

Abstract
Microglia-mediated neuroinflammation is one of the hallmark pathological features following traumatic brain injury (TBI) that contributes to aggravated brain damage and cognitive deficits. These pathologies require novel effective treatments to improve prognosis. Trametinib, a mitogen-activated protein kinase inhibitor approved by the Food and Drug Administration in treating various malignant tumors, has been shown to exert anti-inflammatory effects. The present study demonstrated that TBI mice treated with trametinib exhibited improved cognitive function. Trametinib treatment rescued oligodendrocytes and decreased infiltrating microglial density in the TBI area. Furthermore, this study revealed that ameliorated lipopolysaccharides (LPS) induced inflammatory reaction in microglial cells. Besides, trametinib attenuated inflammation factors expression during the early stages of TBI. In addition, trametinib inhibited LPS-induced microglial chemotactic activity. In conclusion, the results indicate that trametinib efficiently suppresses microglia-induced neuroinflammation and improves cognitive function of TBI mice, providing a potential therapy strategy for TBI patients.
Keywords: Traumatic brain injury, microglia, neuroinflammation, MEK/ERK, trametinib
Introduction
Traumatic brain injury (TBI) remains a major cause of disability and mortality in trauma patients worldwide [1,2]. Among individuals between 1 and 45 years of age, trauma is the leading cause of death, while TBI constitutes the majority of cases, thus leading to significant socioeconomic burden [1,3]. Therefore, TBI represents an enormous challenge to the society and treatments require continuous improvement.
The pathological time-course of TBI may be divided into primary and secondary injury [3]. Primary injury, such as brain contusion and epidural/subdural hematoma, occurs at the time of injury and induces direct damage to the brain tissue [4,5]. Secondary injury arises from minutes to months following the primary impact, which progressively exacerbates the neurological impairment [6,7]. Increasing evidence has shown that neuroinflammation contributes to TBI secondary injury. In addition, several researchers have demonstrated that severe and continuous neuroinflammation is detrimental to neural cell repair, leading to worse clinical outcome [6-8]. The secretion of inflammatory factors is a critical component of the secondary damage, by promoting inflammation and cell death [8]. Immune activation occurs rapidly and involves several cellular components, including microglia, astrocytes and recruited peripheral immune cells [3,4,9].
Microglia are immune cells of the central nervous system (CNS), detecting and reacting to damage stimuli, which in turn initiate pro-inflammatory responses during the early stages [10,11]. Pro-inflammatory factors, such as tumor necrosis factor alpha (TNF-α) and interleukin (IL)-1β, are highly expressed at focal traumatic sites. These factors have also been reported to negatively associate with TBI cognitive outcome in animal models and TBI patients [9]. Therefore, targeting microglia-induced neuroinflammation may be a potential neural protective strategy.
Mitogen-activated protein kinase (MAPK) is involved in directing cellular responses that regulate a wide range of cellular functions, including cell proliferation and migration, and cytokine release [12-14]. Mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated protein kinase (ERK) are distal and important components of the MAPK signaling pathway [15,16]. ERK serves as the downstream kinase cascade of MEK, which is phosphorylated and activated by MEK. Since MEK/ERK pathway regulates several cellular functions, blocking this pathway provides a feasible way to intervene or modulate cell proliferation. This approach has already provided promising effects in animal models and clinical trials on several diseases, such as melanoma and leukemia [17-20]. Interestingly, it has been shown that MEK/ERK pathway plays an important role in microglial pro-inflammatory responses [14,21]. Trametinib is an effective MEK/ERK inhibitor that has already been tested in mice models and clinical trials on metastatic melanoma, with promising tumor remission effects and low toxic complications [22,23]. Therefore, the current study aimed to investigate the effect of trametinib in modulating neuroinflammation and cognitive protection in microglia and a TBI mouse model.
Materials and methods
Animals
BL6/C57J mice used in this study were purchased from Huazhong Kej, Co. All animals were housed in a controlled environment and fed with standard chow, in the animal facility of Tongji Medical College of Huazhong University of Science and Technology. All the experiments using laboratory animals were approved by the Huazhong University of Science and Technology Committee for the Care of Animals.
Cell culture
The primary microglia cultures from neonatal rodents were established as previously described [24]. Briefly, brains from postnatal day 0-2 (P0-P2) mixed male and female newborns were deprived of blood vessels and meninges, mechanically dissociated into 1 mm3 pieces and digested with 1% trypsin and 0.05% deoxyribonuclease. Subsequently, digested tissues were mechanically dissociated with a fire-polished pipette and washed twice with Hanks’ Balanced Salt Solution (HBSS; Thermo Fisher Scientific, Inc.). Dissociated tissues were plated and cultured for 9-12 days in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum and 1% penicillin and streptomycin (all from Thermo Fisher Scientific, Inc.). Following culture for 7 days, 33% L292-conditioned medium was added to the plates and the microglial cells were isolated by gentle shaking at 37°C for 1 h on a shaker (100 rpm). Freshly isolated microglia were then directly seeded into plates for further experiments. To investigate the effects of lipopolysaccharides (LPS) and trametinib, cells were pre-treated with 5 or 10 nM trametinib for 2 h and subsequently stimulated with 100 ng/ml LPS (InvivoGen, Inc.) for 6 h or 24 h.
