomycin. Ionomycin was used rather than the SERCA inhibitor, thapsigargin, because it is a more reliable assessment of the ER content: thapsigargin releases ER Ca2+ so slowly that other Ca2+ removal processes dampen both its kinetics and amplitude, 17 , 18 , 19 , 20 and whether NPC alters these removal processes is unclear. By contrast, ionomycin releases ER Ca2+ with such rapid kinetics that it is less susceptible to Ca2+ buffering processes. In contrast to GPN‐dependent Ca2+ release, ionomycin‐dependent Ca2+ release from the ER was unaffected by U18666A treatment (Figure 4A,B, P = .1643). Together, these data are consistent with U18666A‐treatment reducing the lysosome (but not ER) Ca2+ content in MEG‐01 cells.
3.6. NPC1 patient platelet counts and volumes cluster at the extremes of the normal ranges
Platelet counts were measured in NPC1 patients. As illustrated in Figure 5A, NPC1 patients had mean platelet count values of approximately 174 (k/μL) clustered at the bottom of the normal range (normal range: 150‐450 k/μL). Additionally, in line with previous studies, 21 the platelet counts decrease in age with the patients >18 years have significantly less platelets than the 0.5 to 2 year old patients (P = .0345). Platelet counts in the NPC1 patients also significantly correlate with severity score, 22 with more severe patients having lower platelet counts then less severe patients (Pearson's correlation coefficient r = −.2361, P = .0111, Figure 5B). When platelet volumes were measured the mean platelet volumes were towards the upper limit (mean value: 10.48 fL) of the normal range (normal range: 7.5‐11.5 fL) (Figure 5C).
FIGURE 5.
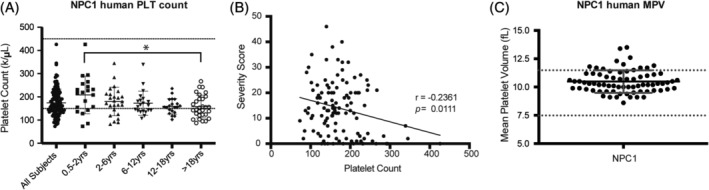
NPC1 patients present at the extremes of normal hematological ranges. A, Platelet count (PLT) was determined using an automated blood analyzer. Dashed lines indicate the estimated normal range (150‐450 k/μL). Patients over 18 years of age had significantly reduced platelet counts as compared to patients age 0.5 to 2 years. n = 116. B, NPC1 patient severity and platelet counts negatively correlate as calculated by Pearson r‐coefficient. Severity scores calculated by scoring nine major domains (ambulation, cognition, eye movement, fine motor, hearing, memory, seizures, speech, and swallowing) and eight minor domains (auditory brainstem response, behavior, gelastic cataplexy, hyperreflexia, incontinence, narcolepsy, psychiatric, and respiratory problems). 22 C, NPC1 patient mean platelet volume (MPV) was determined using an automated blood analyzer. n = 73, dashed lines indicate normal range: 7.5‐11.5. Data shown are mean ± SEM, statistical significance calculated by a one‐way ANOVA, Tukey's multiple comparisons test using GraphPad Prism v5. *P < .05
4. DISCUSSION
We have found an elevated number of circulating platelets, defective platelet aggregation, abnormal platelet morphology and prolonged bleeding times in Npc1 −/− disease mice. Electron microscopy revealed abnormal ultrastructure consistent with lipid storage in Npc1 −/− platelets and in the U18666A‐treated MEG‐01 cell line. Furthermore, U18666A‐treated MEG‐01 cells had reduced levels of acidic store Ca2+ and glycosphingolipid storage. Taken together, these data suggested that megakaryocytes/platelets have functional defects in NPC disease.
We found that Npc1 −/− platelets exhibit functional defects specifically when simulated with thrombin. It has been suggested that platelet lysosome secretion is only activated by potent agonists, such as thrombin. 23 , 24 On the other hand, when Npc1 −/− platelets were stimulated with A23187, a Ca2+ ionophore selectively transporting Ca2+ across cell membranes into the cytoplasm or releasing the divalent cation from intracellular storage sites, the Npc1 −/− platelets did not exhibit aggregation defects. 25 Thrombin activation of platelets is known to affect IP3‐signaling, resulting in an increased intracellular Ca2+ flux. 26 Therefore, this finding suggests the agonists‐mediated aggregation defects occurs in Npc1 −/− platelets due to lysosomal calcium defects.
