

Abstract
Programmed cell death (PCD)—apoptosis, autophagy and programmed necrosis—is any pathological form of cell death mediated by intracellular processes. Ototoxic drugs, ageing and noise exposure are some common pathogenic factors of sensorineural hearing loss (SNHL) that can induce the programmed death of auditory hair cells through different pathways, and eventually lead to the loss of hair cells. Furthermore, several mutations in apoptotic genes including DFNA5, DFNA51 and DFNB74 have been suggested to be responsible for the new functional classes of monogenic hearing loss (HL). Therefore, in this review, we elucidate the role of these three forms of PCD in different types of HL and discuss their guiding significance for HL treatment. We believe that further studies of PCD pathways are necessary to understand the pathogenesis of HL and guide scientists and clinicians to identify new drug targets for HL treatment.
Keywords: apoptosis, autophagy, hearing loss, programmed necrosis
The authors review accumulating evidence pointing out that programmed cell death pathways, including apoptosis, autophagy and necrosis, play key roles in ultimate fates of auditory hair cells, when cells suffer adverse factors. These three forms of PCD may jointly decide occurrence of hearing loss.
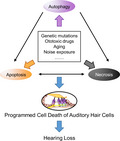
1. INTRODUCTION
Hearing loss (HL) is one of the most common sensory defects in humans. The hearing system is complex and depends on the comprehensive functions of many types of tissues and cells in the inner ear. Therefore, mutations in various genes have been proposed to be the cause of HL. It is estimated that 1‐3 of every 1000 newborn children are deaf, and in nearly half of these cases, HL can be attributed to genetic factors. 1 The well‐known types of acquired HL are ototoxic drug‐induced hearing loss (ODIHL), age‐related hearing loss (ARHL) and noise‐induced hearing loss (NIHL). The pathological characteristics of each type of HL are not the same. The main mechanism of ODIHL is hair cell loss. 2 The loss of hair, spiral ganglion and vascular striated cells is involved in ARHL. 3 Noise‐induced hearing loss is caused by excessive exposure to noise. 4 It involves two main mechanisms, namely mechanical damage and loss of hair cells and spiral ganglia. 4
Programmed cell death (PCD) appears to play a critical role in the development and diseases of the inner ear. When the nucleus of a cell is affected by severe damage, the initiation of PCD leads to irreversible changes, such as metabolic arrest, structural damage and function loss that can balance cell death and normal cell survival. Several forms of PCD have been found in eukaryotes, including apoptosis, autophagy, programmed necrosis, entosis, ferroptosis, lysosome‐dependent cell death and parthanatos. 5 , 6 , 7 , 8 , 9 The contribution of apoptosis in the development of hearing loss has been long studied. Several studies have been conducted to decipher the molecular mechanisms underlying the roles of them in HL. Apoptosis is an ATP‐dependent, enzyme‐mediated, inherently programmed death of cells that are no longer needed or are a threat to the organism. 10 Apoptosis occurs when DNA molecule in a cell is beyond repair, when a cell receives stress signals from other cells, or when misfolded or unfolded proteins accumulate in a cell. The morphological manifestations of apoptosis include chromatin condensation, cell membrane blebbing, cell shrinkage and apoptotic body formation. 10 Autophagy is a conserved process of intracellular material turnover in eukaryotes. It is a key mechanism in the response of cells to extracellular or intracellular stress that aid in their survival under certain circumstances; for instance, autophagy protects cells against NIHL by attenuating oxidative stress. 11 However, overactivation of autophagy may result in cell death. 12 Specialized double‐membrane vesicles, known as autophagosomes, encapsulate degenerating cytoplasmic organelles or cytosol and subsequently degrade them via the fusion with lysosomes. 13 , 14 Necroptosis can be initiated by several factors, and receptor‐interacting proteins (RIPs) 1 and 3 are two key proteins involved in this process. Necroptosis can be morphologically characterized by increased cell volume, swollen organelles, ruptured plasma membrane and the subsequent intracellular content loss. 14 , 15 The contents of a ruptured necrotic cell in the interstitial space may trigger inflammation of the adjacent cells.
The pathology of HL has been studied extensively. 16 , 17 Recent findings suggest that cellular death mediated via PCD is an important mechanism in HL. Apoptosis and programmed necrosis always lead to cell death, whereas autophagy can lead to cell survival or death. In normal cells, there is a delicate balance between apoptosis‐inducing and apoptosis‐inhibiting factors, and it ensures the survival and proliferation of cells. However, under stress, this balance can be disturbed. The activation of autophagy may play a protective role in the early stages of disease progression. Nevertheless, when the imbalance mediated by pathogenic factors becomes prominent, cells may activate the apoptotic or necrotic death process or over‐activate autophagy leading to cell death. Thus, PCD is important in the development and maintenance of multicellular organisms, and the loss of PCD regulation can lead to diseases. Here, we focused on various causes of HL and the well‐characterized cell death mechanisms (apoptosis, autophagy and necrosis) involved in HL.
2. APOPTOTIC PATHWAYS IN HL
Apoptosis is an active and highly ordered cell death process regulated by genes (including Bcl‐2, p53 and c‐Jun) and a series of enzymes (including caspases and endonuclease G (EndoG)) through intrinsic (mitochondrial), extrinsic (death receptor (DR)) and endoplasmic reticulum (ER) pathways. 18 , 19 Apoptosis plays an important role in maintaining the normal growth of an organism. The Bcl‐2 family members are important for the regulation of apoptosis, including Bcl‐2, Bcl‐w, Bax, Bak, Bid and Bad. 20 , 21 The initiation of apoptosis depends on the activation of a series of caspases. Caspases can be divided into the following three categories: initiator (caspases initiator 2, 8, 9 and 10), executioner (caspases 3, 6 and 7) and inflammatory (caspases 1, 4 and 5). 22 , 23 When a cell is exposed to a fatal stress, apoptosis can be triggered by the initiator caspase 9 or 8 via the mitochondrial or DR pathways. Furthermore, the executioner caspases 3 and 7 are activated, causing the fragmentation of DNA, destruction of nuclear proteins, cytoskeleton and protein cross‐linking, and expression of ligands in phagocytic cells. 24 In the caspase‐independent pathway, apoptosis‐inducing factor (AIF) and EndoG are released from the mitochondria, and they migrate to the nucleus to condense the chromatin (Figure 1). 22
FIGURE 1.
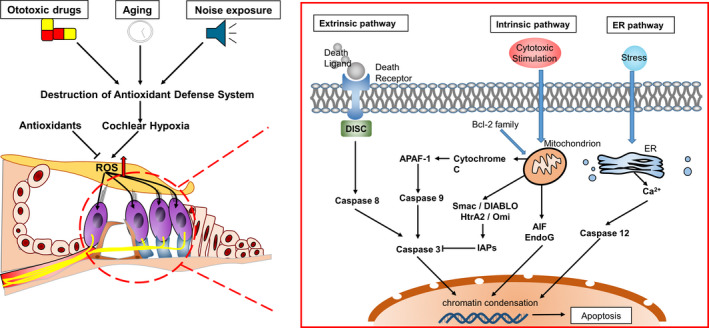
Apoptotic signalling pathways. Factors such as ototoxic drugs, ageing and noise exposure, which lead to hearing loss, damage the antioxidant defence system of the cochlea and cause imbalance of oxidation‐reduction in the inner ear. Reactive oxygen species (ROS) can directly induce the intrinsic apoptosis of cells. Moreover, they can induce the production of cell death ligands to mediate the extrinsic apoptosis process. ROS‐induced intracellular protein damage can cause endoplasmic reticulum stress, which can lead to apoptosis
ER stress is characterized by the accumulation of misfolded and unfolded proteins, and disruption of calcium and redox balances. 25 In multicellular eukaryotes, three upstream signalling proteins (IRE1, PERK and ATF6) act as pressure receptors, and they are activated by the level of unfolded proteins in the organelle cavities. 25 Cells can cope with ER stress by increasing the expression of chaperones and enhancing ER‐associated degradation of misfolded proteins. 26 However, continued damage can lead to apoptosis (Figure 1). Studies have shown that oxidative stress can induce apoptosis via the DR and mitochondrial pathways. 27 Reactive oxygen species (ROS) are oxygen free radicals and non‐radical substances, including hydroxyl radicals (OH‐), superoxide anions (O2‐), hydrogen peroxide (H2O2), ozone (O3) and singlet oxygen (1O2) species. Because these ROS contain unpaired electrons, they have a high chemical reactivity. Reactive oxygen species are considered toxic to cell metabolism. An increase in ROS production and the subsequent apoptosis are related to the development of various HL pathologies. These mechanisms suggest that all these factors individually or interactively lead to apoptosis and cochlear damage.
2.1. Mutations of apoptosis‐related genes leading to monogenic HL
The different chromosomal loci of nonsyndromic hereditary deafness are designated as deafness (DFN); letters A and B represent autosomal dominant inheritance (DFNA) and recessive inheritance (DFNB), respectively. Studies on mutant genes responsible for inherited progressive HL have suggested potential mechanisms underlying hair cell apoptosis. Table 1 lists three mutations in apoptotic genes that cause monogenic HL.
TABLE 1.
Genetic forms of hearing loss (HL)
| Molecular function | Gene symbol | Chromosomal locus | Locus name |
|---|---|---|---|
| Nucleus | GSDME | 7p15 | DFNA5 |
| Tight junctions | TJP2 | 9q21.11 | DFNA51 |
| Mitochondria | MSRB3 | 12q14.2‐15 | DFNB74 |
DFNA5 is targeted at chromosome 7p15 as the fifth DFNA site that leads to progressive HL, which starts at high frequencies. DFNA51 is caused by a tandem inverted genomic duplication of 270 kb at chromosome 9q21.11. DFNB74 is a novel locus on chromosome 12q14.2‐15 that is responsible for autosomal recessive nonsyndromic hearing impairment.