Controlled cortical impact (CCI) model of TBI and drug administration
Surgical procedures were previously described [25]. Briefly, after anesthesia (5% isoflurane in oxygen with a delivery rate of 0.5 L/min for induction and 2% isoflurane for maintaince), mice were subjected to a unilateral, moderately CCI of 2.0 mm depth at 3.5 m/secs and 500 msec dwell time using the TBI 0310 pneumatic impacting device (Precision Systems and Instrumentation) with a hard stop Bimba cylinder (Bimba Manufacturing). The size of the beveled impactor was 5 mm. All craniotomies were performed midway between the bregma and the lambda sutures in the left hemisphere of the skull. Furthermore, 2 h following establishment of TBI model, mice were randomized into the vehicle- and trametinib-treated (Selleck Chemicals) groups. Trametinib was dissolved in sterile 100% DMSO and diluted 1:9 in sterile-filtered 1% carboxymethylcellulose solution supplemented with 0.4% Tween-20 (Sigma Aldrich; Merck KGaA). Finally, vehicle or trametinib were orally administered to mice at doses of 1 mg/kg for 7 consecutive days.
Cognitive assay
Morris water maze test was previously described. Briefly, after TBI surgery, mice were first pre-trained for 5 days. The apparatus used for this test was a circular stainless steel tank filled with water supplemented with skimmed milk (24-26°C) at a depth of 40 cm. Morris water maze test was performed daily for 7 days. Mice were placed in water randomly facing the wall at one of the four starting points of the pool and the time for the mice to successfully find the platform was recorded. The endpoint escape latency, which was defined as the available time for mice to find the platform, was set to 60 sec.
For the eight-arm radial maze test, a previously described method was performed [26]. Briefly, mice were first pre-trained for 7 days prior to the TBI and tests were performed for an additional 7 days. During the 7-days of pre-training period, each mouse was placed in the central starting platform and allowed to explore and consume food pellets. In the spatial working memory task of the eight-arm radial maze, all eight arms were baited with food pellets. This task aimed to record the error arm entries of rodents (the animal enters an arm previously entered). Mice who re-entered into a previously visited arm were recorded for working memory errors (WME), or namely revisiting errors. The test was completed when a mouse obtained all eight food rewards or 15 min had elapsed. Behavioral motor function was unbiased measured daily post TBI for 7 days using rotarod (Ugo Basile Srl) test. Mice with consciousness disorders or severe paralysis were excluded from the rotarod test. Prior to rotarod test, mice were trained on a rod at a rotation speed of 15 rpm for 15 min, followed by three trials with an accelerating rotation speed between 4 and 40 rpm for 5 min. Therefore, baseline latency over the three trials was obtained prior to TBI. Subsequently, the average retention time on rod over three trials for each mouse was recorded.
Quantitative PCR (qPCR)
Total RNA was extracted from primary microglial cells treated with LPS (100 ng/ml) with or without the addition of trametinib (5 or 10 nM) using a RNeasy lipid tissue mini kit (Qiagen, Inc.). Subsequently, cDNA was synthesized with random primers using a reverse transcription kit (Thermo Fisher Scientific, Inc.). The expression levels of M1 phenotype biomarkers, including IL-1β, TNF-α, nitric oxide synthase 2 (NOS2), and M2 biomarkers, IL-10, vascular endothelial growth factor (VEGF) and arginase 1 (Arg1; all from GeneCopoeia, Inc.) were determined using PCR with SYBR green technology on a AB7500 fast real-time thermal cycler (Applied Biosystems; Thermo Fisher Scientific, Inc.). GAPDH was used as a normalization control. Gene expression relative to the control group at 24 h was calculated using the 2-ΔΔCq method. The primer sequences used were as follows: PDGFa forward, 5’-GAGGAAGCCGAGATACCCC-3’, and reverse 5’-TGCTGTGGATCTGACTTCGAG-3’; SOX10 forward, 5’-ACACCTTGGGACACGGTTTTC-3’, and reverse 5’-TAGGTCTTGTTCCTCGGCCAT-3’; CSPG4 forward 5’-GGGCTGTGCTGTCTGTTGA-3’, and reverse 5’-TGATTCCCTTCAGGTAAGGCA-3’; CLDN11 forward 5’-ATGGTAGCCACTTGCCTTCAG-3’, and reverse 5’-AGTTCGTCCATTTTTCGGCAG-3’; TNF-α forward, 5’-TAGTCCTTCCTACCCCAATTTCC-3’ and reverse, 5’-TTGGTCCTTAGCCACTCCTTC-3’; IL-1β forward, 5’-GCAACTGTTCCTGAACTCAACT-3’ and reverse, 5’-ATCTTTTGGGGTCCGTCAACT-3’; NOS2 forward, 5’-GTTCTCAGCCCAACAATACAAGA-3’ and reverse, 5’-GTGGACGGGTCGATGTCAC-3’; IL12 forward, 5’-CTGTGCCTTGGTAGCATCTATG-3’ and reverse, 5’-GCAGAGTCTCGCCATTATGATTC-3’; gm-csf forward, 5’-GGCCTTGGAAGCATGTAGAGG-3’, and reverse, 5’-GGAGAACTCGTTAGAGACGACTT-3’; IL-10 forward, 5’-GCTCTTACTGACT-GGCATGAG-3’ and reverse, 5’-CGCAGCTCTAG-GAGCATGTG-3’; VEGF forward, 5’-GCACATAGAGAGAATGAGCTTCC-3’ and reverse, 5’-CTCCGCTCTGAACAAGGCT-3’; and Arg1 forward, 5’-CTCCAAGCCAAAGTCCTTAGAG-3’ and reverse, 5’-AGGAGCTGTCATTAGGGACATC-3’; and GAPDH forward, 5’-AGGTCGGTGTGAACGGATTTG-3’ and reverse, 5’-TGTAGACCATGTAGTTGAGGTCA-3’.
