

Abstract
Human epidermal growth factor receptor 2 (HER2) amplification occurs in approximately 20% of gastric and gastroesophageal junction cancers in the United States and European Union. Lapatinib, a dual HER2 and epidermal growth factor receptor tyrosine kinase inhibitor, has demonstrated clinical efficacy in HER2‐amplified cancer cells. However, several studies have shown that some cytokines can mediate resistance to lapatinib using their receptor tyrosine kinase (RTK) pathways. One of these, Heregulin1 (HRG1), can confer resistance to lapatinib‐mediated growth inhibition in HER2‐amplified breast cancer cells, but the underlying mechanisms remain unknown. Here, we investigated whether and how HRG1 causes resistance to lapatinib in gastric and gastroesophageal junction cancers in vitro. HER2‐amplified gastric and gastroesophageal junction cancer cell lines were highly sensitive to lapatinib. Exposure to HRG1 together with lapatinib rescued cells from lapatinib‐induced cell cycle arrest and apoptosis. Downregulation of HER3 with siRNA in the presence of HRG1 re‐sensitized HER2‐amplified cancer cells to lapatinib. Immunoblotting analysis indicated that HRG1 re‐activated HER3 and AKT in the presence of lapatinib, which persisted for at least 72 h. Activation of HER3 and downstream AKT was mediated by residual activity of HER2. HRG1‐mediated resistance could be reduced by PI3K/mTOR inhibitors or by complete inhibition of HER2. Thus, we conclude that HRG1 mediates resistance to lapatinib through HER3 and AKT activation, and that this depends on residual HER2 activity. Lapatinib in combination with anti‐PI3K therapies or more potent HER2 inhibitors would improve the efficacy and avoid the emergence of resistant cells.
Here, we investigated whether and how HRG1 causes resistance to lapatinib in gastric and gastroesophageal junction cancers in vitro. Exposure to HRG1 together with lapatinib rescued cells from lapatinib‐induced cell cycle arrest and apoptosis.
Gastric and gastroesophageal junction cancer is one of the leading causes of cancer‐related mortality in the world.1 Despite the recent reported benefits of combination therapies, the prognosis of advanced gastric or gastroesophageal junction cancer remains poor, and new treatments are sorely needed.2, 3 Currently, HER2/neu, a member of the human epidermal growth factor receptor (EGFR) family, has attracted particular attention as a druggable target because it is amplified and/or overexpressed in approximately 20% of gastric and gastroesophageal junction cancers and high levels of HER2 expression are associated with poor patient prognosis.3, 4, 5, 6, 7
Lapatinib (Tykerb, GlaxoSmithKline) is a dual tyrosine kinase inhibitor (TKI) that specifically targets both HER2 and EGFR, thereby inhibiting their activities and their downstream signaling pathways, such as MAPK and AKT.8, 9 Treatment of gastric cancer patients having HER2‐amplified tumors with lapatinib has shown promising results in phase II trials and is currently being tested in phase III trials.10 However, many patients who initially respond to TKIs eventually develop resistance. Several resistance mechanisms against TKIs, including acquired gene mutation, gene amplification or the activation of non‐targeted RTKs by cytokines have been reported.11, 12, 13, 14, 15, 16, 17
HRG1/Neuregulin‐1, known to act as a specific ligand for HER3 and HER4, induces noncovalent heterodimeric complexes of HER3 and HER4 with HER2, and activates multiple biological responses in cells.18, 19 HRG1 expressed in breast, lung, colon, head and neck, and endometrial cancer cells has been shown to contribute to cancer development by activating HER2 through autocrine and/or paracrine mechanisms.20, 21, 22, 23, 24 Gastric fibroblasts also express HRG1, which influences the proliferation of gastric epithelial cells, leading to the suggestion that HRG1 from gastric fibroblasts contributes to for proliferation of gastric and gastroesophageal junction cancer cells through paracrine mechanisms.25, 26 Additionally, some reports showed that HRG1 conferred resistance to HER family‐targeted therapy in breast cancer cell lines, but the underlying mechanisms are poorly understood.17, 27
Here, we investigated whether and how HRG1 confers resistance to lapatinib in gastric and gastroesophageal junction cancers. We found that even with lapatinib treatment there is HER3 and residual HER2 activity, which is needed for HRG1‐meditated resistance to lapatinib and re‐activation of HER3 and AKT signaling. Our data provide a rationale for developing HER2‐targeted therapy with PI3K/mTOR inhibitors, or developing more potent HER2 inhibitors.
Material and Methods
Cell lines
NCI‐N87, SK‐BR‐3 and MRC‐5, a human gastric cancer cell line, a human breast cancer cell line and human fetal lung fibroblasts, respectively, were purchased from the American Type Culture Collection (Manassas, VA, USA). ESO26, KYAE‐1, OE19 and SK‐GT‐2, all human gastroesophageal junction cancer cell lines, were purchased from the European Collection of Cell Cultures (Salisbury, Wiltshire, UK). HSC‐3, a human oral squamous cancer cell line, was purchased from the Japanese Collection of Research Bioresources Cell Bank (Ibaraki, Osaka, Japan). CAF‐54, CAF‐55 and CAF‐56, human gastric cancer‐associated fibroblasts, were established by Dr Yashiro. CAF‐54 was established from a 67‐year‐old man with signet ring cell type gastric carcinoma. CAF‐55 was established from a 78‐year‐old man with a poorly differentiated type of gastric carcinoma. CAF‐56 was established from a 76‐year‐old man with moderately differentiated gastric carcinoma. Primary cultures were initiated as previously reported.28 All cell lines were initially grown and multiple aliquots were stored at −180°C for future use as required. Cells were purchased more than 6 months ago and were not further tested or authenticated by the authors. Cell lines were cultured in DMEM supplemented with 10% fetal calf serum, 100 units/mL penicillin and 100 units/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Informed consent for cell preparation was obtained in accordance with the guidelines of the Osaka City University Institutional Review Board authorization for this study.
