

Abstract
Poly (ADP‐ribose) polymerase (PARP) plays a critical role in responding to DNA damage, by activating DNA repair pathways responsible for cellular survival. Inhibition of PARP is used to treat certain solid cancers, such as breast and ovarian cancers. However, its effectiveness with other solid cancers, such as esophageal squamous cell carcinoma (ESCC), has not been clarified. We evaluated the effects of PARP inhibition on the survival of human esophageal cancer cells, with a special focus on the induction and repair of DNA double‐strand breaks. The effects were monitored by colony formation assays and DNA damage responses, with immunofluorescence staining of γH2AX and RAD51. We found that PARP inhibition synergized with cisplatin, and the cells were highly sensitive, in a similar manner to the combination of cisplatin and 5‐fluorouracil (5‐FU). Comparable increases in RAD51 foci formation were observed after each combined treatment with cisplatin and either 3‐aminobenzamide (3‐AB) or 5‐FU in three human esophageal cancer cell lines, TE11, TE14, and TE15. In addition, decreasing the amount of RAD51 by RNA interference rendered the TE11 cells even more hypersensitive to these treatments. Our findings suggested that the homologous recombinational repair pathway may be involved in the synergism between cisplatin and either 3‐AB or 5‐FU, and that 3‐AB and 5‐FU may similarly modify the cisplatin‐induced DNA damage to types requiring the recruitment of RAD51 proteins for their repair. Understanding these mechanisms could be useful for improving the clinical outcome of ESCC patients who suffer from aggressive disease that presently lacks effective treatment options.
Genomic integrity is maintained by the close cooperation of several DNA repair pathways. Any failure in these pathways can lead to unrepaired DNA lesions, which cause cell‐cycle arrest and cell death, either directly or following DNA replication during the S phase of the cell cycle.1, 2 Therefore, the therapeutic effects of DNA‐damaging agents may be enhanced by the inhibition of DNA repair. This feature makes DNA repair mechanisms a promising target for novel cancer treatment regimens.
In recent years, poly (ADP‐ribose) polymerase (PARP) inhibitors have emerged as a novel class of chemotherapeutic agents. An abundant nuclear protein that catalyzes the formation of PAR polymers from NAD+, PARP is attached primarily to glutamic acid residues on acceptor proteins.3 It participates in maintaining genomic integrity, as it is a DNA damage‐sensing protein that binds to DNA single‐strand breaks (SSBs).4, 5 In addition, PARP plays a role in restarting stalled replication forks, by attracting Mre11 to these sites.6, 7 Therefore, the inhibition of PARP generates DNA damage, and the obstructed replication forks can be converted to replication‐associated DNA double‐strand breaks (DSBs), which lead to cell cycle arrest and cell death unless they are repaired by the homologous recombinational repair pathway (HR).8, 9 Recently, the PARP inhibitors in clinical use have been shown to trap the PARP1 and PARP2 enzymes at damaged DNA.10 The trapped PARP–DNA complexes are more cytotoxic than the unrepaired SSBs caused by PARP inactivation, as the complexes require other genetic repair pathways, such as postreplication repair and the Fanconi anemia pathway, in addition to HR, for their repair.10
Double‐strand breaks are potentially lethal, and are generally considered to be the most toxic DNA lesions.11, 12 Direct DSBs are mainly repaired by the non‐homologous end joining pathway,13 whereas replication‐associated DSBs are repaired by the HR and related replication repair pathways.9 The HR and PARP are intricately linked, because the loss of PARP results in an increase in the recombinogenic lesions normally repaired by HR.14, 15, 16, 17 Therefore, tumor cells defective in HR show extremely high sensitivity to PARP inhibitors.18, 19, 20, 21, 22, 23, 24 In addition, it was recently reported that PARP inhibition sensitizes even HR‐proficient tumor cells to ionizing radiation or alkylating agents, such as methyl methanesulfonate, when treated in combination for a short time.25
Esophageal squamous cell carcinoma (ESCC) is one of the most lethal malignant diseases, especially in the USA and Europe.26, 27 Based on biochemical modulation studies,28, 29 combined therapy with cisplatin and 5‐fluorouracil (5‐FU) has recently shown encouraging results, by exerting a synergistic cytotoxic effect. However, the clinical outcomes and the overall survival rates of ESCC patients remain poor.30 The present study was carried out to evaluate the effects of PARP inhibition on the cellular survival and the DNA damage response in human esophageal cancer cells, with a special focus on DSB induction and repair. We found that PARP inhibition synergized with cisplatin, and strongly increased the percentage of cells bearing nuclear foci of RAD51, a key protein in the HR pathway. This combined therapy was as efficient as the combined treatment with cisplatin and 5‐FU, as compared to that with each drug alone. Importantly, RAD51 depletion significantly sensitized the cells to these combined treatments. Our data suggested that HR may be involved in the synergism between cisplatin and either a PARP inhibitor or 5‐FU in human esophageal cancer cells. In addition, the PARP inhibitor and 5‐FU may similarly modify the cisplatin‐induced DNA damage to types requiring the recruitment of RAD51 proteins for their repair.