2.1.1. DFNA5
DFNA5 is one of the mutated genes related to PCD that leads to sensorineural HL. So far, only intronic mutations have been reported to cause exon 8 skipping in patients with DFNA5‐related HL. 28 , 29 , 30 , 31 The protein encoded by DFNA5 belongs to the gasdermin superfamily as it contains a gasdermin domain. A previous study reported that wild‐type DFNA5 (wtDFNA5) had no effect on yeast cells, whereas mutant DFNA5 (mutDFNA5) led to cell cycle arrest. 32 In mammalian cells, the transfection of mutDFNA5 led mutDFNA5 to cell death, whereas the transfection of wtDFNA5wtDFNA5 could not. 33 Thus, HL caused by mutDFNA5 can be attributed to functional mutations. In a mutant DFNA5 cell line, the upregulation of different cytochrome c oxidase (COX) genes was found to be associated with cell death mechanisms under oxidative stress. 34 In the same research model, the downregulation of protein sorting‐ and folding‐related mechanisms indicated that ER stress has a potential role in cell death induced by DFNA5 (Figure 2A). 34
FIGURE 2.
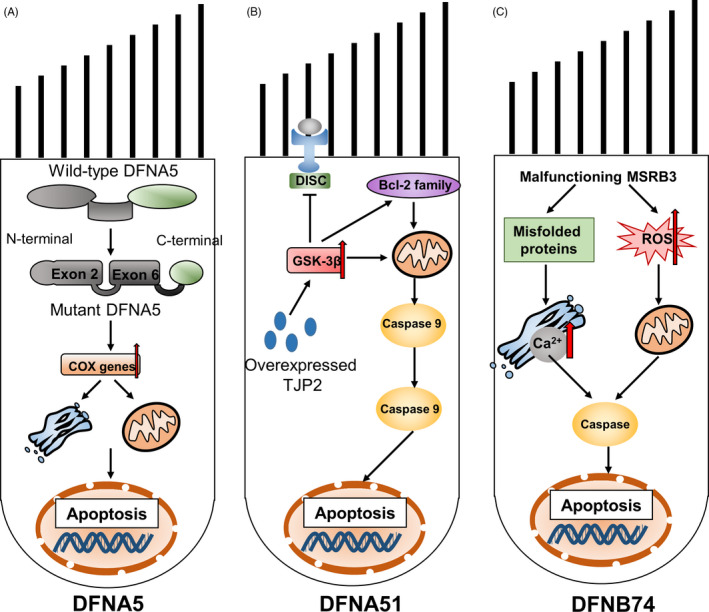
Schematics of three mutations that lead to monogenic hearing loss. A, DFNA5: the apoptosis‐inducing region of DFNA5 is located in exons 2 and 6 of the N‐terminal domain. Skipping exon 8 can change and shorten the C‐terminal domain of DFNA5, reveal the apoptosis‐inducing region and lead to apoptosis. B, DFNA51: overexpression of TJP2 induces apoptosis by activating glycogen synthase kinase 3β (GSK‐3β). C, DFNB74: malfunctioning MSRB3 leads to the accumulation of oxidative damage proteins and reactive oxygen species (ROS), ultimately leading to endoplasmic reticulum stress and subsequent activation of endogenous apoptotic pathways
2.1.2. DFNA51
DFNA51 is an inverted genomic duplication of 270‐kb DNA, including the entire wild‐type TJP2 that encodes the tight junction protein (ZO‐2). ZO‐2 belongs to the membrane‐associated family of guanylate kinase homologs, and it contains 3 PDZ domains, 1 SH3 domain and 1 GUK domain. 35 ZO‐2 binds to the C‐terminal of the connective transmembrane protein and then connects to the actin in the cytoskeleton and regulates the location of different subtypes of cells by interacting with the signal transduction pathway molecules. 36 TJP2 is mainly expressed between the hair and supporting cells in the organ of Corti, and helps maintain the barrier between ductus perilymphaticus and ductus endolymphaticus. Its expression decreases with age. The pathogenic mutation gene is an inverted genomic duplication of TJP2 that results in the overexpression of TJP2, which leads to autosomal dominant nonsyndromic HL. The expression of TJP2 mRNA in patients with duplicate genes was approximately 1.7‐fold higher than that in normal controls. The overexpression of TJP2 in vitro leads to a decrease in the phosphorylation and activation of glycogen synthase kinase 3β (GSK‐3β). GSK‐3β promotes cell death via the mitochondrial intrinsic apoptotic pathway, but it inhibits the DR‐mediated extrinsic apoptotic pathway. 37 The results of real‐time fluorescent quantitative polymerase chain reaction showed that even a slight increase in the expression of Bcl‐w altered the expression of other Bcl‐2 family members and the 18‐kDa translocator protein (TSPO) may shift the overall steady‐state balance towards apoptosis and thus result in HL. 38 However, the complete loss of TJP2 can lead to embryonic death; thus, TJP2 knockout was found to be lethal in mice (Figure 2B). 39
2.1.3. DFNB74
The mutations c.265 T > G and c.55 T > C in methionine sulfoxide reductase B3 (MSRB3) are related to autosomal recessive HL. The mutated gene is also known DFNB74. The gene has four isoforms. Isotype A is located in the ER, and the other three isotypes (B, C and D) are located in the mitochondria. MSRB3 encodes a methionine sulfoxide reductase that is involved in the repair of oxidative damage proteins. In the organ of Corti in mice, the expression of MSRB3 is upregulated in the inner and outer hair cells, but it is downregulated in the supporting cells. MSRB3 mutations lead to the disruption of protein functions. This in turn leads to the accumulation of oxidative damage‐related proteins and ROS, activation of caspase and initiation of apoptosis or programmed necrosis. MSRB3 deficiency can also increase the level of cytosolic calcium. The disruption of calcium homeostasis can trigger ER stress and activate Bcl‐2‐like protein 11 (also known as BIM) molecules that promote apoptosis, and thus lead to HL (Figure 2C). 40 , 41
2.2. Ototoxic drug‐induced hearing loss (ODIHL)
2.2.1. Aminoglycoside antibiotics
Aminoglycosides are broad‐spectrum antibiotics, but their potential ototoxicity needs to be closely monitored. 42 The ototoxicity of aminoglycosides is irreversible because the hair cells of the cochlea cannot proliferate and recover. Aminoglycosides may damage the hair cells of the cochlea and type I sensory cells of the vestibule by triggering different apoptotic signals, thus resulting in HL and vertigo. 43 Mitochondrial pathways play a key role in aminoglycoside‐induced apoptosis and may be the main target of these drugs. Aminoglycosides tend to accumulate in the mitochondria of hair cells. 44 Gentamicin directly inhibits protein synthesis in mitochondrial ribosomes and triggers the opening of mPTP that leads to the release of apoptotic factors. 44 , 45 In addition, L‐carnitine promotes mitochondrial function, which can prevent the damage of the outer hair cells after the administration of gentamicin. 46
ROS has been identified as the main cause of HL mediated by aminoglycosides. 47 ROS induces mitochondrial damage, thereby leading to the activation of various pathways that lead to apoptosis. Aminoglycosides can accelerate the nonenzymatic formation of ROS by redox active iron complex, and induce the intracellular enzymatic reaction. 48 Additionally, a previous study demonstrated that dexamethasone, melatonin (MLT) and tacrolimus decrease the levels of ROS in GM‐exposed explants. 49 Interestingly, the c‐Jun‐NH‐terminal kinase (JNK) cascade reaction combines oxidative stress with apoptosis. 50 Through in vivo experiments, it has been shown that administration of an aminoglycoside leads to the activation of the JNK pathway, which triggers the apoptosis of cochlear cells. Accordingly, JNK cascade inhibitors, such as CEP‐1347 and estradiol, can reduce the loss of hair cells after the administration of gentamicin. 51 , 52 However, Kalinec et al reported that gentamicin ototoxicity is mediated by the inhibition of the JNK pathway. 46 Evidently, there is no consensus on whether the signalling enzyme is activated by gentamicin ototoxicity.
Hair cells are not the only drug targets. Aminoglycosides also have effects on stria vascularis, including thinning of the tissue and reduction of marginal cells. 53 , 54 The degeneration of spiral ganglion cells after aminoglycoside treatment may be attributed to the loss of hair cells innervated by the ganglion cells. 55 However, some studies have shown that the spiral ganglion can be affected without obvious damage to the hair cells. 56 , 57 This suggests the complexity of the damage pattern of aminoglycoside antibiotics.
