

Abstract
The mitochondrial receptor protein FUN14 domain-containing-1 (FUNDC1) can induce mitophagy under hypoxic conditions, as well as playing important roles in normal metabolism and intracellular homeostasis. Exercise not only elevates mitochondrial biosynthesis, but also exerts a significant impact on mitochondrial fission, integration and mitophagy. However, it is still not clear whether FUNDC1 plays a regulatory role in this context. Electrical pulse stimulation (EPS) of cultured myotubes is widely used as an in vitro model of muscle contraction. We simulated the contraction of C2C12 myotubes by EPS (15 V, 1 Hz, 2 ms, 1 h) to examine the role of FUNDC1 in mitophagy. EPS was found to induce mitophagy by activating the AMPK-ULK1 pathway to an even greater extent than AICAR and FUNDC1 is involved in the associated mitophagy. However, when AMPK is inhibited, other pathways may regulate mitophagy. Our findings indicate that mitophagy helps maintain the normal functions of mitochondria. EPS of C2C12 myotubes results in contraction, induction of mitophagy and potential activation of the AMPK-ULK1 pathway that promotes the expression of FUNDC1.
Keywords: FUNDC1, electrical pulse stimulation, mitophagy, AMPK-ULK1 pathway
Introduction
FUN14 domain containing-1 (FUNDC1), a novel receptor protein in the membrane of mammalian mitochondria, consists of 155 amino acids and accumulates under anaerobic conditions, interacting with new calnexin (CANX) in the endoplasmic reticulum [1]. Upon binding DNM1L, this receptor promotes mitochondrial fragmentation as part of the mitophagic response to hypoxia. Mitophagy is also influenced by the YxxL (LC3-Interacting Region, LIR) motif [2] of the microtubule-associated protein light chain (LC3).
Under normoxic conditions the tyrosine protein kinase SRC can phosphorylate FUNDC1 at Tyr-18 in its LIR motif, although FUNDC1 normally inactivates and dephosphorylates SRC kinase prior to hypoxia-induced mitophagy [3]. Dephosphorylated FUNDC1 demonstrates an elevated affinity for LC3, recruiting this factor to form autophagosomes [4]. In addition, FUNDC1 can be phosphorylated at Ser-13 [4] by the serine/threonine protein kinase CK2. Thus, under normoxia Ser-13 is phosphorylated by CK2, while in response to hypoxia the mitochondrial phosphoglycerate mutase PGAM5 dephosphorylates this residue, thereby preventing the interaction with LC3 and facilitating mitophagy.
Reactive oxygen species (ROS), a by-product of mitochondrial electron transport, can cause mitochondrial dysfunction, various forms of structural damage and even cell death [5]. When mitochondria are damaged beyond repair, they are removed via mitophagy, an important form of the quality control [6] required for normal cell homeostasis and function. At the same time, mitophagy is one mechanism by which exercise can influence health [7]. Regulation of mitochondria in mammalian cells occurs in two different ways: the first mechanism involves PTEN-induction of the putative kinase 1 (PINK1) together with the Parkinson protein 2 (Parkin) an E3 ubiquitin ligase [8], and the other mechanism depends on the mitochondrial receptor protein [9].
Appropriate exercise can improve mitochondrial function and inhibit apoptosis of skeletal muscle cells and cardiomyocytes by promoting mitophagy to remove erroneously folded proteins [10,11] and damaged organelles, senescent effects of the exercise itself. However, excessive exercise can injure skeletal muscle cells and cardiomyocytes, lead to fatigue, and even attenuate immunological responses and induce autophagic cell death [12]. When the level of AMP rises and ATP levels falls during exercise, the energy sensor AMPKα is activated [13], to maintain energy homeostasis. AMPK not only phosphorylates TSC2 [14] and Raptor [15], but also reduces the activity of mTORC1. Under conditions of glucose starvation it is also possible to phosphorylate the autophophase priming kinase ULK1 at Ser-317 [16] and thereby promote autophagy by subsequent phosphorylation of the ULK1 and PI3K complexes [17].
Electrical stimulation is an important tool for rehabilitation of skeletal muscle cramps [18], muscle impairment [19], and fatigue [20]. Electrical pulse stimulation (EPS) of myotubes in culture is widely utilized as an in vitro model of muscle contraction [21]. In C2C12 myotubes, EPS can reduce the electrochemical Na+-K+ gradient [22], promote translocation of the GLUT4 glucose transporter and stimulate AMPK phosphorylation [23]. Although exercise can clearly induce mitophagy through the AMPK pathway, it remains to be seen whether contraction of C2C12 myotubes in response to EPS has an analogous effect.
Although many reports have focused on the role of FUNDC1 in connection with hypoxia-induced mitophagy, the potential regulatory role of FUNCDC1 during exercise-induced mitophagy is not yet known in detail. Accordingly, this study was designed to determine whether FUNDC1 participates in the mitophagy induced by electrical pulse stimulation of C2C12 mytotubes and to investigate its relationship with the AMPK-ULK1 pathway in this context.
Methods and materials
Reagents and antibodies
Mouse myoblasts (C2C12) (the Peking Union Medical College Cell Bank, Chinese Academy of Sciences, Beijing, China); fetal bovine serum and horse serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA); penicillin/streptomycin (HyClone (HyClone, USA)); AICAR, Compound C and FK506 (Sigma Aldrich, St. Louis, MO, USA); the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide (MTT) kit (Beyotime Institute of Biotechnology, Shanghai, China); the Citrate synthase ELISA kit CS0720 (Sigma Aldrich, St. Louis, MO, USA); the BCA-200 protein assay kit (Thermo Fisher Scientific, USA); and antibodies against AMPKα, p-AMPKα (Thr172, ULK1, p-ULK1 (Ser555, LC3, p62, PGC-1α, COX-I, PGAM5, FUNDC1, and GAPDH (Abcam, Cambridge, UK))) were obtained from the sources indicated.