Materials and Methods
Cells and chemicals
Three human esophageal cancer cell lines, TE11, TE14, and TE15, were obtained from the Cell Resource Center for the Biomedical Research Institute of Development, Aging, and Cancer (Tohoku University, Sendai, Japan). Both TE11 and TE14 are moderately differentiated squamous cell carcinomas, and TE15 is a well‐differentiated squamous cell carcinoma. These cell lines were routinely grown in RPMI‐1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS, and incubated at 37°C in a humidified atmosphere of 5% CO2 in air. The PARP inhibitor 3‐aminobenzamide (3‐AB) was obtained from Sigma‐Aldrich (#A0788; St. Louis, MO, USA). Cisplatin (Nippon Kayaku, Tokyo, Japan) and 5‐FU (Kyowa Hakko Kogyo, Tokyo, Japan) were dissolved in PBS at 1 mM.
Detailed experimental procedures are also provided in the supplementary experimental procedures (Data S1).
Results
Inhibition of PARP in ESCC cell lines
To examine whether PARP inhibition could be efficacious in the treatment of ESCC, we first tested three ESCC cell lines, TE11, TE14, and TE15, for their sensitivity to PARP inhibition. We confirmed the inactivation of PARP by a PARP inhibitor, 3‐AB, and the depletion of PARP1 by siRNAs, using immunoblotting analyses (Figs S1,S2A), and then measured the cell viability by a colony formation assay. This assay showed that 3‐AB did not decrease the viability of these ESCC cells, as compared to the untreated controls (Fig. 1a).
Figure 1.
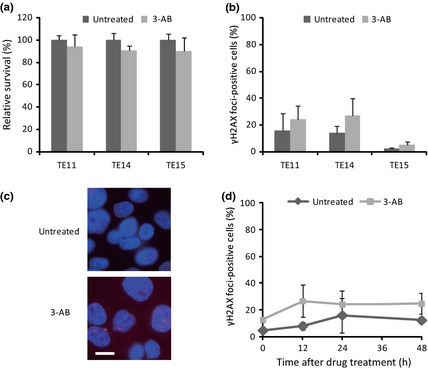
Poly (ADP‐ribose) polymerase (PARP) inhibition by 3‐aminobenzamide (3‐AB) in human esophageal cancer cell lines. (a) Survival of TE11, TE14, and TE15 cells after treatment with 3‐AB. (b) Induction of double‐strand breaks, indicating γH2AX focus formation, at 24 h after PARP inhibition by 3‐AB in TE11, TE14, and TE15 cells. (c) DNA (blue) and γH2AX foci (red) were visualized at 24 h after treatment of TE11 cells. Scale bar = 10 μm. (d) Kinetics of γH2AX foci formation at the indicated periods up to 48 h, after 3‐AB pretreatment for 48 h. Cells with 10 or more foci were counted as positive. At least 200 nuclei were counted for each experiment. The average and SD from at least three experiments are shown.
As PARP inhibition increases the collapse of unresolved SSBs into DSBs at replication forks,31, 32 we investigated the induction of DSBs by PARP inhibition in these ESCC cells. Thus, we carried out immunofluorescence staining for γH2AX, as a marker of DSBs, after the treatment of the ESCC cell lines with 3‐AB. As a result, mild increases in the percentages of γH2AX foci‐positive cells were observed in all of these cells after the 3‐AB treatment (Figs 1b–d,S3A). As the inhibition of PARP by 3‐AB treatment did not impair the colony forming activity, most of the DSBs generated by the PARP inhibition might be exactly repaired, and thus not induce cell cycle arrest or cell death. This notion was supported by similar findings obtained by experiments using PARP1‐depleted cells in place of 3‐AB treatment (Fig. S2A,B).