2.2.2. Cisplatin
The ototoxicity of cisplatin is well known. 58 The ototoxic effects of cisplatin can be divided into two categories. The first is the reversible inhibition of conduction current, voltage‐dependent calcium current in hair cells and the current response in stria vascularis. 59 Another persistent toxic reaction induces irreversible changes in cochlear morphology, thus resulting in irreversible, bilateral, high‐frequency HL. 60 Compared with the inner hair cells, the outer hair cells, 61 vascular marginal cells and spiral ganglion cells are more easily degenerated. 62 , 63 Cisplatin ototoxicity occurs through the formation of ROS in the cochlear tissue, accompanied by changes in potassium conductivity, which lead to cell death. 64 , 65 The cochlea has an effective antioxidant defence system. This system includes antioxidants, such as vitamin C, vitamin E and glutathione (GSH), as well as several antioxidant enzymes that are expressed in the cochlea, such as superoxide dismutase (SOD), GSH peroxidase and catalase. Cochleas extracted from cisplatin‐treated animals demonstrated consumption of GSH, reduction of antioxidant enzyme activity and an increase in lipid peroxidation. 66 , 67
Cisplatin increased the production of ROS in the inner ear. 68 It seems that one of the important sources of these ROS is nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 3, which is a type of superoxide that produces NADPH oxidase. It is highly expressed in the Corti organ, 69 and its level increases after cisplatin treatment. 69 , 70 Other NADPH oxidases are also important in the production of ROS in response to cisplatin ototoxicity. 71 Excessive ROS production may damage the antioxidant defence capacity of cochlear cells. p53 is activated in response to oxidative stress to regulate the expression of genes (eg Bax) that control DNA repair and cell death. 72 Bax can interact with voltage‐dependent ion channels in mitochondria, which mediate the release of cytochrome c and have the effect of apoptosis. 70 , 73 The corresponding targeted drugs have been proved to be suitable for the protection of cisplatin ototoxicity. For example, antioxidant administration in the early stage of cisplatin‐mediated ototoxicity can prevent ROS from having an additional downstream role in the cell death cascade reaction and in the function of reagents. Many of these antioxidants are mercaptan compounds with high‐affinity for platinum, such as N‐acetylcysteine (NAC), 74 sodium thiosulfate 75 and D‐methionine. 75 , 76 Other antioxidants that are resistant to cisplatin include ebselen, lipoic acid, diethyldithiocarbamate and 4‐methylthiobenzoic acid. 77 The application of the p53 inhibitor pifithrin‐α in cisplatin‐exposed cochlear organotypic cultures decreased hair cell injury. This was related to the decreased expression of p53 and caspase 3. 78 Specific caspase 9 and caspase 3 inhibitors can protect auditory hair cells from cisplatin‐induced apoptosis and HL. 79
Other potential apoptotic pathways in the stria vascularis lateralis or spiral ganglia include increased Bax levels, decreased bcl‐2 expression, 61 activation of NF‐κB, 80 , 81 formation of inducible nitric oxide synthase, 62 , 82 activation of the high‐mobility group 1 83 and production of 4‐hydroxynonenal (4‐HNE). 84
2.3. Age‐related Hearing Loss (ARHL)
The prevalence of ARHL is expected to rise with the increase in the ageing population. 85 , 86 , 87 Although many factors have been studied, including environmental, genetic and medical factors, 88 , 89 the precise mechanism of ARHL is unclear. At present, it is generally believed that ARHL is the result of a combination of genetic predispositions and various insults to the inner ear that accumulate during daily activities. ARHL is not the first mock examination in histopathology and pathophysiology. It may be accompanied by the degeneration and loss of sensory hair cells, spiral ganglion cells, stria vascularis cells and basement membrane with age. 90 Apoptosis in ARHL is mediated via exogenous and endogenous pathways. Exogenous pathways are triggered by ligands that bind to cell surface receptors and may be related to environmental and medical factors. 91 , 92 The endogenous pathway is mitochondrial‐dependent and is triggered by the loss of the mitochondrial membrane potential. The prevention of ARHL after the deletion of mitochondrial apoptotic gene (eg Bcl‐2 family member Bak) indicates that the endogenous apoptotic pathway is necessary for progression of ARHL. 93 It has been demonstrated that in ARHL models of mice, rats and gerbils, apoptosis occurs through the caspase‐dependent pathway, and involves the Bcl‐2 family proteins. 94 , 95 , 96 Immunohistochemical analysis of the cochlea of ageing CBA/J mice showed an increase in the phosphorylation (ie activation) of JNK and p38 MAPK in outer hair cells. 94 In addition, the same study also proved the release of cytochrome c, activation of caspase 9 and translocation of Endo G in the hair cells of ageing mouse.
ROS play an important role in ARHL. 97 They may cause DNA damage that leads to the upregulation of p53, which, in turn, leads to the chronic activation of the mitochondrial Bak pathway, finally resulting in apoptosis. 98 Consistent with this process, in all cell types of Corti organs, levels of antioxidant defence factors such as mitochondrial SOD 2 (SOD2) have been observed to decrease significantly with age, thus indicating that oxidative imbalance leads to ARHL. 99 Therefore, the role of antioxidant supplementation in ARHL has been studied. In Fischer 344 rats, vitamin C, vitamin E, MLT and lazaroid treatment yielded better results in the maintenance of auditory sensitivity and reduction of the number of mitochondrial DNA (mtDNA) deletions compared to those observed with the placebo. 100 In addition, C57BL/6J mice provided an antioxidant diet (α‐lipoic acid, coenzyme Q10, NAC) had a much lower ABR hearing threshold than that of control mice. 93 Nevertheless, Sha et al kept CBA/J mice on a long‐term diet from 10 to 22 months of age and claimed that foods rich in vitamins A, C, E and alpha lipoic acid did not delay or reduce ARHL. 94 Accordingly, Keithley et al found that transgenic mice expressing SOD2 yielded outcomes that were contrary to expectations, while the HL in mice who were 20 months old was more prominent than that in the parent strain of B6 mice. 101 These results suggest that mitochondrial ROS may be a factor in ARHL, but there are other factors involved as well.
Mitochondria are particularly susceptible to the accumulation of genetic or environmental damages because—unlike the nucleus—mtDNA is regularly replicating independent of cell cycle replication and lacks an effective DNA repair system and protective histones. Therefore, the total number of its DNA mutations is higher than that of the accumulation of mitochondrial DNA mutations are thought to cause age‐related degenerative diseases, 102 and an increase in mitochondrial DNA mutations in human cochlear tissue has also been observed. 103 The same mechanism was proposed in the mouse ARHL model. 104 , 105 The main mutation of mitochondrial DNA occurs in the gene encoding the mitochondrial oxygen phosphorus complex and leads to abnormal oxygen phosphorus activity and mitochondrial dysfunction, and increases in intracellular free calcium. Calpain and cathepsin are released from lysosomes in response to the increased intracellular calcium. They are calcium‐dependent proteases that activate downstream pathways through the proteolysis of target proteins. They are part of cell death signals that are independent of cystatin and are involved in apoptosis and necrotic cell death. 94 However, the exact pathway of apoptotic activation caused by ARHL has not been clearly defined. In fact, it is possible to activate multiple pathways at the same time because ARHL is the product of a multifactorial process.
2.4. Noise‐induced HL (NIHL)
Noise is also the main cause of HL. 106 After noise exposure, there are two main mechanisms that cause cochlear damage. The first is direct mechanical damage that leads to the loss of hair cells through the mechanical destruction of cilia, and to the damage of supporting and sensory cells. 107 Another mechanism involves the biochemical pathway that leads to cell death through apoptosis or necrosis. It has been shown that apoptosis is the key mediator of noise‐induced HL. After noise exposure, an increase in chromatin condensation and levels of apoptosis markers 108 , 109 , 110 , 111 , 112 such as caspase 3, 8 or 9, 113 tumour necrosis factor receptor, 111 and Bcl‐2‐associated death promoters (Bad) 114 was observed in cochlear cells of guinea pigs, chinchillas and rats models. Wang et al showed that the application of riluzole (inhibitor of apoptosis and necrosis) in the cochlea can protect the cochlea from hearing impairment. 115 , 116 These results indicate the importance of the apoptotic pathway in NIHL.
Several ways of inducing apoptosis in NIHL have been studied in animal models. After noise exposure in a guinea pig model, AIF and EndoG entered the cytoplasm of the cochlear cell 117 and were then transferred to the nucleus, triggering apoptosis in a caspase‐independent manner. Noise exposure triggers the activation of caspase 8 and caspase 9 by exogenous and endogenous pathways, and both apoptosis markers are associated with signalling pathways that lead to caspase 3 activation. 113 A variety of agents can attenuate NIHL, including iron chelators, antioxidants and vasoactive factors. 118 , 119 However, these factors individually have limited protective efficacy. These observations are consistent with the suggestion that noise may induce cell death via both caspase‐dependent and caspase‐independent apoptosis. 117
After noise exposure ends, ROS or other similar reactive species levels are generally increased. 120 ROS were observed to be present in the cochlea for an extended period of time following noise exposure. 121 These species were responsible for the morphological observations of delayed and sustained damages. 121 Consistent with the hypothesis of ROS formation, antioxidant molecules can be used for protection, such as water‐soluble coenzyme Q10, 122 NAC, 119 , 123 D‐methionine 124 and GSH, 125 which will decrease the amount of apoptosis in hair cells after noise exposure. ROS formation can activate the JNK signalling pathway. 126 In the noise‐damaged guinea pig model, JNK mediates apoptosis, 110 and blocking of the JNK pathway has a protective effect on noise. Mice exposed to noise contained fewer apoptotic cells than those in the control group, when they were fed with JNK inhibitors such as all transretinoic acid and CEP‐1347 (small molecules derived from indole‐carbazole K252a). 127 , 128 , 129 Another study reported that blocking the JNK pathway with locally delivered D‐JNK‐1 through the round window membrane can prevent hair cell death and permanent NIHL. 110
In addition, the increase of free Ca2+ in the outer hair cells, or the activation of Ca2+ and calmodulin‐controlled calcineurin may trigger the apoptosis or necrosis pathway without ROS. 130 , 131 Calcium metabolic disorders and free radicals can also cause ER stress. Severe ER stress is more likely to induce the expression of CHOP 132 and would lead to ER stress‐related apoptosis based on the downregulation of the expression of antiapoptotic proteins such as Bcl‐xL. 133 Glucocorticoid‐induced leucine zipper protects the cochlea from ER stress‐induced apoptosis following noise exposure by reducing chop and regulating ER stress‐related apoptosis proteins. 134
3. AUTOPHAGIC PATHWAYS IN HL
Autophagy is a protective mechanism triggered to limit pathological changes. In all eukaryotic cells, autophagy processes unnecessary or dysfunctional cell components such as damaged organelles and misfolded proteins 135 through highly regulated processes. 136 , 137 Autophagy is a process that mediates the formation of a bimembrane autophagic body that surrounds the damaged organelle or other cytoplasmic components. 138 The core molecule of autophagy regulation is the rapamycin kinase mammalian target (mTOR). The pathways activating mTOR such as the Akt and MAPK signalling pathways inhibit autophagy, while the pathways negatively regulating mTOR such as the adenosine 5’‐monophosphate activated protein kinase (AMPK) and p53 signalling pathways promote autophagy. 139 Under adverse conditions, AMPK is activated and mTOR is inactivated to enter the autophagy pathway. Multiple autophagy‐related (ATG) proteins are involved in this process (Figure 3).
FIGURE 3.
