

Abstract
Objective: To explore the role and mechanism of oxidative stress injury in the diabetic foot. Methods: Immunohistochemistry and staining were used to detect changes in diabetic foot tissue, and the CCK-8 method was used to measure high glucose effect on cell viability. The DCFH-DA assay was used to detect the intracellular ROS content, and colorimetric methods were used to detect the activities of the CAT and SOD enzymes and the NO and MDA content in tissues and cells. In addition, the protein expression levels of PKCβ, p66shc, eNOS, ICAM-1 and NF-κB in tissues and cells were detected by Western blotting, and the distribution of p66shc and eNOS was observed by immunofluorescence. Results: The results of clinical specimens experiments showed that the DFU group exhibited disordered morphology and increased glucose metabolism, decreased activities of the enzymes CAT and SOD in tissues, and increased MDA and NO contents compared to those in the CON group. Furthermore, protein levels of the p-PKCβ, p-p66shc, ICAM-1, and p-NF-κB were increased, and eNOS protein level was decreased; these results were consistent in clinical specimens and in vitro experiments. Conclusions: High glucose levels may induce oxidative stress injury in cells and tissues by activating the PKCβ-p66shc signaling pathway.
Keywords: Diabetic foot ulcers, ROS, PKCβ, P66shc, vascular endothelial cells
Introduction
Diabetes mellitus (DM) is an inflammatory metabolic disease that influences more than 0.17 billion people worldwide [1,2]. DE’s prevalence has risen sharply regardless of new medications’ clinical treatments. According to the International Diabetes Federation Diabetes Atlas, the population of patients with DM is expected to grow by 5% annually, predominantly as a result of the increasing prevalence of type 2 diabetes mellitus (T2DM) [3]. According to the World Health Organization, diabetic foot, which is defined as “ulceration of the foot (distally from the ankle and including the ankle) associated with neuropathy and different grades of ischemia and infection”, is a severe complication of diabetes. Diabetes causes diabetic foot with different degrees of lower extremity vascular lesions and neuropathy, preventing wound healing for a long time. However, there are still no effective standard prevention and treatment methods for the serious complications of diabetic foot ulcer (DFU). In cases of DFU, most patients undergo amputation only after suffering from sensory damage, microcirculatory disturbances, difficulty in wound healing, wound infections and gangrene [4-6]. In the past, management of DFU has mainly relied on blood sugar control, a reduction in limb weight-bearing, wound dressing, topical external antibiotics, etc. [7,8], but these strategies do not reduce the risk of amputation, which seriously affects the patient’s physical and mental health [9,10]. Therefore, we are eager to conduct research on the pathological molecular mechanism of DFU to identify possible targets for early intervention to alleviate the progression of this disease and increase the chance for a good outcome.
DM leads to the development of several microvascular and macrovascular complications, including DFU [11]. Recent studies have shown that patients with diabetes are at higher risk of atherosclerotic disease than nondiabetic patients with other analogous risk factors. Diabetic microvasculature changes alter organ perfusion, especially affecting organs that are primarily relying on microvasculature supply [12]. T2DM is diagnosed by the presence of hyperglycemia [13]. Many early pathologic responses to hyperglycemia are manifested in vascular cells, including endothelial cells and vascular smooth muscle cells, which directly suffer from high glucose [14]. In addition, hyperglycemia contributes to vascular complications via various mechanisms, such as glycotoxins production and elevated oxidative stress which promotes both macro- and micro-vascular complications [15]. Changes in the structure and function of vascular endothelial cells are the common pathological basis of a variety of cardiovascular diseases, and these changes are also important causes of various complications of diabetes [16].
In diabetes, hyperglycemia increases ROS via xanthine oxidases [17], lipoxygenases, cyclooxygenases, nitric oxide synthases [18], and peroxidases. In addition, hyperglycemia itself can stimulate oxidative stress, which has been strongly implicated as a driving force in atherosclerosis [19,20]. Diabetic complications elevate ROS and oxidative stress, causing cell death [21,22] and ultimately tissue damage [23]. Various models showed that hyperglycemia-induced complications are due to ROS-imbalance-caused oxidative stress and cell death [24]. Hence, ROS suppression is of great importance in controlling diabetic complications, especially vascular disorders [25]. Therefore, exploring the molecular pathways leading to elevated ROS levels in the pathological mechanism of DFU is an essential step in the search for early therapeutic targets.
Hyperglycemia can activate “dangerous metabolic route in diabetes”, which involves the diacylglycerol (DAG)/protein kinase C (PKC) pathway and ROS production [26]. Studies also showed that PKCβ2 expression is preferably enhanced in diabetic myocardium, which promotes cardiomyocyte hypertrophy, and over-expression or activation of PKCβ2 contributes to diabetic cardiomyopathy [27]. Furthermore, PKCβ suppression improved diabetic animals’ cardiac function [28]. However, the molecular mechanism of the PKC-p66shc pathway in the development of DFU has not been reported in detail. Therefore, we asked the following question: what role does PKCβ assume in the pathophysiological process of DFU? Our results demonstrated that PKCβ-p66shc pathway activation increased ROS and enhanced the expression of pro-inflammatory proteins in DFU in clinical specimens and in vitro experiments. Inhibition of PKCβ by LY333531 protected oxidative stress-induced injury and endothelial dysfunction caused by high glucose, indicating that PKCβ might be a potential target for DFU treatment.
Materials and methods
Reagents and antibodies
Glucose was obtained from Sigma-Aldrich (Shanghai, China). Superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GSH-Px), and malondialdehyde (MDA) kits were bought from the Jiancheng Bio. (Nanjing, Jiangsu). Anti- PKCβ (ab32026) and anti-p66shc (ab54518) antibodies were bought from Abcam (Branford, CT). Antibodies against endothelial nitric oxide synthase (eNOS) (35362), ICAM-1 (4915), nuclear factor-kB (NF-κB) (49D7), p-NF-κB (108D2), and β-actin (13E5) were bought from Cell Signaling Technology (Shanghai, China). Antibody against p-PKCβ (sc-11760) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA.). A Cell Counting Kit-8 (CCK-8) was bought from Abcam (Shanghai, China). ROS assay kits were bought from Beyotime (Shanghai, China).