As anticipated, 27 platelet count levels towards the lower end of the normal range was observed in our clinical hematological studies in NPC1 disease patients; however, thrombocytosis was observed in the Npc1 −/− mice. The reason for this difference is unknown and could be a species‐specific difference or reflect the fact that the mouse model is completely null for Npc1 whereas the patients typically have some residual NPC1 expression leading to less extreme cellular phenotypes. Another possible explanation is that there could be a platelet clearance defect and altered physiological distribution of platelets in NPC1 disease, since enlargement and fibrosis of the spleen occurs in NPC1 patients and Npc1 −/− mice. 28 Platelets may be trapped in the spleen and affect circulating platelets numbers. Therefore, in vivo platelet clearance assays could be helpful to clarify these species‐specific platelet dysregulation phenotypes.
However, we still cannot exclude a potential platelet production defect due to impaired megakaryocyte function in NPC1, and in fact it has been suggested that NPC1 plays a role in the regulation of thrombocyte formation in zebrafish. 14 Sphingosine‐1‐phosphate (S1P) and its associated receptors, especially S1P1 and S1P4 receptors, have also been suggested to be involved in the regulation of megakaryopoiesis, pro‐platelet formation and shedding in vivo and in vitro models. 29 Furthermore, S1P4 receptor‐null megakaryocytes exhibited abnormal cellular morphology, which was characterized by cytoplasmic vacuolation and nuclear‐ploidy. 30 Interestingly, abnormal platelet formation and megakaryopoiesis defects have been reported in a few clinical cases of NPC1 and it has been suggested that this could be associated with cholesterol storage in NPC1 disease. 14 Since the platelet lipid compositions and granule contents are mainly determined during megakaryopoiesis and platelet shedding, the abnormal Npc1 −/− platelet phenotypes could also be associated with abnormal intracellular lipid trafficking and storage phenotypes during megakaryopoiesis which could impair down‐stream lipid‐mediated intracellular signaling. 31
Npc1 −/− platelets functional defects could also primarily be associated with acidic compartment Ca2+ signaling defects. Although the physiological functions and activation mechanisms of platelet LROs remain incompletely understood, our current studies highlight the importance of acidic store Ca2+ signaling in the regulation of platelet function. NAADP is a novel Ca2+‐mobilizing second messenger, which selectively targets acidic organelles. 32 Thrombin and collagen‐related‐peptides stimulated human platelet aggregation and activation, including fibrinogen binding and granule release, is highly dependent on NAADP‐mediated Ca2+ signaling, which suggest a crucial role of NAADP and acidic store Ca2+ release during platelet activation. 33 , 34 Furthermore, NPC1 patients were shown to have reduced platelet aggregation responses to low concentration of collagen and an absent secondary response to epinephrine, 14 consistent with the agonist mediated platelet activation defects in the NPC1 mouse model. Therefore, our hypothesis is that Npc1 −/− platelet functional defects are primarily associated with impaired NAADP‐mediated acidic store Ca2+ signaling.
Furthermore, similar observations of abnormal platelet granule morphology and platelet functional defects have been reported in other lysosomes/LROs dysfunctional diseases, such as Tangier disease, Chediak‐Higashi syndrome and Hermansky‐Pudlak syndrome. 35 , 36 , 37 Very significantly Tangier disease has recently been found to share cell biological and biochemical features of NPC disease, including reduced lysosomal Ca2+ storage. Additionally, one patient with Tangier disease treated with miglustat showed a clinical response to this NPC therapeutic suggesting pathogenic convergence. 38 , 39 Collagen and thrombin induced aggregation defects were also observed in a mouse model of Tangier disease and individuals with Tangier disease, suggesting agonists‐mediated platelet activation signaling defects in Tangier disease. 37
One of the common features of these lysosomes/LROs dysfunction disorders is that they all have intracellular Ca2+ homeostasis defects, either in the ER or LE/Lys Ca2+ stores. 11 , 37 , 40 Since intracellular Ca2+‐mediated signaling is crucial for intracellular vesicle fusion and fission events, acidic compartment‐mediated Ca2+ homeostasis defects could lead to abnormal intracellular vesicle trafficking and storage in these diseases. Therefore, it would be reasonable to hypothesize that acidic stores Ca2+ homeostasis defects in NPC1 disease could impair subcellular trafficking, affect granules content secretion and lead to the activation/aggregation defects observed in Npc1 −/− megakaryocytes/platelets. Better understanding the role of calcium signaling in platelets is important, as both NPC1 and Tangier patients have decreased plasma HDL‐C cholesterol levels, which are associated with an increased risk for all forms of atherosclerotic diseases, including myocardial infarction and stroke. 41 , 42
Taken together, our current studies have demonstrated that Npc1 −/− platelets have functional defects in a murine model of NPC1 disease. Furthermore, our studies suggest that Npc1 −/− platelet functional defects could be associated with impaired acidic stores Ca2+ signaling in NPC1 disease model and implicates acidic store Ca2+ to be further examined in better understanding the homeostasis of LROs.