Western blot analysis
Cultured microglial samples were lysed using RIPA lysis buffer (Beyotime Institute of Biotechnology). In addition, cell lysates from murine TBI tissues were harvested using RIPA lysis buffer. Protein concentration was determined using a protein assay dye (Bio-Rad Laboratories, Inc.). Protein electrophoresis and protein transferring onto membranes were performed as described previously [27]. Following blocking with 1X phosphate buffered saline (PBS) supplemented with 5% w/v non-fat dried milk for 1 h at room temperature, the membranes were then incubated with specific antibodies against β-actin (dilution, 1:2,000, Cat#sc-47778), myelin basic protein (MBP; dilution, 1:500, Cat#ab62631, Abcam), Myelin oligodendrocyte glycoprotein (MOG; dilution, 1:1000, Cat#AF2439), phospho-ERK (dilution, 1:1,000, Cat#4370S), total-ERK (dilution, 1:1,000, Cat#4965S), phospho-nuclear factor-κB (p-NF-κB; dilution, 1:1,000, Cat#3033S), NLR family pyrin domain containing 3 (NLRP3; dilution, 1:1000, Cat#13158S), myeloid differentiation factor 88 (MyD88; dilution, 1:1,000, Cat#4283S; all from Cell Signaling Technology, Inc.). Following incubation with primary antibodies at 4°C overnight and washing step, membranes were further incubated with a goat anti-rabbit or goat anti-mouse secondary antibody (Cat#115-035-008, Cat#111-035-003, Jackson ImmunoResearch Labs, Inc.) for 1 h at room temperature. The relative protein expression levels were quantified by measuring the density of the immunoreactive bands on the X-ray films using a GS-700 Imaging Densitometer (Bio-Rad Laboratories, Inc.). The intensity of the bands was determined using the ImageJ software and the results were presented as relative expression of specific target markers normalized to β-actin expression in each sample.
Enzyme-linked immunosorbent assay (ELISA)
IL-1β, TNF-α, Granulocyte-macrophage colony-stimulating factor (GM-CSF) secretion levels were measured using an ELISA kit (R&D Systems, Inc.). All reagents were prepared according to the manufacturer’s protocol. Supernatants from primary cultured microglia treated with LPS (100 ng/ml) or LPS plus trametinib (5 or 10 nM) for 24 h were collected and were subsequently measured. Standard diluting solution was added to each well in 96-well plates and then standard solution or microglial supernatants were also added to each well. The plates were incubated for 2 h at room temperature. Following washing, murine IL-1β, TNF-α or GM-CSF conjugate solution was supplemented into each well and plates were incubated for an additional 2 h at room temperature. Finally, a substrate solution was added to each well and plates were incubated for 30 min at room temperature in dark. The optical density of each well was determined at 450 nm using a microplate reader and the results were evaluated using a standard curve.
Immunofluorescence staining
Experimental mice were transcardially perfused and fixed with PBS followed by 4% paraformaldehyde (PFA). Murine brains were excised and sectioned into 40 μm-thick sections. The slices were rinsed three times with tris-buffered saline (TBS) for 5 min. Subsequently, following blocking with 5% donkey serum (Sigma-Aldrich; Merck KGaA) in TBS supplemented with 0.1% Triton-X (Sigma-Aldrich; Merck KGaA) for 1 h in room temperature, slices were then incubated overnight with primary antibodies against allograft inflammatory factor 1 (Iba1; dilution, 1:500, Cat#ab178846), oligo2 (dilution, 1:200, Cat#ab109186; both from Abcam), CD86 (dilution, 1:100, Cat#ab119859) and IL-1β (dilution, 1:100, Cat#NB600-633) in 5% donkey serum in TBS at 4°C. After washing for three times with TBS, slices were then incubated with a secondary antibody (cy3-conjugated goat anti-rabbit antibody, Cat#A10520, and 4’,6-diamidino-2-phenylindole (DAPI); all from Thermofisher) in 5% donkey serum in TBS for 2 h. Finally, tissue samples were washed again for three times and cells were photographed and counted in four random fields for each sample using confocal microscopy. The number of labelled cells was quantified using the ImageJ software.