Chemicals and growth factors
Lapatinib and erlotinib were purchased from Sequoia Research Products (Pangbourne, Berkshire, UK). LY294002, everolimus, crizotinib, NVP‐BGJ398, AZD0530, PF‐562271 and BIBW2992 were purchased from Selleck Chemicals (Houston, TX, USA). Trastuzumab was purchased from Roche (Basel, Switzerland). Human HRG1‐α and HRG1‐β were purchased from R&D Systems (Minneapolis, MN, USA).
Quantitative PCR
For the analysis of HER2 gene amplification, Taqman probe Hs00223580 for the HER2 gene was obtained from Applied Biosystems (Carlsbad, CA, USA). Quantitative PCR was performed with TaqMan Copy Number Assays (Applied Biosystems) following the manufacturer′s protocol. Genomic DNA was isolated from each cell line using Blood & Cell Culture DNA Kits (Qiagen, Hilden, NRW, Germany).
For the analysis of HRG1 mRNA expression, Taqman probes for HRG1‐α and HRG1‐β were obtained from Applied Biosystems (Hs01103794_m1 and Hs00247624_m1, respectively). Quantitative PCR was performed with Taqman Gene Expression Assays (Applied Biosystems) following the manufacturer′s protocol. A beta‐actin probe (Applied Biosystems) was used as the internal control. Total RNA was isolated from each cell line using RNeasy Plus Mini Kits (Qiagen). cDNA was synthesized from RNA using High Capacity RNA‐to‐cDNA kits (Applied Biosystems).
Western blotting
Cells were treated as indicated in serum‐supplemented medium. After removal of medium, the tissue culture plates were placed on ice and the cells washed twice with ice‐cold PBS. Cells were then scraped off and placed in ice‐cold RIPA lysis buffer (Sigma, St Louis, MO, USA) containing protease and phosphatase inhibitor cocktails (Roche). After sonication, homogenates were centrifuged at 20 000g for 15 min and the lysates stored at −80°C until further use. For Western blotting, equal amounts of protein (20 μg) were boiled in Laemmli buffer for 5 min, resolved by 7.5–10% SDS‐PAGE (Bio‐Rad Laboratories, Hercules, CA, USA), and electrophoretically transferred onto polyvinylidene difluoride membranes (Bio‐Rad Laboratories). After blocking nonspecific binding sites with 4% Block‐ace (DS Pharma Biomedical, Suita, Osaka, Japan) in TBS + 0.05% Tween 20 (TBS‐T), the membrane was incubated with the respective antibodies overnight at 4°C. After three washes with TBS‐T, the membrane was incubated for 30 min at room temperature with a horseradish peroxidase (HRP)‐linked secondary antibody, followed by several washes with TBS‐T. The immunocomplexes were visualized with the ECL Plus Western Blotting detection system (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Antibodies
Epidermal growth factor receptor, HER2, AKT, ERK, PARP, S6 ribosomal protein, phospho‐EGFR (Tyr1068), phospho‐HER2 (Tyr1248), phospho‐HER3 (Tyr1289), phospho‐AKT (Ser473), phospho‐ERK (Thr202 and Tyr204), phospho‐S6 ribosomal protein (Ser235/236) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). HER3 antibody was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). α‐tubulin and actin antibodies were purchased from SIGMA.
Cell proliferation assay
Cell proliferation was measured by WST‐8 kits (Kishida Chemical, Osaka, Japan). Briefly, the cells were seeded in quadruplicates in flat‐bottomed 96‐well plates at 3000 cells per well and allowed to adhere for 24 h. Thereafter, the cells were treated as indicated. After incubation with WST‐8 reagent for 2–7 h, absorbance at 450 and 650 nm was measured. IC50 values were calculated using Excel 2010 software (Microsoft, Redmond, WA, USA).
Cell‐cycle analysis
Cells were seeded into 100 mm dishes at a density of 1 × 106 per dish and allowed to adhere for 24 h. The cells were first cultured with or without lapatinib for 30 min, then with or without HRG1 for a further 72 h. Cells were harvested and stained with live/dead fixable dead cell stain (Invitrogen, Carlsbad, CA, USA). After fixation in 4% paraformaldehyde solution, cells were permeabilized with 0.2% TritonX‐100. Nuclear staining was then performed with FxCycle Far Red stain (Invitrogen) with RNaseA. Quantification of staining intensity for cell‐cycle distribution analysis was done using a MACSquant Analyzer (Miltenyi Biotech, Bergisch Gladbach, NRW, Germany).
Apoptosis assay
Apoptosis was measured using Annexin V‐APC Apoptosis Detection Kits (eBioscience, San Diego, CA, USA). Briefly, after harvesting with trypsin‐EDTA, 1 × 106 cells in 200 μL of binding buffer were stained directly with Annexin V‐APC. After incubating for 15 min at room temperature, the samples were incubated with PI. Data were acquired using a MACSquant Analyzer.
Small interfering RNA
For small interfering RNA (siRNA) experiments, cells were seeded in quadruplicate in 96‐well plates at 3 000 cells per well in antibiotic‐free complete medium. Thereafter the cells were transfected with 1 nmol/L of silencer HER3 siRNA (s4778 and s4779 from Applied Biosystems) or nontargeting siRNA using Lipofectamine RNAiMAX (Invitrogen). After 24 h, the cells were treated as indicated. Cell proliferation was determined after 72 h of incubation.