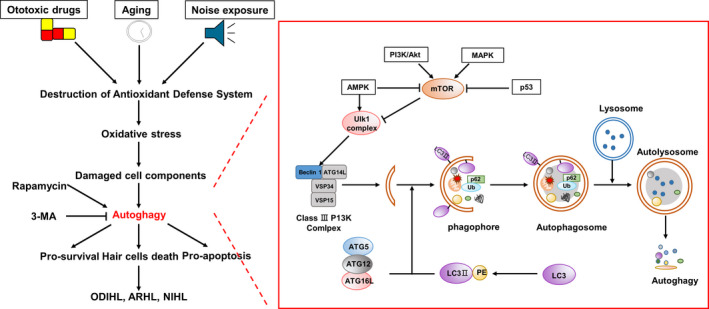
Signalling pathway of autophagy in HL. Unc‐51‐like autophagy activating kinase 1 (ULK1) is phosphorylated upon the activation of AMPK and inactivation of mTOR. The activated ULK1 complex and class III phosphoinositide 3 kinase (PI3K) complex form phosphors. Microtubule‐associated protein 1 light‐chain 3 (LC3) protein can be coupled with phosphatidylethanolamine to form LC3Ⅱ. The complex of ATG5, ATG12, ATG16L and LC3Ⅱ can stimulate the elongation of phagocytes, which provide a platform for the formation of phagosomes. On approaching the ubiquitin protein binding to p60 and LC3Ⅱ, the phagosome closes to form autophagosomes. Further, autophagosomes fuse with lysosomes to form autolysosomes in which the contents are degraded
Autophagy is a common cell response to starvation or other stresses. Basic autophagy is important in controlling the cytoplasmic composition and homeostasis of various mitotic cells. 140 Autophagy is involved in the development of normal cochlea. Based on real‐time PCR, Rodríguez et al found that the peak timepoints of ATG4b, ATG5, ATG9a and Beclin1 in the mouse cochlea were the same as that at which the cochlear was fully functional. 141 Basic autophagy plays an important role in the maintenance of hair cell morphology and hearing ability. Chisato et al found that ATG5 knockout in mice resulted in the degeneration of auditory hair cells and severe congenital HL. In the hair cells of autophagy‐deficient mice, accumulation of polyubiquitin and p62/SQSTM1 (autophagy matrix) as inclusion bodies was observed at the first week of life. 140 Therefore, impaired autophagy function can have adverse effects on auditory hair cells. Tsuchihashi et al used low‐dose H2O2 to construct an auditory cell model; increased phosphorylation of 4EBP1 following H2O2 treatment led to impaired autophagy function, which in turn resulted in oxidative stress‐induced premature ageing. 142
Some studies showed that autophagy plays a role in the prevention of hearing impairment, such as NIHL and ODIHL. 11 , 143 Previous studies have shown that ROS has the ability to induce autophagy in auditory cells. 11 , 144 Yuan et al reported that autophagy reduced NIHL by reducing oxidative stress. 11 Rapamycin can increase autophagy activity, inhibit ROS accumulation and prevent cell death induced by H2O2. It significantly increased the expression of microtubule‐associated protein 1 light‐chain 3Ⅱ (LC3Ⅱ), decreased the levels of 4‐HNE and 3‐nitrotyrosine (3‐NT) and reduced NIHL and loss of hair cells. In contrast, LC3B reduction by the autophagy inhibitor 3‐methyladenine (3‐MA) or LC3Ⅱ small interfering RNA increased the levels of 3‐NT in outer hair cells and promoted hair cell loss and NIHL. He et al found that autophagy activity was significantly increased, including enhanced autophagosome‐lysosome fusion, in both cochlear hair cells and HEI‐OC‐1 cells after neomycin or gentamicin injury, suggesting that autophagy might be correlated with aminoglycoside‐induced cell death. 144 Rapamycin treatment reduced ROS levels and hair cells death induced by aminoglycosides (including cisplatin, gentamicin and neomycin), 144 , 145 , 146 while 3‐MA treatment or ATG5 deletion increased ROS levels and apoptosis. 144 Chika et al demonstrated that oral administration of low‐dose rapamycin induced autophagy activation in cochlear outer sulcus cells, suggesting that rapamycin could be a feasible drug to manipulate inner ear cells. 147 Phosphatase and tensin homolog‐induced putative kinase 1 also protected hair cells from cisplatin‐induced ototoxicity following induction of autophagy. 148
Yin et al found that autophagic activation of HEI‐OC1 cells increased the expression of the nuclear binding domain and leucine‐rich repeat containing family member X1 (NLRX1) in cisplatin‐induced injury. 12 NLRX1 overexpression led to the amount of accumulation of autophagosomes in HEI‐OC1 cells in normal condition and a higher activation of autophagy concurrent with cell injury in HEI‐OC1 cells treated with cisplatin. These findings suggest that decrease the activation level of autophagy concurrent with increased cell viability in HEI‐OC1 cells treated with cisplatin, while overactivation of autophagy can lead to pathological changes in response to cisplatin exposure. 12 MicroRNA‐96 is the first microRNA mutation that has been reported to be related to human deafness. 149 The decreased expression of microRNA‐96 may directly upregulate the expression of ATG7. Excessive activation of autophagy induces degeneration and death of neurons. 150
4. THE PROGRAMMED NECROSIS PATHWAYS IN HL
Programmed necrosis is a type of regulatory cell death caused by microenvironmental disorders inside and outside the cells and is detected by specific DRs. The DR is usually a type‐1 tumour necrosis factor receptor, 151 besides suicide‐related factors (eg Fas) and pathogen recognition receptors (eg toll‐like receptors 3), that can also mediate programmed cell necrosis. 152 , 153 RIP1 and RIP3 can mediate the activation of programmed necrosis pathway through physical and functional interactions 154 , 155 , 156 , 157 (Figure 4).
FIGURE 4.
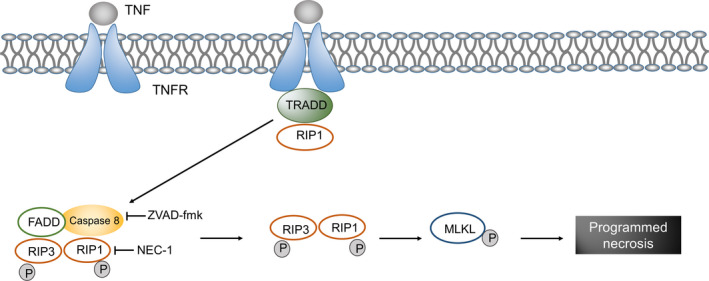
Signalling pathway of programmed necrosis in hearing loss. When a ligand binds to tumour necrosis factor receptor (TNFR), a combination of TNFR‐associated death domain (TRADD) and receptor‐interacting protein (RIP) 1 increases the level of RIP3 and induces self‐ and transphosphorylation, which is followed by the oligomerization of phosphorylated RIP3. Active RIP3 catalyses the phosphorylation of the mixed lineage kinase domain‐like protein (MLKL), thus resulting in the formation of MLKL oligomers and in translocation to the plasma membrane. Through the reversal mechanism, specific phosphatidylinositol phosphates are combined that lead to plasma membrane permeability and eventually cell necrosis
Zheng et al reported that the pan caspase inhibitor ZVAD blocked noise‐induced caspase 8 activation and reduced apoptosis in outer hair cells but stimulated the accumulation of RIP1 and RIP3 levels, resulting in the depletion of adenosine triphosphate and necrosis of cells. 158 These findings suggest that a balance between apoptosis and necrosis is required for noise‐induced death of outer hair cells, which is regulated by caspase 8 and RIP kinase. Choi et al found that treatment with Nec‐1, a selective RIP1 inhibitor, significantly inhibited cisplatin‐induced cell death in HEI‐OC1 cells, while the use of ZVAD did not change cisplatin‐induced cell death in HEI‐OC1 cells. 159 Their results suggested that RIP3‐dependent cell necrosis may mediate cisplatin ototoxicity. 159 Douglas et al studied the ototoxicity of aminoglycosides and cisplatin in a murine model 160 and suggested that the main form of hair cell death induced by aminoglycosides and cisplatin in vitro was mediated by caspase‐dependent apoptosis, without any effects from necrosis. In vivo, Nec‐1 was used to inhibit RIP1‐mediated necrotic disease and reduce HL induced by kanamycin and cisplatin. 160 These results suggest that the harmful factors (ototoxic drugs, ageing and noise exposure) can induce cell death via different PCD pathways. However, the crosstalk between these types of death pathways need to be investigated in future studies.
5. CONCLUSIONS
The auditory system is a complex system in which the failure of one component may lead to HL. HL is the most common disease associated with sensory defects, and it affects daily communications, the quality of life of patients and their psychological and mental activities. Although the pathology of HL is very complex, extensive genetic and molecular biological studies have provided considerable insights into the mechanisms underlying hair cell death. PCD involves the traditional death module apoptosis and various other cell death pathways, including autophagy, programmed necrosis, entosis, ferroptosis, lysosome‐dependent cell death and parthanatos. The goal of this review is to provide a general overview of the current knowledge relating to the contribution of PCD in the pathology of hearing impairment, including apoptosis, autophagy and programmed necrosis. This will provide researchers with a summary of the three forms of PCD in HL and allow them to compare and contrast between them. The substantial increase in studies related to PCD, especially those focusing on apoptosis pathways, has contributed to a wealth of knowledge that can facilitate a better understanding of HL pathogenesis and therapeutics. An increased understanding of autophagy and programmed necrosis in recent decades has led to the development of clinical therapies for HL. The occurrence of different types of HL may involve different PCD pathways. Audiological biologists have been trying to understand how these pathways could be mapped and integrated with each other, what global properties are beginning to emerge from interactome network models, and how these properties may relate to HL and its treatment. Therefore, in‐depth studies on the interconnected pathway network comprising the three main functional modules (apoptosis, autophagy and programmed necrosis) are warranted to better understand the pathogenesis and treatment of HL. It is anticipated that with the research conducted on the effect of related target genes, related gene therapy will become the current research hotspot. All these provide new insights to improve global HL.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
SY and YZ designed the study; JW wrote the first draft of the manuscript; WK critically revised the manuscript; JY prepared the figures. All authors approved the final version of the manuscript for submission.