Agarose spot and Boyden chamber assays
Agarose spot assay was performed as previously described [28]. Briefly, low-melting point agarose was diluted into 0.5% agarose solution with PBS. Subsequently, LPS or trametinib was mixed with 0.5% agarose solution at a final concentration of 100 ng/ml and 10 nM, respectively. The mixed agarose solution was then added onto 35 mm glass bottomed dishes (MatTek Corporation) and four spots were pipetted in each dish, two containing PBS as negative control and two with the selected solutions. Subsequently, primary cultured microglia were plated into the dishes and incubated at 37°C for 3 h. Cells in each spot were counted under microscope. The results were presented as the number of cells in the selected group normalized to the PBS group.
Boyden chamber assay was also performed as previously described [29]. Briefly, the upper and lower wells of the chamber were separated with a polycarbonate filter (8 µm pore size; Poretics Corp.). Culture medium supplemented with 100 ng/ml LPS or LPS plus 10 nM trametinib was pipetted in the lower chamber. Subsequently, primary cultured microglia were seeded into the upper chamber and both chambers were then incubated at 37°C for 3 h. Following incubation, the upper side of membrane was wiped with cotton and cells were stained with crystal violet solution (Beyotime Institute of Biotechnology). Finally, the rate of microglial migration was calculated by counting cells in four random fields from each well under bright-field microscope with 20× objective lens.
Statistical analysis
All experiments were performed independently at least in triplicate. The data are presented as mean ± standard error of the mean (SEM). For all the other datasets comparing between groups, one-way ANOVA with post-hoc Tukey’s test was performed. P<0.05 was considered to indicate a statistically significant difference.
Results
Trametinib improves cognitive function in a TBI mouse model
Cognitive function deficits, including short-term memory loss and body control impairment, are major clinical symptoms of TBI, resulting to significant disease burden [30]. To investigate whether trametinib could improve the cognitive function following TBI, a murine TBI model was established using the CCI method. Therefore, the Morris water maze and eight-arm radial maze tests, and rotarod test were performed, to assess the behavioral memory, and motor deficits following TBI and the effect of trametinib, respectively [31]. Mice were divided into three groups, namely normal, vehicle- and trametinib-treated groups. In the first group, mice did not receive any treatment, while in the second and third group mice were treated daily with vehicle PBS or trametinib, respectively. The treatment was administrated 2 h following TBI. From day 1 after TBI, Morris water maze, rotarod and eight-arm radial maze tests were performed daily to evaluate the behavior of mice following TBI. Although, following TBI, mice had more possibilities of revisiting errors, eight-arm radial maze assay showed that mice treated with trametinib exhibited dramatically reduced revisiting error time compared with that noted to the vehicle-treated group (Figure 1B). Furthermore, in the Morris water maze test, mice in the treated group showed a significantly shortened escape latency to find the platform (Figure 1C). In terms of rotarod assay, TBI mice treated with trametinib demonstrated a significantly longer latency to fall time compared with the TBI vehicle-treated group (Figure 1D).
Figure 1.

Trametinib improves the cognitive function in a murine TBI model. (A) Trametinib (1 mg/kg/day) was administrated to mice by oral gavage 2 h after TBI. Following 24 h, mice were then subjected to (B) eight-arm radial maze test, (C) Morris water maze test and (D) rotarod test. *P<0.05, **P<0.01. TBI, traumatic brain injury.
Recent evidence suggests that oligodendrocytes or oligodendrocyte progenitor cells (OPCs) play vital roles in neural repairing or remyelination in injured CNS, which strongly associate with the cognitive outcome post TBI [32-35]. Therefore, oligodendrocytes or OPCs of the white matter close to the TBI zone were examined by labeling cells with an oligo2 antibody following trametinib treatment. The number of oligo2-positive cells in the focal injury site was significantly decreased compared with the TBI vehicle and normal control groups. In addition, indeed, trametinib rescued the number of oligo2-positive cells on both 3 and 7 days following TBI (Figure 2A). Furthermore, the density of oligo2-positive cells in the trametinib-treated group was increased after 7 days compared with 3 days. However, significant increase was also observed in the TBI vehicle-treated group (Figure 2A). Additionally, the effect of trametinib in remyelination was investigated. Total proteins were extracted from murine brain tissues at 3- and 7-days post TBI. Subsequently, western blot assay was performed to evaluate myelin basic protein (MBP) and myelin oligodendrocyte glycoprotein (MOG) protein expression levels. Indeed, MBP and MOG expression were significantly increased in the trametinib-treated group compared with that noted to the vehicle-treated group (Figure 2B), indicating that trametinib induced remyelination following TBI. To further validate trametinib induced remyelination, we assessed the expression of oligodendrocyte specific genes including platelet-derived growth factor (PDGFa), SRY-Box Transcription Factor 10 (SOX10), Chondroitin sulfate proteoglycan 4 (CSPG4), and Claudin-11 (CLDN11) in the murine brain tissues at 7 days post TBI. Up-regulation of these markers was observed in trametinib treated TBI mice compared to controls (Figure 2C).
Figure 2.