Cell cultures
C2C12 cells were cultured in 35-mm plates in the presence of 10% fetal bovine serum and 1% penicillin/streptomycin in DMEM-High Glucose medium until 80% confluence was reached, at which point this medium was replaced with the same medium containing 2% horse serum instead of bovine serum. All culturing was performed in an incubator at 37°C under a humidified atmosphere containing 5% CO2, with replacement of the medium every other day. These cultured cells were treated with electrical pulse stimulation (C-Pace EP) and exposed to AICAR, Compound C, and FK506.
Cytotoxicity assay
The cells collected in PBS during log-phase growth, were placed onto 96-well plates (102-103 cells per well) and incubated under 5% CO2 at 37°C until a monolayer covered the bottom of the well, at which time triplicate wells were exposed to various concentrations of the drug. After this exposure, 20 µl MTT (5 mg/ml) was added to each well and culturing continued for another 4 h. Incubation was terminated by adding 150 ul dimethyl sulfoxide to each well and shaking at low speed for 10 min, until the crystals were fully dissolved and, finally, the absorbance of each well at 490 nm was measured.
Citrate synthase assay
After determining the protein concentration with the BCA kit, an equal amount of protein (10 µg for CS, 20 µg for β-HAD) was loaded in triplicate into wells of 96-well microtiter plates. To assay CS at 25°C, 8 µL sample was diluted with 190 µL reaction buffer (186 μl 1 X Assay Buffer, 2 μl 30 mM Acetyl CoA Solution, 2 μl 10 mM DTNB; 10 μl 10 mM OAA Solution added to start the reaction; and absorbance at 412 nm read every 10 seconds for 1.5 minutes. The citrate synthase activity was calculated employing the equation provided with the kit (Sigma-Aldrich, CS0720, UK).
Units (µmole/ml/min) = ((ΔA412)/min × V(ml) × dil)/(εmM × L(cm) × Venz(ml))
Immunofluorescence microscopy
The cells were first grown to 60%-70% confluence on a glass dish and then treated. Subsequently, the cells were washed 3 times with PBS; fixed in freshly prepared 4% formaldehyde at 37°C for 20 min; and then washed 3 more times with PBS for 5 minutes each time. To ensure the accessibility of antigens to antibodies, membranes were ruptured with 0.2% Triton X-100. After blocking with 5% sheep serum for 30 min, the cells were incubated with primary antibodies overnight at 4°C; washed with PBS 3 times for 5 min each; stained with the secondary antibody for 1 h in a dark room; and then stained with DAPI. Images were captured with a TCS SPF5 II Leica confocal microscope.
Western blotting
After each treatment, the cells were washed with cold PBS and then lysed with RIPA buffer containing protease inhibitor and phosphatase inhibitors. After determination of the protein concentration with the BCA-200 kit, 10 μg protein was added to each lane and then separated by sodium dodecylsulphate polyacrylamide gel electrophoresis (10% SDS-PAGE) and transferred to membranes at 300 mA for 1.5 h in a PVDcam apparatus (UK) Thereafter, the membranes were blocked with 5% BSA for 30 minutes at room temperature and incubated with primary antibodies (diluted 1:1000, with the exception of GAPDH, which was diluted 1:2000) overnight at 4°C, subsequently, with horseradish peroxidase-conjugated secondary antibody at room temperature for 1.5 h. After being maintained, the specific protein signals were detected by enhanced chemiluminescence (ECL).
Statistical analyses
All values presented are means ± standard deviations (SD). Two-way ANOVA was used to analyze effects and interactions at various drug concentrations (low, medium, high) and treatment times (1 h, 2 h, 6 h). When the main effect was significant, a post-multiple test was performed; and when the interaction was significant, the simple effect test was performed. The independent-sample t test was used to compare the effects between the individual groups. One-way ANOVA was used for multiple comparisons. A P-value <0.05 was considered to be statistically significant.
Results
Electrical pulse stimulation (EPS) leads to mitophagy in C2C12 cells
The level of citrate synthase (CS) activity is an ideal indicator of mitochondrial number, since mitochondrial dysfunction, such as suppression of respiratory chain or loss of mitochondrial DNA, does not affect this activity [24,25]. An hour ‘s treatment of C2C12 cells with EPS (15 V, 1 Hz, 2 ms) did not reduce the number of mitochondria in the C2C12 cells, in contrast to what is observed following heavy exercise (Figure 1A). At the same time, the levels of PGC-1α and COX-I protein, routinely used to assess the function and biosynthesis of mitochondria, were elevated (Figure 1B).
Figure 1.