Combination of PARP inhibition with cisplatin or 5‐FU in ESCC cell lines
Cisplatin and 5‐FU are effective chemotherapeutic agents used with ESCC patients.28, 29 We wished to examine whether PARP inhibition acts synergistically with either cisplatin or 5‐FU against esophageal cancer cells. Thus, we treated TE11, TE14, and TE15 cells with either cisplatin or 5‐FU, with or without the inhibition of PARP, and then carried out a colony formation assay to assess the cellular survival after these treatments (Fig. S4). The colony assay revealed that 3‐AB sensitized all of these cell lines to cisplatin (Fig. 2a). The synergistic inhibition of cell growth was observed by the combined treatment of TE11 cells with 5 μM cisplatin and 5 mM 3‐AB (Fig. S5, Combination Index = 0.471). In stark contrast, no synergism between 3‐AB and 5‐FU was observed (Fig. 2a).
Figure 2.
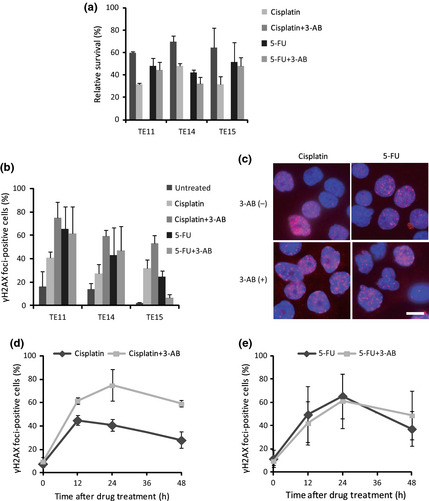
Combination of a poly (ADP‐ribose) poly‐merase (PARP) inhibitor 3‐aminobenzamide (3‐AB) with cisplatin or 5‐fluorouracil (5‐FU) in human esophageal cancer cell lines. (a) Survival of TE11, TE14, and TE15 cells after treatment with either 5 μM cisplatin for 1 h or 3 μM 5‐FU for 24 h, with or without pretreatment with 3 mM (TE14) or 5 mM (TE11 and TE15) 3‐AB for 48 h. (b) Evaluation of the γH2AX focus formation at 24 h after treatment of TE11, TE14, and TE15 cells with either cisplatin or 5‐FU, with or without pretreatment with 3‐AB. (c) DNA (blue) and γH2AX foci (red) were visualized at 24 h after treatment of TE11 cells. Scale bar = 10 μm. (d,e) TE11 cells were treated with either cisplatin or 5‐FU, with or without pretreatment with 3‐AB. Cells with 10 or more foci were counted as positive. At least 200 nuclei were counted for each experiment. The average and SD from at least three experiments are shown.
To explore the reason for this distinct sensitization of cells to cisplatin and 5‐FU by 3‐AB, we next studied the levels of γH2AX focus formation generated by each treatment. The combined treatment with cisplatin and 3‐AB induced significantly higher percentages of γH2AX focus formation compared to the single treatment with cisplatin (Figs 2b,c,S3A). The γH2AX focus formation of TE11 cells peaked at 24 h, and 60% of cells remained foci‐positive even at 48 h after treatment (Fig. 2d). Combined treatment with 5‐FU and 3‐AB induced γH2AX focus formation with similar increases and kinetics as the single treatment with 5‐FU, and it peaked at 24 h after treatment (Figs 2b,c,e,S3A). Although the TE15 cells treated with 5‐FU and 3‐AB showed a lower percentage of γH2AX foci‐positive cells, as compared to the 5‐FU single treatment, the average numbers of γH2AX foci per cell generated by these treatments were similar to those of the other cell lines (Figs 2b,S3A). Similar findings were obtained by the depletion of PARP1 using siRNA, instead of 3‐AB treatment (Fig. S2C). Therefore, the significantly increased induction of DSBs by PARP inhibition could enhance the cytotoxic effects of cisplatin treatment of these ESCC cells.
Modulation of ESCC cell sensitivity to combined treatment with cisplatin and 5‐FU by PARP inhibition
Having established that the PARP inhibitor could sensitize ESCC cells to cisplatin, we next compared the anticancer effects between the combined treatments with cisplatin/3‐AB and cisplatin/5‐FU, the most standard chemotherapy for ESCC. We treated the ESCC cells with cisplatin and 5‐FU in concurrent combinations, and then measured the cell viability by a colony formation assay. As a result, all of the ESCC cell lines treated with cisplatin plus 5‐FU showed similar high degrees of sensitivity to the cisplatin plus PARP inhibition (Figs 2a,3a,S2B).
Figure 3.