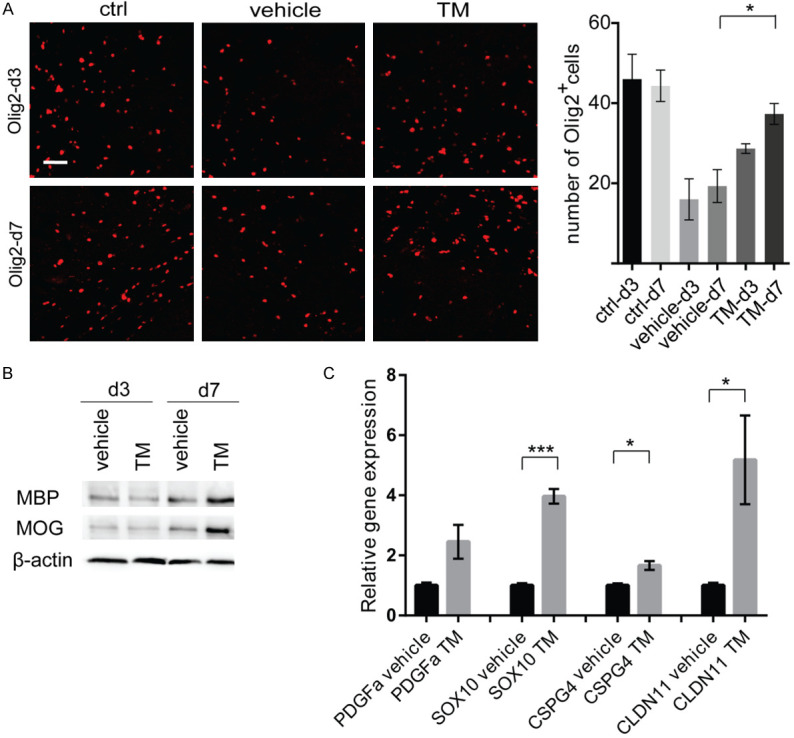
Trametinib increases the density of oligodendrocytes in the traumatic area of TBI mice. (A) On the left, red signal indicates the immunofluorescence staining of oligodendrocyte marker oligo2 in the traumatic area of normal control, TBI control and trametinib-treated (1 mg/kg/day) TBI mice at days 3 and 7 post TBI. On the right, bar graph shows the quantification of oligo2-positive cells within the traumatic area. *P<0.05. Scale bar =50 μm. (B) Western blot analysis of MBP and MOG protein expression levels in murine TBI brain tissues (same treatment as A) at days 3 and 7 posts TBI. (C) Relative gene expression of oligodendrocyte specific genes in murine TBI brain tissues (same treatment as A) at days 7 post TBI using qPCR. *P<0.05, ***P<0.001. TM, trametinib; MBP, myelin basic protein; MOG, Myelin oligodendrocyte glycoprotein; PDGFa, Platelet Derived Growth Factor Subunit A; SOX10, SRY-Box Transcription Factor 10; CSPG4, Chondroitin sulfate proteoglycan 4; CLDN11, Claudin 11.
Trametinib ameliorates neuroinflammation and reduces microglial density post TBI
Microglial activation plays a crucial role in neuroinflammation during the early TBI, which aggravates cognitive impairment [7,9]. To determine microglial density and neuro-inflammation after TBI, first, the microglial cell number was measured in the traumatic zone of the murine brain cortex on days 3 and 7. The density of microglia in the TBI vehicle-treated group was dramatically increased approximately 3-folds compared with control mice after 3 days, while in the -treated TBI mice the density of microglia was decreased compared with the TBI vehicle-treated group (Figure 3A). In addition, following 7 days, in the trametinib-treated TBI mice the density of microglia was decreased to similar levels to the control mice group (Figure 3A). Up-regulation of inflammatory factors, such as IL-1β, is extremely detrimental to post TBI cognitive function [36,37]. Therefore, using immunofluorescence staining, pro-inflammatory markers IL-1β and CD86 were determined in the TBI mice. We observed that trametinib treatment significantly ameliorated the IL-1β and CD86 expression levels in the traumatic mice brains (Figure 3B). To further investigate whether the expression of inflammatory factors was suppressed, total RNA were extracted from murine brain tissues and the mRNA expression levels of IL-1β, TNF-α, NOS2, IL12, and GM-CSF, which are important inflammation-associated cytokines, were determined using qRT-PCR analysis. The results showed that the protein expression levels of IL-1β, TNF-α, NOS2, IL12, and GM-CSF were remarkably down-regulated in the trametinib-treated TBI mice compared with those of the TBI control mice after 3 days (Figure 3C). Similarly, trametinib decreased the expression of inflammatory factors 7 days following TBI (Figure 3C).
Figure 3.