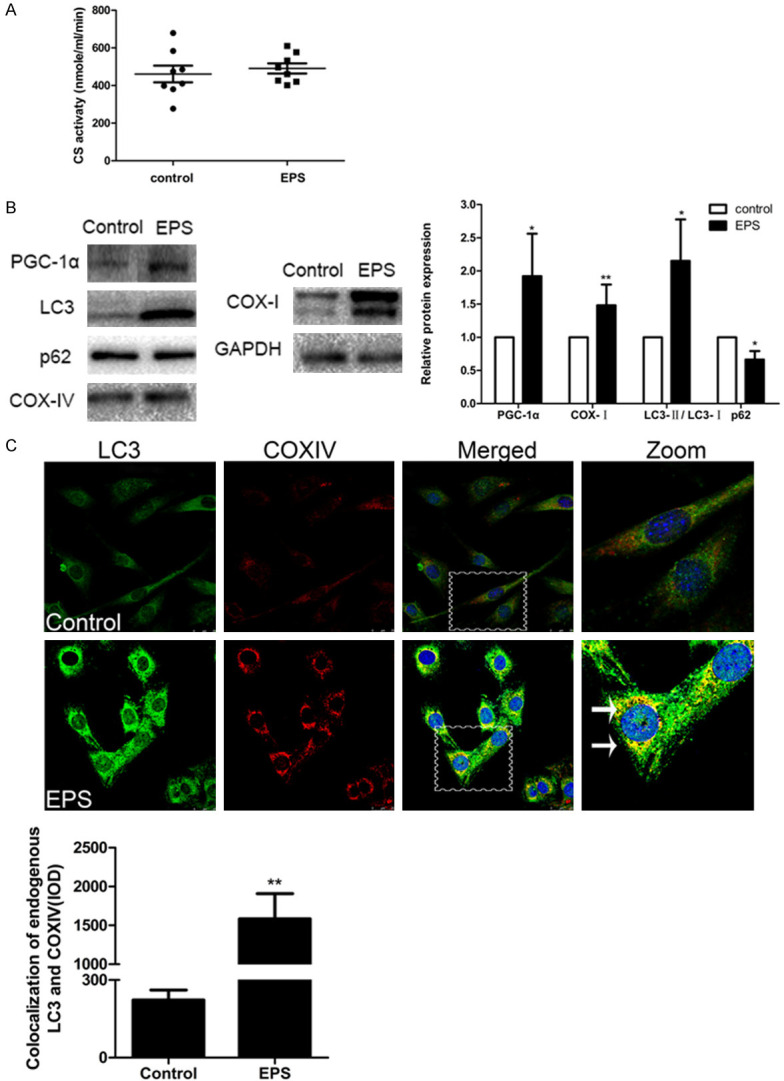
EPS causes mitophagy in C2C12 cells. A. The activity of CS in control C2C12 cells and cells treated by EPS. Means ± SD (control 485±101, EPS 491±67.9) and the 8 individual values for each group are shown; P>0.05. B. Western blotting of the indicators of mitochondrial biosynthesis PGC-1α and COX-I and of the autophagy-related factors LC3 and p62. The optical densities of the different protein bands (means ± SD) are shown at the right. Data are presented as *P<0.05 or **P<0.01 in comparison to the control group (n=6). C. Immunofluorescent staining reveals that COXIV-labeled (red) mitochondria co-localize with LC3 (green) (white arrows). Nuclei are stained blue with DAPI. Scale bar, 25 µm. Quantitative analysis of the co-localization is shown beneath (means ± SD; n=6; **P<0.01 in comparison to the control group).
Furthermore, the LC3-II/LC3-I ratio was enhanced by EPS, in contrast to the reduction in the level of the p62 protein (Figure 1B). In agreement, immunofluorescent double-staining for LC3 and COXIV also revealed increases in the levels of these proteins in C2C12 cells following EPS (Figure 1C). These findings confirmed that electrical stimulation can cause mitophagy in C2C12 cells.
EPS induced mitophagy is activated by AMPK-ULK1
To determine whether EPS induces mitophagy by activating the AMPK-ULK1 pathway, Western blotting was performed, revealing that EPS promoted activating phosphorylation of AMPKα2 at Thr172. Moreover, this activation led to increased levels of the downstream ULK1 and p-ULK1 (at Ser555) (Figure 2A). Thus, EPS induces mitophagy in C2C12 cells via AMPK-ULK1.
Figure 2.
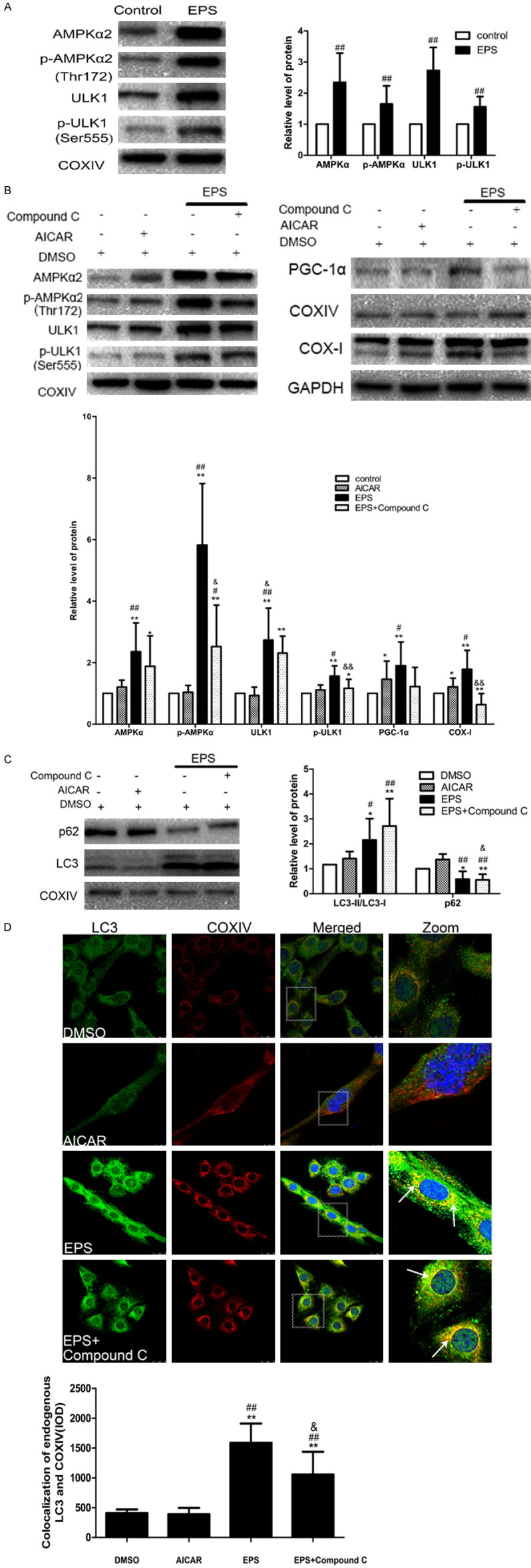
The mitophagy induced in C2C12 cells by EPS is mediated by AMPK-ULK1. A. Western blotting of AMPK, ULK1 and their phosphorylated forms, with the optical densities of the protein bands being shown to the right (means ± SD (n=6), ##P<0.01 in comparison to the control DMSO group). B. Western blotting of AMPK, ULK1 and their phosphorylated forms and of the indicators of mitochondrial biosynthesis PGC-1α and COX-I in cells exposed to DMSO alone, AICAR alone, or EPS with or without Compound C. C. Western blotting analysis of the indicators of autophagy LC3 and p62. The graph shows means ± SD (n=6). *P<0.05, **P<0.01 in comparison to the control with DMSO group; #P<0.05, ##P<0.01 in comparison to AICAR alone; &P<0.05, &&P<0.01 in comparison to EPS alone; D. Immunofluorescent double-staining shows that COXIV-labeled mitochondria (red) co-localized with LC3 (green) in all of the groups (white arrows). Nuclei are stained blue with DAPI. Scale bar: 25 µm. The quantitative data are means ± SD (n=6). *P<0.05, **P<0.01 in comparison to the control DMSO group; n=6; #P<0.05, ##P<0.01 in comparison to AICAR alone; &P<0.05, &&P<0.01 in comparison to EPS alone.
