

Abstract
Hepatocellular carcinoma is the fourth leading cause of cancer-related deaths due to its high rate of recurrence and metastasis. All-trans-retinoic acid (ATRA) can inhibit the malignant behaviors of hepatocarcinoma cells. Autophagy is reportedly involved in the migration and metastasis of various cancer cells. This study aimed to investigate the effect of autophagy on the function of ATRA on hepatocarcinoma cells, and to explore its possible underlying mechanism. Hepatocarcinoma cell lines, Hepa1-6 and HepG2, were treated with ATRA and autophagy inhibitors, including 3-methyladenine (3-MA) and Bafilomycin (Baf). Transmission electron microscopy, laser scanning, western blot, and real-time PCR demonstrated that ATRA induces autophagy in hepatocarcinoma cells. Trypan blue staining, a wound healing assay, and a transwell assay showed that 3-MA and Baf reverses the inhibitory functions of ATRA on the proliferation, migration, and invasion of hepatocarcinoma cells. Flow cytometry, Hoechst staining, periodic acid-Schiff staining, and indocyanine green uptake validated that 3-MA and Baf reverses the function of ATRA on apoptosis and the differentiation of hepatocarcinoma cells. Real-time PCR, western blot, and an immunofluorescence assay demonstrated that the reversal of the epithelial-mesenchymal transition (EMT) process by ATRA is weakened when autophagy is inhibited. Additionally, we confirmed that Bcl-2 is associated with the induction of ATRA-induced autophagy instead of the PI3K/Akt/mTOR pathway. These findings suggest that ATRA induces autophagy and autophagic cell death through the Bcl-2/Beclin1 pathway. Furthermore, ATRA-induced autophagy is involved in the inhibitory effect of ATRA on the malignant behaviors of hepatocarcinoma cells by reversing the EMT process.
Keywords: Hepatocellular carcinoma, all-trans-retinoic acid, autophagy, epithelial-mesenchymal transition, Bcl-2
Introduction
Hepatocellular carcinoma (HCC) is the most common type of liver cancer, accounting for 90% of all primary hepatic malignancies [1,2]. Additionally, it is the fourth leading cause of cancer-related deaths, largely due to its high recurrence and metastasis rates [3,4]. At present, the clinical treatment of hepatocarcinoma primarily focuses on surgical therapy, supplemented with radiotherapy and chemotherapy. However, the effects of this treatment are not ideal.
All-trans-retinoic acid (ATRA) is an active metabolite of vitamin A, which plays a vital role in cell growth, differentiation, apoptosis, and other cell functions. In recent years, ATRA has been involved in the clinical treatment of various cancers due to its role in inhibiting the proliferation and invasion of cancer cells, including leukemia, osteosarcoma, pancreatic cancer, gastric cancer, breast cancer, and liver cancer [5-10]. Our previous studies demonstrated that ATRA can inhibit the malignant behaviors of Hepa1-6 cells [11]. The regulation of ATRA activity is expected to provide a new therapeutic approach for patients with HCC; however, its potential pathogenesis and molecular mechanism remain poorly understood. Therefore, elucidating the underlying molecular mechanism of ATRA’s effect on HCC would be useful for further clinical applications in the treatment strategy for liver cancer.
Autophagy is an exceedingly complex process of degrading modified, superfluous, or damaged cellular macromolecules and whole organelles to recycle and maintain intracellular homeostasis [12]. Autophagy plays a critical role in cell development, differentiation, and cell survival under nutrient deficiency conditions [13]. Previous studies demonstrated that ATRA can upregulate cell autophagy and promote the differentiation of leukemia cancer cells [14-16], but the detailed mechanism has not been fully investigated in other diseases.
This study aimed to investigate the effect of autophagy on ATRA-regulated behaviors of hepatocarcinoma cells and to explore its potential underlying mechanism. We found that ATRA increases the autophagy level of Hepa1-6 and HepG2 cells and that the downregulation of autophagy by 3-methyladenine (3-MA) and bafilomycin (Baf) reverses the behaviors of ATRA-treated hepatocarcinoma cells, likely by mediating the epithelial-mesenchymal transition (EMT). These findings demonstrated that ATRA inhibits the proliferation, migration, and invasion, as well as stimulates the apoptosis and differentiation, of hepatocarcinoma cells. A possible mechanism may be that ATRA not only induces apoptosis but also increases autophagic cell death by regulating Bcl-2 and Beclin1. Our study provides a novel theoretical basis for the potential future application of ATRA to treat patients with liver cancer.
Materials and methods
Cell culture and treatment
The mouse hepatocarcinoma cell line Hepa1-6 and human hepatocarcinoma cell line HepG2 were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in complete Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco) with 5% CO2 at 37°C.
Firstly, Hepa1-6 cells were treated with different concentrations of ATRA (0.1, 1 and 10 μmol/L) to test the autophagy level. Then both Hepa1-6 and HepG2 cells were divided into 4 groups: control group, ATRA group (10 μmol/L), 3-MA (4 mmol/L) +ATRA group, and Baf (50 nmol/L) +ATRA group. In the last two groups, cells were treated with 3-MA or Baf for 3 h in advance, then the medium was changed to DMEM medium containing 10 μmol/L of ATRA. ATRA and all inhibitors were purchased from Sigma Aldrich (St. Louis, MO, USA).
Western blot analysis
Cells were lysed with radioimmunoprecipitation (RIPA) lysis buffer containing phenylmethanesulfonyl fluoride (PMSF; Beyotime Biotechnology, Shanghai, China) and phosphatase inhibitor (Beyotime). Equal amounts of protein of each group were separated by 10% or 15% sodium dodecyl sulfate-polyacrylamide gel (Beyotime), and subsequently transferred to polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA). After blocking in the Quick-Block blocking buffer (Beyotime) for 15 min, the membranes were then incubated overnight at 4°C with different primary antibodies respectively against LC3 (1:1000), Beclin1 (1:1000), P62 (1:1000), β-actin (1:2000), Snail (1:1000), Vimentin (1:1000), E-cadherin (1:1000), N-cadherin (1:1000), Twist (1:1000), p-PI3K (including p85 and p110, 1:1000), p-Akt (1:1000), mTOR (1:1000), p-mTOR (1:1000), Bcl-2 (1:500), p-Bcl-2 (1:1000), JNK (1:1000) and p-JNK (1:1000). Antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), Sigma Aldrich (St. Louis, MO, USA), Wanleibio (Wuhan, China) and Bioss (Beijing, China). After being washed by Tris-Buffered Saline and Tween 20 (TBST), the membranes were incubated with appropriate second antibodies (1:2000, ZSGB-BIO, Beijing, China) for 1 h at room temperature. Finally, the blots were visualized by using enhanced chemiluminescent substrate (Bio-Rad, CA, USA) and exposed he ChemiDoc Touch Imaging System (Bio-Rad).