CONFLICT OF INTEREST
The authors have no conflict of interest.
INFORMED CONSENT
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. 5 Informed consent was obtained from all patients for being included in the study.
ANIMAL RIGHTS
All institutional and national guidelines for the care and use of laboratory animals were followed.
AUTHOR CONTRIBUTIONS
O.C.‐W.C., A.C., L.C.D., F.N.K., and A.O.S. developed methods, devised, and performed experiments; O.C.‐W.C., A.C., L.C.D., and F.N.K., analyzed the data and performed statistical analysis; D.A.S. and L.M. helped with animal studies; P.T. and E.E. performed the transmission electron microscopy. F.D.P. and N.Y.F. obtained ethical permission, collected clinical samples, and provided data; O.C.‐W.C., A.O.S., G.C.C., A.G., F.D.P., and F.M.P. devised and designed the research; O.C.‐W.C., A.C., and F.M.P. wrote the article.
ETHICS APPROVAL
NPC1 patients were enrolled in a longitudinal observational study (NCT00344331) at the National Institutes of Health (Bethesda, Maryland, USA). The NICHD Institutional Review Board approved this study. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. All animal studies were conducted to comply with the ARRIVE guidelines using protocols approved by the UK Home Office for the conduct of regulated procedures under license (Animal scientific Procedures Act, 1986).
PATIENT CONSENT
No identifiable personal data is included in this article.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
ACKNOWLEDGMENTS
We thank Dr Jackie Sharpe and Dr Jackie Sloane for help with hematological analysis, and Dr Alistair Poole and Dr Chris Williams for helpful discussions. This work was supported by grants to F.M.P. from CLIMB (Children Living with Inherited Metabolic Diseases, United Kingdom), Niemann‐Pick Research Foundation (NPRF) and the John Fell Foundation (Oxford University Press, University of Oxford). F.M.P. is a Royal Society Wolfson Research Merit Award holder and a Wellcome Trust Investigator in Science. O.C.‐W.C. was funded by the Clarendon Fund (Clarendon Foundation, University of Oxford) the Jason Hu scholarship (Balliol College, University of Oxford). A.C. was funded by Action Medical Research. F.N.K. was supported by funding from the Lister Institute of Preventative Medicine. D.S. was funded by SOAR‐NPC and NPRF. F.D.P. received support from the Ara Parseghian Medical Research Foundation (N.Y.), the Therapeutics for Rare and Neglected Diseases program, a Bench to Bedside award from the Office of Rare Diseases, and the intramural research program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Chen OCW, Colaco A, Davis LC, et al. Defective platelet function in Niemann‐Pick disease type C1 . JIMD Reports. 2020;56:46–57. 10.1002/jmd2.12148
Communicating Editor: Carla E. Hollak
Oscar C. W. Chen and Alexandria Colaco contributed equally to this work.