To confirm this finding, C2C12 cells were exposed to varying concentrations of AICAR (100 μM, 500 μM, 1 mM) and Compound C (2 μM, 10 μM, 50 μM) (both first dissolved in DMSO and then diluted 1,000-fold with medium) for 1, 2 or 6 hours. AICAR is taken up by the adenosine transporter and phosphorylated by adenosine kinase to form the AMP analogue ZMP, which activates the AMPK signaling pathway [26,27]. Compound C is the only cell-permeable inhibitor of AMPK [28]. As seen in (Supplementary Figure 1), cell viabilities (as assessed with the MTT assay) decreased gradually only with periods of exposure longer than 1 hour. Therefore, we chose 1 hour of exposure to 1 mM or 50 μM Compound C for subsequent experiments.
We found that EPS not only increased the levels of AMPKα2, p-AMPKα2, ULK1 and p-ULK1, but also enhanced the expression of AMPK to a greater extent than AICAR (Figure 2B). Compound C attenuated these effects of EPS somewhat, but the protein levels were still higher than after exposure to AICAR alone. These observations demonstrate clearly that the AMPK-ULK1 pathway mediates these effects of EPS. At the same time, neither AICAR nor EPS influenced the expression of PGC-1α or COX-I (Figure 2B), confirming that mitochondrial function remained intact.
Finally, with respect to mitophagy, although AICAR did not stimulate the expression of LC3, in agreement with previous studies, EPS significantly elevated the extent of mitophagy (Figure 2C). Then, to determine whether the AMPK-ULK1 pathway plays a role in this mitophagy, we showed that the LC3-II/LC3-I ratio in the presence of EPS and Compound C group was between that with AICAR or EPS alone. A similar observation was made with immunofluorescence microscopy (Figure 2D).
FK506 reduces the expression of FUNDC1
PGAM5, which can induce mitophagy by dephosphorylating FUNDC1 at Ser13, is inhibited by FK506. To first determine the optimal period of exposure, we treated C2C12 cells with 6 μM FK506 (dissolved in DMSO and then diluted 1,000-fold with medium) for 24, 48 and 72 hours and examined the expression of FUNDC1 (Supplementary Figure 2). Since such treatment for 48 h attenuated this expression maximally, without altering cell viability (as assessed by the MTT assay) (Supplementary Figure 3), we selected these conditions for further experiments. Clearly, FK506 prevented FUNDC1 dephosphorylation effectively by inhibiting the expression of PGAM5 (Figure 3).
Figure 3.
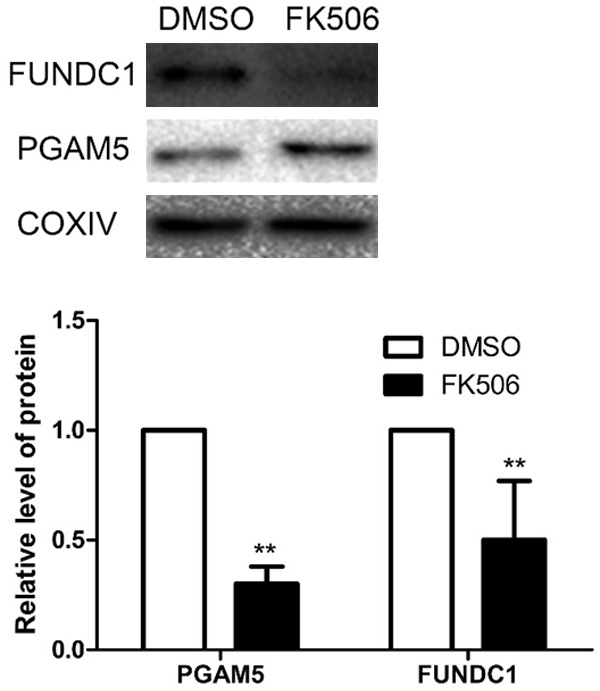
FK506 reduces the expression of FUNDC1 by C2C12 cells. After treating the cells with 6 μM FK506 for 48 h, the levels of PGAM5 and FUNDC1 were determined by Western blotting. The values presented are means ± SD (n=6). **P<0.01 in comparison to the control DMSO.