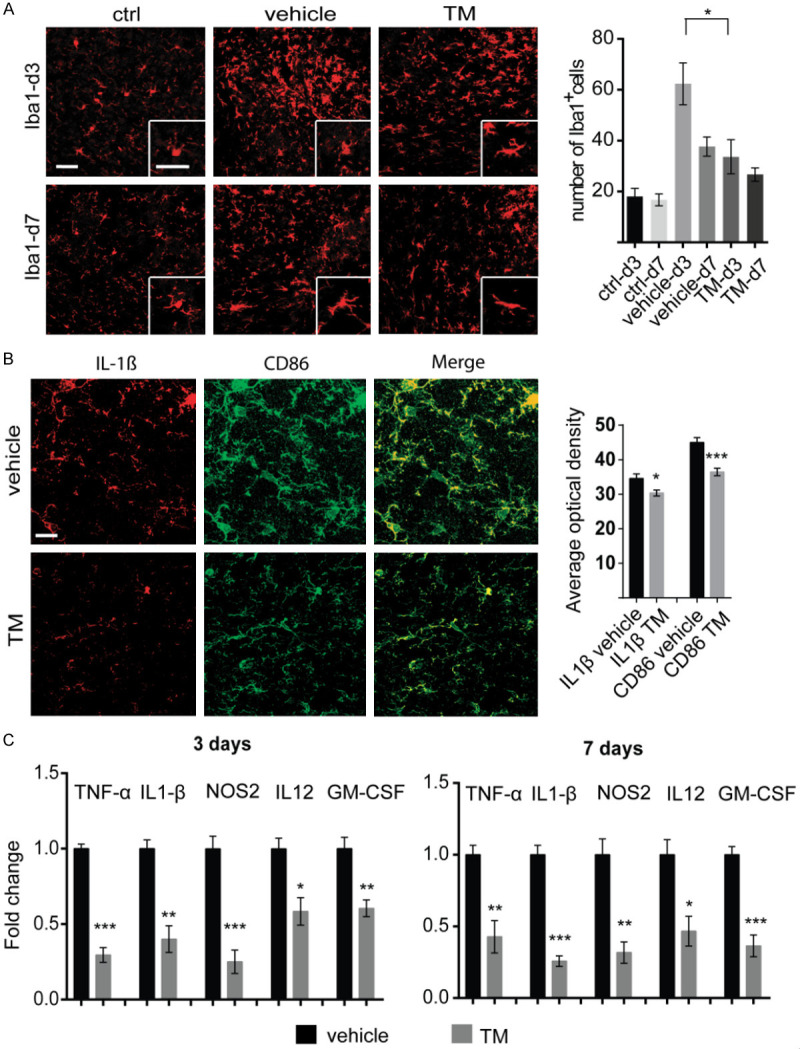
Trametinib ameliorates neuro-inflammation and reduces microglial density post TBI. (A) On the left, red signal indicates the of microglial specific marker Iba1 in the traumatic area of normal control, TBI control and trametinib-treated (1 mg/kg/day) TBI mice at days 3 and 7 post TBI. On the right, bar graph shows the quantification of Iba1-positive cells within the traumatic area. *P<0.05. Scale bar =50 μm. (B) On the left, representative figures of IL-1β and CD86 immunofluorescence staining on TBI mice brains at days 3 posts TBI (same treatment as A). On the right, bar graph shows the quantification of Average optical density (AOD) of each marker. *P<0.05, ***P<0.001. (C) Relative expression of the pro-inflammatory genes, including TNF-α, IL-1β, NOS2, IL12 and GM-CSF in TBI murine tissues at days 3 and 7 post TBI. *P<0.05, **P<0.01, ***P<0.001. TM, trametinib; Iba1, allograft inflammatory factor 1; TNF-α, tumor necrosis factor alpha; IL-1β, interleukin 1β; NOS2, oxide synthase 2; IL-12, interleukin 12; GM-CSF, granulocyte-macrophage colony-stimulating factor.
Trametinib attenuates LPS-induced microglial activation and cell migration
Microglial cells are rapidly activated following TBI and release several inflammatory factors, which in turn participate actively in the neural damage during the early stages of TBI. The above results demonstrated that trametinib suppressed pro-inflammatory factors protein expression after TBI, therefore, the direct effect of trametinib in microglia was then investigated. Primary microglia were pre-treated or not with 5 or 10 nM trametinib for 2 h and subsequently stimulated with 100 ng/ml LPS for 6 h. IL-1β, TNF-α and NOS2 mRNA levels were measured using qPCR. The results showed that upregulation of IL-1β, TNF-α and NOS2 evoked by LPS was attenuated by both 5 and 10 nM trametinib treatment. However, mRNA expression levels of anti-inflammatory factors Arg1, VEGFA and IL-10 were not affected by trametinib in both concentrations (Figure 4A). The aforementioned findings were also confirmed using ELISA. Following pre-treatment or not with trametinib for 2 h, cells were treated with LPS for 24 h and supernatants ware harvested to measure IL-1β, TNF-α, GM-CSF and Nitric Oxide (NO) secretion levels. Consistent with the previous qPCR results, IL-1β, TNF-α, GM-CSF and NO secretion evoked by LPS was also notably alleviated by trametinib (Figure 4B).
Figure 4.