Wu J, Ye J, Kong W, Zhang S, Zheng Y. Programmed cell death pathways in hearing loss: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2020;53:e12915 10.1111/cpr.12915
Contributor Information
Shouyue Zhang, Email: shouyue.zhang@outlook.com.
Yun Zheng, Email: 1141679315@qq.com, Email: shouyue.zhang@outlook.com.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analysed in this study.
REFERENCES
- 1. Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, Nance WE. Genetic epidemiological studies of early‐onset deafness in the U.S. school‐age population. Am J Med Genet Part A. 1993;46(5):486‐491. 10.1002/ajmg.1320460504 [DOI] [PubMed] [Google Scholar]
- 2. Huth ME, Ricci AJ, Cheng AG. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. International J Otolaryngol. 2011;2011:937861 10.1155/2011/937861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen H, Tang J. The role of mitochondria in age‐related hearing loss. Biogerontology. 2014;15(1):13‐19. 10.1007/s10522-013-9475-y [DOI] [PubMed] [Google Scholar]
- 4. Hong O, Kerr MJ, Poling GL, Dhar S. Understanding and preventing noise‐induced hearing loss. Disease‐a‐month. 2013;59(4):110‐118. 10.1016/j.disamonth.2013.01.002 [DOI] [PubMed] [Google Scholar]
- 5. Green DR, Llambi F. Cell death signaling. Cold Spring Harbor Perspect Biol. 2015;7(12). 10.1101/cshperspect.a006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Overholtzer M, Mailleux AA, Mouneimne G, et al. A nonapoptotic cell death process, entosis, that occurs by cell‐in‐cell invasion. Cell. 2007;131(5):966‐979. 10.1016/j.cell.2007.10.040 [DOI] [PubMed] [Google Scholar]
- 7. Yang W, SriRamaratnam R, Welsch M, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317‐331. 10.1016/j.cell.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Serrano‐Puebla A, Boya P. Lysosomal membrane permeabilization as a cell death mechanism in cancer cells. Biochem Soc Trans. 2018;46(2):207‐215. 10.1042/bst20170130 [DOI] [PubMed] [Google Scholar]
- 9. Virág L, Robaszkiewicz A, Rodriguez‐Vargas JM, Oliver FJ. Poly(ADP‐ribose) signaling in cell death. Mol Aspects Med. 2013;34(6):1153‐1167. 10.1016/j.mam.2013.01.007 [DOI] [PubMed] [Google Scholar]
- 10. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide‐ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239‐257. 10.1038/bjc.1972.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuan HU, Wang X, Hill K, et al. Autophagy attenuates noise‐induced hearing loss by reducing oxidative stress. Antioxid Redox Signal. 2015;22(15):1308‐1324. 10.1089/ars.2014.6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yin H, Yang Q, Cao Z, et al. Activation of NLRX1‐mediated autophagy accelerates the ototoxic potential of cisplatin in auditory cells. Toxicol Appl Pharmacol. 2018;343:16‐28. 10.1016/j.taap.2018.02.007 [DOI] [PubMed] [Google Scholar]
- 13. Yu L, Lenardo MJ, Baehrecke EH. Autophagy and caspases: a new cell death program. Cell Cycle. 2004;3(9):1124‐1126. [PubMed] [Google Scholar]
- 14. Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3‐11. 10.1038/cdd.2008.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146(1):3–15. [PMC free article] [PubMed] [Google Scholar]
- 16. Dinh CT, Goncalves S, Bas E, Van De Water TR, Zine A. Molecular regulation of auditory hair cell death and approaches to protect sensory receptor cells and/or stimulate repair following acoustic trauma. Front Cell Neurosci. 2015;9 10.3389/fncel.2015.00096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Böttger EC, Schacht J. The mitochondrion: A perpetrator of acquired hearing loss. Hear Res. 2013;303:12‐19. 10.1016/j.heares.2013.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Igney FH, Krammer PH. Death and anti‐death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2(4):277‐288. 10.1038/nrc776 [DOI] [PubMed] [Google Scholar]
- 19. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep. 2006;7(9):880‐885. 10.1038/sj.embor.7400779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life‐or‐death switch. Nat Rev Cancer. 2002;2(9):647‐656. 10.1038/nrc883 [DOI] [PubMed] [Google Scholar]
- 21. Cory S, Huang DC, Adams JM. The Bcl‐2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22(53):8590‐8607. 10.1038/sj.onc.1207102 [DOI] [PubMed] [Google Scholar]
- 22. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495‐516. 10.1080/01926230701320337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(Pt 1):1‐16. 10.1042/bj3260001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14(3):166‐180. 10.1038/nri3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519‐529. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 26. Fu XL, Gao DS. Endoplasmic reticulum proteins quality control and the unfolded protein response: the regulative mechanism of organisms against stress injuries. BioFactors. 2014;40(6):569‐585. 10.1002/biof.1194 [DOI] [PubMed] [Google Scholar]
- 27. Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria‐dependent and mitochondria‐independent pathways of apoptosis. Arch Toxicol. 2013;87(7):1157‐1180. 10.1007/s00204-013-1034-4 [DOI] [PubMed] [Google Scholar]
- 28. Laer LV, Huizing EH, Verstreken M, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20(2):194‐197. 10.1038/2503 [DOI] [PubMed] [Google Scholar]
- 29. Cheng J, Han DY, Dai P, et al. A novel DFNA5 mutation, IVS8+4 A>G, in the splice donor site of intron 8 causes late‐onset non‐syndromic hearing loss in a Chinese family. Clin Genet. 2007;72(5):471‐477. 10.1111/j.1399-0004.2007.00889.x [DOI] [PubMed] [Google Scholar]
- 30. Yu C, Meng X, Zhang S, Zhao G, Hu L, Kong X. A 3‐nucleotide deletion in the polypyrimidine tract of intron 7 of the DFNA5 gene causes nonsyndromic hearing impairment in a Chinese family. Genomics. 2003;82(5):575‐579. 10.1016/s0888-7543(03)00175-7 [DOI] [PubMed] [Google Scholar]
- 31. Bischoff AM, Luijendijk MW, Huygen PL, et al. A novel mutation identified in the DFNA5 gene in a Dutch family: a clinical and genetic evaluation. Audiology Neuro‐Otology. 2004;9(1):34‐46. 10.1159/000074185 [DOI] [PubMed] [Google Scholar]
- 32. Gregan J, Van Laer L, Lieto LD, Van Camp G, Kearsey SE. A yeast model for the study of human DFNA5, a gene mutated in nonsyndromic hearing impairment. Biochem Biophys Acta. 2003;1638(2):179‐186. 10.1016/s0925-4439(03)00083-8 [DOI] [PubMed] [Google Scholar]
- 33. Van Laer L, Vrijens K, Thys S, et al. DFNA5: hearing impairment exon instead of hearing impairment gene? J Med Genet. 2004;41(6):401‐406. 10.1136/jmg.2003.015073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Van Rossom S, Op de Beeck K, Hristovska V, Winderickx J, Van Camp G. The deafness gene DFNA5 induces programmed cell death through mitochondria and MAPK‐related pathways. Front Cellular Neurosci. 2015;9:231 10.3389/fncel.2015.00231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gonzalez‐Mariscal L, Bautista P, Lechuga S, Quiros M. ZO‐2, a tight junction scaffold protein involved in the regulation of cell proliferation and apoptosis. Ann NY Acad Sci. 2012;1257:133‐141. 10.1111/j.1749-6632.2012.06537.x [DOI] [PubMed] [Google Scholar]
- 36. Bauer H, Zweimueller‐Mayer J, Steinbacher P, Lametschwandtner A, Bauer HC. The dual role of zonula occludens (ZO) proteins. J Biomed Biotechnol. 2010;2010:402593 10.1155/2010/402593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beurel E, Jope RS. The paradoxical pro‐ and anti‐apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurogibol. 2006;79(4):173‐189. 10.1016/j.pneurobio.2006.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walsh T, Pierce SB, Lenz DR, et al. Genomic duplication and overexpression of TJP2/ZO‐2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am J Hum Genet. 2010;87(1):101‐109. 10.1016/j.ajhg.2010.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu J, Kausalya PJ, Phua DC, Ali SM, Hossain Z, Hunziker W. Early embryonic lethality of mice lacking ZO‐2, but Not ZO‐3, reveals critical and nonredundant roles for individual zonula occludens proteins in mammalian development. Mol Cell Biol. 2008;28(5):1669‐1678. 10.1128/mcb.00891-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ahmed ZM, Yousaf R, Lee BC, et al. Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am J Hum Genet. 2011;88(1):19‐29. 10.1016/j.ajhg.2010.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kwak GH, Kim HY. MsrB3 deficiency induces cancer cell apoptosis through p53‐independent and ER stress‐dependent pathways. Arch Biochem Biophys. 2017;621:1‐5. 10.1016/j.abb.2017.04.001 [DOI] [PubMed] [Google Scholar]
- 42. Lopez‐Novoa JM, Quiros Y, Vicente L, Morales AI, Lopez‐Hernandez FJ. New insights into the mechanism of aminoglycoside nephrotoxicity: an integrative point of view. Kidney Int. 2011;79(1):33‐45. 10.1038/ki.2010.337 [DOI] [PubMed] [Google Scholar]
