

Abstract
Objective: This study aims to report the clinical features and gene mutation of a rare MODY10 patient in China. Methods: This study summarizes the clinical data of a MODY10 child in the Endocrine Department of our hospital and an analysis and discussion of the results of the gene sequencing of the child. Results: The child was a two-year-old boy. The main reason for his visit to our hospital was “founding hyperglycemia for 3 days”. The fasting blood glucose was between 8.1-10.7 mmol/L, and two-hour postprandial blood glucose was between 10.6-12.6 mmol/L. Glycosylated hemoglobin was 8.5%, fasting C-peptide was 0.6 ng/mL, fasting insulin was 2.9 μIU/mL, and the islet antibody series were all negative. Whole-genome/exon sequencing results: Exon 3 of the insulin gene in the child carried a c.309-314del CCAGCT insGCGC heterozygous mutation. The mutation was a nonsense mutation, and family sequencing showed that the mutation originated from the mother of the child. The mother of the child was diagnosed with diabetes when she was a year old and developed bilateral fundus hemorrhage and right retinal detachment at the age of 23. Conclusion: Among Chinese children, the insulin gene c.309-314del CCAGCT insGCGC mutation may induce MODY10. For diabetic children with a negative islet autoantibody, gene detection and analysis is helpful for the diagnosis and typing of MODY.
Keywords: MODY10, INS genes, second-generation sequencing, nonsense mutation, single gene mutation
Introduction
Maturity-onset diabetes of the young (MODY) is a special type of diabetes caused by a single gene mutation. It belongs to the most common type of single gene diabetes [1-4]. With the development of molecular genetics, to date, 14 MODY-related pathogenic genes have been discovered [5-7]. Maturity-onset diabetes of the young 10 (MODY10) caused by the mutation of the insulin gene (INS) is a rare type of MODY and was first discovered and reported in 2008 by Edgill et al. Patients with this type of disease often have early onset, and have a large age span of onset, i.e., patients can develop this disease from infancy through to childhood and adulthood. The clinical manifestations are different. In patients who are mildly affected, the disease can be controlled by a diabetes diet combined with exercise. Some patients with manifestations similar to type 1 diabetes should be treated with insulin for life.
MODY10 is extremely rare, up to now. There is no literature or reports on its occurrence in Chinese children. This study aims to report the clinical features and gene mutation of a patient in China with the rare MODY10 condition and to review related literature in order to improve clinicians’ understanding of the disease.
Materials and methods
The current study meets the requirements of the Declaration of Helsinki of the World Medical Association. The Ethics Committee of our hospital has approved this study, and all patients provided signed informed consent.
History of present illness
The child was a two-year-old boy. The main reason for his visit to our hospital was “found hyperglycemia for 3 days”. The child was G1P1, birthed through cesarean section at a gestational age of 38+2 weeks. The birth weight was 2.95 kg, and the birth height was 50 cm. The child had jaundice in the neonatal period, and this subsided ten days after orally taking Artemisia Capillaris Granules. Blood glucose was monitored several times during the first six months after birth, and all were found to be within the normal range. The child usually had a poor appetite. Blood glucose was monitored due to urine viscosity three days before admission. The results revealed that the fasting blood glucose was between 8.1-10.7 mmol/L, and two-hour postprandial blood glucose was between 10.6-12.6 mmol/L. The patient was admitted to our hospital for further diagnosis and treatment.
Family history
The mother of the child was diagnosed with diabetes as a one-year-old and treated with insulin after the diagnosis indefinitely. She developed hypoglycemia many times due to an irregular diet and developed bilateral fundus hemorrhage and right retinal detachment at the age of 23 years (six years ago). She underwent vitrectomy and retinal reattachment of the right eye, then developed turbid right cornea and was treated with insulin as part 30 injection. At present, her fasting blood glucose is between 6-7 mmol/L. Both the grandfather and grandmother of the child were healthy. The great-grandmother of the child had type 2 diabetes, which developed after the age of 60, and she died at the age of 84. The father and grandfather of the child had hypertension, but no history of diabetes themselves or in their family history.