Cell culture
Human umbilical vein endothelial cells (HUVECs) were generously provided by the Genetic Research Department, 3rd Military Medical University (Chongqing, China). A human environment was simulated using Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS, Invitrogen, Shanghai, China) and 1% antibiotics (Beyotime, Shanghai, China). Cells were cultured at 37°C with 5% carbon dioxide.
Cell viability assay
5 × 103 cells were loaded to each well of 96-well plates following the manufacturer’s protocol. After 24 h of attachment, the medium was replaced by fresh medium containing glucose at indicated concentration, followed by another 24 h incubation. Then, cell viability was detected with the CCK-8 kit. Briefly, 10 µL of CCK-8 reagent was mixed with 100 µL of the medium mixture in each well. The plates were incubated at 37°C for 2 h. An automatic microplate reader (BioTek, Winooski, VT) was used to measure the absorbance at 450 nm, which was directly proportional to cell viability. All experiments were repeated three times.
ROS assay
Take 3 × 105 HUVECs and inoculate them in a 6-well plate. After the cells adhere to the wall, the cells are loaded with 2.0 mmol/L glucose for 24 h. Use the DCFH method to detect ROS according to the instructions. Observe the fluorescence intensity of DCFH under the fluorescence microscope.
Western blotting
Cells were separated, PBS-washed, and re-suspended in lysis buffer containing 1% phenylmethanesulfonyl fluoride and phosphatase inhibitors Beyotime. After incubation on ice for 30 minutes, supernatants were harvested by centrifugation at 12,000 rpm for 15 min at 4°C, and protein concentrations were measured by a bicinchoninic acid (BCA) kit (Beyotime, Shanghai, China). Samples containing same amounts of proteins were resolved by sodium lauryl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membrane (Elabscience, Houston, TX). The membrane was blocked with 5% skim milk for two hours, incubated with first antibodies at 4°C overnight, rinsed 3 times with Tris-buffered saline with Tween-20 (TBST), and incubated with secondary antibody for 1 h at 25°C. Protein bands were measured with enhanced chemiluminescence (ECL) solution (Amersham Biosciences, Piscataway, NJ) and quantified by an image analyzer (FujiFilm Medical Systems, Stamford, CT).
Immunofluorescence staining
Cells (3 × 104) were cultured on glass slides for twenty-four hours, treated in the presence or absence of glucose for another twenty-four hours, fixed paraformaldehyde (4%) for thirty minutes at 25°C, washed two times and permeabilized using 0.5% Triton X-100 in PBS for twenty minutes. Cells were then blocked by goat serum albumin (1%) for one hour and incubated with anti-P66shc and anti-eNOS antibo and dies at 4°C overnight. After three times of PBS-washing, cells were incubated with FITC-conjugated secondary antibodies (CST, Danvers, MA) for 2 h at 37°C. 4’,6-diamidino-2-phenylindole (DAPI) was added to the staining solution 5 minutes before the end of staining to stain the nuclei. Slices were observed under a fluorescence microscope (Leica, Buffalo Grove, IL).
Measurement of oxidation-related biological parameters
Normal saline containing heparin (0.16 mg/mL) was used to get rid of the blood from collected tissues (n≥3 per group). An appropriate amount of precooled PBS was added to each tissue sample for homogenization in an ice bath and centrifuged at 4°C. The supernatants were collected, and protein concentrations of the supernatants were measured with a BCA kit as mentioned above. MDA contents and activities of SOD, GSH-Px, and CAT were measured following the manufacturer’s protocols. All experiments were repeated three times.
Histological analysis of liver steatosis
A piece of fresh tissue was fixed in formaldehyde and embedded in paraffin. The 6-μm-thick sections were subjected to hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), and Masson trichrome staining, followed by observation under a microscope.
Statistical analysis
Data and figures in the present study are representatives of at least three independent experiments. The data were analyzed using Statistical Product and Service Solutions (SPSS) 22.0 (IBM, Armonk, NY) and expressed as mean ± SD (standard deviation), and Student’s t-test was used to compare the significance of differences between groups, which is indicated by *P<0.05, **P<0.01.
Results
Tissue changes in DFU
We processed and collected DFU tissues after surgery. Figure 1A shows necrotic parts of the feet of diabetic foot patients. The red box shows the central tissue of the DFU. The green box shows the marginal tissue of DFUs in the control (CON) group used in subsequent experiments. A pathological sectioning technique was performed to detect tissue changes. Under a light microscope, the healthy control (HC) group showed a complete tissue structure, regular morphology, a small number of normal blood vessels distributed between the tissues, regular cell alignment, tight connections, and normal gaps. The structure of the marginal tissue of DFUs was slightly disordered, and a large number of vascular rings with different diameters were visible. The cell connections were somewhat loose, and the cells were slightly swollen. The tissue structure of the center of the DFU was disordered with few vascular rings. The nuclei were polymorphous and aggregated or located at the edges of the cells (Figure 1B). Masson staining was used to detect fibrotic changes in necrotic tissue from the diabetic foot, with collagen fibers stained blue and muscle fibers stained red. The staining results showed small blood vessel-like hyperplasia evident in the marginal tissue of DFUs (Figure 1C). PAS staining was used to detect changes in glucose metabolism in diabetic foot necrotic tissue. Periodic acid oxidizes the hydroxyl groups on two adjacent carbon atoms of the sugar to aldehyde groups, and Schiff reagent reacts with the aldehyde groups, forming a purplish red color. As shown in Figure 1D, glucose metabolism in the marginal tissue in the CON group was elevated.
Figure 1.