- 43. Cheng AG, Cunningham LL, Rubel EW. Hair cell death in the avian basilar papilla: characterization of the in vitro model and caspase activation. J Ass Res Otolaryngol. 2003;4(1):91‐105. 10.1007/s10162-002-3016-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Bottger EC. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci USA. 2008;105(52):20888‐20893. 10.1073/pnas.0811258106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dehne N, Rauen U, de Groot H, Lautermann J. Involvement of the mitochondrial permeability transition in gentamicin ototoxicity. Hear Res. 2002;169(1–2):47‐55. 10.1016/s0378-5955(02)00338-6 [DOI] [PubMed] [Google Scholar]
- 46. Kalinec GM, Fernandez‐Zapico ME, Urrutia R, Esteban‐Cruciani N, Chen S, Kalinec F. Pivotal role of Harakiri in the induction and prevention of gentamicin‐induced hearing loss. Proc Natl Acad Sci USA. 2005;102(44):16019‐16024. 10.1073/pnas.0508053102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abi‐Hachem RN, Zine A, Van De Water TR. The injured cochlea as a target for inflammatory processes, initiation of cell death pathways and application of related otoprotectives strategies. Recent Pat CNS Drug Discovery. 2010;5(2):147‐163. 10.2174/157488910791213121 [DOI] [PubMed] [Google Scholar]
- 48. Priuska EM, Schacht J. Formation of free radicals by gentamicin and iron and evidence for an iron/gentamicin complex. Biochem Pharmacol. 1995;50(11):1749‐1752. 10.1016/0006-2952(95)02160-4 [DOI] [PubMed] [Google Scholar]
- 49. Bas E, Van De Water Tr, Gupta C, Dinh J, Vu L, Martínez‐Soriano F, Láinez Jm, Marco J. Efficacy of three drugs for protecting against gentamicin‐induced hair cell and hearing losses. British J Pharmacol. 2012;166(6):1888–1904. 10.1111/j.1476-5381.2012.01890.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mielke K, Herdegen T. JNK and p38 stresskinases – degenerative effectors of signal‐transduction‐cascades in the nervous system. Prog Neurogibol. 2000;61(1):45‐60. 10.1016/s0301-0082(99)00042-8 [DOI] [PubMed] [Google Scholar]
- 51. Ylikoski J, Xing‐Qun L, Virkkala J, Pirvola U. Blockade of c‐Jun N‐terminal kinase pathway attenuates gentamicin‐induced cochlear and vestibular hair cell death. Hear Res. 2002;166(1–2):33‐43. 10.1016/s0378-5955(01)00388-4 [DOI] [PubMed] [Google Scholar]
- 52. Nakamagoe M, Tabuchi K, Uemaetomari I, Nishimura B, Hara A. Estradiol protects the cochlea against gentamicin ototoxicity through inhibition of the JNK pathway. Hear Res. 2010;261(1–2):67‐74. 10.1016/j.heares.2010.01.004 [DOI] [PubMed] [Google Scholar]
- 53. Ruedi L, Furrer W, Luthy F, Nager G, Tschirren B. Further observations concerning the toxic effects of streptomycin and quinine on the auditory organ of guinea pigs. The Laryngoscope. 1952;62(4):333‐351. 10.1288/00005537-195204000-00001 [DOI] [PubMed] [Google Scholar]
- 54. Hawkins E Jr. Ototoxic mechanisms. A working hypothesis. International J Audiol. 1973;12(5):383‐393. 10.3109/00206097309071652 [DOI] [PubMed] [Google Scholar]
- 55. Johnsson LG, Hawkins JE Jr, Kingsley TC, Black FO, Matz GJ. Aminoglycoside‐induced cochlear pathology in man. Acta oto‐laryngologica Supplementum. 1981;383:1‐19. [PubMed] [Google Scholar]
- 56. Hinojosa R, Lerner SA. Cochlear neural degeneration without hair cell loss in two patients with aminoglycoside ototoxicity. J Infect Dis. 1987;156(3):449‐455. 10.1093/infdis/156.3.449 [DOI] [PubMed] [Google Scholar]
- 57. Sone M, Schachern PA, Paparella MM. Loss of spiral ganglion cells as primary manifestation of aminoglycoside ototoxicity. Hear Res. 1998;115(1–2):217‐223. 10.1016/s0378-5955(97)00191-3 [DOI] [PubMed] [Google Scholar]
- 58. Rybak LP, Ramkumar V. Ototoxicity. Kidney Int. 2007;72(8):931‐935. 10.1038/sj.ki.5002434 [DOI] [PubMed] [Google Scholar]
- 59. Kimitsuki T, Nakagawa T, Hisashi K, Komune S, Komiyama S. Cisplatin blocks mechano‐electric transducer current in chick cochlear hair cells. Hear Res. 1993;71(1–2):64‐68. 10.1016/0378-5955(93)90021-r [DOI] [PubMed] [Google Scholar]
- 60. Musial‐Bright L, Fengler R, Henze G, Hernaiz DP. Carboplatin and ototoxicity: hearing loss rates among survivors of childhood medulloblastoma. Child's Nervous Sys. 2011;27(3):407‐413. 10.1007/s00381-010-1300-1 [DOI] [PubMed] [Google Scholar]
- 61. Alam SA, Ikeda K, Oshima T, et al. Cisplatin‐induced apoptotic cell death in Mongolian gerbil cochlea. Hear Res. 2000;141(1–2):28‐38. 10.1016/s0378-5955(99)00211-7 [DOI] [PubMed] [Google Scholar]
- 62. Lee JE, Nakagawa T, Kita T, et al. Mechanisms of apoptosis induced by cisplatin in marginal cells in mouse stria vascularis. ORL. 2004;66(3):111‐118. 10.1159/000079329 [DOI] [PubMed] [Google Scholar]
- 63. Lee JE, Nakagawa T, Kim TS, et al. A novel model for rapid induction of apoptosis in spiral ganglions of mice. The Laryngoscope. 2003;113(6):994‐999. 10.1097/00005537-200306000-00015 [DOI] [PubMed] [Google Scholar]
- 64. Clerici WJ, Hensley K, DiMartino DL, Butterfield DA. Direct detection of ototoxicant‐induced reactive oxygen species generation in cochlear explants. Hear Res. 1996;98(1–2):116‐124. 10.1016/0378-5955(96)00075-5 [DOI] [PubMed] [Google Scholar]
- 65. García‐Berrocal JR, Nevado J, Ramírez‐Camacho R, et al. The anticancer drug cisplatin induces an intrinsic apoptotic pathway inside the inner ear. Br J Pharmacol. 2007;152(7):1012‐1020. 10.1038/sj.bjp.0707405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ravi R, Somani SM, Rybak LP. Mechanism of cisplatin ototoxicity: antioxidant system. Pharmacol Toxicol. 1995;76(6):386‐394. 10.1111/j.1600-0773.1995.tb00167.x [DOI] [PubMed] [Google Scholar]
- 67. Rybak LP, Husain K, Morris C, Whitworth C, Somani S. Effect of protective agents against cisplatin ototoxicity. Am J Otology. 2000;21(4):513‐520. [PubMed] [Google Scholar]
- 68. Kopke RD, Liu W, Gabaizadeh R, et al. Use of organotypic cultures of Corti's organ to study the protective effects of antioxidant molecules on cisplatin‐induced damage of auditory hair cells. Am J Otology. 1997;18(5):559‐571. [PubMed] [Google Scholar]
- 69. Banfi B, Malgrange B, Knisz J, Steger K, Dubois‐Dauphin M, Krause KH. NOX3, a superoxide‐generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279(44):46065‐46072. 10.1074/jbc.M403046200 [DOI] [PubMed] [Google Scholar]
- 70. Mukherjea D, Whitworth CA, Nandish S, Dunaway GA, Rybak LP, Ramkumar V. Expression of the kidney injury molecule 1 in the rat cochlea and induction by cisplatin. Neuroscience. 2006;139(2):733‐740. 10.1016/j.neuroscience.2005.12.044 [DOI] [PubMed] [Google Scholar]
- 71. Kim H‐J, Lee J‐H, Kim S‐J, et al. Roles of NADPH oxidases in cisplatin‐induced reactive oxygen species generation and ototoxicity. J Neurosci. 2010;30(11):3933‐3946. 10.1523/jneurosci.6054-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393‐405. 10.1038/nrm4007 [DOI] [PubMed] [Google Scholar]
- 73. Rybak LP, Mukherjea D, Ramkumar V. Mechanisms of cisplatin‐induced ototoxicity and prevention. Seminars in hearing. 2019;40(2):197‐204. 10.1055/s-0039-1684048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dickey DT, Muldoon LL, Doolittle ND, Peterson DR, Kraemer DF, Neuwelt EA. Effect of N‐acetylcysteine route of administration on chemoprotection against cisplatin‐induced toxicity in rat models. Cancer Chemother Pharmacol. 2008;62(2):235‐241. 10.1007/s00280-007-0597-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wimmer C, Mees K, Stumpf P, Welsch U, Reichel O, Suckfüll M. Round window application of d‐methionine, sodium thiosulfate, brain‐derived neurotrophic factor, and fibroblast growth factor‐2 in cisplatin‐induced ototoxicity. Otology Neurotol. 2004;25(1):33‐40. 10.1097/00129492-200401000-00007 [DOI] [PubMed] [Google Scholar]
- 76. Korver KD, Rybak LP, Whitworth C, Campbell KM. Round window application of D‐methionine provides complete cisplatin otoprotection. Otolaryngology–Head Neck Surgery. 2002;126(6):683‐689. 10.1067/mhn.2002.125299 [DOI] [PubMed] [Google Scholar]
- 77. Rybak LP, Whitworth C, Somani S. Application of antioxidants and other agents to prevent cisplatin ototoxicity. Laryngoscope. 1999;109(11):1740‐1744. 10.1097/00005537-199911000-00003 [DOI] [PubMed] [Google Scholar]
- 78. Zhang M, Liu W, Ding D, Salvi R. Pifithrin‐alpha suppresses p53 and protects cochlear and vestibular hair cells from cisplatin‐induced apoptosis. Neuroscience. 2003;120(1):191‐205. 10.1016/s0306-4522(03)00286-0 [DOI] [PubMed] [Google Scholar]
- 79. Wang J, Ladrech S, Pujol R, Brabet P, Van De Water TR, Puel JL. Caspase inhibitors, but not c‐Jun NH2‐terminal kinase inhibitor treatment, prevent cisplatin‐induced hearing loss. Can Res. 2004;64(24):9217‐9224. 10.1158/0008-5472.Can-04-1581 [DOI] [PubMed] [Google Scholar]