Transmission electron microscopy detection
Cells were washed by PBS twice and digested by 0.25% Trypsin. Then the cell suspension was centrifuged with the rapid of 1000 r/min for 10 min. The cell pellet samples were subsequently fixed, rinsed, dehydrated, soaked and embedded. The ultrathin sections were observed by TEM in the electron microscope laboratory of the Institute of Life Sciences, Chongqing Medical University.
Laser scanning confocal microscope to measure the autophagic flux
The dual-fluorescene mRFP-GFP-LC3 plasmid (ptfLC3, Addgene, USA) were transfected into the Hepa1-6 cells with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). After cells were treated respectively as described before, the laser scanning confocal microscope (Nikon, Tokyo, Japan) was used to dynamically measure the autophagic flux at indicated times.
Real-time PCR analysis
Real-time PCR assays were performed to assess the mRNA levels of autophagy and EMT related genes. Primer sequences designed by using the Primer3.0 program are listed in Table 1. Total RNA of each group was extracted by RNA Extraction kit (Bioteke, Beijing, China) and subsequently reverse transcribed into cDNA by using Superscript II reverse transcription kit (Takara, Dalian, China). The gene expressions were quantified by using the SYBR Green assay (Takara). All data were normalized by the expression of β-actin.
Table 1.
RT-PCR Primers (5’-3’)
| Forward | Reverse | |
|---|---|---|
| β-actin | AGGGAAATCGTGCGTGAC | CGCTCGTTGCCAATAGTGA |
| BECN1 | AACCAATGTCTTCAATGCCA | TTTCATTCCACTCCACAGGA |
| LC3 | CCTTCTTCCTGCTGGTCAAC | TAGATGTCAGCGATGGGTGT |
| CK18 | CTGGGCTCTGTGCGAACT | ACAGAGCCACCCCAGACA |
| ALB | CCAGACATTCCCCAATGC | CAAGTTCCGCCCTGTCAT |
| Snail | AAACCCACTCGGATGTGAAG | GAAGGAGTCCTGGCAGTGAG |
| Vimentin | CAGATGCGTGAGATGGAAGA | TCCAGCAGCTTCCTGTAGGT |
| Twist | TTCGCCGACCGCTGTAA | CTGCGTGGGGTTGAGTTG |
| N-cadherin | CCCCAAGTCCAACATTTC | CGCCGTTTCATCCATACC |
| E-cadherin | GAGGCCAAGCAGCAATAC | CTCCGCAGGCATAAAGAT |
Cell viability assay
Trypan blue staining was performed to measure cell viability. Cells were first treated with 3-MA and Baf as before and then re-plated in the 12-well plates with 10 μmol/L ATRA (5 × 104 cells for Hepa1-6 cells and 1 × 105 cells for HepG2 cells per well). The entire cell suspensions were collected and mixed with 0.4% trypan blue buffer (Solarbio, Beijing, China) at indicated time points. Cell mixture (10 μL) were counted in the hemocytometer under the microscope (Nikon), while blue-stained cells were identified as dead cells. Three independent experiments were carried out in duplicate.
Cell apoptosis assay
Flow cytometry and Hoechst staining were used to evaluate cell apoptosis. An Annexin V-FITC apoptosis detection kit (BD, New Jersey, USA) was used in flow cytometry. the apoptosis rates of different groups were tested following the instructions of the kit and the samples were subsequently analyzed by flow cytometry. For Hoechst staining, briefly, cells were fixed with 4% paraformaldehyde at room temperature for 30 min, and stained with Hoechst 33258 (Solarbio) for 10 min and observed under a fluorescence microscope (Nikon).
Wound-healing assay
Cells were seeded into 6-well plates and exposed to the treatment as described above. After cells reached to 100% confluent monolayer, a consistent linear wound was created by a pipette tip across the cell surface. The same scratched sections were photographed at 0, 1, 2, 3 and 5 days to calculate the wound healing rate. The assay was independently repeated three times in triplicate.
Transwell assay for cell migration and invasion
After 3 days of treatment as described before, cells of each group were resuspended with serum-free DMEM and then 200 μL cell suspension (5 × 104 per well for migration and 1 × 105 per well for invasion) with 10 μmol/L ATRA was seeded into the upper transwell chambers (8.0 μm, Corning, NY, USA). For the invasion assay, the chambers were pre-coated with 50 mg/L Matrigel (BD). The DMEM containing 10% FBS were added into the lower chambers. After 48 h of incubation, cells in the upper chamber were carefully removed, and the remaining cells that have migrated and invaded to the lower surface were fixed in 4% paraformaldehyde at room temperature for 30 min and stained with 0.1% crystal violet (Beyotime) for 15 min. Five independent fields of view were quantified under a microscope (Nikon). The stain was dissolved by absolute ethanol and the absorbance was detected at 570 nm with a micro plate reader (Thermo Scientific, MA, USA). The experiments were performed three times with triplicate independently.
Periodic acid-Schiff staining and indocyanine green uptake
Hepatic functions of cells were assessed by periodic acid-Schiff staining and indocyanine uptake and release. For periodic acid-Schiff staining, cells were treated as described above for 10 days and fixed in 4% paraformaldehyde for 30 min, then cells were stained with 0.5% periodic acid for 5 min and Schiff’s solution for 15 min (Solarbio). The whole procedure was carried out at room temperature and cells were washed with PBS gently after each step. More than 10 non-overlapping fields of view were captured under a light microscope (Nikon), and purple stained cells were identified as positive. For indocyanine uptake and release, cells were cultured in complete DMEM with 1 mg/mL freshly prepared indocyanine solution (Sigma) at 37°C for 1 h. More than 10 non-overlapping fields of view were captured under a light microscope (Nikon), and green stained cells were identified as positive. Cells were then incubated at 37°C for another 6 h to detect the indocyanine release. Both of the procedures were performed at least three times in duplicate.
Immunofluorescence assay
Cells were fixed in 4% paraformaldehyde for 30 min, followed by permeabilized with 0.3% Triton X-100 (Solarbio) for 15 min and blocked with 5% albumin from bovine serum (BSA; Solarbio) for 20 min at room temperature. Then the cells were incubated with primary antibodies against Albumin (ALB; 1:200), Alpha fetoprotein (AFP; 1:200), Cytokeratin 18 (CK18; 1:200), Bcl-2 (1:200) and p-Bcl-2 (1:200) at 4°C overnight. ALB, AFP and CK18 primary antibodies were purchased from Proteintech (Chicago, Illinois, USA), Bcl-2 antibody was purchased from Wanleibio, and p-Bcl-2 was purchased from Bioss. After gently washing with PBS, cells were subsequently incubated with appropriate secondary fluorescent antibodies (1:200; ZSGB-Bio) for 1 h at room temperature. The nuclei were stained with Hoechst 33258. The presences of the proteins were ascertained under the fluorescence microscope (Nikon).