Results
HRG1 rescues HER2‐amplified gastric and gastroesophageal junction cancer cells from lapatinib‐induced growth inhibition
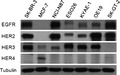
The gastric and gastroesophageal junction cancer cell lines selected for this study, NCI‐N87, ESO26, KYAE‐1, OE19 and SK‐GT‐2, had varying degrees of HER2 gene amplification and HER family protein expression as determined by quantitative PCR and Western blotting (Fig. 1a,b). NCI‐N87 and OE19 overexpressed and ESO26, KYAE‐1 and SK‐GT‐2 moderately expressed the HER2 protein. Epidermal growth factor receptor was overexpressed in NCI‐N87 and SK‐GT‐2. Only SK‐GT‐2 did not express HER3, but none of the cell lines expressed HER4.
Figure 1.
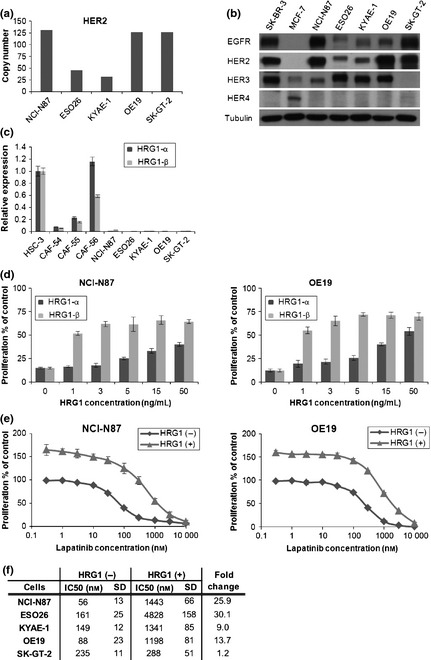
HRG1 induces resistance to lapatinib‐mediated growth inhibition. (a) DNA from five cell lines was subjected to quantitative polymerase chain reaction (PCR) analysis. Mean HER2 copy number is relative to the copy number of MRC‐5 human fetal lung fibroblasts, as 2. RNaseP was used for calibration. (b) Western blotting was done with whole‐cell lysate from each cell line and HER family expression analyzed. SK‐BR‐3 and MCF‐7, breast cancer cell lines, were used as the positive control. Tubulin was used as the loading control. (c) Quantitative PCR was performed for each HRG1 isoform with a specific probe. HSC‐3, a human oral squamous cancer cell line, was used as the positive control and reference.23 (d) NCI‐N87 and OE19 were first incubated with 1 μmol/L lapatinib for 30 min, then cultured with titrated doses of HRG1‐α1 or HRG1‐β1 for 72 h. Cell proliferation was analyzed with an MTT‐based method. All cell proliferation data were averaged from quadruplicates of three independent experiments. (e) NCI‐N87 and OE19 were incubated with titrated doses of lapatinib for 30 min, then cultured with or without 5 ng/mL HRG1 for 72 h. Cell proliferation was analyzed as in (d). (f) IC50s of cells treated with or without 5 ng/mL HRG1 and titrated doses of lapatinib for 72 h. Cell proliferation was analyzed as in (d). Bars, SD.
HRG1‐α and HRG1‐β are expressed in human gastric fibroblasts.25 Here, we examined the expression of HRG1 isoforms in gastric and gastroesophageal junction cancer cells and gastric cancer‐associated fibroblasts. Interestingly, both HRG1‐α and HRG1‐β were more highly expressed in cancer‐associated fibroblasts than in cancer cells (Fig. 1c). To determine whether HRG1 can rescue HER2‐amplified gastric cancer cells from lapatinib‐induced growth inhibition, NCI‐N87 was treated with 1 μmol/L lapatinib and increasing concentrations of HRG1‐α or β for 72 h. The results showed that both forms of HRG1 rescued cells from lapatinib‐induced growth inhibition in a dose‐dependent manner and that HRG1‐β was more potent than HRG1‐α in this respect (Fig. 1d). Therefore, we decided to use HRG1‐β for further experiments.
Next, we assessed whether HRG1 can also rescue gastroesophageal junction cancer cell lines from lapatinib‐induced growth inhibition. It was found that all cell lines except for SK‐GT‐2, which did not express HER3, acquired resistance to lapatinib (Fig. 1e,f).
HRG1 abrogates cell cycle arrest and inhibits apoptosis induced by lapatinib treatment
Lapatinib induces G0‐G1 cell cycle arrest and apoptosis in HER2‐amplified cancer cells.4 To confirm that HRG1 confers resistance to lapatinib by facilitating escape from cell‐cycle arrest and inhibition of apoptosis, NCI‐N87 and OE19 were incubated for 72 h with or without lapatinib ± HRG1 and cell cycle analysis was performed by flow cytometry. Figure 2(a) shows that the addition of HRG1 to lapatinib‐treated cells reversed cell‐cycle from G0‐G1 to S and G2/M. We also observed that HRG1 decreased the percentage of apoptotic cells on lapatinib treatment, as assessed by Annexin V/PI staining and the presence of cleaved PARP proteins (Fig. 2b,c). These results indicate that HRG1 reverses lapatinib‐mediated cell cycle arrest and decreases apoptosis.
Figure 2.
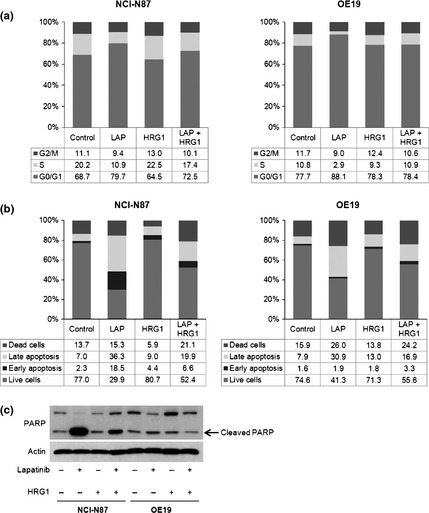
Lapatinib‐mediated G0‐G1 arrest and apoptosis is abrogated by HRG1. (a) NCI‐N87 and OE19 were incubated with or without 1 μmol/L lapatinib ± 5 ng/mL HRG1 for 72 h. Cell cycle analysis was done as described in Material and Methods. Representative results of three independent experiments. (b) NCI‐N87 and OE19 were incubated with or without 1 μmol/L lapatinib ± ng/mL HRG1 for 72 h. Apoptosis detection was done with Annexin V/PI staining. Flow cytometric analysis was as described in Material and Methods. Representative results of three independent experiments. (c) NCI‐N87 and OE19 were incubated with or without 1 μmol/L lapatinib ± 5 ng/mL HRG1 for 72 h. Whole‐cell lysates were collected and protein lysate (20 μg) subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blotting with anti‐PARP antibody. Actin was used as the loading control.