Funding information Eunice Kennedy Shriver National Institute of Child Health and Human Development; Therapeutics for Rare and Neglected Diseases program; Ara Parseghian Medical Research Foundation; SOAR‐NPC; Lister Institute of Preventative Medicine; Action Medical Research; Clarendon Fund (Clarendon Foundation, University of Oxford); John Fell Foundation; Niemann‐Pick Research Foundation (NPRF); CLIMB (Children Living with Inherited Metabolic Diseases, United Kingdom)
REFERENCES
- 1. Hodivala‐Dilke KM, McHugh KP, Tsakiris DA, et al. β3‐integrin‐deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103:229‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clarke MCH, Savill J, Jones DB, Noble BS, Brown SB. Compartmentalized megakaryocyte death generates functional platelets committed to caspase‐independent death. J Cell Biol. 2003;160:577‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Te Vruchte D, Speak AO, Wallom KL, et al. Relative acidic compartment volume as a lysosomal storage disorder‐associated biomarker. J Clin Invest. 2014;124:1320‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vruchte D t, Lloyd‐Evans E, Veldman RJ, et al. Accumulation of Glycosphingolipids in Niemann‐Pick C disease disrupts endosomal transport. J Biol Chem. 2004;279:26167‐26175. [DOI] [PubMed] [Google Scholar]
- 5. Morgan AJ, Platt FM, Lloyd‐Evans E, Galione A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem J. 2011;439:349‐374. [DOI] [PubMed] [Google Scholar]
- 6. Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A. NAADP mobilizes Ca(2+) from reserve granules, lysosome‐related organelles, in sea urchin eggs. Cell. 2002;111:703‐708. [DOI] [PubMed] [Google Scholar]
- 7. Ruas M, Rietdorf K, Arredouani A, et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca2+ signaling and endolysosomal trafficking. Curr Biol. 2010;20:703‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morgan AJ, Davis LC, Wagner SK, et al. Bidirectional Ca(2)(+) signaling occurs between the endoplasmic reticulum and acidic organelles. J Cell Biol. 2013;200:789‐805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Atakpa P, Thillaiappan NB, Mataragka S, Prole DL, Taylor CW. IP3 receptors preferentially associate with ER‐2+ lysosome contact sites and selectively deliver Ca to lysosomes. Cell Rep. 2018;25:3180‐3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marks MS, Heijnen HF, Raposo G. Lysosome‐related organelles: unusual compartments become mainstream. Curr Opin Cell Biol. 2013;25:495‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lloyd‐Evans E, Morgan AJ, He X, et al. Niemann‐Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247‐1255. [DOI] [PubMed] [Google Scholar]
- 12. Vanier MT. Niemann‐Pick disease type C. Orphanet J Rare Dis. 2010;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Speak AO, Te Vruchte D, Davis LC, et al. Altered distribution and function of natural killer cells in murine and human Niemann‐Pick disease type C1. Blood. 2014;123:51‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Louwette S, Régal L, Wittevrongel C, et al. NPC1 defect results in abnormal platelet formation and function: studies in Niemann–Pick disease type C1 patients and zebrafish. Hum Mol Genet. 2013;22:61‐73. [DOI] [PubMed] [Google Scholar]
- 15. Lu F, Liang Q, Abi‐Mosleh L, et al. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife. 2015;4:e12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morgan AJ, Yuan Y, Patel S, Galione A. Does lysosomal rupture evoke Ca2+ release? A question of pores and stores. Cell Calcium. 2020;86:102139. [DOI] [PubMed] [Google Scholar]
- 17. Huang Y, Putney JWJ. Relationship between intracellular calcium store depletion and calcium release‐activated calcium current in a mast cell line (RBL‐1). J Biol Chem. 1998;273:19554‐19559. [DOI] [PubMed] [Google Scholar]
- 18. Toescu EC, Petersen OH. The thapsigargin‐evoked increase in Ca2+i involves an InsP3‐dependent Ca2+ release process in pancreatic acinar cells. Pflugers Arch. 1994;427:325‐331. [DOI] [PubMed] [Google Scholar]
- 19. Cavarra MS, Assef YA, Kotsias BA. Effects of ionomycin and thapsigargin on ion currents in oocytes of Bufo arenarum . J Exp Zool. 2003;297A:130‐137. [DOI] [PubMed] [Google Scholar]