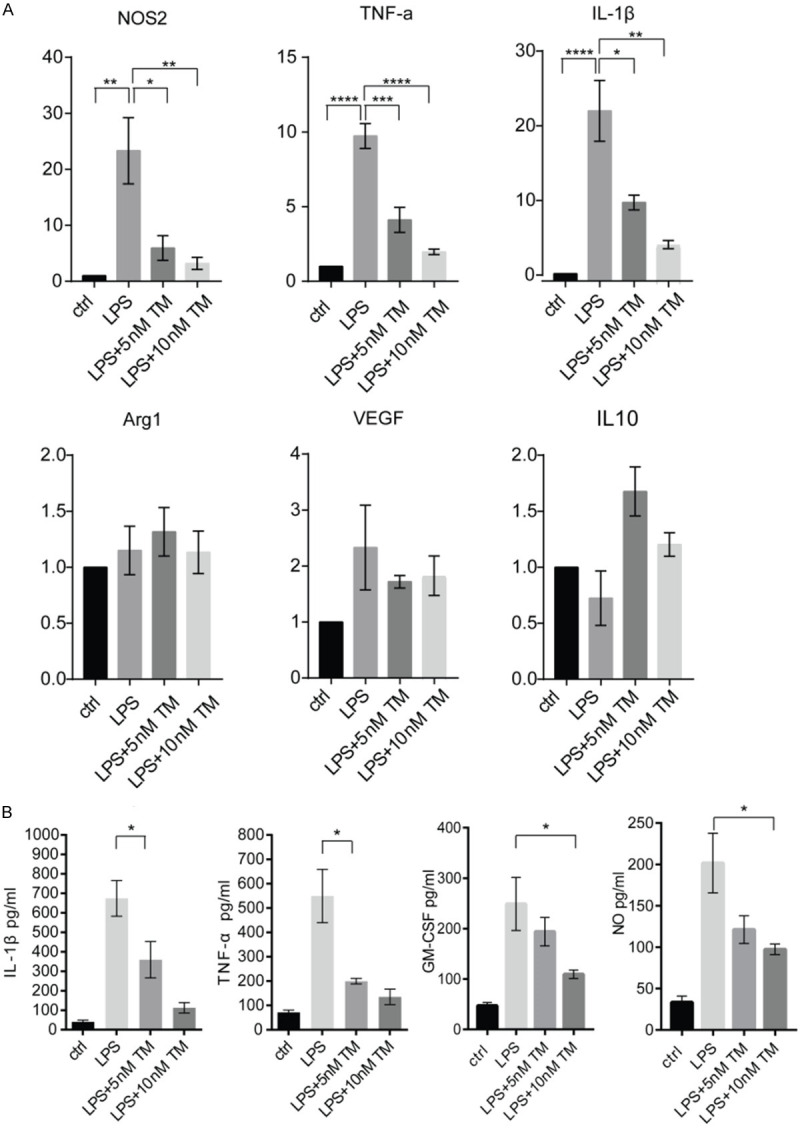
Trametinib attenuates LPS-induced microglial activation. A. Cultured microglia was treated with 100 ng/ml LPS supplemented or not with 5 or 10 nM trametinib for 6 h. Subsequently, the mRNA expression levels of NOS2, TNF-α, IL-1β, Arg1, VEGF and IL-10 were determined by qPCR, using β-actin as a house-keeping gene. The results were normalized to the control group. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. B. Cultured microglia were pre-treated with or without 5 or 10 nM trametinib for 2 h and subsequently stimulated with 100 ng/ml LPS for 24 h. Supernatant was collected and the secreting levels of IL-1β, TNF-α, NOS2 and GM-CSF were detected via ELISA. *P<0.05. LPS, lipopolysaccharide; TM, trametinib; Arg1, arginase 1; VEGF, vascular endothelial growth factor.
Since the suppression of microglial infiltration by in murine TBI model was confirmed, the present study further investigated the effects of LPS stimulation with or without trametinib in microglial chemotactic activity using agarose spot and Boyden chamber assays. Both chemotaxis models demonstrated that LPS induced microglial migration, which was attenuated by tramtetinib treatment (Figure 5A and 5B). This finding may explain the changes in the density of infiltrating microglia in TBI mice following treatment with trametinib.
Figure 5.
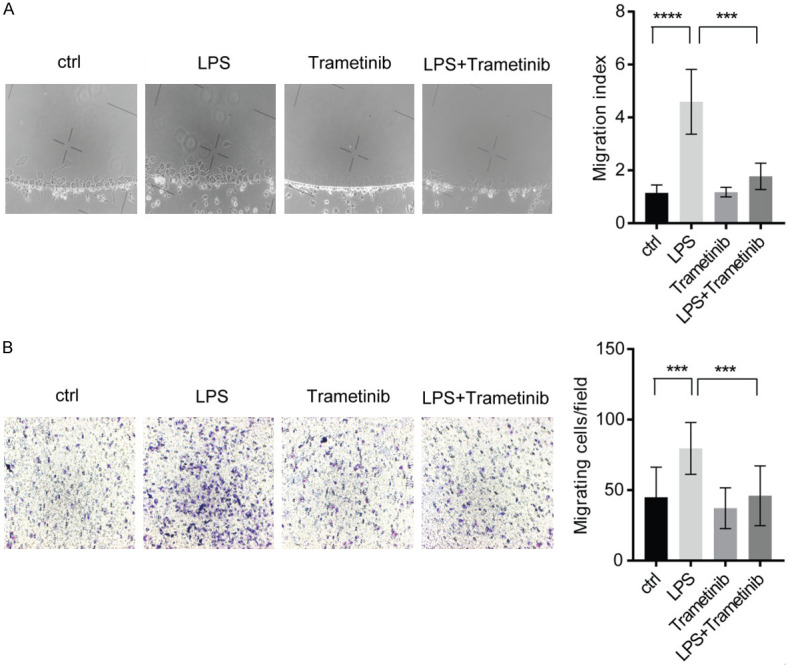
Trametinib attenuates LPS-induced microglial migration. A. Chemotactic activity of cultured microglia was determined by Agarose spot assay. Representative figures are shown on the left. On the right, the number of cells migrated into the agarose spot was normalized to the number of cells in the inner control PBS group. ***P<0.001, ****P<0.0001. B. Chemotactic activity of cultured microglia was determined by Boyden chamber assay. The representative figures are on the left and the quantification of cells migrating into the down chamber is presented on the right. ***P<0.001. LPS, lipopolysaccharide; PBS, phosphate buffered saline.