Admission physical examination
Body temperature: 36.3°C, heart rate: 96 times/minute, breath: 21 times/minute. Height: 89 cm (P10-25), weight: 11.5 kg (P3-10), body mass index (BMI): 14.5 kg/m2. The mental state was lucid, the child was not upset, the breath was stable, and the skin elasticity was acceptable. No abnormality was found in the cardiopulmonary physical examination. The abdomen was soft, and the liver and spleen were not enlarged. No abnormalities were found by physical examination of the nervous system. The external genitalia of the boy was normal.
Adjuvant examinations
Fasting blood glucose: 8.54 mmol/L (normal value: 3.9-6.1 mmol/L), glycosylated hemoglobin: 8.5% (reference value: 4%-6%), fasting C-peptide: 0.6 ng/mL (reference value: 1.1-5.0 ng/mL), fasting insulin; 2.9 μIU/mL (reference value: 6.0-27 μIU/mL). Anti-islet cell antibody (ICA), anti-insulin autoantibody (IAA), and anti-glutamic acid decarboxylase antibody (GADA) were all negative. The routine blood test results were normal. Routine urine test: urine glucose (+-), ketone body (-), and remainder (-). Blood gas analysis and blood electrolytes were normal. Blood lipid, liver, and kidney function were normal. Insulin-like growth factor-1 (IGF-1) and five items of thyroid function were normal. Anti-thyroglobulin antibodies and anti-thyroid peroxidase antibodies were negative. The blood trace elements were roughly normal. Abdominal ultrasonography revealed that no abnormalities were found in the echo of the pancreatic parenchyma, the thickness of the pancreas was about 1.1 cm, no dilated pancreatic ducts were found, no obvious abnormalities were found in the liver, gallbladder, spleen, and kidney, enlargement of the mesenteric lymph nodes was observed, and no abnormality was found in morphology and echo.
Results
Results of molecular genetics test
After obtaining informed consent, 3 mL of ethylenediaminetetraacetic acid (EDTA) anticoagulant blood was drawn from the child and parents, and whole-genome/exon sequencing analysis was performed. The technical method of whole-genome/exon sequencing analysis detection: gene DNA was extracted from the peripheral blood of the subjects to construct a genomic library. Biotin-labeled probes (developed independently by Beijing MyGenostics Medical Laboratory, in which the total exon probe was P039-Exon) were hybridized with library DNA under certain conditions. The probe labeled with biotin was covalently bound with streptavidin-modified magnetic beads to capture the target gene. Finally, magnetic beads carrying the target gene were absorbed by a magnetic frame, and the target gene was eluted, purified, and enriched. The enriched target gene was sequenced using NextSeq 500 high throughput sequencing (Illumina). Sequencing data were compared with the reference sequence of human genome hg19 using BWA software, and the comparison files were sorted, filtered, and compared with local multi-sequence, to eliminate false positives. Sanger sequencing was performed to validate the genetic mutations that were clearly or possibly associated with the clinical phenotype of the patients. The above-mentioned tests were completed by Beijing MyGenostics Medical Laboratory.
Gene sequencing results
A c.309-314del CCAGCT insGCGC heterozygous mutation was detected in exon 3 of the insulin gene in the child. A 6-base (CCAGCT) deletion and a 4-base (GCGC) insertion were detected in the coding region 309-314, resulting in the 103rd bit of the coding product becoming the termination codon (Figure 1). The amino acid coding after tyrosine (Y) was completely terminated (X) (p.Y103X). The mutation was a nonsense mutation, and family sequencing showed that the mutation originated from the mother of the child (Figure 2). The genotype of this locus was normal in the father of the child (Figure 3). The mutation was not found in the normal population database, suggesting that the mutation is a low-frequency mutation. Bioinformatics protein function prediction software SIFT, PolyPhen_2, and REVEL were used to predict the protein function. The results were unknown, unknown, and unknown, respectively. Furthermore, none of the other 13 gene mutations that have been reported to cause MODY were detected. According to ASMG guidelines, the mutation was judged as a suspected pathogenic mutation. On the basis of the maternal family history of diabetes, it was considered that this mutation was a heterozygous pathogenic mutation leading to diabetes in children, and the type was MODY10. This mutation has not been reported in any previous literature.