Statistical analysis
All data are presented as the means ± standard deviation and were analyzed using SPSS 19.0 software (IBM Corp., Armonk, NY, USA). The two-tailed Student’s t-test was performed to evaluate the difference between two groups. One-way ANOVA and Student-Newman-Keuls post hoc test were used to measure significant differences among three or more groups. A p value less than 0.05 was considered to indicate a statistically significant difference.
Results
ATRA induces autophagy in hepatocarcinoma cells
Previously, we found that 10 µmol/L ATRA induces autophagy in Hepa1-6 cells [17]. In this study, we further evaluated the function of different concentrations of ATRA on autophagy in Hepa1-6 cells and also validated this assumption in HepG2 cells. The results of transmission electron microscopy demonstrated that the groups of varying ATRA concentrations had more autophagic vacuoles than the control group. Meanwhile, the 10 μmol/L ATRA group showed the most autophagosomes and autophagy lysosomes in the cytoplasm (Figure 1A).
Figure 1.

ATRA-induced autophagy in a dose-dependent manner in Hepa1-6 cells. A. Cells treated with varying concentrations of ATRA had more autophagic vacuoles than the control group using Transmission electron microscopy; B. After ptfLC3 transfection, autophagy of cells treated with varying concentrations of ATRA increased and autophagic flux was unobstructed using a laser scanning confocal microscope. Scale bar = 20 μm; C. Western blots detecting the autophagy-related marker protein LC3 in cells treated with varying concentrations of ATRA for 3 days, using β-actin as a control.
Hepa1-6 cells were then transfected with ptfLC3, GFP, and RFP co-expressing particles that represented LC3-II formation and autophagosomes. The only RFP expressing particles represented autophagy lysosomes formation. As shown in Figure 1B, the basic level of autophagy in Hepa1-6 cells was low, exhibiting only dispersive co-expression of GFP and RFP. More co-expressing particles were found in the ATRA-treated group, and many RFP alone expressing particles were seen in the 10 μmol/L ATRA group, suggesting that autophagy increased and autophagic flux was unobstructed. In addition, western blot analysis verified that the ratio of LC3-II/LC3-I increased in a dose-dependent manner in Hepa1-6 cells with ATRA treatment (Figure 1C). These results indicate ATRA induces the autophagy of Hepa1-6 cells in a concentration-dependent manner. Furthermore, similar results were observed in HepG2 cells (Figure 3D) after treatment with 10 μmol/L ATRA and showed an increase in the expression of autophagy-related protein LC3 and Beclin1. The above results indicate that ATRA induces autophagy in hepatocarcinoma cells.
Figure 3.

Autophagy was successfully inhibited by 3-MA and Baf. Hepa1-6 and HepG2 cells were pretreated with 3-MA and Baf for 3 h before exposure to 10 μmol/L of ATRA. (A) After ptfLC3 transfection, the autophagic flux of Hepa1-6 cells was dynamically observed using a laser scanning confocal microscope after 48 h and 72 h of ATRA treatment. Scale bar = 20 μm; (B) After ATRA treatment, the mRNA expression of the BECN1 was upregulated and decreased in the presence of 3-MA and Baf, whereas LC3 showed no significant change among these groups in Hepa1-6 cells using real-time PCR. All results were obtained from three independent experiments. *P<0.05 vs. control group; #P<0.05 vs. ATRA group; (C, D) After ATRA treatment, the autophagy-related marker proteins LC3, Beclin1, and P62 were detected using western blot with β-actin normalization in Hepa1-6 cells (C) and HepG2 cells (D).
3-MA and Baf inhibit ATRA-induced autophagy
Thus, we found that ATRA-induced autophagy in hepatocarcinoma cells and that a 10 μmol/L concentration of ATRA exhibits the strongest effect. To inhibit the level of autophagy, 3-MA and Baf were first used to treat hepatocarcinoma cells together with 10 μmol/L ATRA. Transmission electron microscopy and laser scanning confocal microscopy, confirmed that the autophagy level induced by ATRA was reversed by 3-MA and Baf, respectively, in Hepa1-6 cells. The 3-MA+ATRA group revealed a reduction of autophagosomes, yet the Baf+ATRA group exhibited more autophagosomes than the ATRA-only treated group (Figure 2). As shown in Figure 3A, the 3-MA+ATRA group had fewer co-expressing particles or RFP alone expressing particles. In this same figure, the Baf+ATRA group had more GFP expressing particles and fewer RFP expressing particles. In addition, the mRNA expression of the autophagy-related gene, BECN1, was upregulated by ATRA and decreased in the presence of 3-MA and Baf (P<0.05), whereas LC3 showed no significant change among these groups (Figure 3B). These results were further verified by western blot (Figure 3C and 3D). In Hepa1-6 and HepG2 cells, the protein expression of Beclin1 was downregulated by 3-MA and Baf compared with the ATRA-treated group.
Figure 2.
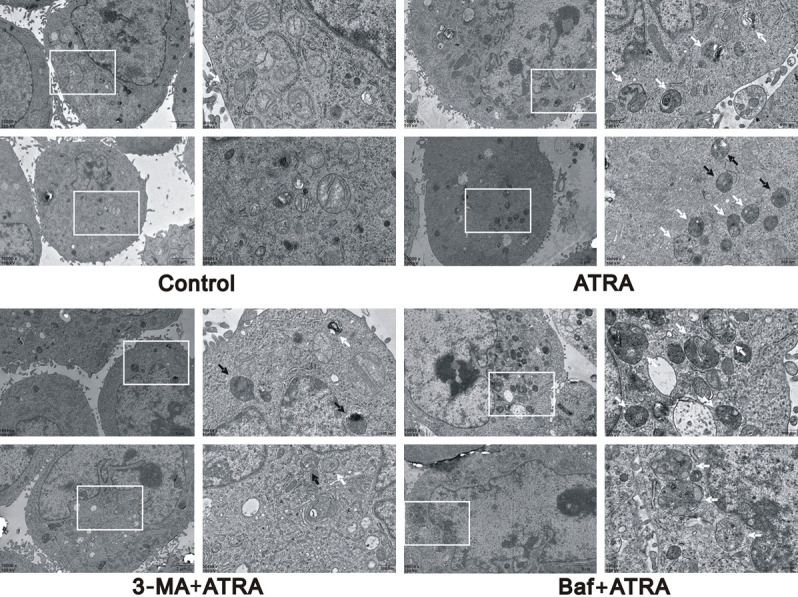
The detection of autophagosomes in different treatment groups. The 3-MA+ATRA group revealed a reduction of autophagosomes, yet the Baf+ATRA group exhibited more autophagosomes than the ATRA-only treated group in Hepa1-6 cells under transmission electron microscopy.