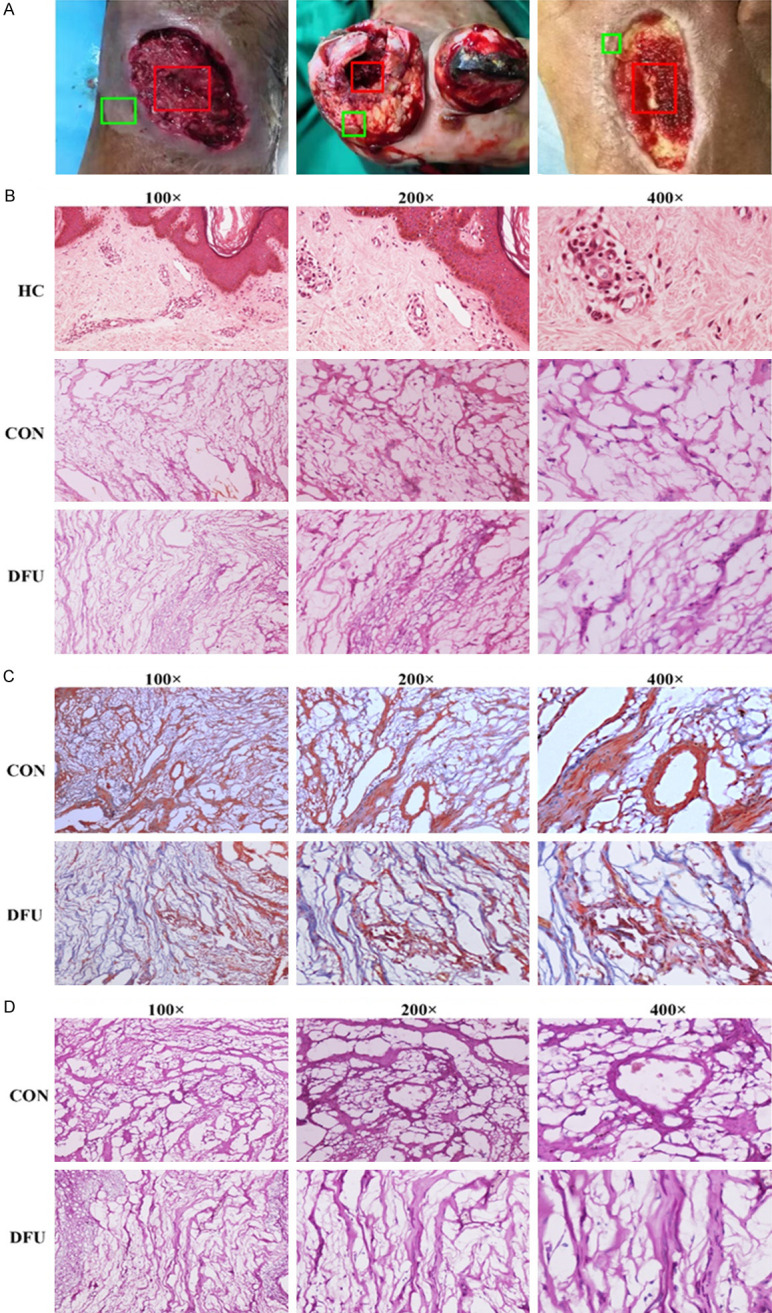
Tissue changes in diabetic foot ulcer. A. The necrotic parts on the feet of diabetic patients. B. Hematoxylin and eosin staining of diabetic foot ulcer tissues. C. Masson staining of diabetic foot ulcer tissues. D. PAS staining of diabetic foot ulcer tissues.
Induced oxidative stress damage in diabetic foot tissue
Figure 2 showed the activities of SOD, CAT, and GSH-Px and MDA content in both the DFU and CON groups compared with those in the CON group, activities of SOD, GSH-Px, and CAT were dramatically decreased (Figure 2A-C), and the MDA content was significantly higher (Figure 2D) in the DFU group, which suggested the weakened antioxidative ability of the diabetic foot tissue, inducing oxidative stress damage.
Figure 2.
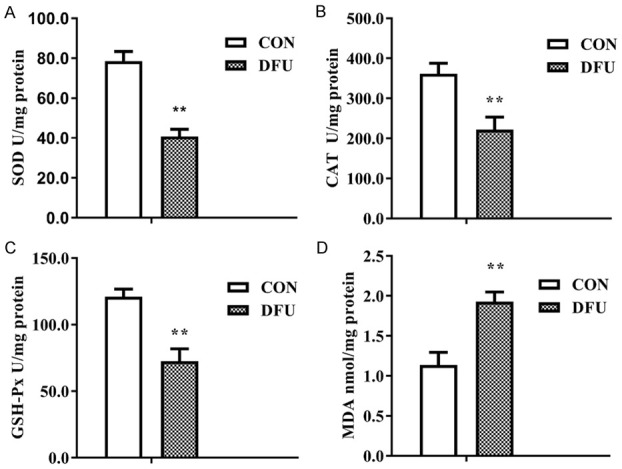
Induced oxidative stress damage in diabetic foot ulcer tissue. A. Decreased SOD activity (P<0.01). B. Decreased CAT activity (P<0.01). C. Decreased GSH-Px activity (P<0.01). D. Increased MDA content (P<0.01). n≥3 per group, with the experiment repeated three times. Values are the means and standard errors (*P<0.05 and **P<0.01 versus CON).
Expression changes in the PKCβ-p66shc pathway and inflammation-related proteins in DFU
As mentioned above, PKCβ plays an important role in the development of the diabetic myocardium by regulating the activity of the adaptor protein p66shc, which is translocated into mitochondria and causes oxidative stress damage. Thus, we examined the expression of PKCβ-p66shc pathway members and inflammation-related proteins in DFU tissue. Figure 3 showed that the expression of p-PKCβ, p-P66shc, NF-κB, and ICAM-1 was dramatically elevated, while eNOS expression was substantially decreased in the DFU group compared to the CON group.
Figure 3.
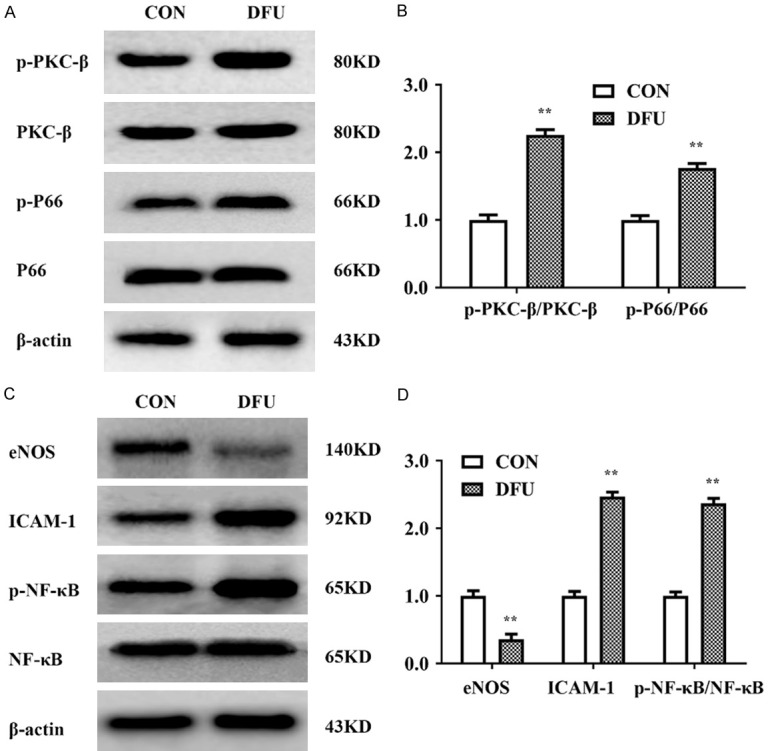
Expression changes in PKCβ/p66shc and inflammation-related proteins in diabetic foot ulcer. A, B. Western blot analysis showed significantly increased expression levels of p-PKCβ and p-P66shc in the DFU group vs. the CON group (P<0.01). C, D. Western blot analysis showed decreased protein levels of eNOS and significantly increased expression levels of ICAM-1 and p-NF-κB in the DFU group (P<0.01). n≥3 per group, with the experiment repeated three times. Values are the means and standard errors (*P<0.05 and **P<0.01 versus CON).