Figure 1.
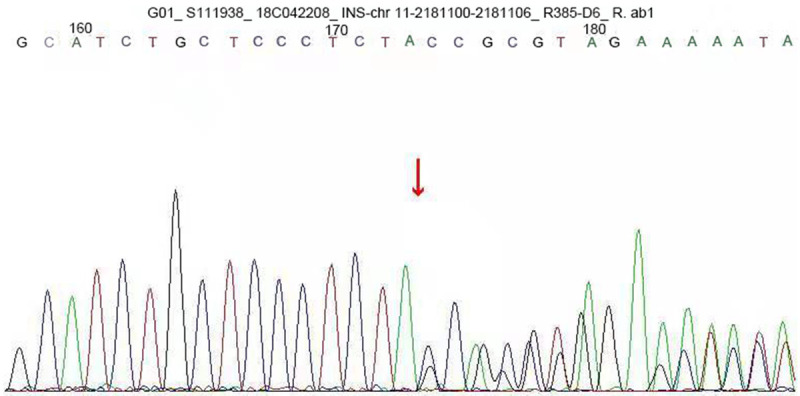
INS gene sequence map of the child. The results showed that exon 3 carried a c.309-314del CCAGCT ins GCGC heterozygous mutation. A 6-base (CCAGCT) deletion and a 4-base (GCGC) insertion were detected in the coding region 309-314, resulting in the 103rd bit of the coding product becoming the termination codon. The amino acid coding after tyrosine (Y) was completely terminated (X) (p.Y103X). The mutation was a nonsense mutation.
Figure 2.
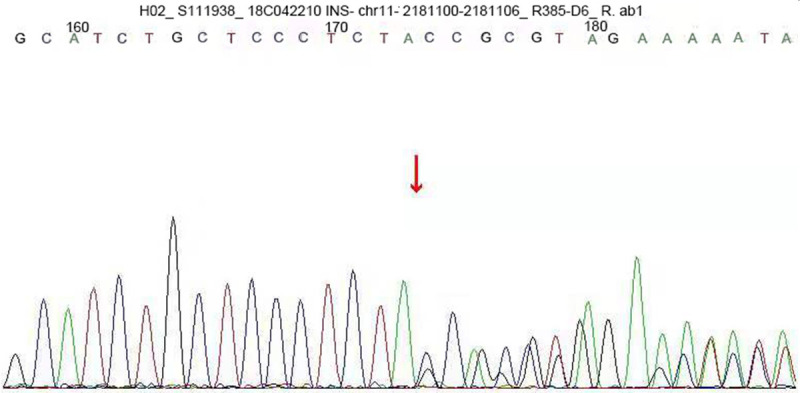
INS gene sequence map of the child’s mother. The exon 3 carried a c.309-314del CCAGCT insGCGC heterozygous mutation, indicating that the mutation of the gene originated from the child’s mother.
Figure 3.
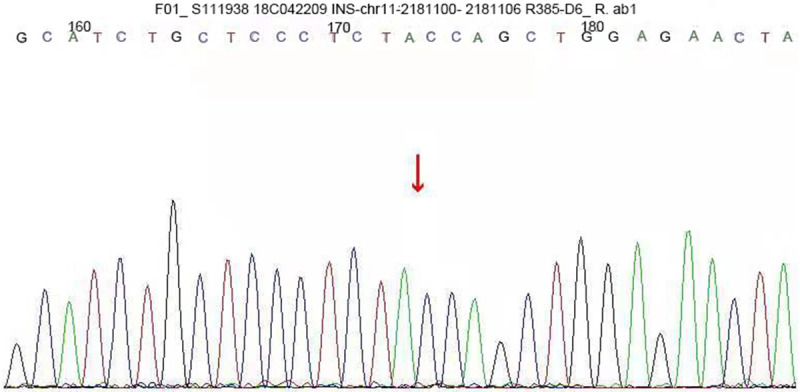
INS gene sequence map of the child’s father. The genotype of this locus was normal in the father of the child.