HER3 is necessary for HRG1‐mediated resistance to lapatinib
HRG1 is a known ligand for HER3 and HER4. None of the cell lines that we used expressed HER4, so to determine whether HER3 is responsible for lapatinib resistance caused by HRG1 treatment, NCI‐N87 and OE19 were transfected with nonsilencing siRNA, HER3‐targeting siRNA, or no siRNA. We confirmed that cells transfected with nonsilencing siRNA showed no significant decrease in HER3 protein compared with cells not transfected with any siRNA. In contrast, HER3 protein was significantly downregulated in both NCI‐N87 and OE19 following transfection with HER3 siRNA (Fig. 3a). When both transfected groups of NCI‐N87 and OE19 cells were additionally treated with lapatinib and HRG1, the HRG1‐mediated resistance to lapatinib was now suppressed (Fig. 3b). These results indicate that HER3 is responsible for HRG1‐mediated lapatinib resistance.
Figure 3.
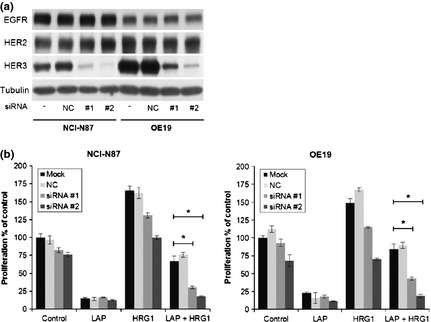
HER3 siRNA restores lapatinib‐mediated growth inhibition. (a) NCI‐N87 and OE19 were transfected with negative control siRNA or HER3‐targeting siRNA, or not transfected, for 72 h. Whole‐cell lysate was collected and protein lysates (20 μg) subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blotting with the indicated antibodies. (b) Transfected cells were first incubated with dimethylsulfoxide (DMSO) or 1 μmol/L lapatinib for 30 min, then treated with or without 5 ng/mL HRG1 for 72 h. Cell proliferation was analyzed as described in Material and Methods. All cell proliferation data were averaged from quadruplicates of three independent experiments. Bars, SD. *t‐test, P < 0.001.
PI3K/mTOR signaling, not MAPK signaling, is required for HRG1‐mediated lapatinib resistance
Lapatinib dephosphorylated EGFR, HER2 and HER3, as well as MAPK and AKT in NCI‐N87 and OE19 cells (Fig. 4a and data not shown). The addition of HRG1 to lapatinib‐treated cancer cells resulted in phosphorylated HER3 and AKT, but not MAPK, within 2 h. Phosphorylated HER3 and AKT were increased in a time‐dependent manner. HER3 protein was upregulated especially in lapatinib‐treated cells, consistent with the recent report by Garrett et al. in breast cancer cell lines.15 Interestingly, phosphorylated HER2 was slightly restored at 24 h regardless of HRG1 treatment, while HRG1 increased phosphorylated HER2 more. Epidermal growth factor receptor expression was unexpectedly decreased in both groups in a time‐dependent manner. The underlying mechanisms for this are not known.
Figure 4.
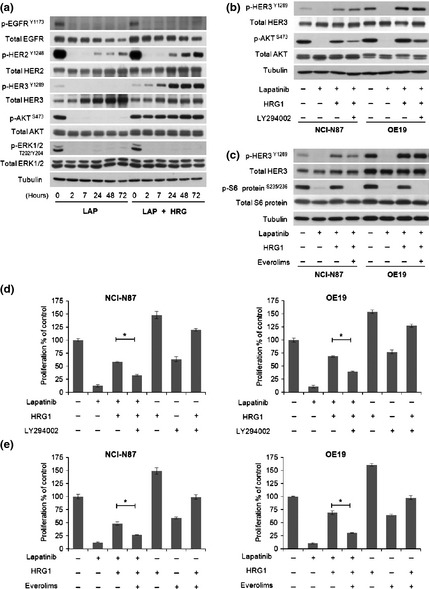
PI3K/mTOR inhibitors restore lapatinib‐mediated growth inhibition.(a) NCI‐N87 and OE19 were first incubated with 1 μmol/L lapatinib for 30 min, then treated with or without 5 ng/mL HRG1 for the time indicated. Whole cell lysates were subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blotting with the indicated antibodies. Tubulin was used as the loading control. (b and c) NCI‐N87 and OE19 were first incubated with 1 μmol/L lapatinib ± 10 μmol/L LY294002 or 1 μmol/L everolimus for 30 min, then treated with or without 5 ng/mL HRG1 for 2 h. Whole‐cell lysate was collected and protein lysates (20 μg) subjected to SDS‐PAGE and Western blotting with the indicated antibodies. Tubulin antibody was used as the loading control. (d and e) NCI‐N87 and OE19 were first incubated with 1 μmol/L lapatinib ± 10 μmol/L LY294002 or 1 μmol/L everolimus for 30 min, then treated with or without 5 ng/mL HRG1 for 72 h. Cell proliferation was analyzed as in (d) bars, SD. *t‐test, P < 0.001.