High glucose-induced morphological changes and increased ROS levels in HUVECs
The results above showed decreased antioxidative stress ability in DFU tissue, activation of the PKCβ/p66shc pathway, decreased expression of eNOS and increased expression of ICAM-1. To confirm the pathological role of the PKCβ/P66shc pathway in endothelial cell dysfunction, we performed further in vitro experiments.
We first measured the antiproliferative effect of glucose on HUVECs. Cell viability in HUVECs treated with glucose at different concentrations (0, 10, 20, and 40 mM) for 24 h was investigated by the CCK-8 assay. Figure 4A, 4B indicated that the viability of HUVECs was dramatically decreased after treatment with 40 mM glucose. The results of a timed CCK-8 assay indicated that high glucose suppression of cell proliferation did not continue after 24 h. Thus, 20 mM glucose incubation with 20 mM glucose for 24 h was used in subsequent experiments. As shown in Figure 4C, under light microscopy, the cell morphology was intact in HUVECs treated with 20 mM glucose-containing medium, but the HUVECs became constricted and round when treated with 40 mM glucose-containing medium. To further confirm that glucose-induced ROS generation was p-P66shc-dependent, we preincubated HUVECs with 5 nM LY333531, a specific inhibitor of PKCβ1 and PKCβ2, for two hours before administration of glucose (20 mM). The intracellular ROS content was measured by the DCFH-DA assay, and the cells were then observed with a fluorescence microscope. As shown in Figure 4D, 4E, DCFH-DA staining suggested that as the concentration of glucose (0, 10, 20 mM) in the medium increased, the cellular ROS level increased accordingly, and ROS were reduced to the normal level by the application of LY333531.
Figure 4.
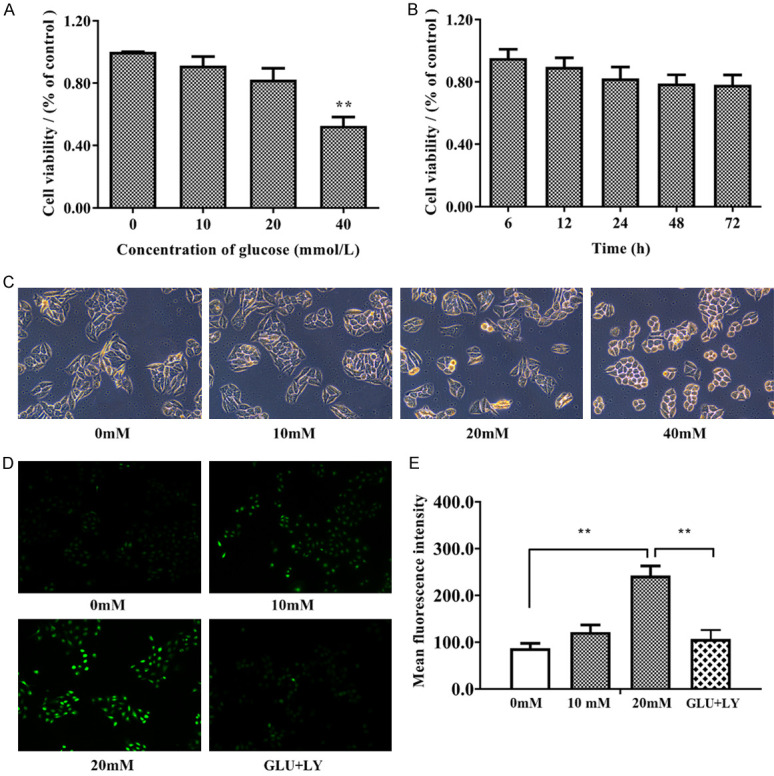
High glucose induced morphological changes and increased ROS levels in HUVECs. A. HUVECs were incubated with glucose at increasing concentrations (0, 10, 20, and 40 mM) for 24 h. The Cell Counting Kit-8 (CCK-8) assay was performed to detect the cytotoxic effect of glucose. B. HUVECs were incubated with 20 mM glucose for the indicated time. The Cell Counting Kit-8 (CCK-8) assay was performed to detect the cytotoxic effect of glucose. C. The morphology of the cells under light microscopy shows that 40 mM glucose changed the morphology of HUVECs. D, E. The results of the DCFH-DA assay under a fluorescence microscope. Software analysis of the fluorescence density. n≥3 per group, with the experiment repeated three times. Values are the means and standard errors (*P<0.05 and **P<0.01 versus control).
High glucose affects oxidative and antioxidative activities in HUVECs
Due to an imbalance between oxidative and antioxidative systems in DFU, we measured the levels of CAT, SOD, GSH-Px, and MDA in HUVECs treated with normal medium, high-glucose medium and high-glucose medium with LY333531. Figure 5 indicated that the activities of CAT, SOD, and GSH-Px were significantly inhibited, and the MDA content was significantly increased in 24 h glucose treatment group. However, the decreased SOD, CAT, and GSH-Px activities and increased MDA content were rescued in HUVECs pretreated with LY333531 before exposure to high glucose. All of these results suggest that the PKCβ inhibitor LY333531 could alleviate oxidative stress caused by high glucose and that PKCβ has an important biological function in the development of DFU.
Figure 5.