Discussion
This study aims to report the clinical features and gene mutation of a patient in China with rare MODY10. The patient was an approximately two-year-old male child. The main reason for his visit to our hospital was “found hyperglycemia for 3 days”. Whole-genome/exon sequencing results revealed exon 3 of the insulin gene in the child carried a c.309-314del CCAGCT insGCGC heterozygous mutation. The mutation was a nonsense mutation, and family sequencing showed that the mutation originated from the mother of the child. The mother of the child was diagnosed with diabetes at the age of one and developed bilateral fundus hemorrhage and right retinal detachment at the age of 23.
MODY is a group of autosomal dominant hereditary diseases and is induced by the genetic defect of islet β cell function caused by single gene mutation, and is non-insulin resistant and non-autoimmune. In Europe, MODY patients account for 1%-2% of the total diabetic population. Traditional MODY diagnostic criteria mainly include the following: (1) ≥2 patients in the family have developed diabetes before 25 years; (2) at least three generations of the family have diabetes; (3) insulin therapy is not needed within five years after diabetes diagnosis; (4) diabetic patients have dysfunction of islet β cells. However, with constant research, relevant information revealed that the clinical manifestations of MODY tend to be more diversified. Several MODY cases do not fully conform to the above manifestations, and there are also significant differences in treatment options. Therefore, strictly following the old diagnostic criteria will miss some actual MODY patients.
With the development of molecular genetics, to date, 14 MODY-related pathogenic genes have been discovered [8]. Specifically, they include the following: hepatocyte nuclear factor 4α (HNF-4α, MODY1), glucokinase (GCK, MODY2), hepatocyte nuclear factor 1α (HNF-1α, MODY3), insulin promoter factor-1 (IPF-1, MODY4), hepatocyte nuclear factor-1β (HNF1-β, MODY5), neuron differentiation factor (NEUROD-1, MODY6), transcription factor Krüppel-like factor 11 (KLF-11, MODY7), carboxyl ester lipase (CEL, MODY8), paired-homeodomain transcription factor-4 (PAX-4, MODY9), insulin gene (INS, MODY10), B-lymphocyte kinase (BLK, MODY11), ATP-binding cassettes, sub-family C member 8, (ABCC8, MODY12), potassium inward-rectifying channel (sub-family J, member 11, KCAJ-11, MODY13) and adaptor protein, phosphotyrosine interacting PH domain and leucine zipper 1 (APPL1, MODY14). The above-mentioned known pathogenic gene mutations are not detected in about half of the patients with the MODY phenotype. The result suggests that MODY has complex pathogenesis. With constant research, new pathogenic genes and genetic types may be discovered.
Among the 14 types of MODY mentioned above, MODY1, MODY2, and MODY3 are the most common. Other types are relatively rare. Some studies reveal that the distribution of pathogenic MODY genes in different countries and races is different. MODY2 and MODY3 are the most common types in Caucasians, accounting for more than 80% of MODY patients, while in the Asian population, these two mutations account for only 7.5%-10.3% [9]. The research on MODY in China began relatively late; the specific epidemic situation and gene distribution of the disease remain unclear. The clinical manifestations of the above-mentioned types of MODY are different, and although they have their own clinical characteristics, clinical classification is difficult. Therefore, the final diagnosis and clear type need genetic examination.
MODY10 is a rare type in MODY patients and is caused by INS gene mutation. The INS gene is located on chromosome 11p 15.5 and is a gene that directly encodes insulin protein, containing three exons and two introns. Exon 2 and exon 3 are the main peptide coding regions of insulin. Exon 2 is the coding region of the signal peptide B chain, and parts of C-peptides and exon 3 are responsible for coding another part of C-peptides and A-chain.