AKT is phosphorylated in the PI3K pathway and activates downstream signaling pathways. To confirm that PI3K/AKT/mTOR signaling is responsible for HRG1‐mediated resistance to lapatinib, we treated cells with combinations of lapatinib and PI3K/AKT/mTOR signaling inhibitors, namely, LY294002 (PI3K inhibitor) and everolimus (mTOR inhibitor, Affinitor, Roche), in the presence of HRG1. Both inhibitors suppressed the phosphorylation of downstream signaling pathways, AKT for PI3K and S6 ribosomal protein for mTOR respectively (Fig. 4b,c), and reduced the HRG1‐mediated resistance to lapatinib (P < 0.001, Fig. 4d,e). These results indicate that HRG1 induces resistance to lapatinib in association with activation of the PI3K/AKT/mTOR pathway.
HRG1‐mediated resistance to lapatinib depends on residual HER2 activity
HER3 was phosphorylated in cancer cells after 2 h of HRG1 treatment in the presence of lapatinib. Some tyrosine kinases have been reported to associate with and phosphorylate HER3.29, 30, 31, 32 Therefore, by using small‐molecule inhibitors, we investigated whether tyrosine kinases other than HER2 can potentially be involved in HER3 phosphorylation and HRG1‐mediated resistance to lapatinib. Kinases that have been implicated as associated with HER3 include EGFR, Src, c‐MET, FGFR2 and PYK2. We treated cells with erlotinib (Tarceva, Roche), AZD0530, crizotinib (Xalkori, Pfizer), NVP‐BGJ398 or PF‐562271, which are TKIs of EGFR, Src, c‐MET, FGFR and PYK2, respectively, in the presence of lapatinib. After 30 min, HRG1 was additionally added to the cells. Immunoblotting showed that none of these small molecules dampened the recovery of HER3 and AKT phosphorylation (Fig. S1). We also evaluated whether these inhibitors abrogate HRG‐mediated lapatinib resistance, but found that none reversed the resistance (Fig. S2).
Higher concentrations of lapatinib inhibited the proliferation of HRG1‐treated cells as well as untreated cells (Fig. 1e). We hypothesized that incomplete inhibition of HER2 activity by the dose of 1 μmol/L lapatinib used here might explain the recovery of HER3 phosphorylation and HRG1‐mediated resistance. To test this, we first examined whether increasing concentrations of lapatinib block HER3 phosphorylation. When we exposed the immunoblotting film for longer periods, residual HER2 phosphorylation was observed at lower concentrations of lapatinib, while higher concentrations inhibited residual HER2 phosphorylation and decreased HER3 and AKT phosphorylation (Fig. 5a). Next, we tested whether combinations of other HER2 inhibitors, BIBW2992 and trastuzumab (Herceptin, Roche), can block HER3 phosphorylation. BIBW2992 is a small‐molecule inhibitor that covalently binds Cys805 in HER2. Because of the different mechanisms of HER2 inhibition used by lapatinib and BIBW2992, this combination has used to inhibit HER2 more completely in vitro.15 Trastuzumab blocks the phosphorylation of HER3 that results from ligand‐independent but not ligand‐dependent HER2–HER3 interactions. Lapatinib in combination with BIBW2992 inhibited the phosphorylation of HER2, HER3 and AKT and reduced the HRG1‐mediated rescue effects more than each inhibitor alone (Fig. 5b,c), indicating that residual HER2 activity is involved in HRG1‐mediated resistance to lapatinib. Although lapatinib in combination with trastuzumab slightly inhibited HER3 and AKT phosphorylation only in NCI‐N87, the rescue effects were not similar (Fig. S3A,B).
Figure 5.
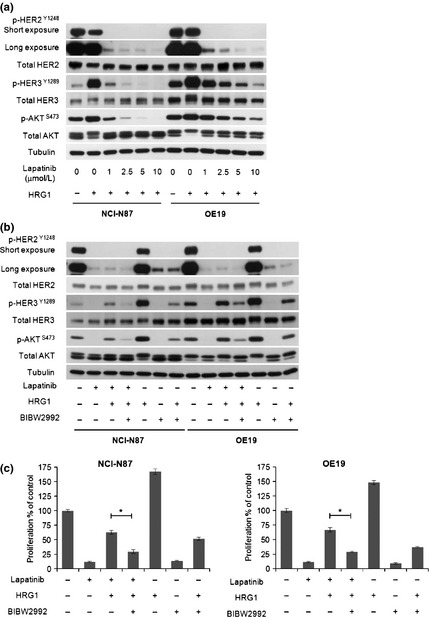
HRG1‐mediated resistance to lapatinib depends on residual HER2 activity. (a) NCI‐N87 and OE19 were first incubated with titrated doses of lapatinib for 30 min, then cultured with 5 ng/mL HRG1 for 2 h. Whole cell lysates were subjected to sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blotting with the indicated antibodies. Tubulin antibody was used as the loading control. (b) NCI‐N87 and OE19 were first incubated with 1 μmol/L lapatinib ± 25 nmol/L BIBW2992 for 30 min, then cultured with or without 5 ng/mL HRG1 for 2 h. Whole cell lysates were Western blotted as in (a). (c) NCI‐N87 and OE19 were first incubated with 1 μmol/L lapatinib ± 25 nmol/L BIBW2992 for 30 min, then cultured with or without 5 ng/mL HRG1 for 72 h. Cell proliferation was analyzed as described in Material and Methods. All cell proliferation data were averaged from quadruplicates of three independent experiments. Bar, SD. *t‐test, P < 0.001.