FUNDC1 is involved in the induction of mitophagy in C2C12 cells by EPS
Since EPS can induce mitophagy in C2C12 cells and FK506 reduces expression of FUNDC1, we next measured the levels of FUNDC1, LC3 and p62 and demonstrated that after EPS, the expression of FUNDC1 and level of mitophagy were markedly higher than upon treatment with FK506 (Figure 4A). Moreover, examination by immunofluorescence microscopy confirmed the co-localization of LC3, FUNDC1 and COXIV (Figure 4C). Clearly, EPS leads to mitophagy by attenuating FUNDC1 expression.
Figure 4.
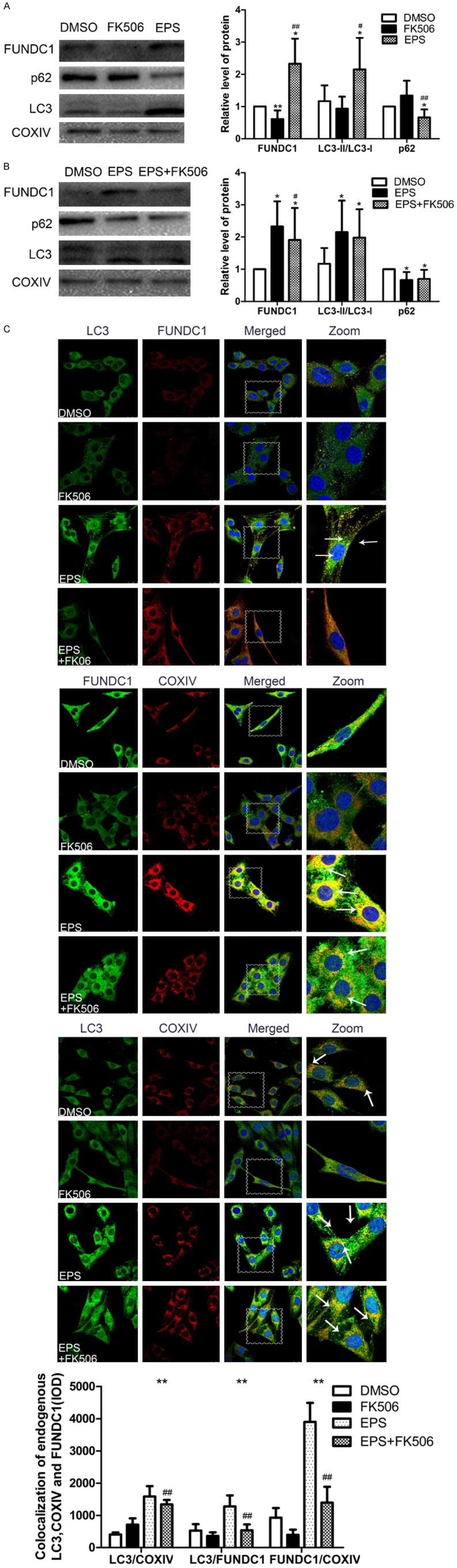
FUNDC1 is involved in the induction of mitophagy by EPS. A. Western blotting of and the indicators related to autophagy LC3 and p62 in C2C12 cells treated with EPS or FK506. The values presented are means ± SD (n=6). *P<0.05, **P<0.01 in comparison to the control DMSO group; #P<0.05, ##P<0.01 in comparison to treatment with FK506 alone; B. Western blotting of these same proteins in cells treated with EPS with and without FK506. The values presented are means ± SD (n=6). *P<0.05, **P<0.01 in comparison to the control DMSO group; #P<0.05, ##P<0.01 in comparison with EPS alone; C. Immunofluorescent staining of COXIV in mitochondria (red) and co-localized LC3 (green) and FUNDC1 (green or red). Nuclei are stained blue with DAPI. (Scale bar: 25 µm) and quantitative analysis of these co-localizations (means ± SD; n=6). *P<0.05, **P<0.01 in comparison to treatment with FK506 alone; #P<0.05, ##P<0.01 in comparison to treatment with EPS alone.
Moreover, the levels of FUNC1 and mitophagy induced by EPS declined upon co-treatment with FK506 (Figure 4B), with no change in co-localization (Figure 4C). These observations further indicate that FUNDC1 plays an important role in EPS-induced mitophagy.
FUNDC1 participates in EPS-induced mitophagy, at least in part, by activating the AMPK-ULK1 pathway
We showed above that EPS can activate AMPK-ULK1 pathway (Figure 2). When expression of AMPK in C2C12 cells was prevented with Compound C, the level of FUNDC1 after EPS also declined, but the LC3-II/LC3-I ratio was elevated (Figure 5), with the usual co-localization of LC3, FUNDC1 and COXIV (Figure 5C). Compound C alone elevated the level of AMPK (Figure 5B). Thus, EPS-induced mitophagy involves enhancing the activity of the AMPK-ULK1 pathway, which in turns influences FUNDC1 expression, although other mechanisms may also be involved.
Figure 5.