In 2008, Edgill et al. [10] screened 296 patients who met the diagnostic criteria for MODY. The age of these patients ranged from 5 to 77 years. The age at diagnosis of diabetes of these patients ranged from 3 to 25 years, and the BMI of these patients ranged from 13-30 kg/m2. The proportion of males was about 36%. After excluding HNF4α (MODY1), GCK (MODY2), and HNF1α (MODY3) gene mutations through INS gene sequencing, a heterozygous mutation of the INS gene c.16C > T was first discovered in a MODY family in the United Kingdom. The mutation resulted in amino acid variation of the coding product P.Arg 6 Cys (R6C), and the type was MODY10. In this family, the clinical characteristics of three affected family members were consistent with that of MODY. The proband and the mother and grandmother of the proband were diagnosed with diabetes at the age of 15, 15, and 65, respectively. Their BMI was 24.1 kg/m2, 26.9 kg/m2 and 29.3 kg/m2, respectively. All of them presented with non-insulin-dependent diabetes. After the diagnosis, the proband was treated with diet control and oral medication, and blood glucose control was acceptable. After ten years, the proband began to receive low-dose insulin (0.2 IU/kg. d), and the mother of the proband began to take oral hypoglycemic drugs after 40 years of diet control. The grandmother of the proband insisted on diet control and achieved stable blood glucose eight years after diagnosis.
The follow-up studies reported 14 MODY10 families successively. One of these was a Chinese family. Data of a study revealed that in patients with MODY10, the onset age had a large span and ranged from eight months (c.265C > T, R89C) to 65 years (C, 16C > T, R6C), and the birth weight was between 2770-4500 g. The difference in clinical phenotype was also large; simple diabetes, light diabetes that can be controlled by diet, exercise, and oral medicine, insulin-dependent diabetes, and ketoacidosis have been reported. In the present study, the proband had a normal birth weight, developed the disease at two years, had an occult onset, and mild clinical symptoms. He had a family history of diabetes, and the mother of the child was diagnosed with diabetes at one year. This is in line with the clinical characteristics of MODY10 reported in the literature.
With constant research being undertaken on MODY10, so far, at least 11 INS gene mutations have been reported. Gene mutations that have been discovered are mainly missense mutations. Specifically they include the following: R6C (Edghill et al. 2008) [10]; R55C, R46Q (Mollend et al. 2008) [11]; G32S (Bonfanti et al. 2009) [12]; G32S, R89C (O, Rubio et al. 2009) [13]; R6H, R46Q (Boesgaard et al. 2010) [14]; L30M, R55C (Meur et al. 2010) [15]; V42A (Piccini et al. 2016) [16]; and A2T (Jing Yan et al. 2017) [17]. In addition, Dusatkova et al. (2015) [18] found the INS gene heterozygous frameshift mutation in a Czech family, INS gene c.233del A, resulting in an amino acid substitution of p.Gln78fs. An intron mutation c.188-31G > A was also found.
A2T is a MODY10 mutation reported for the first time by the Chinese, reported by Jing Yan et al. in 2017. In the inpatient database of the Shanghai Institute of Diabetes Mellitus, 31 patients with low genetic and environmental risk who were clinically diagnosed with type 2 diabetes were selected for whole-genome/exon sequencing and analysis, and a heterozygous missense mutation, INS gene c.4G > A was detected in one patient. The mutation can cause a p.Ala 2 Thr (A2T) mutation. The patient was diagnosed with type 2 diabetes at the age of 31, and the islet autoantibodies were all negative. The fasting blood glucose was 16 mmol/L. The patient was treated with oral hypoglycemic drugs, gliclazide, and acarbose. Molecular functional studies reveal that p.A2T mutation can lead to the substitution of hydrophilic amino acid threonine (Thr) in the signal peptide for hydrophobic amino acid alanine (Ala). It can also produce abnormal proinsulin and affect the intracellular transport of insulin, resulting in endoplasmic reticulum stress, but cannot cause apoptosis cascade reaction. A total of 6,523 patients with type 2 diabetes were studied, and the results revealed that the mutation rate of the p.A2T gene was 0.09%. Therefore, p.A2T is considered to be a hot spot mutation in the Chinese type 2 diabetic population.
In the current study, a c.309-314del CCAGCT insGCGC heterozygous mutation was detected in exon 3 of the insulin gene in the child. A 6-base- (CCAGT) deletion and a 4-base (GCGC) insertion were detected in the coding region 309-314, resulting in the 103rd bit of the coding product becoming the termination codon. The amino acid coding after tyrosine (Tyr) was completely terminated (X) (p.Y103X). The mutation was a nonsense mutation, and family sequencing showed that the mutation originated from the mother of the child. The mutation was not found in the normal population database. The result suggests that the mutation was a low-frequency mutation. Furthermore, none of the other 13 gene mutations that have been reported to cause MODY were detected. According to ASMG guidelines, the mutation was judged as a suspected pathogenic mutation. On the basis of the maternal family history of diabetes, it was considered that this mutation was a heterozygous pathogenic mutation leading to diabetes in children and was an autosomal dominant inheritance. This mutation has not been reported in any previous literature.