- 80. Watanabe K, Inai S, Jinnouchi K, et al. Nuclear‐factor kappa B (NF‐kappa B)‐inducible nitric oxide synthase (iNOS/NOS II) pathway damages the stria vascularis in cisplatin‐treated mice. Anticancer Res. 2002;22(6c):4081‐4085. [PubMed] [Google Scholar]
- 81. Nagy I, Monge A, Albinger‐Hegyi A, Schmid S, Bodmer D. NF‐kappaB is required for survival of immature auditory hair cells in vitro. J Assoc Res Otolaryngol. 2005;6(3):260‐268. 10.1007/s10162-005-0006-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kelly TC, Whitworth CA, Husain K, Rybak LP. Aminoguanidine reduces cisplatin ototoxicity. Hear Res. 2003;186(1–2):10‐16. 10.1016/s0378-5955(03)00303-4 [DOI] [PubMed] [Google Scholar]
- 83. Li G, Liu W, Frenz D. Cisplatin ototoxicity to the rat inner ear: a role for HMG1 and iNOS. Neurotoxicology. 2006;27(1):22‐30. 10.1016/j.neuro.2005.05.010 [DOI] [PubMed] [Google Scholar]
- 84. Lee JE, Nakagawa T, Kim TS, et al. Role of reactive radicals in degeneration of the auditory system of mice following cisplatin treatment. Acta Otolaryngol. 2004;124(10):1131‐1135. 10.1080/00016480410017521 [DOI] [PubMed] [Google Scholar]
- 85. Yamasoba T, Someya S, Yamada C, Weindruch R, Prolla TA, Tanokura M. Role of mitochondrial dysfunction and mitochondrial DNA mutations in age‐related hearing loss. Hear Res. 2007;226(1–2):185‐193. 10.1016/j.heares.2006.06.004 [DOI] [PubMed] [Google Scholar]
- 86. Bowl MR, Dawson SJ. Age‐related hearing loss. Cold Spring Harbor Perspectives Med. 2019;9(8). 10.1101/cshperspect.a033217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Roth TN, Hanebuth D, Probst R. Prevalence of age‐related hearing loss in Europe: a review. European Archives of Oto‐Rhino‐Laryngology. 2011;268(8):1101–1107. 10.1007/s00405-011-1597-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gopinath B, Rochtchina E, Wang JJ, Schneider J, Leeder SR, Mitchell P. Prevalence of age‐related hearing loss in older adults: Blue Mountains Study. Arch Intern Med. 2009;169(4):415‐416. 10.1001/archinternmed.2008.597 [DOI] [PubMed] [Google Scholar]
- 89. Gopinath B, Schneider J, Rochtchina E, Leeder SR, Mitchell P. Association between age‐related hearing loss and stroke in an older population. Stroke. 2009;40(4):1496‐1498. 10.1161/strokeaha.108.535682 [DOI] [PubMed] [Google Scholar]
- 90. Gates GA, Mills JH. Presbycusis. Lancet. 2005;366(9491):1111–1120. 10.1016/S0140-6736(05)67423-5 [DOI] [PubMed] [Google Scholar]
- 91. Lindsten T, Ross AJ, King A, et al. The combined functions of proapoptotic Bcl‐2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6(6):1389‐1399. 10.1016/s1097-2765(00)00136-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Youle RJ, Strasser A. The BCL‐2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47‐59. 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- 93. Someya S, Xu J, Kondo K, et al. Age‐related hearing loss in C57BL/6J mice is mediated by Bak‐dependent mitochondrial apoptosis. Proc Natl Acad Sci USA. 2009;106(46):19432‐19437. 10.1073/pnas.0908786106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sha SH, Chen FQ, Schacht J. Activation of cell death pathways in the inner ear of the aging CBA/J mouse. Hear Res. 2009;254(1–2):92‐99. 10.1016/j.heares.2009.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Alam SA, Oshima T, Suzuki M, Kawase T, Takasaka T, Ikeda K. The expression of apoptosis‐related proteins in the aged cochlea of Mongolian gerbils. Laryngoscope. 2001;111(3):528‐534. 10.1097/00005537-200103000-00026 [DOI] [PubMed] [Google Scholar]
- 96. Hu BH, Yang WP, Bielefeld EC, Li M, Chen GD, Henderson D. Apoptotic outer hair cell death in the cochleae of aging Fischer 344/NHsd rats. Hear Res. 2008;245(1–2):48‐57. 10.1016/j.heares.2008.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Darrat I, Ahmad N, Seidman K, Seidman MD. Auditory research involving antioxidants. Current Opinion Otolaryngol Head Neck Surg. 2007;15(5):358‐363. 10.1097/MOO.0b013e3282efa641 [DOI] [PubMed] [Google Scholar]
- 98. Someya S, Prolla TA. Mitochondrial oxidative damage and apoptosis in age‐related hearing loss. Mechanisms Ageing Development. 2010;131(7–8):480‐486. 10.1016/j.mad.2010.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jiang H, Talaska AE, Schacht J, Sha SH. Oxidative imbalance in the aging inner ear. Neurobiol Aging. 2007;28(10):1605‐1612. 10.1016/j.neurobiolaging.2006.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Seidman MD. Effects of dietary restriction and antioxidants on presbyacusis. Laryngoscope. 2000;110(5 Pt 1):727‐738. 10.1097/00005537-200005000-00003 [DOI] [PubMed] [Google Scholar]
- 101. Le T, Keithley EM. Effects of antioxidants on the aging inner ear. Hear Res. 2007;226(1–2):194‐202. 10.1016/j.heares.2006.04.003 [DOI] [PubMed] [Google Scholar]
- 102. Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1(8639):642‐645. 10.1016/s0140-6736(89)92145-4 [DOI] [PubMed] [Google Scholar]
- 103. Bai U, Seidman MD, Hinojosa R, Quirk WS. Mitochondrial DNA deletions associated with aging and possibly presbycusis: a human archival temporal bone study. Am J Otology. 1997;18(4):449‐453. [PubMed] [Google Scholar]
- 104. Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481‐484. 10.1126/science.1112125 [DOI] [PubMed] [Google Scholar]
- 105. Crawley BK, Keithley EM. Effects of mitochondrial mutations on hearing and cochlear pathology with age. Hear Res. 2011;280(1–2):201‐208. 10.1016/j.heares.2011.05.015 [DOI] [PubMed] [Google Scholar]
- 106. Nelson DI, Nelson RY, Concha‐Barrientos M, Fingerhut M. The global burden of occupational noise‐induced hearing loss. Am J Ind Med. 2005;48(6):446‐458. 10.1002/ajim.20223 [DOI] [PubMed] [Google Scholar]
- 107. Slepecky N. Overview of mechanical damage to the inner ear: noise as a tool to probe cochlear function. Hear Res. 1986;22:307‐321. 10.1016/0378-5955(86)90107-3 [DOI] [PubMed] [Google Scholar]
- 108. Hu BH, Henderson D, Nicotera TM. Involvement of apoptosis in progression of cochlear lesion following exposure to intense noise. Hear Res. 2002;166(1–2):62‐71. 10.1016/s0378-5955(02)00286-1 [DOI] [PubMed] [Google Scholar]
- 109. Ohinata Y, Miller JM, Altschuler RA, Schacht J. Intense noise induces formation of vasoactive lipid peroxidation products in the cochlea. Brain Res. 2000;878(1–2):163‐173. 10.1016/s0006-8993(00)02733-5 [DOI] [PubMed] [Google Scholar]
- 110. Wang J, Ruel J, Ladrech S, Bonny C, van de Water TR, Puel JL. Inhibition of the c‐Jun N‐terminal kinase‐mediated mitochondrial cell death pathway restores auditory function in sound‐exposed animals. Mol Pharmacol. 2007;71(3):654‐666. 10.1124/mol.106.028936 [DOI] [PubMed] [Google Scholar]
- 111. Hu BH, Cai Q, Manohar S, et al. Differential expression of apoptosis‐related genes in the cochlea of noise‐exposed rats. Neuroscience. 2009;161(3):915‐925. 10.1016/j.neuroscience.2009.03.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Niu X, Shao R, Canlon B. Suppression of apoptosis occurs in the cochlea by sound conditioning. NeuroReport. 2003;14(7):1025‐1029. 10.1097/01.wnr.0000070830.57864.32 [DOI] [PubMed] [Google Scholar]
- 113. Nicotera TM, Hu BH, Henderson D. The caspase pathway in noise‐induced apoptosis of the chinchilla cochlea. J Assoc Res Otolaryngol. 2003;4(4):466‐477. 10.1007/s10162-002-3038-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vicente‐Torres MA, Schacht J. A BAD link to mitochondrial cell death in the cochlea of mice with noise‐induced hearing loss. J Neurosci Res. 2006;83(8):1564‐1572. 10.1002/jnr.20832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Lang‐Lazdunski L, Heurteaux C, Mignon A, et al. Ischemic spinal cord injury induced by aortic cross‐clamping: prevention by riluzole. European J Cardio‐Thoracic Surg. 2000;18(2):174‐181. 10.1016/s1010-7940(00)00430-9 [DOI] [PubMed] [Google Scholar]
- 116. Wang J, Dib M, Lenoir M, et al. Riluzole rescues cochlear sensory cells from acoustic trauma in the guinea‐pig. Neuroscience. 2002;111(3):635‐648. 10.1016/s0306-4522(02)00004-0 [DOI] [PubMed] [Google Scholar]
- 117. Yamashita D, Miller JM, Jiang HY, Minami SB, Schacht J. AIF and EndoG in noise‐induced hearing loss. NeuroReport. 2004;15(18):2719‐2722. [PubMed] [Google Scholar]
- 118. Yamasoba T, Schacht J, Shoji F, Miller JM. Attenuation of cochlear damage from noise trauma by an iron chelator, a free radical scavenger and glial cell line‐derived neurotrophic factor in vivo. Brain Res. 1999;815(2):317‐325. 10.1016/s0006-8993(98)01100-7 [DOI] [PubMed] [Google Scholar]
- 119. Ohinata Y, Miller JM, Schacht J. Protection from noise‐induced lipid peroxidation and hair cell loss in the cochlea. Brain Res. 2003;966(2):265‐273. 10.1016/s0006-8993(02)04205-1 [DOI] [PubMed] [Google Scholar]
- 120. Ohlemiller KK, Wright JS, Dugan LL. Early elevation of cochlear reactive oxygen species following noise exposure. Audiology Neuro‐Otology. 1999;4(5):229‐236. 10.1159/000013846 [DOI] [PubMed] [Google Scholar]