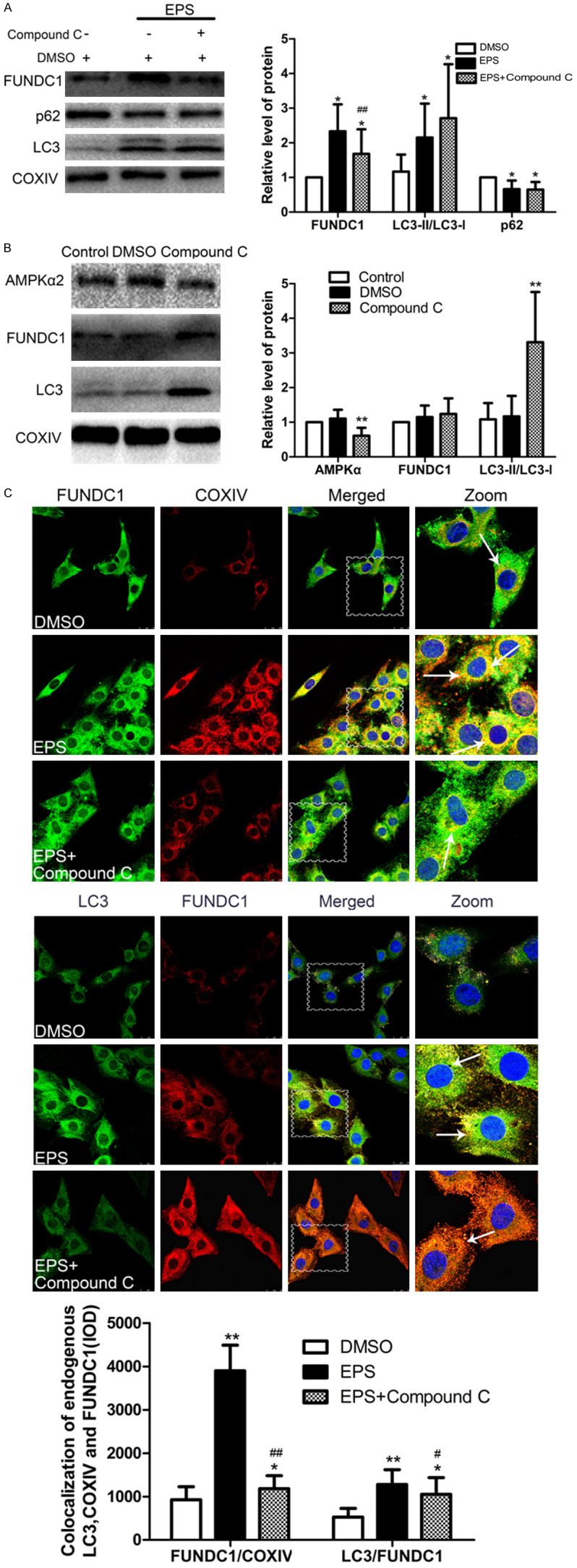
FUNDC1 participates in EPS-induced mitophagy through the AMPK-ULK1 pathway. A. Western blotting of FUNDC1 and the indicators related to autophagy LC3 and p62. The values presented are means ± SD (n=6). *P<0.05, **P<0.01 in comparison to the control DMSO group; #P<0.05, ##P<0.01 in comparison with EPS alone. B. Western blotting of AMPKα2, FUNDC1 and LC3 in C2C12 cells with and without exposure to Compound C. The values presented are means ± SD (n=6). *P<0.05, **P<0.01, in comparison to the control DMSO group. C. Immunofluorescent staining of the FUNDC1-labeled outer mitochondria membrane (green or red) and co-localized with LC3 (green) and COXIV (red) (white arrows). Nuclei are stained blue with DAPI. (Scale bar: 25 µm). Quantitative IOD analysis of the co-localization of LC3, FUNDC1 and COXIV (means ± SD; (n=6). *P<0.05, **P<0.01 in comparison to the control DMSO group; #P<0.05, ##P<0.01 in comparison to treatment with EPS alone).
Discussion
Our present findings reveal that electrical pulse stimulation can induce mitophagy, FUNDC1 is involved in this induction and in this connection FUNDC1 may be activated by the AMPK-ULK1 pathway.
One key to effective exercise is appropriate alteration of energy metabolism, in which mitochondria play the central role, with AMP levels increasing and ATP levels decreasing, and the energy sensor AMPK being activated [13] Feng [29] showed that within a certain range the higher the concentration of AICAR, the more pronounced the expression of AMPK. After 1 h of exposure to 1 mM AICAR, here the level of AMPK expression in C2C12 cells was elevated, while the levels of FUNDC1 and the autophagy-associated protein LC3 tended to increase as well.
In addition, Evers-van Gogh found [30] that phosphorylation of AMPK and secretion of myokine by muscles are largely dependent on EPS-induced contraction. Here, when C2C12 myotubes were subjected to EPS, the levels of AMPK and its phosphorylated form were enhanced even more than by treatment with AICAR In addition, we found similar results to previous studies,increasing in the levels of cytochrome c-oxide subunit I (COX I) [31], and a key regulator of mitochondrial biosynthesis or the transcriptional co-activator of peroxisome proliferator-activated receptor γ, coactivator 1α (PARGC1A, also known as PGC-1α) [32]. However, there are still limitations to the use of electrical impulse stimulation. Although such stimulation can cause muscle cells to contract, the specific mode and intensity of exercise being modelled cannot be determined. Therefore, as a next step, we will examine the various parameters of such stimulation in greater detail.