The INS gene regulates β cell development and insulin secretion. Data of a study revealed that different mutation sites and ways of the INS gene could cause diabetes through different molecular mechanisms. The mutation R46Q results in glutamine replacing arginine at the residue 22 of the B chain, destroying a key hydrogen bond, which results in insulin precursor synthesis disorder [7]. Heterozygous single nucleotide deletion (c.233del A) can cause INS gene frameshift mutations (Gln78 fs). This mutation can alter and prolong the amino acid sequence and lose the intrinsic structure of C-peptide and A-chain, producing abnormal proinsulin. Proinsulin contains six cysteine residues, to form three evolutionarily conserved disulfide bonds: B7-A7, B19-A20 and A6-A11. Proper disulfide bond pairing is the key factor in determining whether the proinsulin molecule can reach its natural folding structure. Many INS gene mutations can destroy the disulfide bonds of normal proinsulin by producing unpaired cysteine residues by introducing or eliminating cysteine residue in the polypeptide chain, affecting the folding of insulin molecules and leading to diabetes [19].
In the current study, exon 3 of the INS gene in the child carried a c.309-314del CCAGCT insGCGC heterozygous mutation, resulting in the 103rd bit of the coding product becoming the termination codon. The amino acid coding after tyrosine (Y) was completely terminated (X) (p.Y103X). This mutation can lead to an abnormal A-chain structure in the coding product. The last eight amino acids, including the cysteine residue of A20, are all deleted, thus, resulting in the disulfide bond of B19-A20 disappearing, accordingly affecting the molecular folding of proinsulin. Its specific pathogenesis needs to be elucidated through a further study of functional expression.
Among patients with MODY10 who have the same mutation in the same family, most of the clinical phenotypes have high reproducibility, and a few of them can show a significant gap. If the same mutation occurs in different races, the clinical phenotype may be more different. It is reported in studies that the R46Q mutation is carried in two families from Norway and Czechoslovakia. In the Norwegian family, the proband with the R46Q mutation, the father, and the aunt of the proband developed diabetes at 20, 18, and 17 years, respectively. The proband and the aunt of the proband can control the disease through diet treatment only, while the father of the proband was initially treated with diet treatment and began to take oral sulfonylureas 20 years later. He was then treated with low-dose insulin a few years later [7]. In the Czech family, the patient with the R46Q mutation, and the mother and the grandmother of the proband were diagnosed with diabetes at 13, 14, and 35 years old, respectively. All of them needed insulin treatment immediately after the onset of the disease [10]. These results suggest that epigenetics, environmental factors, or other factors may play an important role in the pathogenesis of MODY10. In this current study, the clinical manifestation of the mother of the child was serious, similar to type 1 diabetes mellitus. Since being diagnosed with diabetes at one year of age, she has been receiving insulin treatment up until now. The result suggests that although both carry the same mutation, the clinical phenotype of the child was significantly different from that of the mother of the child.
The treatment of MODY10 is similar to that of other types of MODY, mainly by diabetes diet, exercise, and insulin. Some of the patients achieve effective results by taking insulin-secreting agents. Patients with the main clinical manifestation, similar to type 1 diabetes, mainly receive insulin treatment. Some patients need to upgrade treatment gradually during the course of the disease. In the present study, 11 months after the diagnosis of diabetes, the child had been treated with a diabetes diet and exercise, and blood glucose control was acceptable. After regular follow-ups, fasting blood glucose was between 4.2-6.2 mmol/L, and two-hour postprandial blood glucose was between 5.7-11.2 mmol/L, and glycosylated hemoglobin decreased from 8.5% to 7.6%. These met the clinical characteristics of patients with mild MODY10. The child was asked to have regular follow-up visits and was given drug treatment if necessary.