High glucose affected oxidative and antioxidative activities in HUVECs. A-C. LY333531 rescued the decreased activities of CAT, SOD, and GSH-Px in HUVECs treated with glucose. D. LY333531 rescued the glucose-induced increase in the MDA content in HUVECs. Each group was a mixture of cells collected three times (n = 3), and the experiment was repeated three times. Values are the means and standard errors (*P<0.05 and **P<0.01 versus CON).
LY333531 inhibits the activity of the PKCβ-p66shc signaling caused by high glucose in HUVECs
To verify that glucose-induced oxidative stress is PKCβ-dependent, we pre-treated HUVECs with LY333531 (5 nM) for two hours prior to glucose treatment. Expression levels of PKCβ-p66shc pathway members were detected by Western blotting. As shown in Figure 6A-D, LY333531 rescued the increased expression of p-PKCβ, p-P66shc, ICAM-1, and NF-κB and the decreased expression of eNOS in HUVECs treated with glucose. Subsequently, by immunofluorescence, we further observed the distribution of p-P66shc and eNOS in HUVECs treated with 20 mM glucose for 24 h. After treatment with glucose, the green fluorescent signal of p-P66shc was significantly increased, and p-P66shc had translocated from the cytoplasm into the nucleus, unlike the reduced cytoplasmic expression of eNOS in the HUVECs. However, in HUVECs pretreated with LY333531, the increase in the expression of p-p66shc was reduced, with a weak green fluorescent signal maintained in the cytoplasm, and the reduced levels of eNOS were remarkably restored (Figure 6E-H). These findings indicate that LY333531 can rescue activation of the PKCβ-P66shc pathway and the inhibitory activity of eNOS caused by high glucose.
Figure 6.
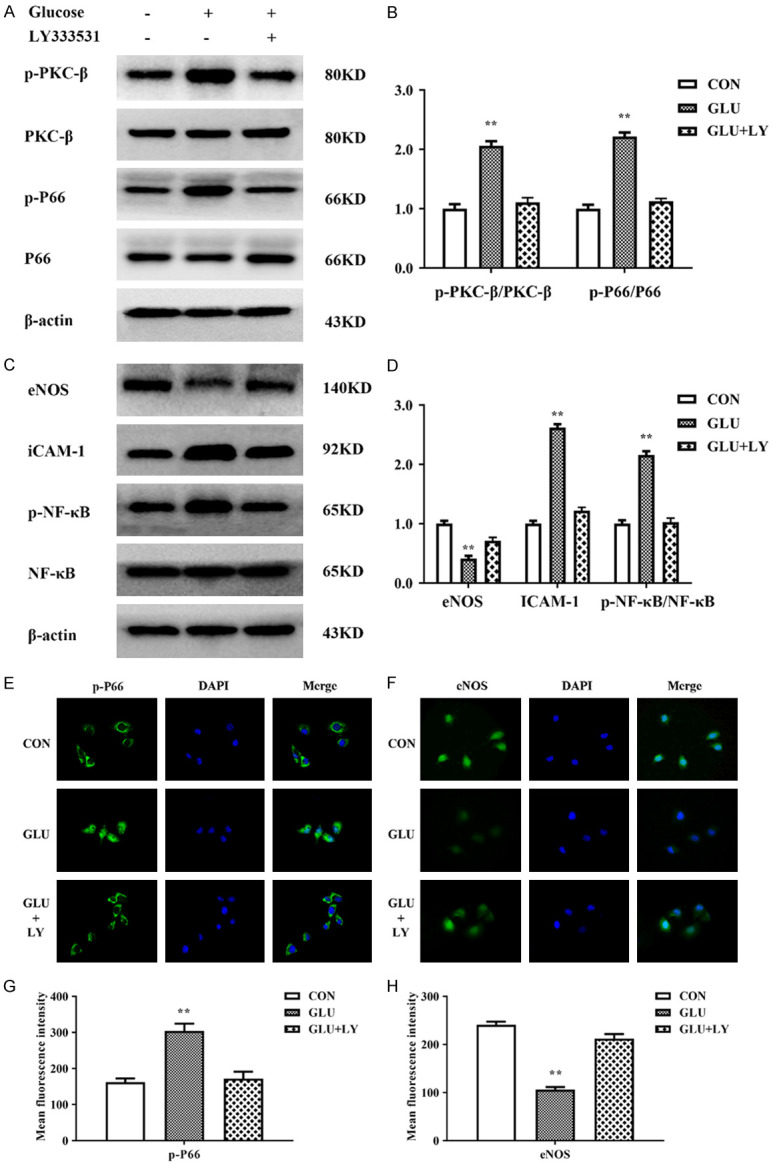
LY333531 inhibited changes in the activity of the PKCβ-p66shc pathway caused by high glucose in HUVECs. A, B. LY333531 rescued the increased levels of p-PKCβ and p-P66shc in HUVECs treated with glucose. C, D. LY333531 rescued the increased levels of ICAM-1 and p-NF-κB and the inhibition of eNOS in HUVECs treated with glucose. E, F. Immunofluorescence showed that LY333531 could restore the expression levels and distribution of p-P66shc and restore the eNOS level in HUVECs treated with high glucose. G, H. The mean fluorescence intensity of p-P66shc and eNOS was quantified and is presented as the means ± SDs. Each group was a mixture of cells collected three times (n = 3), and the experiment was repeated three times. Values are the means and standard errors (*P<0.05 and **P<0.01 versus CON).
Discussion
In diabetic patients, lower limb protection is reduced due to neuropathy, and large vessel and microvascular lesions cause ulceration and gangrene due to insufficient arterial perfusion [29]. DFU is a serious complication of diabetes and an important cause of disability and death in diabetic patients [30]. It not only brings pain to patients but also creates a very large economic burden. The amputation rate of patients with diabetic foot disease is 15 times greater than that of nondiabetic patients, and approximately 50% of amputated patients each year have diabetes [31]. The incidence of peripheral vascular disease in diabetic patients is significantly higher than that in nondiabetic patients. According to the WHO Diabetes Complications Study in London, 3% to 20% of DFUs present only lower limb ischemia, and 46% of amputations are associated with lower limb ischemia. Peripheral vascular arteriosclerosis leads to lower limb ischemia and severe gangrene [32]. Abnormal vascular structure and sclerosis, accompanied by advanced dysfunction and aggravation of tissue ischemia and hypoxia, promote tissue necrosis and ulceration, causing foot ulcers to remain unhealed for a long time [33]. As shown in our results, marginal tissue exhibited less vascular proliferation and metabolic enhancement compared with the central tissue of DFUs. At this time, interventions to reverse or delay ulceration of the diabetic foot may be effective.