Previous studies have demonstrated that regulation of autophagy involves activation of AMPK. Such activation in response to glucose starvation promotes autophagy by directly phosphorylating ULK1 at Ser317 and Ser777 [17]. In contrast, under rich nutrient conditions, high mTOR activity prevents ULK1 activation by phosphorylating this protein at Ser757, thereby disrupting the interaction between ULK1 and AMPK. In addition, phosphorylation of ULK1 regulates, in turn, AMPK or mTORC1 in a feedback loop [33-35]. Therefore, phosphorylation of ULK1 determines its involvement in autophagy. Under hypoxic conditions, AMPK phosphorylates ULK1 primarily at Ser555 to induce autophagy [36]. Furthermore, exercise can enhance AMPK activity.
Following exercise, mitochondria in skeletal muscle are of different sizes, accumulate under the sarcolemma and appear to be abnormal or injured. Normally, damaged mitochondria regain their functions through fusion with neighboring, intact mitochondria [37], but when the extent of damage exceeds this capacity, mitophagy occurs [7]. Here, electrical pulse stimulation of C2C12 cells elevated their levels of AMPK and LC3, as well as of ULK1 and its phosphorylated form, at the same as LC3 and COXIV (a marker for mitochondria) became more co-localized. These findings demonstrate that like other stress conditions of stress, simulation of exercise by EPS stimulates AMPK-ULK1 and promotes mitophagy.
Interestingly, when we prevented expression of AMPK with Compound C, the extent of mitophagy was still very high, overturning our previous hypothesis. However, Amanda [38] found a similar situation., knockdown of AMPKα2 in mouse heart markedly elevated the marker of autophagy LC3-II, perhaps because the lowered level of AMPK activated other pathways that lead to mitophagy. This also indicates that the role played by AMPK in regulating mitophagy requires further elucidation.
The affinity of FUNDC1, a mitochondrial membrane protein that is also a connexin for mitophagy, for LC3 is enhanced by dephosphorylation of the former at Tyr18 [3], located in the LIR motif, and Ser13 [4], playing an important role in hypoxia-induced mitophagy [1]. Hypoxia elevates phosphorylation of AMPK at Thr172 and phosphorylation of ULK1 at Ser555 [17,39]. Our results show that EPS of C2C12 cells increased the levels of the mitochondrial proteins FUNDC1 and LC3 and at the same time, enhanced the co-localization of COXIV-labeled mitochondria with these two proteins, confirming the involvement of FUNDC1 in EPS-induced mitophagy. The co-localization of COXIV and FUNDC1 is inherently high since both of them are mitochondrial membrane proteins. Therefore, we compared co-localization of FUNDC1 and LC3 before and after EPS and observed it enhanced binding further confirming that EPS-induced mitophagy involves FUNDC1.
As a downstream effector of AMPK, ULK1 belongs to the serine/threonine kinase family of factors required for induction of autophagy [40-42], which plays an important role in the clearance of mitochondria after FCCP treatment [43]. ULK1 can be up-regulated and recruit fragmented mitochondria under hypoxic conditions or in the presence of FCCP, and the translocated ULK1 interacts with its substrate FUNDC1. However, mitophagy will be inhibited when FUNDC1 binds to ULK1 or when knockdown of FUNDC1 affects the transfer of ULK1 to fragmented mitochondria [44]. Our present observations reveal that upon EPS of C2C12 cells AMPK activates ULK1 through phosphorylation at Ser555, as also occurs under conditions of hypoxia, triggering mitochondrial translocation and transport [36]. Moreover, the trend towards more extensive phosphorylation of p-ULK1 at Ser555 was consistent with the increased level of FUNDC1, indicating that interaction between these two was involved in the induction of mitophagy.
In summary, the present study demonstrates that electrical pulse stimulation of C2C12 cells induces the mitochondrial outer membrane protein FUNDC1 through the AMPK-ULK1 pathway, thereby initiating mitophagy.
Acknowledgements
This work was supported financially by the Natural Science Foundation of China (31500964), The Fok Ying Tung Educational Foundation (151095) and special scientific research business funds.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Wu WX, Li W, Chen H, Jiang L, Zhu RZ, Feng D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy. 2016;12:1675–1676. doi: 10.1080/15548627.2016.1193656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schweers RL, Zhang J, Randall MS, Loyd MR, Li WM, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu L, Feng D, Chen G, Chen M, Zheng QX, Song PP, Ma Q, Zhu CZ, Wang R, Qi WJ, Huang L, Xue P, Li BW, Wang XH, Jin HJ, Wang J, Yang FQ, Liu PS, Zhu YS, Sui SF, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 4.Chen G, Han Z, Feng D, Chen YF, Chen LB, Wu H, Huang L, Zhou CQ, Cai XY, Fu CY, Duan LW, Wang XH, Liu L, Liu XQ, Shen YQ, Zhu YS, Chen Q. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54:362–377. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 5.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 6.Mao K, Wang K, Liu X, Klionsky DJ. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell. 2013;26:9–18. doi: 10.1016/j.devcel.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campello S, Strappazzon F, Cecconi F. Mitochondrial dismissal in mammals, from protein degradation to mitophagy. Biochim Biophys Acta. 2014;1837:451–460. doi: 10.1016/j.bbabio.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14:133–139. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng ZH, Bai LY, Yan J, Li Y, Shen WL, Wang Y, Wertz K, Weber P, Zhang Y, Chen Y, Liu JK. Mitochondrial dynamic remodeling in strenuous exercise- induced muscle and mitochondrial dysfunction: regulatory effects of hydroxytrosol. Free Radic Biol Med. 2011;50:1437–1446. doi: 10.1016/j.freeradbiomed.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Golbidi S, Laher I. Molecular mechanisms in exercise-induced cardioprotection. Cardiol Res Pract. 2011:972807. doi: 10.4061/2011/972807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270:299–304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 14.Inoki K, Ouyang H, Zhu TQ, Lindvall C, Wang Y, Zhang XJ, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 15.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behringer M, Harmsen JF, Fasse A, Mester J. Effects of neuromuscular electrical stimulation on the frequency of skeletal muscle cramps: a prospective controlled clinical trial. Neuromodulation. 2018;21:815–822. doi: 10.1111/ner.12728. [DOI] [PubMed] [Google Scholar]