- 20. Pizzo P, Fasolato C, Pozzan T. Dynamic properties of an inositol 1,4,5‐trisphosphate– and thapsigargin‐insensitive calcium pool in mammalian cell lines. J Cell Biol. 1997;136:355‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balduini CL, Noris P. Platelet count and aging. Haematologica. 2014;99:953‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yanjanin NM, Vélez JI, Gropman A, et al. Linear clinical progression, independent of age of onset, in Niemann‐Pick disease, type C. Am J Med Genet B Neuropsychiatr Genet. 2011;153B:132‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silverstein R, Febbraio M. Identification of lysosome‐associated membrane protein‐2 as an activation‐dependent platelet surface glycoprotein. Blood. 1992;80:1470‐1475. [PubMed] [Google Scholar]
- 24. Febbraio M, Silverstein RL. Identification and characterization of LAMP‐1 as an activation‐dependent platelet surface glycoprotein. J Biol Chem. 1990;265:18531‐18537. [PubMed] [Google Scholar]
- 25. White JG, Rao GH, Gerrard JM. Effects of the lonophore A23187 on blood platelets I. Influence on aggregation and secretion. Am J Pathol. 1974;77:135‐149. [PMC free article] [PubMed] [Google Scholar]
- 26. Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341‐2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Crocker AC, Farber S. Niemann‐Pick disease: a review of eighteen patients. Medicine (Baltimore). 1958;37:1‐95. [DOI] [PubMed] [Google Scholar]
- 28. Nicholson AG, Florio R, Hansell DM, et al. Pulmonary involvement by Niemann–Pick disease. A report of six cases. Histopathology. 2006;48:596‐603. [DOI] [PubMed] [Google Scholar]
- 29. Zhang L, Orban M, Lorenz M, et al. A novel role of sphingosine 1‐phosphate receptor S1pr1 in mouse thrombopoiesis. J Exp Med. 2012;209:2165‐2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Golfier S, Kondo S, Schulze T, et al. Shaping of terminal megakaryocyte differentiation and proplatelet development by sphingosine‐1‐phosphate receptor S1P4. FASEB J. 2010;24:4701‐4710. [DOI] [PubMed] [Google Scholar]
- 31. Hla T, Galvani S, Rafii S, Nachman R. S1P and the birth of platelets. J Exp Med. 2012;209:2137‐2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Galione A, Parrington J, Funnell T. Physiological roles of NAADP‐mediated Ca2+ signaling. Sci China Life Sci. 2011;54:725‐732. [DOI] [PubMed] [Google Scholar]
- 33. Coxon CH, Lewis AM, Sadler AJ, et al. NAADP regulates human platelet function. Biochem J. 2012;441:435‐442. [DOI] [PubMed] [Google Scholar]
- 34. Dionisio N, Albarran L, Lopez JJ, et al. Acidic NAADP‐releasable Ca(2+) compartments in the megakaryoblastic cell line MEG01. Biochim Biophys Acta. 2011;1813:1483‐1494. [DOI] [PubMed] [Google Scholar]
- 35. Li W, Rusiniak ME, Chintala S, Gautam R, Novak EK, Swank RT. Murine Hermansky–Pudlak syndrome genes: regulators of lysosome‐related organelles. Bioessays. 2004;26:616‐628. [DOI] [PubMed] [Google Scholar]
- 36. Dell'Angelica EC, Mullins C, Caplan S, Bonifacino JS. Lysosome‐related organelles. FASEB J. 2000;14:1265‐1278. [DOI] [PubMed] [Google Scholar]
- 37. Nofer J‐R, Herminghaus G, Brodde M, et al. Impaired platelet activation in familial high density lipoprotein deficiency (Tangier disease). J Biol Chem. 2004;279:34032‐34037. [DOI] [PubMed] [Google Scholar]
- 38. Sechi A, Dardis A, Zampieri S, et al. Effects of miglustat treatment in a patient affected by an atypical form of Tangier disease. Orphanet J Rare Dis. 2014;9:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Colaco A, Kaya E, Adriaenssens E, et al. Mechanistic convergence and shared therapeutic targets in Niemann‐Pick disease. J Inherit Metab Dis. 2020;43: 574‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lloyd‐Evans E, Platt FM. Lysosomal Ca2+ homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium. 2011;50:200‐205. [DOI] [PubMed] [Google Scholar]
- 41. Afzali M, Hashemi M, Tabatabaei SP, Fakheri KT, Nakhaee A. Association between the rs1805081 polymorphism of Niemann‐Pick type C1 gene and cardiovascular disease in a sample of an Iranian population. Biomed Rep. 2017;6:83‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yu XH, Jiang N, Yao PB, Zheng XL, Cayabyab FS, Tang CK. NPC1, intracellular cholesterol trafficking and atherosclerosis. Clin Chim Acta. 2014;15:69‐75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.