Diabetic foot is a multifactorial disease with complicated causes and underlying mechanisms [34]. However, there is increasing evidence that endothelial dysfunction could be one primary mechanism of its pathogenesis [35,36]. Hyperglycemia in diabetic patients has been shown to cause metabolic disorders and oxidative stress in endothelial cells, leading to miscellaneous physiological and pathological effects, such as permeability increase, impairment of endothelial dilation and decreased NO utilization [37,38]. Therefore, endothelial cells may become potential therapeutic targets in the treatment of diabetic foot. This study focused on the mechanism and influence of endothelial cells in the progression of diabetic foot and could provide new ideas for diabetic foot treatment and targeted drugs development. Increased production of ROS and the loss of endothelial NO bioavailability are key features of vascular disease in DM [38]. ROS are byproducts of normal oxygen metabolism and play essential roles in cell signaling and homeostasis [39]. ROS can be finely regulated and constitutes a physiological signaling pathway [40]. Although ROS originate from different subcellular sources, mitochondria are their primary important source [41]. ROS, a normal component of oxidative phosphorylation generated by the electron transport chain, plays an important role in regulating cell signaling, growth, and differentiation [42,43]. However, a portion of O2 molecules (1-3%) can be converted into superoxide anion radicals by complexes I and III [44]. The radical can then be transferred to hydrogen peroxide (H2O2) and hydroxyl radicals. These oxidizing substances are normal metabolic byproducts continuously produced by mitochondria and kept in check by endogenous antioxidant systems (such as SOD (which generate H2O2 and O2 from superoxide), CAT, GSH-Px, and peroxidase) [45]. Imbalanced ROS generation and cellular antioxidant defense system will lead to oxidative stress [46]. Consistent with this view, our experimental results show that the activities of SOD, CAT, and glutathione peroxidase were significantly inhited in DFU tissues compared with the tissues surrounding the DFU. The results in endothelial cells after high-glucose loading were consistent with the results in DFU tissues, and the activities of SOD, CAT, and GSH-Px were dramatically enhanced, and MDA contents were decreased in cells pretreated with LY333531. This finding indicates that the antioxidant capacity of tissues or cells under high-glucose conditions is weakened, but antioxidant capacity was remarkably restored after the use of LY333531.
Chronic hyperglycemia results in PKC activation, which promotes diabetic cardiovascular complications [47]. Moreover, PKCβ2-activation has been involved in diabetic abnormalities through inhibition of eNOS, and restoration of Akt-eNOS-NO signaling was shown to be attenuated in diabetic cardiomyopathy and myocardial dysfunction. Some studies assessed PKCβ-mediated regulation of p-p66shc and showed that PKCβ-phosphorylation is key for P66shc activation. Furthermore, studies showed that P66shc is essential for regulating intracellular oxidative stress levels and redox balance. Increasing evidence support that cells lacking P66shc has less free radicals. Indeed, ROS leads to upregulation and the nuclear translocation of NF-κB subunit p65 [48], resulting in the transcription of proinflammatory genes encoding monocyte chemoattractant protein-1 (MCP-1), vascular cell adhesion molecule-1 (VCAM-1), and intracellular cell adhesion molecule-1 (ICAM-1) [49]. In addition, PKCβ-mediated p66shc phosphorylation at serine 36 has been considered as a critical step preceding mitochondrial translocation, ROS production, and cell death, and thus PKCβ may serve as a therapeutic target [50]. As shown by our experimental results, PKCβ expression in tissues and cells under hyperglycemia (vs. controls) was indeed upregulated, and p-p66shc expression was also increased, whereas the expression of eNOS was decreased, and the expression of ICAM-1 was elevated. However, after the addition of LY333531, these proteins levels returned to normal.
In summary, we have demonstrated in clinical specimens and in vitro experiments that high glucose affects endothelial cells by the PKCβ-p66shc pathway. After the expression of PKCβ-p66shc was increased, p66 affected the level of ROS in tissues or cells, resulting in the activation of NF-κB, increased expression of ICAM-1, and decreased expression of eNOS. LY333531 rescued changes in the expression of PKCβ-p66shc pathway members, NF-κB, ICAM-1, and eNOS, which suggests inhibition of PKC protein expression as a key link in inhibiting oxidative stress damage and vascular endothelial damage in tissues and cells of the diabetic foot.
Acknowledgements
This work was supported by Science and Technology Projects of Yuzhong Direct of Chongqing (20170123) and National Natural Science Foundation of China (81770738, 81672230).
Disclosure of conflict of interest
None.