- 121. Yamashita D, Jiang HY, Schacht J, Miller JM. Delayed production of free radicals following noise exposure. Brain Res. 2004;1019(1–2):201‐209. 10.1016/j.brainres.2004.05.104 [DOI] [PubMed] [Google Scholar]
- 122. Fetoni AR, Piacentini R, Fiorita A, Paludetti G, Troiani D. Water‐soluble Coenzyme Q10 formulation (Q‐ter) promotes outer hair cell survival in a guinea pig model of noise induced hearing loss (NIHL). Brain Res. 2009;1257:108‐116. 10.1016/j.brainres.2008.12.027 [DOI] [PubMed] [Google Scholar]
- 123. Lorito G, Giordano P, Prosser S, Martini A, Hatzopoulos S. Noise‐induced hearing loss: a study on the pharmacological protection in the Sprague Dawley rat with N‐acetyl‐cysteine. Acta Otorhinolaryngol Ital. 2006;26(3):133‐139. [PMC free article] [PubMed] [Google Scholar]
- 124. Campbell KCM, Meech RP, Klemens JJ, et al. Prevention of noise‐ and drug‐induced hearing loss with D‐methionine. Hear Res. 2007;226(1–2):92‐103. 10.1016/j.heares.2006.11.012 [DOI] [PubMed] [Google Scholar]
- 125. Hight NG, McFadden SL, Henderson D, Burkard RF, Nicotera T. Noise‐induced hearing loss in chinchillas pre‐treated with glutathione monoethylester and R‐PIA. Hear Res. 2003;179(1–2):21‐32. 10.1016/s0378-5955(03)00067-4 [DOI] [PubMed] [Google Scholar]
- 126. Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c‐Jun NH2‐terminal kinases. J Biological Chem. 1996;271(26):15703‐15707. 10.1074/jbc.271.26.15703 [DOI] [PubMed] [Google Scholar]
- 127. Ahn JH, Kang HH, Kim YJ, Chung JW. Anti‐apoptotic role of retinoic acid in the inner ear of noise‐exposed mice. Biochem Biophys Res Comm. 2005;335(2):485‐490. 10.1016/j.bbrc.2005.07.114 [DOI] [PubMed] [Google Scholar]
- 128. Shim HJ, Kang HH, Ahn JH, Chung JW. Retinoic acid applied after noise exposure can recover the noise‐induced hearing loss in mice. Acta Otolaryngol. 2009;129(3):233‐238. 10.1080/00016480802226155 [DOI] [PubMed] [Google Scholar]
- 129. Pirvola U, Xing‐Qun L, Virkkala J, et al. Rescue of hearing, auditory hair cells, and neurons by CEP‐1347/KT7515, an inhibitor of c‐Jun N‐terminal kinase activation. J Neurosci. 2000;20(1):43‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Fridberger A, Flock A, Ulfendahl M, Flock B. Acoustic overstimulation increases outer hair cell Ca2+ concentrations and causes dynamic contractions of the hearing organ. Proc Natl Acad Sci USA. 1998;95(12):7127‐7132. 10.1073/pnas.95.12.7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Minami SB, Yamashita D, Schacht J, Miller JM. Calcineurin activation contributes to noise‐induced hearing loss. J Neurosci Res. 2004;78(3):383‐392. 10.1002/jnr.20267 [DOI] [PubMed] [Google Scholar]
- 132. Xue Q, Li C, Chen J, Guo H, Li D, Wu X. The Protective effect of the endoplasmic reticulum stress‐related factors BiP/GRP78 and CHOP/Gadd153 on noise‐induced hearing loss in guinea pigs. Noise & health. Sep‐Oct. 2016;18(84):247‐255. 10.4103/1463-1741.192481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Jager R, Bertrand MJ, Gorman AM, Vandenabeele P, Samali A. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol Cell. 2012;104(5):259‐270. 10.1111/boc.201100055 [DOI] [PubMed] [Google Scholar]
- 134. Jia H, Yu Z, Ge X, Chen Z, Huang X, Wei Y. Glucocorticoid‐induced leucine zipper protects noise‐induced apoptosis in cochlear cells by inhibiting endoplasmic reticulum stress in rats. Med Mol Morphol. 2020;53(2):73‐81. 10.1007/s00795-019-00232-7 [DOI] [PubMed] [Google Scholar]
- 135. Boya P, González‐Polo R‐A, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(3):1025‐1040. 10.1128/mcb.25.3.1025-1040.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kobayashi S. Choose delicately and reuse adequately: the newly revealed process of autophagy. Biological Pharmaceutical Bulletin. 2015;38(8):1098‐1103. 10.1248/bpb.b15-00096 [DOI] [PubMed] [Google Scholar]
- 137. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323‐335. 10.1038/nature09782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Berliocchi L, Russo R, Maiaru M, Levato A, Bagetta G, Corasaniti MT. Autophagy impairment in a mouse model of neuropathic pain. Molecular Pain. 2011;7:83 10.1186/1744-8069-7-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451‐460. 10.1016/j.cell.2008.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Fujimoto C, Iwasaki S, Urata S, et al. Autophagy is essential for hearing in mice. Cell Death Dis. 2017;8(5):e2780. 10.1038/cddis.2017.194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. de Iriarte RR, Pulido S, Rodríguez‐de la Rosa L, Magariños M, Varela‐Nieto I. Age‐regulated function of autophagy in the mouse inner ear. Hearing Res. 2015;330:39–50. 10.1016/j.heares.2015.07.020 [DOI] [PubMed] [Google Scholar]
- 142. Tsuchihashi NA, Hayashi K, Dan K, et al. Autophagy through 4EBP1 and AMPK regulates oxidative stress‐induced premature senescence in auditory cells. Oncotarget. 2015;6(6):3644‐3655. 10.18632/oncotarget.2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Vernon PJ, Tang D. Eat‐me: autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid Redox Signal. 2013;18(6):677‐691. 10.1089/ars.2012.4810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. He Z, Guo L, Shu Y, et al. Autophagy protects auditory hair cells against neomycin‐induced damage. Autophagy. 2017;13(11):1884‐1904. 10.1080/15548627.2017.1359449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Fang B, Xiao H. Rapamycin alleviates cisplatin‐induced ototoxicity in vivo. Biochem Biophys Res Comm. 2014;448(4):443‐447. 10.1016/j.bbrc.2014.04.123 [DOI] [PubMed] [Google Scholar]
- 146. Guo S, Xu N, Chen P, et al. Rapamycin protects spiral ganglion neurons from gentamicin‐induced degeneration in vitro. J Assoc Res Otolaryngol. 2019;20(5):475‐487. 10.1007/s10162-019-00717-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Saegusa C, Hosoya M, Nishiyama T, et al. Low‐dose rapamycin‐induced autophagy in cochlear outer sulcus cells. Laryngoscope Investigative Otolaryngol. 2020;5(3):520‐528. 10.1002/lio2.392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Yang Q, Sun G, Yin H, et al. PINK1 protects auditory hair cells and spiral ganglion neurons from cisplatin‐induced ototoxicity via inducing autophagy and inhibiting JNK signaling pathway. Free Radic Biol Med. 2018;120:342‐355. 10.1016/j.freeradbiomed.2018.02.025 [DOI] [PubMed] [Google Scholar]
- 149. Mencía Á, Modamio‐Høybjør S, Redshaw N, et al. Mutations in the seed region of human miR‐96 are responsible for nonsyndromic progressive hearing loss. Nat Genet. 2009;41(5):609‐613. 10.1038/ng.355 [DOI] [PubMed] [Google Scholar]
- 150. Bandyopadhyay U, Nagy M, Fenton WA, Horwich AL. Absence of lipofuscin in motor neurons of SOD1‐linked ALS mice. Proc Natl Acad Sci USA. 2014;111(30):11055‐11060. 10.1073/pnas.1409314111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. 2014;35:24‐32. 10.1016/j.semcdb.2014.02.006 [DOI] [PubMed] [Google Scholar]
- 152. Kaiser WJ, Sridharan H, Huang C, et al. Toll‐like receptor 3‐mediated necrosis via TRIF, RIP3, and MLKL. J Biological Chem. 2013;288(43):31268‐31279. 10.1074/jbc.M113.462341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Vercammen D, Brouckaert G, Denecker G, et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188(5):919‐930. 10.1084/jem.188.5.919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Vanlangenakker N, Vanden Berghe T, Krysko DV, Festjens N, Vandenabeele P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med. 2008;8(3):207‐220. 10.2174/156652408784221306 [DOI] [PubMed] [Google Scholar]
- 155. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700‐714. 10.1038/nrm2970 [DOI] [PubMed] [Google Scholar]
- 156. Hitomi J, Christofferson DE, Ng A, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135(7):1311‐1323. 10.1016/j.cell.2008.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Trichonas G, Murakami Y, Thanos A, et al. Receptor interacting protein kinases mediate retinal detachment‐induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA. 2010;107(50):21695‐21700. 10.1073/pnas.1009179107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Zheng HW, Chen J, Sha SH. Receptor‐interacting protein kinases modulate noise‐induced sensory hair cell death. Cell Death Dis. 2014;5(5):e1262. 10.1038/cddis.2014.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Choi MJ, Kang H, Lee YY, et al. Cisplatin‐induced ototoxicity in rats is driven by RIP3‐dependent necroptosis. Cells. 2019;8(5):409 10.3390/cells8050409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ruhl D, Du T‐T, Wagner EL, et al. Necroptosis and apoptosis contribute to cisplatin and aminoglycoside ototoxicity. J Neurosci. 2019;39(15):2951‐2964. 10.1523/jneurosci.1384-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.