Complications of MODY10, such as retinopathy, neuropathy, and microalbuminuria, have been reported [11,14], but obesity, hypertension, and hyperlipidemia, have not been reported. In the current study, the mother of the child suffered from a fundus hemorrhage and retinal detachment at the age of 23. At present, there is no report that patients with MODY10 die of diabetes-related complications. The reason may be related to the prolonged time of carrying out gene diagnosis, the shorter course of the disease and follow-up time.
Conclusion
Among Chinese children, the insulin gene c.309-314del CCAGCT insGCGC mutation may induce MODY10. In the clinic, the second-generation sequencing technology should be used in time for the diagnosis of the children with MODY characteristics and phenotype of type 1 diabetes but negative relevant antibodies.
Acknowledgements
This project is supported by the capital clinical characteristic applied research project of Beijing Science and Technology Commission (Z141107002514142).
Disclosure of conflict of interest
None.
References
- 1.Giuffrida FMA, Moises RS, Weinert LS, Calliari LE, Manna TD, Dotto RP, Franco LF, Caetano LA, Teles MG, Lima RA, Alves C, Dib SA, Silveiro SP, Dias-da-Silva MR, Reis AF Brazilian Monogenic Diabetes Study Group (BRASMOD) Maturity-onset diabetes of the young (MODY) in Brazil: establishment of a national registry and appraisal of available genetic and clinical data. Diabetes Res Clin Pract. 2017;123:134–142. doi: 10.1016/j.diabres.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 2.Billings LK, Jablonski KA, Warner AS, Cheng YC, McAteer JB, Tipton L, Shuldiner AR, Ehrmann DA, Manning AK, Dabelea D, Franks PW, Kahn SE, Pollin TI, Knowler WC, Altshuler D, Florez JC Diabetes Prevention Program Research Group. Variation in maturity-onset diabetes of the young genes influence response to interventions for diabetes prevention. J Clin Endocrinol Metab. 2017;102:2678–2689. doi: 10.1210/jc.2016-3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunerova L, Rahelić D, Ceriello A, Broz J. Use of oral antidiabetic drugs in the treatment of maturity-onset diabetes of the young: a mini review. Diabetes Metab Res Rev. 2018;34:10. doi: 10.1002/dmrr.2940. [DOI] [PubMed] [Google Scholar]
- 4.Johnson SR, Leo PJ, McInerney-Leo AM, Anderson LK, Marshall M, McGown I, Newell F, Brown MA, Conwell LS, Harris M, Duncan EL. Whole-exome sequencing for mutation detection in pediatric disorders of insulin secretion: maturity onset diabetes of the young and congenital hyperinsulinism. Pediatr Diabetes. 2018;19:656–662. doi: 10.1111/pedi.12638. [DOI] [PubMed] [Google Scholar]
- 5.Griscelli F, Feraud O, Ernault T, Oudrihri N, Turhan AG, Bonnefond A, Froguel P, Bennaceur-Griscelli A. Generation of an induced pluripotent stem cell (iPSC) line from a patient with maturity-onset diabetes of the young type 13 (MODY13) with a the potassium inwardly-rectifying channel, subfamily J, member 11 (KCNJ11) mutation. Stem Cell Res. 2017;23:178–181. doi: 10.1016/j.scr.2017.07.023. [DOI] [PubMed] [Google Scholar]
- 6.Ovsyannikova AK, Rymar OD, Ivanoshchuk DE, Mikhailova SV, Shakhtshneider EV, Orlov PS, Malakhina ES, Voevoda MI. A case of maturity onset diabetes of the young (MODY3) in a family with a novel HNF1A gene mutation in five generations. Diabetes Ther. 2018;9:413–420. doi: 10.1007/s13300-017-0350-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eeckhoute J, Formstecher P, Laine B. Maturity-onset diabetes of the young Type 1 (MODY1)-associated mutations R154X and E276Q in hepatocyte nuclear factor 4alpha (HNF4alpha) gene impair recruitment of p300, a key transcriptional co-activator. Mol Endocrinol. 2001;15:1200–1210. doi: 10.1210/mend.15.7.0670. [DOI] [PubMed] [Google Scholar]