Discussion
Gastric and gastroesophageal junction cancer represents a major global health care problem. Despite recent improvements in combination therapies, the prognosis of advanced gastric and gastroesophageal junction cancer remains poor, and new and better treatments are urgently needed. Currently, HER2 has attracted particular attention as a drug target because approximately 20% of gastric and gastroesophageal junction cancer patients have amplified and/or overexpressed HER2, and high levels of HER2 are associated with worse clinical outcome.3, 4, 5, 6, 7 In 2010, the first HER2‐targeting drug, trastuzumab, was approved for HER2‐positive metastatic gastric and gastroesophageal junction cancer. Following the success of trastuzumab, lapatinib has been assessed in phase III clinical trials comparing first‐line therapy with or without this drug in HER2‐positive gastric, gastroesophageal junction and esophageal cancer. The results of this trial indicate that Asian may benefit more from the combination with lapatinib than North Americans.33
Molecular targeted therapies are effective in patients whose tumors express the appropriate targets. However, the emergence of resistance to molecular targeting therapies is a serious problem. Numerous studies have reported several resistance mechanisms against TKIs, including acquired gene mutation, gene amplification or the activation of different RTKs by cytokines.11, 12, 13, 14, 15, 16, 17 HRG1 has been shown to induce resistance to molecular therapies targeting members of the HER family of RTKs in breast cancer cells, but the underlying mechanisms are unknown.17, 27 The expression of HRG1 has been shown in breast, lung, colon, head and neck, and endometrial cancer cells and also in gastric fibroblasts.21, 22, 23, 24, 25 Here, we assessed whether HRG1 confers a growth advantage on and resistance to lapatinib treatment in HER2‐positive gastric and gastroesophageal junction cancer cells and investigated the underlying mechanisms of such HRG1‐mediated resistance.
Our results showed that gastric cancer‐associated fibroblasts expressed HRG1 at higher levels than HER2‐amplifed gastric and gastroesophageal junction cancer cell lines. HRG1 reversed lapatinib‐mediated growth inhibition in the cancer cell lines, with the exception of the HER3‐negative line SK‐GT‐2. Cell cycle arrest and apoptosis induced by lapatinib were reduced by HRG1 treatment of both NCI‐N87 and OE19, albeit apoptosis was more obvious in the former. The different mechanisms of action of lapatinib and HRG1 might depend on cell type or protein expression levels of HER2 and HER3.9
Downregulation of HER3 expression with siRNA abrogated the rescue effect of HRG1 and completely restored lapatinib‐mediated growth inhibition. These results indicated that HER3 is responsible for HRG1‐mediated resistance to lapatinib. HER3 overexpression is significantly associated with advanced TNM stage and strongly associated with tumor progression and poor prognosis in gastric cancer.34 This suggests that HER3 overexpression might correlate with lapatinib resistance in cancer patients.
Recently, Garrett et al. found that lapatinib induces transcriptional and posttranslational upregulation of HER3 and pHER3 was recovered after 24 h of lapatinib treatment in breast cancer cell lines.15, 35 Although HER3 was upregulated in gastric and gastroesophageal junction cancer cell lines by lapatinib, pHER3 was not recovered without HRG1. This discrepancy might depend on cell type and derivation. Additionally, exposure to HRG1 and lapatinib together induced less upregulation of HER3 than lapatinib alone. Because inhibition of AKT activity induces HER3 expression via feedback mechanisms,36 HRG1‐mediated AKT activation might suppress this feedback. Interestingly, pHER2 was slightly restored by 24 h of lapatinib treatment regardless of the presence of HRG1. Lapatinib treatment results in the accumulation of HER2 on the cell surface; thus, this phosphorylation effect might depend on homodimerization of surface HER2.37, 38 We also discovered that lapatinib reduced EGFR expression in a time‐dependent manner. Epidermal growth factor receptor activity is necessary for its src‐mediated degradation39 but EGFR is one of the targets of lapatinib and was dephosphorylated in our experiments. Dephosphorylated EGFR might be degraded by some other unknown mechanism.
pAKT, but not MAPK, recovered under the influence of HRG1 in the presence of lapatinib. LY294002 and everolimus, PI3K and mTOR inhibitors, respectively, reduced the rescue effects. This result indicates that the PI3K/AKT/mTOR pathway is a pathway for HRG1‐mediated resistance to lapatinib. Everolimus already finished testing in phase III clinical trials for advanced gastric cancer in 2012, but failed to meet the primary endpoint of improved overall survival.40 Our data suggest that combination therapy with lapatinib and everolimus in HER2‐amplified gastric and gastroesophageal junction cancer may improve efficacy and prevent the emergence of resistant cells.
pHER3 was recovered by HRG1 treatment even in the presence of lapatinib. Although EGFR, Src, c‐MET, FGFR2 and PYK2, are known to associate with HER3, none of the inhibitors of these kinases suppressed the HRG1‐mediated phosphorylation of HER3 and AKT, and did not restore lapatinib‐mediated growth inhibition. We therefore hypothesized that residual HER2 activity despite the presence of lapatinib is responsible for HRG1‐mediated HER3 phosphorylation. Although we could not clearly demonstrate direct cross‐talk through HER2/HER3 heterodimers by HRG1 in the presence of lapatinib by coimmunoprecipitation experiments (data not shown), higher concentrations of lapatinib or a combination of lapatinib with BIBW2992 (but not trastuzumab), further suppressed phosphorylation of HER2, pHER3 and pAKT, and reduced HRG1‐mediated rescue effects. HER3 is thought to be an inactive pseudokinase, although Shi et al. recently reported that it can have low level kinase activity and can trans‐autophosphorylate its intracellular region. These investigators also found that lapatinib was unable to inhibit trans‐phosphorylation of HER3 in vitro.41 Our results and theirs support the notion that residual HER2 phosphorylates HER3 in the presence of HRG1. Higher concentrations than we used in vitro are not achievable in patients, at least not in breast cancer, using the current daily schedule.15 Although we have not formally confirmed the lapatinib concentration achievable in gastric and gastroesophageal junction cancer patients, it is unlikely that higher concentrations could be achieved in these patients either.