Comparison of standard combined trea‐tments for esophageal squamous cell carcinoma using cisplatin and 5‐fluorouracil (5‐FU) with or without poly (ADP‐ribose) polymerase (PARP) inhibition . (a) Survival of TE11, TE14, and TE15 cells after concurrent combined treatments with cisplatin and 5‐FU, with or without pretreatment with 3‐aminobenzamide (3‐AB). (b) Evaluation of the γH2AX focus formation at 24 h after combined treatment of TE11, TE14, and TE15 cells with cisplatin and 5‐FU, with or without a pretreatment with 3‐AB. (c) DNA (blue) and γH2AX foci (red) were visualized at 24 h after treatment of TE11 cells. Scale bar = 10 μm. (d) TE11 cells were treated with cisplatin and 5‐FU in concurrent combination, after pretreatment with 3‐AB. Cells with 10 or more foci were counted as positive. At least 200 nuclei were counted for each experiment. The average and SD from at least three experiments are shown.
To clarify the reason for the high sensitivity of the ESCC cells to the combined treatment with cisplatin and 5‐FU, we next carried out an immunofluorescence assay for γH2AX proteins in these cell lines. This assay revealed that the γH2AX focus formation following the cisplatin plus 5‐FU treatment was quite consistent with that following the cisplatin plus 3‐AB treatment (Figs 2b,d,3b,d,S3A). Thus, we hypothesized that 3‐AB and 5‐FU might play analogous roles in the increased numbers of DSBs formed in combination with cisplatin, resulting in the similar sensitivities of the cells to combined treatments with cisplatin and either 3‐AB or 5‐FU.
To confirm our hypothesis, we next investigated the cytotoxic effect of the triple combination of 3‐AB, cisplatin, and 5‐FU against ESCC cells. First, we inhibited PARP by 3‐AB or depleted it by siRNAs, and then treated the cells concurrently with cisplatin and 5‐FU. The cellular survival was confirmed by a colony formation assay. This assay showed that the triple treatment did not cause a further decrease in the survival of the cells, as compared to the combined treatment with cisplatin and either 3‐AB or 5‐FU (Figs 2a,3a).
Next, we examined the γH2AX focus formation, to assess the induction of DSBs by the triple treatment of the ESCC cells. Immunofluorescence staining analysis of γH2AX revealed that the level of γH2AX focus formation induced by the triple treatment was not significantly different from that induced by the combined treatment with cisplatin and either 3‐AB or 5‐FU in these cells (Figs 2b–d,3b–d,S3A). Similar findings were obtained by PARP1 depletion instead of 3‐AB treatment (Fig. S2B,C). Therefore, these findings indirectly supported our hypothesis that PARP inhibition and 5‐FU increase the sensitivity of ESCC cells to cisplatin, by disturbing the same pathway involving the induction or repair of DNA damage.
Validation of HR, indicating RAD51 foci formation in ESCC cells
Poly (ADP‐ribose) polymerase inhibition induces DSBs, which require HR for their repair.31, 32 Thus, we next examined the involvement of HR after the induction of DSBs by PARP inhibition, to understand the mechanisms underlying the synergism between cisplatin and either 3‐AB or 5‐FU in ESCC cells. We assessed RAD51 focus formation, as a hallmark of ongoing HR, after treatments with 3‐AB, cisplatin, and 5‐FU alone and in combination.
Immunofluorescence staining of RAD51 revealed that the 3‐AB treatment significantly increased the percentage of cells with RAD51 foci, as compared to the untreated controls (Figs 4a–c,S3B). As 3‐AB did not disturb the colony formation activity of the ESCC cells (Fig. 1a), this finding suggested that the 3‐AB‐induced DNA damage might be exactly repaired by HR.
Figure 4.

Validation of the homologous reco‐mbinational repair pathway in esophageal squamous cell carcinoma cells, indicating RAD51 focus formation. (a) Evaluation of the RAD51 focus formation 24 h after treatment of TE11, TE14, and TE15 cells with cisplatin or 5‐fluorouracil (5‐FU) alone and in combination, with or without a pretreatment with 3‐aminobenzamide (3‐AB). (b) Representative images of DNA (blue) and RAD51 foci (green) 24 h after the treatment of TE11 cells. Scale bar = 10 μm. (c) TE11 cells were treated with or without 3‐AB. (d,e) TE11 cells were treated with either cisplatin or 5‐FU, with or without pretreatment with 3‐AB. (f) TE11 cells were treated concurrently with cisplatin and 5‐FU, after pretreatment with 5 mM 3‐AB for 48 h. Cells with five or more foci were counted as positive. At least 200 nuclei were counted for each experiment. The average and SD from three experiments are shown.