A marker of downstream autophagic flow is P62 (also known as SQSTM1). Compared with the ATRA group, the expression of LC3-II/LC3-I and P62 was decreased after 3-MA treatment but increased after Baf treatment. These findings indicate that 3-MA and Baf inhibit autophagy in a variety of ways: 3-MA inhibited the formation of autophagosome, whereas Baf blocked the process of autophagosome and lysosome fusion.
Autophagy is involved in the function of ATRA on cell proliferation and apoptosis of hepatocarcinoma cells
As presented in the cell growth curve from Figure 4A and 4B, ATRA obviously inhibited the proliferation of cells, and this inhibition was attenuated after the cells were exposed to autophagy inhibitors 3-MA and Baf (P<0.05). Consistent with the proliferation, ATRA induced the apoptosis rate of both Hepa1-6 and HepG2 cells, whereas the 3-MA+ATRA and Baf+ATRA groups had significantly lower apoptotic rates than the ATRA-only treated group (Figure 4C-F; P<0.05). These results suggest that autophagy plays a role in inhibiting the proliferation and apoptosis of ATRA-treated hepatocarcinoma cells.
Figure 4.

ATRA-induced autophagy regulated cell proliferation apoptosis of hepatocarcinoma cells. Cell proliferation of Hepa1-6 cells (A) and HepG2 cells (B) were inhibited after ATRA treatment and attenuated by 3-MA and Baf from the results of trypan blue staining. Cell apoptosis of Hepa1-6 cells (C, E) and HepG2 cells (D, F) were induced after ATRA treatment and reversed by 3-MA and Baf from the results of flow cytometry and Hoechst staining. *P<0.05 vs. control group; #P<0.05 vs. ATRA group. Scale bar = 200 μm.
Autophagy plays a role in ATRA-inhibited cell migration and in the invasion of hepatocarcinoma cells
The wound healing, transwell migration, and invasion assays were performed to detect the parallel and vertical migration, in addition to the invasion capacity of the cells. As shown in Figure 5, we found that ATRA significantly inhibits the migration and invasion of Hepa1-6 and HepG2 cells. However, after autophagy was inhibited by 3-MA and Baf, the hepatocarcinoma cells healed faster and the number of migrating and invasive cells in transwell were markedly increased. This indicates that autophagy is involved in the process of ATRA-inhibited cell migration and invasion.
Figure 5.

ATRA-induced autophagy regulated cell migration and invasion in hepatocarcinoma cells. The migration and invasion of Hepa1-6 cells (A, C, E) and HepG2 cells (B, D, F) were inhibited after ATRA treatment and markedly increased in the presence of 3-MA and Baf from the results of wound healing assay and crystal violet staining. The absorbance was measured using a microplate reader at 570 nm. *P<0.05 vs. control group; #P<0.05 vs. ATRA group. Scale bar = 200 μm.
Autophagy plays a role in the ATRA-induced cell differentiation of hepatocarcinoma cells
To investigate the effect of autophagy on the differentiation induced by ATRA, Hepa1-6 and HepG2 cells were treated with ATRA in the presence and absence of 3-MA and Baf. Cells treated with ATRA exhibited more periodic acid-Schiff and indocyanine green positive cells than the control group. In contrast, the 3-MA+ATRA and Baf+ATRA groups had less positive-stained cells (Figure 6A and 6B; P<0.05).
Figure 6.
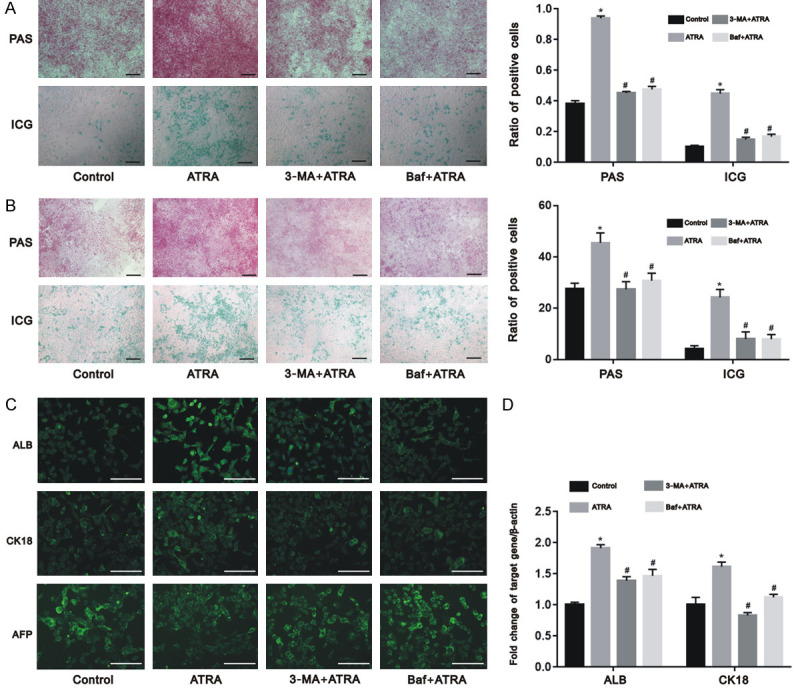
ATRA-induced autophagy regulated cell differentiation of hepatocarcinoma cells. The differentiation of Hepa1-6 cells (A) and HepG2 cells (B) were induced after ATRA treatment and significantly reversed by 3-MA and Baf from the results of wound periodic acid-Schiff staining and indocyanine green uptake assay; The 3-MA+ATRA and Baf+ATRA groups exhibited lower protein expression (C) and mRNA expression (D) of ALB and CK18 than the ATRA group in Hepa1-6 cells while the expression of AFP was inhibited by ATRA and reversed by 3-MA and Baf using immunofluorescence assay and real-time PCR. *P<0.05 vs. control group; #P<0.05 vs. ATRA group. Scale bar = 200 μm.
ALB and CK18 are two markers of the mature hepatocyte. Alpha fetoprotein (AFP) is the specific marker of hepatoma carcinoma cells. As shown in Figure 6C and 6D, the 3-MA+ATRA and Baf+ATRA groups exhibited lower expression of ALB and CK18 than the ATRA group in Hepa1-6 cells (P<0.05). Otherwise, the expression of AFP was inhibited by ATRA and reversed by 3-MA and Baf (Figure 6C). These findings suggest that ATRA-induced cell differentiation in hepatocarcinoma cells occurs by upregulating autophagy.