PI3K/mTOR inhibition and complete HER2 inhibition significantly reduced HRG1‐mediated rescue effects, but neither treatment completely reversed growth inhibition. HER3 siRNA experiments indicate that interactions between HER3 and unknown targets may be involved in these rescue effects. Recently, anti‐HER3 therapies such as antibody treatment and use of antisense oligonucleotides were reported to improve the efficacy of lapatinib in vitro and in vivo.33, 42 Combinations of anti‐HER2 and anti‐HER3 therapies with lapatinib might be effective for overcoming HRG1‐mediated resistance.
In summary, HRG1 induces resistance to lapatinib through interactions between HRG1 and HER3, residual HER2 activity and the PI3K/AKT/mTOR pathway. PI3K/mTOR inhibition or more complete HER2 inhibition abrogated the HRG1‐mediated rescue effect. These results indicate that lapatinib in combination with anti‐PI3K therapies or more potent HER2 inhibitors might be more potent strategies for treating HER2‐positive gastric and gastroesophageal junction cancer patients.
Disclosure Statement
Yuji Sato is an employee of Shionogi pharmaceutical.
Supporting information
Fig. S1. Other inhibitors cannot block HRG1‐mediated phosphorylation of HER3 and AKT.
Fig. S2. Other inhibitors cannot abrogate HRG1‐mediated resistance.
Fig. S3. Trastuzumab cannot inhibit HRG1‐mediated phosphorylation of HER3 and AKT and HRG1‐mediated resistance to lapatinib.
(Cancer Sci 2013; 104: 1618–1625)
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63: 11–30. [DOI] [PubMed] [Google Scholar]
- 2. Cunningham D, Starling N, Rao S et al Capecitabine and oxaliplatin for advanced esophagogastric cancer. New Engl J Med 2008; 358: 36–46. [DOI] [PubMed] [Google Scholar]
- 3. Bang YJ, Van Cutsem E, Feyereislova A et al Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet 2010; 376: 687–97. [DOI] [PubMed] [Google Scholar]
- 4. Wainberg ZA, Anghel A, Desai AJ et al Lapatinib, a dual EGFR and HER2 kinase inhibitor, selectively inhibits HER2‐amplified human gastric cancer cells and is synergistic with trastuzumab in vitro and in vivo. Clin Cancer Res 2010; 16: 1509–19. [DOI] [PubMed] [Google Scholar]
- 5. Park D, Yun JW, Park JH et al HER‐2/neu amplification is an independent prognostic factor in gastric cancer. Dig Dis Sci 2006; 51: 1371–9. [DOI] [PubMed] [Google Scholar]
- 6. Zhang XL, Yang YS, Xu DP et al Comparative study on overexpression of HER2/neu and HER3 in gastric cancer. World J Surg 2009; 33: 2112–8. [DOI] [PubMed] [Google Scholar]
- 7. De Vita F, Giuliani F, Silvestris N, Catalano G, Ciardiello F, Orditura M. Human epidermal growth factor receptor 2 (HER2) in gastric cancer: a new therapeutic target. Cancer Treat Rev 2010; 36 (Suppl. 3): S11–5. [DOI] [PubMed] [Google Scholar]
- 8. Xia W, Mullin RJ, Keith BR et al Anti‐tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 2002; 21: 6255–63. [DOI] [PubMed] [Google Scholar]
- 9. Kim JW, Kim HP, Im SA et al The growth inhibitory effect of lapatinib, a dual inhibitor of EGFR and HER2 tyrosine kinase, in gastric cancer cell lines. Cancer Lett 2008; 272: 296–306. [DOI] [PubMed] [Google Scholar]
- 10. Iqbal S, Goldman B, Fenoglio‐Preiser CM et al Southwest Oncology Group study S0413: a phase II trial of lapatinib (GW572016) as first‐line therapy in patients with advanced or metastatic gastric cancer. Ann Oncol 2011; 22: 2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jänne PA. Challenges of detecting EGFR T790M in gefitinib/erlotinib‐resistant tumors. Lung Cancer 2008; 60 (Suppl. 2): s3–9. [DOI] [PubMed] [Google Scholar]
- 12. Pao W, Miller VA, Politi KA et al Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2(3): e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu L, Greger J, Shi H et al Novel mechanism of lapatinib resistance in HER2‐positive breast tumor cells: activation of AXL. Cancer Res 2009; 69: 6871–8. [DOI] [PubMed] [Google Scholar]
- 14. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 15. Garrett JT, Olivares MG, Rinehart C et al Transcriptional and posttranslational up‐regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci USA 2011; 108: 5021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen CT, Kim H, Liska D, Gao S, Christensen JG, Weiser MR. MET activation mediates resistance to lapatinib inhibition of HER2‐amplified gastric cancer cells. Mol Cancer Ther 2012; 11: 660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilson TR, Fridlyand J, Yan Y et al Widespread potential for growth‐factor‐driven resistance to anticancer kinase inhibitors. Nature 2012; 487: 505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horan T, Wen J, Arakawa T et al Binding of Neu differentiation factor with the extracellular domain of Her2 and Her3. J Biol Chem 1995; 270: 24604–8. [DOI] [PubMed] [Google Scholar]
- 19. Hijazi MM, Thompson EW, Tang C et al Heregulin regulates the actin cytoskeleton and promotes invasive properties in breast cancer cell lines. Int J Oncol 2000; 17: 629–41. [DOI] [PubMed] [Google Scholar]
- 20. Li Q, Ahmed S, Loeb JA. Development of an autocrine neuregulin signaling loop with malignant transformation of human breast epithelial cells. Cancer Res 2004; 64: 7078–85. [DOI] [PubMed] [Google Scholar]