The treatment of cells with cisplatin or 5‐FU alone caused only a slight increase in the percentage of RAD51 foci‐positive cells up to 48 h after treatment. However, the combined treatment of these cells with cisplatin and either 3‐AB or 5‐FU markedly increased the percentage of cells with RAD51 foci, which reached a maximum at 24 h after each treatment (Figs 4,S3B). When used in combination with cisplatin, both 3‐AB and 5‐FU vigorously promoted the production of DSBs, which require the recruitment of RAD51 proteins for their repair in these ESCC cells.
The PARP‐inhibited ESCC cells treated with or without 5‐FU showed similar levels of RAD51 focus formation after each treatment (Figs 4a–c,e,S3B). Moreover, the addition of 5‐FU did not change the kinetics of RAD51 focus formation by the combined treatment with cisplatin and 3‐AB (Figs 4a,b,d,f,S3B). These data suggested that 5‐FU might not affect the RAD51 focus formation induced by 3‐AB, with or without cisplatin, in these ESCC cells. Similar results were obtained by the depletion of PARP1 in place of 3‐AB treatment (Fig. S2D). Thus, the validation of HR additionally supported our hypothesis that the enhancement of the anticancer effect of cisplatin, by either PARP inhibition or 5‐FU, might be attributed to similar mechanisms involving HR repair in ESCC cells.
Depletion of RAD51 enhances 3‐AB‐ or 5‐FU‐mediated sensitization of ESCC cells to cisplatin
As RAD51 plays crucial and well‐established roles in HR,33, 34, 35 we hypothesized that the critical role of HR would be the underlying reason for the synergistic effect of cisplatin and either 3‐AB or 5‐FU in ESCC cells. Thus, we examined how RAD51 depletion affected the sensitivity of TE11 cells to these combined treatments. The depletion of RAD51 by siRNAs was confirmed by an immunoblotting analysis, using anti‐RAD51 antibodies (Figs 5a,S6A).
Figure 5.

Knockdown of RAD51 protein renders esophageal squamous cell carcinoma TE11 cells hypersensitive to combinations of cisplatin and either 3‐aminobenzamide (3‐AB) or 5‐fluorouracil (5‐FU). (a) Immunoblotting analysis of RAD51 (top) and α‐tubulin (bottom) in TE11 cells, after RAD51 siRNA or non‐targeting (NT) siRNA depletion for 24 h. (b) Relative survival of TE11 cells treated with 3‐AB, cisplatin, and 5‐FU alone and in combination, under the same experimental conditions after RAD51 siRNA or NT siRNA depletion for 24 h, confirmed by colony formation assay. The average and SD from at least three experiments are shown. Values marked with asterisks are statistically significant, as compared with each control (*P < 0.05).
We treated RAD51‐knockdown cells with either 3‐AB, cisplatin, and 5‐FU alone or in combination, under the same experimental conditions as described above, and then the cellular survival was measured by a colony formation assay (Figs 5b,S6B). As expected, RAD51 depletion caused a drastic increase in the cellular sensitivity to 3‐AB. In contrast, RAD51 knockdown showed neither a synergistic nor an additive effect on the sensitivity to cisplatin or 5‐FU alone (Figs 5b,S6B). These findings suggested that HR might play a major role in the repair of DNA damage induced by treatment with 3‐AB, but not with cisplatin or 5‐FU alone. In contrast to the treatment with cisplatin or 5‐FU alone, RAD51 repression significantly sensitized the TE11 cells to cisplatin in combination with either 3‐AB or 5‐FU. Considering our finding that the addition of 3‐AB to treatment with 5‐FU or cisplatin/5‐FU did not enhance the γH2AX focus formation (Figs 2b,e,3b,d,S3A), these data supported our hypothesis that the conversion of cisplatin‐induced DNA damage to the types requiring HR for their repair could play an important role in TE11 cells sensitization to cisplatin by both 3‐AB and 5‐FU.
Discussion
Our data showed that the inhibition of PARP exerts a synergistic tumor‐cell killing effect in combination with cisplatin, but not 5‐FU, against three ESCC cell lines, TE11, TE14, and TE15, by the increased induction of DNA damage requiring HR for repair. Moreover, in the sensitization of cells to cisplatin, PARP inhibition by 3‐AB and 5‐FU may function by similar mechanisms involving HR.