References
- 1.Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9:119. doi: 10.1038/s41419-017-0135-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Diabetes Association. Classification and diagnosis of diabetes. Diabetes Care. 2015;38(Suppl):S8–S16. doi: 10.2337/dc15-S005. [DOI] [PubMed] [Google Scholar]
- 3.Bommer C, Sagalova V, Heesemann E, Manne-Goehler J, Atun R, Bärnighausen T, Davies J, Vollmer S. Global Economic burden of diabetes in adults: projections from 2015 to 2030. Diabetes Care. 2018;41:963–970. doi: 10.2337/dc17-1962. [DOI] [PubMed] [Google Scholar]
- 4.Perez-Favila A, Martinez-Fierro ML, Rodriguez-Lazalde JG, Cid-Baez MA, Zamudio-Osuna MJ, Martinez-Blanco MDR, Mollinedo-Montaño FE, Rodriguez-Sanchez IP, Castañeda-Miranda R, Garza-Veloz I. Current therapeutic strategies in diabetic foot ulcers. Medicina (Kaunas) 2019;55:714. doi: 10.3390/medicina55110714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parker CN, Van Netten JJ, Parker TJ, Jia L, Corcoran H, Garrett M, Kwok CF, Nather A, Que MT, Srisawasdi G, Wraight P, Lazzarini PA. Differences between national and international guidelines for the management of diabetic foot disease. Diabetes Metab Res Rev. 2019;35:e3101. doi: 10.1002/dmrr.3101. [DOI] [PubMed] [Google Scholar]
- 6.Wang GY, Wang LY, Wang Y, Xu XY. Multidisciplinary approach to scheduling surgery for diabetic foot: a case report. BMC Musculoskelet Disord. 2019;20:168. doi: 10.1186/s12891-019-2522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turns M. Diabetic foot ulcer management: the podiatrist’s perspective. Br J Community Nurs. 2013;(Suppl):S14, S16–9. doi: 10.12968/bjcn.2013.18.sup12.s14. [DOI] [PubMed] [Google Scholar]
- 8.Lavery LA, Hunt NA, Lafontaine J, Baxter CL, Ndip A, Boulton AJ. Diabetic foot prevention: a neglected opportunity in high-risk patients. Diabetes Care. 2010;33:1460–2. doi: 10.2337/dc10-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen N, Gigi R, Haim A, Salai M, Chechik O. Mortality and reoperations following lower limb amputations. Isr Med Assoc J. 2014;16:83–7. [PubMed] [Google Scholar]
- 10.Hasanadka R, McLafferty RB, Moore CJ, Hood DB, Ramsey DE, Hodgson KJ. Predictors of wound complications following major amputation for critical limb ischemia. J Vasc Surg. 2011;54:1374–82. doi: 10.1016/j.jvs.2011.04.048. [DOI] [PubMed] [Google Scholar]
- 11.Jupiter DC, Thorud JC, Buckley CJ, Shibuya N. The impact of foot ulceration and amputation on mortality in diabetic patients. I: from ulceration to death, a systematic review. Int Wound J. 2016;13:892–903. doi: 10.1111/iwj.12404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goncalves NP, Vaegter CB, Andersen H, Ostergaard L, Calcutt NA, Jensen TS. Schwann cell interactions with axons and microvessels in diabetic neuropathy. Nat Rev Neurol. 2017;13:135–147. doi: 10.1038/nrneurol.2016.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyenihi AB, Langa S, Mukaratirwa S, Masola B. Effects of Centella asiatica on skeletal muscle structure and key enzymes of glucose and glycogen metabolism in type 2 diabetic rats. Biomed Pharmacother. 2019;112:108715. doi: 10.1016/j.biopha.2019.108715. [DOI] [PubMed] [Google Scholar]
- 14.Jorgens K, Stoll SJ, Pohl J, Fleming TH, Sticht C, Nawroth PP, Hammes HP, Kroll J. High tissue glucose alters intersomitic blood vessels in zebrafish via methylglyoxal targeting the VEGF receptor signaling cascade. Diabetes. 2015;64:213–25. doi: 10.2337/db14-0352. [DOI] [PubMed] [Google Scholar]
- 15.D’Apolito M, Du X, Pisanelli D, Pettoello-Mantovani M, Campanozzi A, Giacco F, Maffione AB, Colia AL, Brownlee M, Giardino I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis. 2015;239:393–400. doi: 10.1016/j.atherosclerosis.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoon CH, Choi YE, Cha YR, Koh SJ, Choi J, Kim TW, Woo SJ, Park YB, Chae IH, Kim HS. Diabetes-induced jagged1 overexpression in endothelial cells causes retinal capillary regression in a murine model of diabetes mellitus: insights into diabetic retinopathy. Circulation. 2016;134:233–47. doi: 10.1161/CIRCULATIONAHA.116.014411. [DOI] [PubMed] [Google Scholar]
- 17.Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase-derived reactive species: physiological and pathological effects. Oxid Med Cell Longev. 2016;2016:3527579. doi: 10.1155/2016/3527579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchard KJ, Ackerman AW, Gross ER, Stepp DW, Shi Y, Fontana JT, Baker JE, Sessa WC. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J Biol Chem. 2001;276:17621–4. doi: 10.1074/jbc.C100084200. [DOI] [PubMed] [Google Scholar]
- 19.Dang VT, Zhong LH, Huang A, Deng A, Werstuck GH. Glycosphingolipids promote pro-atherogenic pathways in the pathogenesis of hyperglycemia-induced accelerated atherosclerosis. Metabolomics. 2018;14:92. doi: 10.1007/s11306-018-1392-2. [DOI] [PubMed] [Google Scholar]
- 20.Yuan T, Yang T, Chen H, Fu DL, Hu YY, Wang J, Yuan Q, Yu H, Xu WF, Xie X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019;20:247–60. doi: 10.1016/j.redox.2018.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krakauer T. Inflammasome, mTORC1 activation, and metabolic derangement contribute to the susceptibility of diabetics to infections. Med Hypotheses. 2015;85:997–1001. doi: 10.1016/j.mehy.2015.08.019. [DOI] [PubMed] [Google Scholar]
- 22.Jha JC, Ho F, Dan C, Jandeleit-Dahm K. A causal link between oxidative stress and inflammation in cardiovascular and renal complications of diabetes. Clin Sci (Lond) 2018;132:1811–1836. doi: 10.1042/CS20171459. [DOI] [PubMed] [Google Scholar]