ATRA-induced autophagy plays a role in mediating EMT in hepatocarcinoma cells
To further investigate the underlying mechanisms of ATRA-induced autophagy on hepatocarcinoma cells, we investigated the expression of EMT-related markers. The mesenchymal markers including Snail, Vimentin, Twist, and N-cadherin were all expressed at lower levels in the ATRA-only treated group than in the control group. Nevertheless, the expression levels of the aforementioned markers increased after autophagy was inhibited in the 3-MA+ATRA and Baf+ATRA groups (Figure 7A-C; P<0.05). In contrast, the expression of epithelial markers, such as CK18, was upregulated after ATRA treatment and downregulated after cotreatment with autophagy inhibitors (Figure 6D; P<0.05). The expression of E-cadherin was increased after ATRA treatment but no statistical difference was found among the ATRA, 3-MA+ATRA, and Baf+ATRA groups (P<0.05). Therefore, we postulate that the inhibition of autophagy regulates the EMT reversed by ATRA in hepatocarcinoma cells.
Figure 7.

ATRA-induced autophagy regulated EMT markers in hepatocarcinoma cells and ATRA-induced autophagy via the Bcl-2/Beclin1 pathway independent of mTOR. A. ATRA suppressed the mRNA expression of the mesenchymal markers including Snail, Vimentin, Twist, and N-cadherin and were reversed by 3-MA and Baf. The expression of E-cadherin was increased after ATRA treatment but no statistical difference was found among the ATRA, 3-MA+ATRA, and Baf+ATRA groups in Hepa1-6 cells. The results were confirmed in at least three batches of independent experiments, and representative results are shown. *P<0.05 vs. control group; #P<0.05 vs. ATRA group; B, C. The protein expression of EMT-related proteins were measured by western blot analysis with β-actin normalization in Hepa1-6 and HepG2 cells; D, G. The results of western blot assay showed that the protein expression of PI3K/Akt/mTOR pathway-related markers have no significant variation after ATRA treatment in Hepa1-6 cells and HepG2 cells; E, H. The protein levels of Bcl-2/Beclin1 pathway-related markers were determined using western blot with β-actin normalization in Hepa1-6 cells and HepG2 cells; F, I. An immunofluorescence assay was performed to analyze the protein levels of Bcl-2 and p-Bcl-2 in Hepa1-6 cells and HepG2 cells. Scale bar = 200 μm.
ATRA-induced autophagy may occur by regulating Bcl-2 but not PI3K/Akt/mTOR signaling
The PI3K/Akt/mTOR pathway is an important signal to negatively regulate autophagy. The phosphorylation of PI3K, Akt, and mTOR yields their active forms to exert a biological function. However, no significant change of the protein expression of p-PI3K (including p85 and p110), p-Akt, and p-mTOR was found among the different treatment groups (Figure 7D and 7G). We further found that the expression of Bcl-2 was downregulated after ATRA treatment, and that the expression of phosphorylated Bcl-2, JNK, and phosphorylated JNK were increased (Figure 7E-I). This finding indicates that ATRA-induced autophagy in hepatocarcinoma cells occurs by regulating Bcl-2 expression and its phosphorylation but not the PI3K/Akt/mTOR signaling.
Discussion
The metastasis and recurrence of hepatocellular carcinoma cells is known to restrict the effect of clinical treatments in patients with HCC [18-20]. Recent studies have shown the therapeutic potential of ATRA in several cancers [21-24]. Additionally, it has been used as a clinical therapy to treat acute promyelocytic leukemia [25,26]. Our previous study revealed that ATRA inhibits the malignant behaviors of Hepa1-6 cells [11]. A deeper understanding of the underlying detailed mechanisms of ATRA is necessary to improve its future application as a treatment strategy for hepatocarcinoma.
Autophagy is a highly conserved cellular self-digestion progress that maintains homeostasis and survival. A wealth of evidence has shown that autophagy is involved in the occurrence and development of many types of tumors [27-29]. However, current views on the role of autophagy in cancer are inconsistent. Some studies have demonstrated that autophagy activation suppresses tumor progression and enhances chemotherapy in some cancer cells [30-32]. Conversely, some researchers considered autophagy as a protective mechanism for cancer cells [33-35]. This paradoxical phenomenon indicates that autophagy may be a double-edged sword. The protective or damaging function of autophagy may depend on its stimulating factors, or in the balance of the intracellular environment in health and disease.
In our study, we first investigated the relationship between autophagy and ATRA, followed by demonstrating that ATRA induces cell autophagy in a concentration-dependent manner in hepatocarcinoma cells. Additionally, our results substantiated that ATRA-induced autophagy affects the characteristics and behaviors of hepatocarcinoma cells. Blocking autophagy by specific inhibitors, 3-MA and Baf [36,37], significantly reversed the effects of ATRA on the proliferation, apoptosis, migration, invasion, and differentiation of Hepa1-6 and HepG2 cells. The EMT process is complex and associated with tumor progression. It is well established that EMT contributes to cancer metastasis and invasion [38-41]. During this process, epithelial cells lose their original polarized organization and specific intercellular junctions, by instead adopting characteristics of a mesenchymal cell [42,43]. Our previous studies revealed that the EMT process in Hepa1-6 can be reversed after ATRA treatment; however, the expression of specific epithelial proteins, such as CK18, decreased. Simultaneously, the mesenchymal-specific markers, such as N-cadherin, Vimentin, Snail and Twist, increased when ATRA-induced autophagy was inhibited by 3-MA or Baf. Although ATRA could also upregulate the expression of E-cadherin, no significant change was found after autophagy was inhibited. Therefore, these results indicate that autophagy meditates the effects of ATRA on hepatocarcinoma cells by regulating EMT, particularly with respect to the mesenchymal cell characteristics.
In addition, we explored the possible pathway responsible for ATRA-induced autophagy. Multiple signaling pathways have been reported to regulate autophagy [44-47], and the PI3K/Akt/mTOR pathway is an established autophagic pathway. Unexpectedly, ATRA treatment and treatment with autophagic inhibitors, showed no significant effect on the PI3K/Akt/mTOR pathway, indicating that ATRA induces autophagy independent of mTOR.
ATRA treatment increased the apoptosis of hepatocarcinoma cells. As an anti-apoptotic gene, Bcl-2 plays an important role in promoting cellular survival and inhibits the function of pro-apoptotic proteins [48,49]. Additionally, present studies demonstrated that the interaction between the Bcl-2 and Beclin1 proteins plays a critical role in the initiation stage of autophagosome formation [44,50,51]. The phosphorylation of Bcl-2 can disrupt the binding of the Bcl-2-Beclin1 complex and release Beclin1; the dissociated Beclin1 subsequently integrates with Vps34 (Class III PI3K) and initiates the autophagic progress [44,52-55]. Similarly, our results showed that the expression of Bcl-2 is downregulated by ATRA, and that the phosphorylation level of Bcl-2 is increased by ATRA. In addition, phosphorylation sites of Bcl-2 may be targets of a JNK1-related pathway given that both the JNK and phosphorylated JNK were upregulated after ATRA treatment. These data suggest that Bcl-2 may be associated with the induction of autophagy by ATRA in hepatocarcinoma cells. Autophagy and apoptosis are two major correlated mechanisms of cell death, and the enhancement of lysosome activity can lead to autophagic cell death [56-58]. ATRA promotes apoptosis and autophagy in both Hepa1-6 and HepG2 cells, therefore we believe that ATRA also promoted autophagy-dependent cell death by regulating Bcl-2 and its phosphorylated form.