Poly (ADP‐ribose) polymerase inhibitors were originally developed to selectively target HR‐defective cells, and have been tested as a monotherapy and in combination with an alkylating agent and cisplatin in patients with certain solid tumors.36, 37, 38, 39 In this study, 3‐AB, as a single agent, had minimal cytotoxic efficacy (Fig. 1a), and only modest increases of γH2AX and RAD51 focus formation in response to 3‐AB were observed in the ESCC cells, as compared to the untreated controls (Figs 1b,4a). These data indicated that the DSBs induced by 3‐AB may be exactly repaired by HR, and therefore, these cells are predicted to be proficient in HR repair.40 Thus, consistent with a previous study,25 PARP inhibitors, in combination with certain DNA damaging agents, could be useful in the treatment of even HR‐proficient cancer cells.
Poly (ADP‐ribose) polymerase and RAD51 are required to reactivate replication at stalled DNA forks.6, 7, 41, 42 Therefore, the synergism between cisplatin and either 3‐AB or 5‐FU may be similarly attributed to the failure of replication reactivation at stalled replication forks, due to the inhibition of PARP activity or the incorporation of 5‐FU into replicating DNA in the cisplatin‐induced DNA lesions. This failure may lead to increased RAD51 focus formation, for the efficient restarting of stalled replication forks by HR. The addition of 3‐AB to either the 5‐FU or cisplatin/5‐FU treatment neither facilitated nor repressed the cellular survival and γH2AX/RAD51 focus formation (Figs 2a,b,3a,b,4a), therefore, 3‐AB and 5‐FU may function in an epistatic pathway for the cisplatin‐induced DNA lesion repair in ESCC cells. Our results suggested that a novel regimen, combining cisplatin with a PARP inhibitor, may have similar efficacy to the standard combined chemotherapy of cisplatin and 5‐FU in the treatment of ESCC patients.
In conclusion, we have shown that HR may be involved in the synergism between cisplatin and either PARP inhibition or 5‐FU treatment, in human esophageal cancer cell lines. Our findings provide a platform for extending the potential use of PARP inhibitors to ESCC patients. Poly (ADP‐ribose) polymerase inhibitors could be novel combinational counterparts of cisplatin in the treatment of ESCC. Moreover, cancer cells with decreased RAD51 activity, due to mutations or dysregulation, would be more sensitive to PARP inhibitors than the surrounding HR‐proficient tissue.43, 44 Therefore, considering the therapeutic potential of PARP inhibitors in the treatment of ESCC, such cases would be ideal candidates for PARP inhibitor therapy, and the side‐effects usually seen with classical cytotoxic anticancer drugs could be minimized. Although it may be premature to extrapolate our results from only three cultured cell lines, further investigations of the mechanisms responsible for the increases of RAD51 foci, in combination with cisplatin and either PARP inhibition or 5‐FU treatment, in human cancer cells will provide novel insights into cancer therapies.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Immunoblotting analysis of poly (ADP‐ribose) PAR and poly (ADP‐ribose) polymerase 1 (PARP1) in esophageal squamous cell carcinoma TE11 cells after exposure to 3‐aminobenzamide (3‐AB).
Fig. S2. Treatment of poly (ADP‐ribose) polymerase 1 (PARP1)‐depleted esophageal squamous cell carcinoma TE11 cells with anticancer drugs.
Fig. S3. Numbers of γH2AX and RAD51 foci per nucleus after treatment with anticancer drugs.
Fig. S4. Time schedule of treatments with anticancer drugs.
Fig. S5. Dose–response analysis of the survival of esophageal squamous cell carcinoma TE11 cells treated with cisplatin and 3‐aminobenzamide (3‐AB) in combination.
Fig. S6. Knockdown of RAD51 protein using RAD51 siRNA (#2) also renders TE11 cells hypersensitive to combinations of cisplatin and either 3‐aminobenzamide (3‐AB) or 5‐fluorouracil (5‐FU).
Data S1. Supplementary experimental procedures and discussion.