- 8.Prudente S, Jungtrakoon P, Marucci A, Ludovico O, Buranasupkajorn P, Mazza T, Hastings T, Milano T, Morini E, Mercuri L, Bailetti D, Mendonca C, Alberico F, Basile G, Romani M, Miccinilli E, Pizzuti A, Carella M, Barbetti F, Pascarella S, Marchetti P, Trischitta V, Di Paola R, Doria A. Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet. 2015;97:177–185. doi: 10.1016/j.ajhg.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang M, Zhou JJ, Cui W, Li Y, Yang P, Chen X, Sheng C, Li H, Qu S. Molecular and phenotypic characteristics of maturity-onset diabetes of the young compared with early onset type 2 diabetes in China. J Diabetes. 2015;7:858–863. doi: 10.1111/1753-0407.12253. [DOI] [PubMed] [Google Scholar]
- 10.Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, MacDonald MJ, Støy J, Steiner DF, Philipson LH, Bell GI Neonatal Diabetes International Collaborative Group. Hattersley AT, Ellard S. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57:1034–1042. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molven A, Ringdal M, Nordbø AM, Raeder H, Støy J, Lipkind GM, Steiner DF, Philipson LH, Bergmann I, Aarskog D, Undlien DE, Joner G, Søvik O Norwegian Childhood Diabetes Study Group. Bell GI, Njølstad PR. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes. 2008;57:1131–1135. doi: 10.2337/db07-1467. [DOI] [PubMed] [Google Scholar]
- 12.Bonfanti R, Colombo C, Nocerino V, Massa O, Lampasona V, Iafusco D, Viscardi M, Chiumello G, Meschi F, Barbetti F. Insulin gene mutations as cause of diabetes in children negative for five type 1 diabetes autoantibodies. Diabetes Care. 2009;32:123–125. doi: 10.2337/dc08-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubio-Cabezas O, Edghill EL, Argente J, Hattersley AT. Testing for monogenic diabetes among children and adolescents with antibody-negative clinically defined Type 1 diabetes. Diabet Med. 2009;26:1070–1074. doi: 10.1111/j.1464-5491.2009.02812.x. [DOI] [PubMed] [Google Scholar]
- 14.Boesgaard TW, Pruhova S, Andersson EA, Cinek O, Obermannova B, Lauenborg J, Damm P, Bergholdt R, Pociot F, Pisinger C, Barbetti F, Lebl J, Pedersen O, Hansen T. Further evidence that mutations in INS can be a rare cause of Maturity-Onset Diabetes of the Young (MODY) BMC Med Genet. 2010;11:42. doi: 10.1186/1471-2350-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meur G, Simon A, Harun N, Virally M, Dechaume A, Bonnefond A, Fetita S, Tarasov AI, Guillausseau PJ, Boesgaard TW, Pedersen O, Hansen T, Polak M, Gautier JF, Froguel P, Rutter GA, Vaxillaire M. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes. 2010;59:653–661. doi: 10.2337/db09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piccini B, Artuso R, Lenzi L, Guasti M, Braccesi G, Barni F, Casalini E, Giglio S, Toni S. Clinical and molecular characterization of a novel INS mutation identified in patients with MODY phenotype. Eur J Med Genet. 2016;59:590–595. doi: 10.1016/j.ejmg.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 17.Yan J, Jiang F, Zhang R, Xu T, Zhou Z, Ren W, Peng D, Liu Y, Hu C, Jia W. Whole-exome sequencing identifies a novel INS mutation causative of maturity-onset diabetes of the young 10. J Mol Cell Biol. 2017;9:376–383. doi: 10.1093/jmcb/mjx039. [DOI] [PubMed] [Google Scholar]
- 18.Dusatkova L, Dusatkova P, Vosahlo J, Vesela K, Cinek O, Lebl J, Pruhova S. Frameshift mutations in the insulin gene leading to prolonged molecule of insulin in two families with Maturity-Onset Diabetes of the Young. Eur J Med Genet. 2015;58:230–234. doi: 10.1016/j.ejmg.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J, Arvan P. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab. 2010;21:652–659. doi: 10.1016/j.tem.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]