Notably, although autophagic flux was unobstructed, the expression of P62 increased. P62 is a multifunctional protein of downstream autophagic flux that binds to LC3-II for autophagic degradation of polyubiquitinated substrates, and is degraded when autophagosomes are fused with lysosomes [59-61]. It is commonly believed that the expression of P62 should decrease in the status of autophagy activation; however, some studies determined that the intracellular level of P62 does not always inversely correlate with autophagy activity. This likely depends on the balance of transcriptional regulation and post-translational autophagic degradation [59,61]. Sahani et al found that P62 returns to basal levels due to transcription upregulation under prolonged amino acid starvation, thus reducing P62 levels via autophagic degradation [59]. Additionally, Chu et al demonstrated that both the level of autophagy and P62, increased during 24~72 h following acute spinal cord injury complicated with lung injury [62]. Consistent with the aforementioned results, excessive accumulation of the autophagy substrate, P62, can lead to excessive lysosomal digestion and result in autophagic death.
Conclusion
Our study demonstrated that ATRA induces autophagy and autophagic cell death via the Bcl-2/Beclin1 pathway in hepatocarcinoma cells. ATRA-induced autophagy induces the inhibitory effect of ATRA on the malignant behaviors of hepatocarcinoma cells by reversing the EMT process. These results may provide a novel theoretical basis for the potential application of ATRA to treat patients with liver cancer.
Acknowledgements
The reported work was supported by a scientific and technological research program of Chongqing Municipal Education Commission (No. KJQN201900442 to YB).
Disclosure of conflict of interest
None.
References
- 1.Mou SJ, Yang PF, Liu YP, Xu N, Jiang WW, Yue WJ. BCLAF1 promotes cell proliferation, invasion and drug-resistance though targeting lncRNA NEAT1 in hepatocellular carcinoma. Life Sci. 2020;242:117177. doi: 10.1016/j.lfs.2019.117177. [DOI] [PubMed] [Google Scholar]
- 2.Han J, Han ML, Xing H, Li ZL, Yuan DY, Wu H, Zhang H, Wang MD, Li C, Liang L, Song YY, Xu AJ, Wu MC, Shen F, Xie Y, Yang T. Tissue and serum metabolomic phenotyping for diagnosis and prognosis of hepatocellular carcinoma. Int J Cancer. 2020;146:1741–1753. doi: 10.1002/ijc.32599. [DOI] [PubMed] [Google Scholar]
- 3.Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462. doi: 10.1056/NEJMra1713263. [DOI] [PubMed] [Google Scholar]
- 4.Li Z, Zhu JY. Hepatocellular carcinoma: current situation and challenge. Hepatobiliary Pancreat Dis Int. 2019;18:303–304. doi: 10.1016/j.hbpd.2019.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Wang K, Baldwin GS, Nikfarjam M, He H. Antitumor effects of all-trans retinoic acid and its synergism with gemcitabine are associated with downregulation of p21-activated kinases in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2019;316:632–640. doi: 10.1152/ajpgi.00344.2018. [DOI] [PubMed] [Google Scholar]
- 6.Ni X, Hu G, Cai X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit Rev Food Sci Nutr. 2019;59:71–80. doi: 10.1080/10408398.2018.1509201. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Q, Xian M, Xiang S, Xiang D, Shao X, Wang J, Cao J, Yang X, Yang B, Ying M, He Q. All-trans retinoic acid prevents osteosarcoma metastasis by inhibiting M2 polarization of tumor-associated macrophages. Cancer Immunol Res. 2017;5:547–559. doi: 10.1158/2326-6066.CIR-16-0259. [DOI] [PubMed] [Google Scholar]
- 8.Bouriez D, Giraud J, Gronnier C, Varon C. Efficiency of all-trans retinoic acid on gastric cancer: a narrative literature review. Int J Mol Sci. 2018;19:3388. doi: 10.3390/ijms19113388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu MJ, Kim MR, Chen YS, Yang JY, Chang CJ. Retinoic acid directs breast cancer cell state changes through regulation of TET2-PKCζ pathway. Oncogene. 2017;36:3193–3206. doi: 10.1038/onc.2016.467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 10.Liu H, Chen F, Zhang L, Zhou Q, Gui S, Wang Y. A novel all-trans retinoic acid derivative 4-amino-2-trifluoromethyl-phenyl retinate inhibits the proliferation of human hepatocellular carcinoma HepG2 cells by inducing G0/G1 cell cycle arrest and apoptosis via upregulation of p53 and ASPP1 and downregulation of iASPP. Oncol Rep. 2016;36:333–341. doi: 10.3892/or.2016.4795. [DOI] [PubMed] [Google Scholar]
- 11.Cui JJ, Gong MJ, He Y, Li QL, He TC, Bi Y. All-trans retinoic acid inhibits proliferation, migration, invasion and induces differentiation of Hepa1-6 cells through reversing EMT in vitro. Int J Oncol. 2016;48:349–357. doi: 10.3892/ijo.2015.3235. [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019;177:1682–1699. doi: 10.1016/j.cell.2019.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eskelinen EL. Autophagy: supporting cellular and organismal homeostasis by self-eating. Int J Biochem Cell Biol. 2019;111:1–10. doi: 10.1016/j.biocel.2019.03.010. [DOI] [PubMed] [Google Scholar]
- 14.Schläfli AM, Isakson P, Garattini E, Simonsen A, Tschan MP. The autophagy scaffold protein ALFY is critical for the granulocytic differentiation of AML cells. Sci Rep. 2017;7:12980. doi: 10.1038/s41598-017-12734-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moosavi MA, Djavaheri-Mergny M. Autophagy: new insights into mechanisms of action and resistance of treatment in acute promyelocytic leukemia. Int J Mol Sci. 2019;20:3559. doi: 10.3390/ijms20143559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin J, Britschgi A, Schläfli AM, Humbert M, Shan-Krauer D, Batliner J, Federzoni EA, Ernst M, Torbett BE, Yousefi S, Simon HU, Tschan MP. Low autophagy (ATG) gene expression is associated with an immature AML blast cell phenotype and can be restored during AML differentiation therapy. Oxid Med Cell Longev. 2018;2018:1482795. doi: 10.1155/2018/1482795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang SY, Cui JJ, Gong MJ, He Y, Zhang JF, Bi Y. Changes in autophagy during maturation and differentiation of Hepa1-6 cells induced by all-trans retinoic acid. Nan Fang Yi Ke Da Xue Xue Bao. 2018;38:527–533. doi: 10.3969/j.issn.1673-4254.2018.05.