Acknowledgment
This work was supported by the Grants‐in‐Aid Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
(Cancer Sci 2013; 104: 1593–1599)
References
- 1. Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411: 366–74. [DOI] [PubMed] [Google Scholar]
- 2. Farmer H, McCabe N, Lord CJ et al Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–21. [DOI] [PubMed] [Google Scholar]
- 3. Satoh MS, Lindahl T. Role of poly (ADP‐ribose) formation in DNA repair. Nature 1992; 356: 356–8. [DOI] [PubMed] [Google Scholar]
- 4. Lindahl T, Satoh MS, Poirier GG, Klungland A. Post‐translational modification of poly (ADP‐ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci 1995; 20: 405–11. [DOI] [PubMed] [Google Scholar]
- 5. D′Amours D, Desnoyers S, D′Silva I, Poirier GG. Poly(ADP‐ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 1999; 342: 249–68. [PMC free article] [PubMed] [Google Scholar]
- 6. Bryant HE, Petermann E, Schultz N et al PARP is activated at stalled forks to mediate Mre11‐dependent replication restart and recombination. EMBO J 2009; 28: 2601–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ. Ablation of PARP‐1 does not interfere with the repair of DNA double‐strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004; 23: 3872–82. [DOI] [PubMed] [Google Scholar]
- 8. Schultz N, Lopez E, Saleh‐Gohari N, Helleday T. Poly (ADP‐ribose) polymerase (PARP‐1) has a controlling role in homologous recombination. Nucleic Acids Res 2003; 31: 4959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arnaudeau C, Lundin C, Helleday T. DNA double‐strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 2001; 307: 1235–45. [DOI] [PubMed] [Google Scholar]
- 10. Murai J, Huang SY, Das BB et al Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 2012; 72: 5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hsiang YH, Lihou MG, Liu LF. Arrest of replication forks by drug‐stabilized topoisomerase I‐DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res 1989; 49: 5077–82. [PubMed] [Google Scholar]
- 12. Markovits J, Pommier Y, Kerrigan D, Covey JM, Tilchen EJ, Kohn KW. Topoisomerase II‐mediated DNA breaks and cytotoxicity in relation to cell proliferation and the cell cycle in NIH 3T3 fibroblasts and L1210 leukemia cells. Cancer Res 1987; 47: 2050–5. [PubMed] [Google Scholar]
- 13. Sargent RG, Brenneman MA, Wilson JH. Repair of site‐specific double‐strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol Cell Biol 1997; 17: 267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Magnusson J, Ramel C. Inhibitor of poly (ADP‐ribose)transferase potentiates the recombinogenic but not the mutagenic action of alkylating agents in somatic cells in vivo in Drosophila melanogaster . Mutagenesis 1990; 5: 511–4. [DOI] [PubMed] [Google Scholar]
- 15. Waldman AS, Waldman BC. Stimulation of intrachromosomal homologous recombination in mammalian cells by an inhibitor of poly(ADP‐ribosylation). Nucleic Acids Res 1991; 19: 5943–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schreiber V, Hunting D, Trucco C et al A dominant‐negative mutant of human poly (ADP‐ribose) polymerase affects cell recovery, apoptosis and sister chromatid exchange following DNA damage. Proc Natl Acad Sci USA 1995; 92: 4753–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Semionov A, Cournoyer D, Chow TY. Inhibition of poly (ADP‐ribose)polymerase stimulates extrachromosomal homologous recombination in mouse Ltk‐fibroblasts. Nucleic Acids Res 1999; 27: 4526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donawho CK, Luo Y, Luo Y et al ABT‐888, an orally active poly (ADP‐ribose) polymerase inhibitor that potentiates DNA‐damaging agents in preclinical tumor models. Clin Cancer Res 2007; 13: 2728–37. [DOI] [PubMed] [Google Scholar]
- 19. Penning TD, Zhu GD, Gandhi VB et al Discovery of the Poly (ADP‐ribose) polymerase (PARP) inhibitor 2‐[(R)‐2‐methylpyrrolidin‐2‐yl]‐1H‐benzimidazole‐4‐carboxamide (ABT‐888) for the treatment of cancer. J Med Chem 2009; 52: 514–23. [DOI] [PubMed] [Google Scholar]
- 20. Evers B, Drost R, Schut E et al Selective inhibition of BRCA2‐deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res 2008; 14: 3916–25. [DOI] [PubMed] [Google Scholar]
- 21. Hay T, Matthews JR, Pietzka L et al Poly (ADP‐ribose) polymerase‐1 inhibitor treatment regresses autochthonous Brca2/p53‐mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res 2009; 69: 3850–5. [DOI] [PubMed] [Google Scholar]
- 22. Menear KA, Adcock C, Boulter R et al 4‐[3‐(4‐cyclopropanecarbonylpiperazine‐1‐carbonyl)‐4‐fluorobenzyl]‐2H‐phthalazin‐1‐one: a novel bioavailable inhibitor of poly (ADP‐ribose) polymerase‐1. J Med Chem 2008; 51: 6581–91. [DOI] [PubMed] [Google Scholar]