- 19.Nussbaum EL, Houghton P, Anthony J. Neuromuscular electrical stimulation for treatment of muscle impairment: critical review and recommendations for clinical practice. Physiother Can. 2017;69:1–76. doi: 10.3138/ptc.2015-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanderthommen M, Makrof S, Demoulin C. Comparison of active and electrostimulated recovery strategies after fatiguing exercise. J Sports Sci Med. 2010;9:164–169. [PMC free article] [PubMed] [Google Scholar]
- 21.Brown AE, Jones DE, Walker M, Newton J. Abnormalities of AMPK activation and glucose uptake in cultured skeletal muscle cells from individuals with chronic fatigue syndrome. PLoS One. 2015;10:e0122982. doi: 10.1371/journal.pone.0122982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z, Yue YY, Hu F, Zhang C, Ma XF, Li NN, Qiu LH, Fu ML, Chen LM, Yao Z, Bilan PJ, Klip A, Niu WY. Electrical pulse stimulation induces GLUT4 glucose transporter translocation in C2C12 myotubes that depends on Rab8A, Rab13 and Rab14. Am J Physiol Endocrinol Metab. 2018;314:478–493. doi: 10.1152/ajpendo.00103.2017. [DOI] [PubMed] [Google Scholar]
- 23.Danilov K, Sidorenko S, Milovanova K, Klimanova E, Kapilevich LV, Orlov SN. Electrical pulse stimulation decreases electrochemical Na+ and K+ gradients in C2C12 myotubes. Biochem Biophys Res Commun. 2017;493:875–878. doi: 10.1016/j.bbrc.2017.09.133. [DOI] [PubMed] [Google Scholar]
- 24.Hargreaves IP, Duncan AJ, Wu L, Agrawal A, Land JM, Heales SJR. Inhibition of mitochondrial complex IV leads to secondary loss complex II-III activity: implications for the pathogenesis and treatment of mitochondrial encephalomyopathies. Mitochondrion. 2007;7:284–287. doi: 10.1016/j.mito.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 25.Watts JA, Kline JA, Thornton LR, Grattan RM, Brar SS. Metabolic dysfunction and depletion of mitochondria in hearts of septic rats. J Mol Cell Cardiol. 2004;36:141–150. doi: 10.1016/j.yjmcc.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 26.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 27.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–295. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu XN, Chhipa RR, Nakano I, Dasgupta B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Ther. 2014;13:596–605. doi: 10.1158/1535-7163.MCT-13-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo F, Liu SQ, Gao XH, Zhang LY. AICAR induces AMPK-independent programmed necrosis in prostate cancer cells. Biochem Biophys Res Commun. 2016;474:277–283. doi: 10.1016/j.bbrc.2016.04.077. [DOI] [PubMed] [Google Scholar]
- 30.Evers-van Gogh IJ, Alex S, Stienstra R, Brenkman JB, Kersten K, Kalkhoven E. Electric pulse stimulation of myotubes as an in vitro exercise model: cell-mediated and non-cell-mediated effects. Sci Rep. 2015;5:10944. doi: 10.1038/srep10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorska-Ponikowska M, Kuban-Jankowska A, Eisler SA, Perricone U, Bosco GL, Barone G, Nussberger S. 2-methoxyestradiol affects mitochondrial biogenesis pathway and succinate dehydrogenase complex flavoprotein subunit A in osteosarcoma cancer cells. Cancer Genomics Proteomics. 2018;15:73–89. doi: 10.21873/cgp.20067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promotemetastasis. Nat Cell Biol. 2014;16:992–1003. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M, Campbell DG, Wesselborg S, Alessi DR, Stork B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy. 2011;7:696–706. doi: 10.4161/auto.7.7.15451. [DOI] [PubMed] [Google Scholar]
- 34.Dunlop EA, Hunt DK, Acosta-Jaquez HA. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy. 2011;7:737–747. doi: 10.4161/auto.7.7.15491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Egan D, Kim J, Shaw RJ, Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:643–644. doi: 10.4161/auto.7.6.15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian WL, Li W, Chen YQ, Yan ZM, Huang X, Zhuang HX, Zhong WT, Chen YS, Wu WX, Lin CX, Chen H, Hou XY, Zhang LQ, Sui SF, Zhao B, Hu Z, Li LX, Feng D. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015;589:1847–1854. doi: 10.1016/j.febslet.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 37.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 38.Kaminaris A, Kobayashi S, Mcstay G, Liang QR. AMPK negatively regulates mitophagy in the heart. Published Online. 2017 [Google Scholar]
- 39.Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Mchumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31:3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Wirth M, Joachim J, Tooze SA. Autophagosome formation--the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol. 2013;23:301–309. doi: 10.1016/j.semcancer.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 42.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2010;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci. 2012;125:1488–1499. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- 44.Wu WX, Tian WL, Hu Z, Chen G, Huang L, Li W, Zhang XL, Xue P, Zhou CQ, Liu L, Zhu YS, Zhang XL, Li LX, Zhang LQ, Sui SF, Zhao B, Feng D. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15:566–575. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.