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan L, Huang X, Chen J, Zhang K, Gu YH, Sun J, Cui SY. Long non-coding RNA MALAT1 contributes to sorafenib resistance by targeting miR-140-5p/Aurora-A signaling in hepatocellular carcinoma. Mol Cancer Ther. 2020;19:1197–1209. doi: 10.1158/1535-7163.MCT-19-0203. [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Zheng Y, Dai M, Wang X, Wu J, Yu B, Zhang H, Cui Y, Kong W, Wu H, Yu X. G9a and histone deacetylases are crucial for Snail2-mediated E-cadherin repression and metastasis in hepatocellular carcinoma. Cancer Sci. 2019;110:3442–3452. doi: 10.1111/cas.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meng F, Zhang S, Song R, Liu Y, Wang J, Liang Y, Wang J, Han J, Song X, Lu Z, Yang G, Pan S, Li X, Liu Y, Zhou F, Wang Y, Cui Y, Zhang B, Ma K, Zhang C, Sun Y, Xin M, Liu L. NCAPG2 overexpression promotes hepatocellular carcinoma proliferation and metastasis through activating the STAT3 and NF-κB/miR-188-3p pathways. EBioMedicine. 2019;44:237–249. doi: 10.1016/j.ebiom.2019.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Song Y, Zhang C, Gao P, Huang B, Yang J. The protective role of all-transretinoic acid (ATRA) against colorectal cancer development is achieved via increasing miR-3666 expression and decreasing E2F7 expression. Biomed Pharmacother. 2018;104:94–101. doi: 10.1016/j.biopha.2018.05.015. [DOI] [PubMed] [Google Scholar]
- 22.Jobani BM, Najafzadeh N, Mazani M, Arzanlou M, Vardin MM. Molecular mechanism and cytotoxicity of allicin and all-trans retinoic acid against CD44+ versus CD117+ melanoma cells. Phytomedicine. 2018;48:161–169. doi: 10.1016/j.phymed.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 23.Kozono S, Lin YM, Seo HS, Pinch B, Lian X, Qiu C, Herbert MK, Chen CH, Tan L, Gao ZJ, Massefski W, Doctor ZM, Jackson BP, Chen Y, Dhe-Paganon S, Lu KP, Zhou XZ. Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat Commun. 2018;9:3069. doi: 10.1038/s41467-018-05402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun R, Liu Y, Li SY, Shen S, Du XJ, Xu CF, Cao ZT, Bao Y, Zhu YH, Li YP, Yang XZ, Wang J. Co-delivery of all-trans-retinoic acid and doxorubicin for cancer therapy with synergistic inhibition of cancer stem cells. Biomaterials. 2015;37:405–414. doi: 10.1016/j.biomaterials.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 25.Smitheman KN, Severson TM, Rajapurkar SR, McCabe MT, Karpinich N, Foley J, Pappalardi MB, Hughes A, Halsey W, Thomas E, Traini C, Federowicz KE, Laraio J, Mobegi F, Ferron-Brady G, Prinjha RK, Carpenter CL, Kruger RG, Wessels L, Mohammad HP. Lysine specific demethylase 1 inactivation enhances differentiation and promotes cytotoxic response when combined with all-trans retinoic acid in acute myeloid leukemia across subtypes. Haematologica. 2019;104:1156–1167. doi: 10.3324/haematol.2018.199190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Labrador J, Luño E, Vellenga E, Brunet S, González-Campos J, Chillón MC, Holowiecka A, Esteve J, Bergua J, González-Sanmiguel JD, Gil C, Tormo M, Salamero O, Manso F, Fernández I, de laSerna J, Moreno MJ, Pérez-Encinas M, Krsnik I, Ribera JM, Cervera J, Calasanz MJ, Boluda B, Sobas M, Lowenberg B, Sanz MA, Montesinos P. Clinical significance of complex karyotype at diagnosis in pediatric and adult patients with de novo acute promyelocytic leukemia treated with ATRA and chemotherapy. Leuk Lymphoma. 2019;60:1146–1155. doi: 10.1080/10428194.2018.1522438. [DOI] [PubMed] [Google Scholar]
- 27.Nozima BH, Mendes TB, Pereira GJDS, Araldi RP, Iwamura ESM, Smaili SS, Carvalheira GMG, Cerutti JM. FAM129A regulates autophagy in thyroid carcinomas in an oncogene-dependent manner. Endocr Relat Cancer. 2019;26:227–238. doi: 10.1530/ERC-17-0530. [DOI] [PubMed] [Google Scholar]
- 28.Poillet-Perez L, White E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019;33:610–619. doi: 10.1101/gad.325514.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colella B, Faienza F, Carinci M, D’Alessandro G, Catalano M, Santoro A, Cecconi F, Limatola C, Di Bartolomeo S. Autophagy induction impairs Wnt/β-catenin signalling through β-catenin relocalisation in glioblastoma cells. Cell Signal. 2019;53:357–364. doi: 10.1016/j.cellsig.2018.10.017. [DOI] [PubMed] [Google Scholar]
- 30.You M, Gao J, Jin J, Hou Y. PPARα enhances cancer cell chemotherapy sensitivity by autophagy induction. J Oncol. 2018;2018:6458537. doi: 10.1155/2018/6458537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo W, Chen Z, Chen Z, Yu J, Liu H, Li T, Lin T, Chen H, Zhao M, Li G, Hu Y. Promotion of cell proliferation through inhibition of cell autophagy signalling pathway by Rab3IP is restrained by microRNA-532-3p in gastric cancer. J Cancer. 2018;9:4363–4373. doi: 10.7150/jca.27533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim V, Zhu H, Diao S, Hu L, Hu J. PKP3 interactions with MAPK-JNK-ERK1/2-mTOR pathway regulates autophagy and invasion in ovarian cancer. Biochem Biophys Res Commun. 2019;508:646–653. doi: 10.1016/j.bbrc.2018.11.163. [DOI] [PubMed] [Google Scholar]
- 33.Deng S, Shanmugam MK, Kumar AP, Yap CT, Sethi G, Bishayee A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer. 2019;125:1228–1246. doi: 10.1002/cncr.31978. [DOI] [PubMed] [Google Scholar]