- 23. Rottenberg S, Jaspers JE, Kersbergen A et al High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA 2008; 105: 17079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones P, Altamura S, Boueres J et al Discovery of 2‐{4‐[(3S)‐piperidin‐3‐yl]phenyl}‐2H‐indazole‐7‐carboxamide (MK‐4827): a novel oral poly (ADP‐ribose)polymerase (PARP) inhibitor efficacious in BRCA‐1 and ‐2 mutant tumors. J Med Chem 2009; 52: 7170–85. [DOI] [PubMed] [Google Scholar]
- 25. Löser DA, Shibata A, Shibata AK, Woodbine LJ, Jeggo PA, Chalmers AJ. Sensitization to radiation and alkylating agents by inhibitors of poly (ADP‐ribose) polymerase is enhanced in cells deficient in DNA double‐strand break repair. Mol Cancer Ther 2010; 9: 1775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 27. Shimada H, Nabeya Y, Okazumi S et al Prediction of survival with squamous cell carcinoma antigen in patients with resectable esophageal squamous cell carcinoma. Surgery 2003; 133: 486–94. [DOI] [PubMed] [Google Scholar]
- 28. Sekiguchi H, Akiyama S, Fujiwara M et al Phase II trial of 5‐fluorouracil and low‐dose cisplatin in patients with squamous cell carcinoma of the esophagus. Surg Today 1999; 29: 97–101. [DOI] [PubMed] [Google Scholar]
- 29. Scanlon KJ, Newman EM, Lu Y, Priest DG. Biochemical basis for cisplatin and 5‐fluorouracil synergism in human ovarian carcinoma cells. Proc Natl Acad Sci USA 1986; 83: 8923–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Borghesi S, Hawkins MA, Tait D. Oesophagectomy after definitive chemoradiation in patients with locally advanced oesophageal cancer. Clin Oncol (R Coll Radiol) 2008; 20: 221–6. [DOI] [PubMed] [Google Scholar]
- 31. Bryant HE, Schultz N, Thomas HD et al Specific killing of BRCA2‐deficient tumours with inhibitors of poly (ADP‐ribose) polymerase. Nature 2005; 434: 913–7. [DOI] [PubMed] [Google Scholar]
- 32. Saleh‐Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single‐strand breaks. Mol Cell Biol 2005; 25: 7158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lim DS, Hasty P. A mutation in mouse rad51 results in an early embryonic lethal that is suppressed by a mutation in p53. Mol Cell Biol 1996; 16: 7133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsuzuki T, Fujii Y, Sakumi K et al Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc Natl Acad Sci USA 1996; 93: 6236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sonoda E, Sasaki MS, Buerstedde JM et al Rad51‐deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J 1998; 17: 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer 2004; 4: 814–9. [DOI] [PubMed] [Google Scholar]
- 37. Vesprini D, Narod SA, Trachtenberg J et al The therapeutic ratio is preserved for radiotherapy or cisplatin treatment in BRCA2‐mutated prostate cancers. Can Urol Assoc J 2011; 5: E31–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Plummer R, Jones C, Middleton M et al Phase I study of the poly (ADP‐ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res 2008; 14: 7917–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rajan A, Carter CA, Kelly RJ et al A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res 2012; 18: 2344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mukhopadhyay A, Elattar A, Cerbinskaite A et al Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly (ADP‐ribose) polymerase inhibitors. Clin Cancer Res 2010; 16: 2344–51. [DOI] [PubMed] [Google Scholar]
- 41. Davies SL, North PS, Hickson ID. Role for BLM in replication‐fork restart and suppression of origin firing after replicative stress. Nat Struct Mol 2007; 14: 677–9. [DOI] [PubMed] [Google Scholar]
- 42. Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea‐stalled replication forks become progressively inactivated and require two different RAD51‐mediated pathways for restart and repair. Mol Cell 2010; 37: 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010; 10: 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double‐strand break repair. J Clin Oncol 2008; 26: 3785–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunoblotting analysis of poly (ADP‐ribose) PAR and poly (ADP‐ribose) polymerase 1 (PARP1) in esophageal squamous cell carcinoma TE11 cells after exposure to 3‐aminobenzamide (3‐AB).
Fig. S2. Treatment of poly (ADP‐ribose) polymerase 1 (PARP1)‐depleted esophageal squamous cell carcinoma TE11 cells with anticancer drugs.
Fig. S3. Numbers of γH2AX and RAD51 foci per nucleus after treatment with anticancer drugs.
Fig. S4. Time schedule of treatments with anticancer drugs.
Fig. S5. Dose–response analysis of the survival of esophageal squamous cell carcinoma TE11 cells treated with cisplatin and 3‐aminobenzamide (3‐AB) in combination.
Fig. S6. Knockdown of RAD51 protein using RAD51 siRNA (#2) also renders TE11 cells hypersensitive to combinations of cisplatin and either 3‐aminobenzamide (3‐AB) or 5‐fluorouracil (5‐FU).
Data S1. Supplementary experimental procedures and discussion.