- 21. Gollamudi M, Nethery D, Liu J, Kern JA. Autocrine activation of ErbB2/ErbB3 receptor complex by HRG1‐1 in non‐small cell lung cancer cell lines. Lung Cancer 2004; 43: 135–43. [DOI] [PubMed] [Google Scholar]
- 22. Venkateswarlu S, Dawson DM, St Clair P, Gupta A, Willson JK, Brattain MG. Autocrine heregulin generates growth factor independence and blocks apoptosis in colon cancer cells. Oncogene 2002; 21: 78–86. [DOI] [PubMed] [Google Scholar]
- 23. Wilson TR, Lee DY, Berry L, Shames DS, Settleman J. Neuregulin‐1‐mediated autocrine signaling underlies sensitivity to HER2 kinase inhibitors in a subset of human cancers. Cancer Cell 2011; 20: 158–72. [DOI] [PubMed] [Google Scholar]
- 24. Srinivasan R, Benton E, McCormick F, Thomas H, Gullick WJ. Expression of the c‐erbB‐3/HER‐3 and c‐erbB‐4/HER‐4 growth factor receptors and their ligands, neuregulin‐1 alpha, neuregulin‐1 beta, and betacellulin, in normal endometrium and endometrial cancer. Clin Cancer Res 1999; 5: 2877–83. [PubMed] [Google Scholar]
- 25. Noguchi H, Sakamoto C, Wada K et al Expression of heregulin alpha, erbB2, and erbB3 and their influences on proliferation of gastric epithelial cells. Gastroenterology 1997; 117: 1119–27. [DOI] [PubMed] [Google Scholar]
- 26. Nagata K, Wada K, Tatsuguchi A et al Heregulin‐alpha and heregulin‐beta expression is linked to a COX‐2‐PGE2 pathway in human gastric fibroblasts. Am J Physiol Gastrointest Liver Physiol 2006; 290: G1243–51. [DOI] [PubMed] [Google Scholar]
- 27. Hutcheson IR, Knowlden JM, Hiscox SE et al Heregulin beta1 drives gefitinib‐resistant growth and invasion in tamoxifen‐resistant MCF‐7 breast cancer cells. Breast Cancer Res 2007; 9: R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fuyuhiro Y, Yashiro M, Noda S et al Upregulation of cancer‐associated myofibroblasts by TGF‐β from scirrhous gastric carcinoma cells. Br J Cancer 2011; 105: 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ishizawar RC, Miyake T, Parsons SJ. c‐Src modulates ErbB2 and ErbB3 heterocomplex formation and function. Oncogene 2007; 26: 3503–10. [DOI] [PubMed] [Google Scholar]
- 30. Guo A, Villén J, Kornhauser J et al Signaling networks assembled by oncogenic EGFR and c‐Met. Proc Natl Acad Sci USA 2008; 105: 692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kunii K, Davis L, Gorenstein J et al FGFR2‐amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res 2008; 68: 2340–8. [DOI] [PubMed] [Google Scholar]
- 32. Van der Horst EH, Weber I, Ullrich A. Tyrosine phosphorylation of PYK2 mediates heregulin‐induced glioma invasion: novel heregulin/HER3‐stimulated signaling pathway in glioma. Int J Cancer 2005; 113: 689–98. [DOI] [PubMed] [Google Scholar]
- 33. Hecht JR, Bang YJ, Qin S et al Lapatinib in combination with capecitabine plus oxaliplatin (CapeOx) in HER2‐positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma (AC): the TRIO‐013/LOGiC Trial. J Clin Oncol (Meeting abstracts) 2013; 31: Abstract LBA4001. [DOI] [PubMed] [Google Scholar]
- 34. Hayashi M, Inokuchi M, Takagi Y et al High expression of HER3 is associated with a decreased survival in gastric cancer. Clin Cancer Res 2008; 14: 7843–9. [DOI] [PubMed] [Google Scholar]
- 35. Garrett JT, Sutton CR, Kuba MG, Cook RS, Arteaga CL. Dual blockade of HER2 in HER2‐overexpressing tumor cells does not completely eliminate HER3 function. Clin Cancer Res 2013; 19: 610–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chandarlapaty S, Sawai A, Scaltriti M et al AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011; 19: 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scaltriti M, Verma C, Guzman M et al Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab‐dependent cell cytotoxicity. Oncogene 2009; 28: 803–14. [DOI] [PubMed] [Google Scholar]
- 38. Maruyama T, Mimura K, Izawa S et al Lapatinib enhances herceptin‐mediated antibody‐dependent cellular cytotoxicity by up‐regulation of cell surface HER2 expression. Anticancer Res 2011; 31: 2999–3005. [PubMed] [Google Scholar]
- 39. Schroeder B, Srivatsan S, Shaw A, Billadeau D, McNiven MA. CIN85 phosphorylation is essential for EGFR ubiquitination and sorting into multivesicular bodies. Mol Biol Cell 2012; 23: 3602–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Van Cutsem E, Yah KH, Bang YJ et al Phase 3 trial of everolimus in previously treated patients with advanced gastric cancer: GRANITE‐1. Gastrointestinal Cancers Symposium 2012; Abstract LBA3.
- 41. Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci USA 2010; 107: 7692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu Y, Zhang Y, Wang M et al Downregulation of HER3 by a novel antisense oligonucleotide, EZN‐3920, improves the antitumor activity of EGFR and HER2 tyrosine kinase inhibitors in animal models. Mol Cancer Ther 2013; 12: 427–37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Other inhibitors cannot block HRG1‐mediated phosphorylation of HER3 and AKT.
Fig. S2. Other inhibitors cannot abrogate HRG1‐mediated resistance.
Fig. S3. Trastuzumab cannot inhibit HRG1‐mediated phosphorylation of HER3 and AKT and HRG1‐mediated resistance to lapatinib.