- 34.Walker A, Singh A, Tully E, Woo J, Le A, Nguyen T, Biswal S, Sharma D, Gabrielson E. Nrf2 signaling and autophagy are complementary in protecting breast cancer cells during glucose deprivation. Free Radic Biol Med. 2018;120:407–413. doi: 10.1016/j.freeradbiomed.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci. 2017;18:1279. doi: 10.3390/ijms18061279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Tang D. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy. 2011;7:401–411. doi: 10.4161/auto.7.4.14397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roest G, Hesemans E, Welkenhuyzen K, Luyten T, Engedal N, Bultynck G, Parys JB. The ER stress inducer l-azetidine-2-carboxylic acid elevates the levels of phospho-eIF2α and of LC3-II in a Ca2+-dependent manner. Cells. 2018;7:239. doi: 10.3390/cells7120239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, De Clercq S, Minguijón E, Balsat C, Sokolow Y, Dubois C, De Cock F, Scozzaro S, Sopena F, Lanas A, D’Haene N, Salmon I, Marine JC, Voet T, Sotiropoulou PA, Blanpain C. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463–468. doi: 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 39.Lu W, Kang Y. Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev Cell. 2019;49:361–374. doi: 10.1016/j.devcel.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29:212–226. doi: 10.1016/j.tcb.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 41.Zhou P, Li Y, Li B, Zhang M, Liu Y, Yao Y, Li D. NMIIA promotes tumor growth and metastasis by activating the Wnt/β-catenin signaling pathway and EMT in pancreatic cancer. Oncogene. 2019;38:5500–5515. doi: 10.1038/s41388-019-0806-6. [DOI] [PubMed] [Google Scholar]
- 42.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 43.Shi G, Zheng X, Wu X, Wang S, Wang Y, Xing F. All-trans retinoic acid reverses epithelial-mesenchymal transition in paclitaxel-resistant cells by inhibiting nuclear factor kappa B and upregulating gap junctions. Cancer Sci. 2019;110:379–388. doi: 10.1111/cas.13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao SJ, Kong FQ, Cai W, Xu T, Zhou ZM, Wang ZB, Xu AD, Yang YQ, Chen J, Tang PY, Wang Q, Cheng L, Luo YJ, Zhou Z, Li LW, Huang YF, Zhao X, Yin GY, Xue MX, Fan J. GIT1 contributes to autophagy in osteoclast through disruption of the binding of Beclin1 and Bcl2 under starvation condition. Cell Death Dis. 2018;9:1195. doi: 10.1038/s41419-018-1256-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartolini D, Dallaglio K, Torquato P, Piroddi M, Galli F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl Res. 2018;193:54–71. doi: 10.1016/j.trsl.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Chung SJ, Nagaraju GP, Nagalingam A, Muniraj N, Kuppusamy P, Walker A, Woo J, Győrffy B, Gabrielson E, Saxena NK, Sharma D. ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy. 2017;13:1386–1403. doi: 10.1080/15548627.2017.1332565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Fu YF, Liu X, Feng G, Xiong D, Mu GF, Chen FP. ROS promote Ox-LDL-induced platelet activation by up-regulating autophagy through the inhibition of the PI3K/AKT/mTOR pathway. Cell Physiol Biochem. 2018;50:1779–1793. doi: 10.1159/000494795. [DOI] [PubMed] [Google Scholar]
- 48.Kvansakul M, Caria S, Hinds MG. The Bcl-2 family in host-virus interactions. Viruses. 2017;9:290. doi: 10.3390/v9100290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knight T, Luedtke D, Edwards H, Taub JW, Ge Y. A delicate balance - The BCL-2 family and its role in apoptosis, oncogenesis, and cancer therapeutics. Biochem Pharmacol. 2019;162:250–261. doi: 10.1016/j.bcp.2019.01.015. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Zhang W, Guo X, Ren J, Gao A. The crosstalk between autophagy and apoptosis was mediated by phosphorylation of Bcl-2 and beclin1 in benzene-induced hematotoxicity. Cell Death Dis. 2019;10:772. doi: 10.1038/s41419-019-2004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Z, Li Q, Lv W, Jiang L, Geng C, Yao X, Shi X, Liu Y, Cao J. The interaction of Atg4B and Bcl-2 plays an important role in Cd-induced crosstalk between apoptosis and autophagy through disassociation of Bcl-2-Beclin1 in A549 cells. Free Radic Biol Med. 2019;130:576–591. doi: 10.1016/j.freeradbiomed.2018.11.020. [DOI] [PubMed] [Google Scholar]
- 52.Fernández ÁF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, He C, Ting T, Liu Y, Chiang WC, Marciano DK, Schiattarella GG, Bhagat G, Moe OW, Hu MC, Levine B. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136–140. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu J, Liu W, Lu Y, Tian H, Duan C, Lu L, Gao G, Wu X, Wang X, Yang H. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models. Autophagy. 2018;14:845–861. doi: 10.1080/15548627.2017.1390636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiang WC, Wei Y, Kuo YC, Wei S, Zhou A, Zou Z, Yehl J, Ranaghan MJ, Skepner A, Bittker JA, Perez JR, Posner BA, Levine B. High-throughput screens to identify autophagy inducers that function by disrupting beclin 1/Bcl-2 binding. ACS Chem Biol. 2018;13:2247–2260. doi: 10.1021/acschembio.8b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J, Yao S. JNK-Bcl-2/Bcl-xL-Bax/Bak pathway mediates the crosstalk between matrine-induced autophagy and apoptosis via interplay with beclin 1. Int J Mol Sci. 2015;16:25744–25758. doi: 10.3390/ijms161025744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sachan R, Kundu A, Jeon Y, Choi WS, Yoon K, Kim IS, Kwak JH, Kim HS. Afrocyclamin A, a triterpene saponin, induces apoptosis and autophagic cell death via the PI3K/Akt/mTOR pathway in human prostate cancer cells. Phytomedicine. 2018;51:139–150. doi: 10.1016/j.phymed.2018.10.012. [DOI] [PubMed] [Google Scholar]
- 57.Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26:605–616. doi: 10.1038/s41418-018-0252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bialik S, Dasari SK, Kimchi A. Autophagy-dependent cell death - where, how and why a cell eats itself to death. J Cell Sci. 2018;131:215152. doi: 10.1242/jcs.215152. [DOI] [PubMed] [Google Scholar]
- 59.Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10:431–441. doi: 10.4161/auto.27344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Zhu M, Wang Q, Hou Y, Li L, Weng H, Zhao Y, Chen D, Ding H, Guo J, Li M. Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis. 2018;9:1027. doi: 10.1038/s41419-018-1036-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, Yang C, Liu HF. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett. 2016;21:29. doi: 10.1186/s11658-016-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chu R, Wang J, Bi Y, Nan G. The kinetics of autophagy in the lung following acute spinal cord injury in rats. Spine J. 2018;18:845–856. doi: 10.1016/j.spinee.2018.01.001. [DOI] [PubMed] [Google Scholar]