- 23.Kohnert KD, Freyse EJ, Salzsieder E. Glycaemic variability and pancreatic beta-cell dysfunction. Curr Diabetes Rev. 2012;8:345–54. doi: 10.2174/157339912802083513. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Peng T, Zhu H, Zheng XF, Zhang XS, Jiang N, Cheng XS, Lai XY, Shunnar A, Singh M, Riordan N, Bogin V, Tong NW, Min WP. Prevention of hyperglycemia-induced myocardial apoptosis by gene silencing of Toll-like receptor-4. J Transl Med. 2010;8:133. doi: 10.1186/1479-5876-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez CD, Lee MS, Marchetti P, Pietropaolo M, Towns R, Vaccaro MI, Watada H, Wiley JW. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7:2–11. doi: 10.4161/auto.7.1.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nogueira-Machado JA, Chaves MM. From hyperglycemia to AGE-RAGE interaction on the cell surface: a dangerous metabolic route for diabetic patients. Expert Opin Ther Targets. 2008;12:871–82. doi: 10.1517/14728222.12.7.871. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Lei S, Gao X, Mao XW, Wang TT, Wong GT, Vanhoutte PM, Irwin MG, Xia ZY. PKCbeta inhibition with ruboxistaurin reduces oxidative stress and attenuates left ventricular hypertrophy and dysfunction in rats with streptozotocin-induced diabetes. Clin Sci (Lond) 2012;122:161–73. doi: 10.1042/CS20110176. [DOI] [PubMed] [Google Scholar]
- 28.Wei L, Yin Z, Yuan Y, Hwang A, Lee A, Sun DD, Li F, Di CX, Zhang RQ, Cao F, Wang HC. A PKC-beta inhibitor treatment reverses cardiac microvascular barrier dysfunction in diabetic rats. Microvasc Res. 2010;80:158–65. doi: 10.1016/j.mvr.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Apelqvist JA, Lepantalo MJ. The ulcerated leg: when to revascularize. Diabetes Metab Res Rev. 2012;28(Suppl 1):30–5. doi: 10.1002/dmrr.2259. [DOI] [PubMed] [Google Scholar]
- 30.Abbas ZG, Archibald LK. Epidemiology of the diabetic foot in Africa. Med Sci Monit. 2005;11:RA262–70. [PubMed] [Google Scholar]
- 31.Pascale R, Vitale M, Esposito S, Noviello S. Update on diabetic foot infections. Infez Med. 2012;20:155–68. [PubMed] [Google Scholar]
- 32.Kum S, Huizing E, Schreve MA, Ünlü C, Ferraresi R, Samarakoon LB, Heuvel DA. Percutaneous deep venous arterialization in patients with critical limb ischemia. J Cardiovasc Surg (Torino) 2018;59:665–699. doi: 10.23736/S0021-9509.18.10569-6. [DOI] [PubMed] [Google Scholar]
- 33.Sree VD, Rausch MK, Tepole AB. Linking microvascular collapse to tissue hypoxia in a multiscale model of pressure ulcer initiation. Biomech Model Mechanobiol. 2019;18:1947–1964. doi: 10.1007/s10237-019-01187-5. [DOI] [PubMed] [Google Scholar]
- 34.Kolossvary E, Bansaghi Z, Szabo GV, Jarai Z, Farkas K. Ischemic origin of diabetic foot disease. Epidemiology, difficulties of diagnosis, options for prevention and revascularization. Orv Hetil. 2017;158:203–11. doi: 10.1556/650.2017.30649. [DOI] [PubMed] [Google Scholar]
- 35.Gao J, Zhao G, Li W, Zhang JY, Che YL, Song MY, Gao S, Zeng B, Wang YH. MiR-155 targets PTCH1 to mediate endothelial progenitor cell dysfunction caused by high glucose. Exp Cell Res. 2018;366:55–62. doi: 10.1016/j.yexcr.2018.03.012. [DOI] [PubMed] [Google Scholar]
- 36.Catrina SB, Zheng X. Disturbed hypoxic responses as a pathogenic mechanism of diabetic foot ulcers. Diabetes Metab Res Rev. 2016;32(Suppl 1):179–85. doi: 10.1002/dmrr.2742. [DOI] [PubMed] [Google Scholar]
- 37.Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Aydin S, Bacanli M, Anlar HG, Çal T, Arı N, Ündeğer Bucurgat Ü, Başaran AA, Başaran N. Preventive role of Pycnogenol((R)) against the hyperglycemia-induced oxidative stress and DNA damage in diabetic rats. Food Chem Toxicol. 2019;124:54–63. doi: 10.1016/j.fct.2018.11.038. [DOI] [PubMed] [Google Scholar]
- 39.Ward JPT. From physiological redox signalling to oxidant stress. Adv Exp Med Biol. 2017;967:335–342. doi: 10.1007/978-3-319-63245-2_21. [DOI] [PubMed] [Google Scholar]
- 40.Bitar MS. Diabetes impairs angiogenesis and induces endothelial cell senescence by up-regulating thrombospondin-CD47-dependent signaling. Int J Mol Sci. 2019;20:673. doi: 10.3390/ijms20030673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li S, Sun Y, Qi X, Shi Y, Gao H, Wu Q, Liu XC, Yu HT, Zhang CJ. Protective effect and mechanism of glutaredoxin 1 on coronary arteries endothelial cells damage induced by high glucose. Biomed Mater Eng. 2014;24:3897–903. doi: 10.3233/BME-141221. [DOI] [PubMed] [Google Scholar]
- 42.Scialo F, Sriram A, Fernandez-Ayala D, Gubina N, Lõhmus M, Nelson G, Logan A, Cooper HM, Navas P, Enríquez JA, Murphy MP, Sanz A. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. 2016;23:725–34. doi: 10.1016/j.cmet.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (Review) Int J Mol Med. 2019;44:3–15. doi: 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goncalves RL, Rothschild DE, Quinlan CL, Scott GK, Benz CC, Brand MD. Sources of superoxide/H2O2 during mitochondrial proline oxidation. Redox Biol. 2014;2:901–9. doi: 10.1016/j.redox.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ekoue DN, He C, Diamond AM, Bonini MG. Manganese superoxide dismutase and glutathione peroxidase-1 contribute to the rise and fall of mitochondrial reactive oxygen species which drive oncogenesis. Biochim Biophys Acta Bioenerg. 2017;1858:628–32. doi: 10.1016/j.bbabio.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Marchi E, Baldassari F, Bononi A, Wieckowski MR, Pinton P. Oxidative stress in cardiovascular diseases and obesity: role of p66Shc and protein kinase C. Oxid Med Cell Longev. 2013;2013:564961. doi: 10.1155/2013/564961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng YS, Dai DZ, Ji H, Zhang Q, Dai Y. Sildenafil and FDP-Sr attenuate diabetic cardiomyopathy by suppressing abnormal expression of myocardial CASQ2, FKBP12.6, and SERCA2a in rats. Acta Pharmacol Sin. 2011;32:441–8. doi: 10.1038/aps.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramanan S, Kooshki M, Zhao W, Hsu FC, Robbins ME. PPARalpha ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-kappaB and AP-1 pathways. Free Radic Biol Med. 2008;45:1695–704. doi: 10.1016/j.freeradbiomed.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li M, Wang X, Fu W, He S, Li D, Ke Q. CD4+CD25+Foxp3+ regulatory T cells protect endothelial function impaired by oxidized low density lipoprotein via the KLF-2 transcription factor. Cell Physiol Biochem. 2011;28:639–48. doi: 10.1159/000335759. [DOI] [PubMed] [Google Scholar]
- 50.Haller M, Khalid S, Kremser L, Fresser F, Furlan T, Hermann M, Guenther J, Drasche A, Leitges M, Giorgio M, Baier G, Lindner H, Troppmair J. Novel insights into the PKCbeta-dependent regulation of the oxidoreductase p66Shc. J Biol Chem. 2016;291:23557–23568. doi: 10.1074/jbc.M116.752766. [DOI] [PMC free article] [PubMed] [Google Scholar]