Trametinib effectively inhibits microglial MEK/ERK signaling cascade activation
MAPK (MEK/ERK) undergoes sequential phosphorylation on various types of substrate proteins via a multitude of cytoplasmic and nuclear effectors, including transcription factors, multiple regulators, kinases and phosphatases. These effectors serve a crucial role in regulating immune responses and the inflammation process [21,38]. Furthermore, it has been reported that NF-κΒ phosphorylation regulates microglial cytokine production in different pathological conditions [39-41]. To verify that trametinib inhibited MEK/ERK signaling pathway, primary microglial cells were stimulated with LPS in the presence or not of tramtetinib for 24 h. Subsequently, protein extraction was performed to detect the phosphorylation status of ERK and NF-κΒ. In addition, important mediater of inflammatory NF-κB signialing, NLR family pyrin domain containing 3 (NLRP3) was also determined. The results demonstrated that trametinib treatment significantly inhibited LPS-induced ERK and NF-κΒ phosphorylation, as well as down-regulating NLRP3 expression (Figure 6A). Furthermore, protein lysates from murine TBI tissues treated with or without trametinib were used to examine phosphorylated ERK protein levels. Western blot analysis revealed that the protein expression levels of phosphorylated ERK were drastically increased, while treatment with trametinib efficaciously inhibited TBI-mediated ERK phosphorylation (Figure 6B). These findings indicate that trametinib inhibits TBI-induced neuroinflammation via the MEK/ERK signaling pathway.
Figure 6.
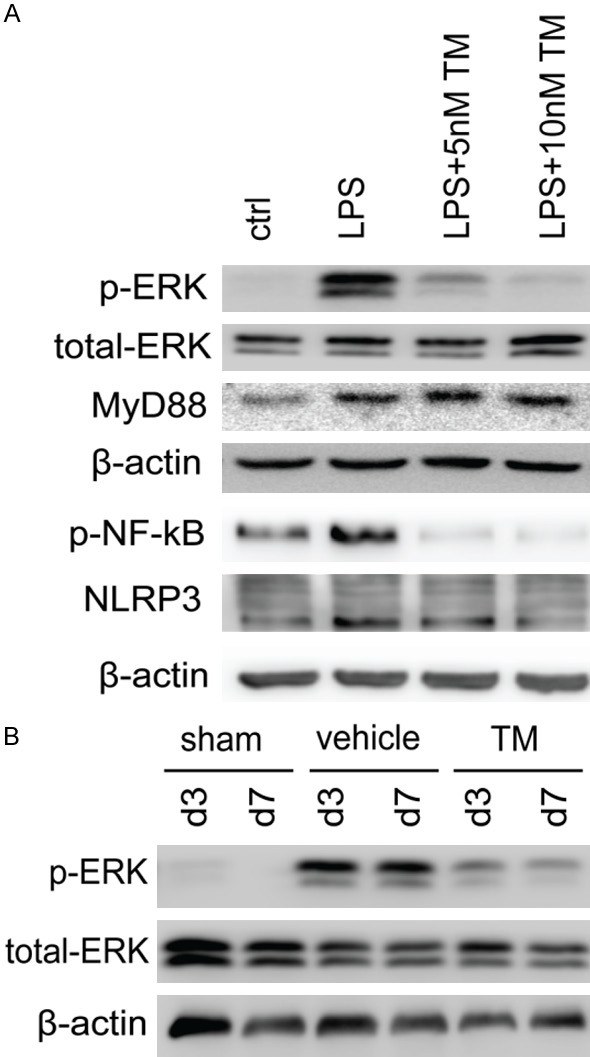
Trametinib effectively inhibits MEK/ERK signaling pathway activation in microglia. A. Cultured microglia were pre-treated with or without 5 or 10 nM trametinib for 2 h and subsequently stimulated with 100 ng/ml LPS for 24 h and the protein levels of p-ERK, total ERK, p-NF-κB, NLRP3, MyD88 and β-actin were determined using western blot analysis. B. The protein expression levels of p-ERK and total-ERK were detected at days 3 and 7 via western blot analysis in protein extracts isolated from TBI mice treated with trametinib (1 mg/kg/day). MEK/ERK, mitogen-activated protein kinase kinase/extracellular signal-regulated protein kinase; LPS, lipopolysaccharide; TM, trametinib; p-ERK, phosphorylated ERK; p-NF-κB, phosphorylated NF-κB; MyD88, myeloid differentiation factor 88; NLRP3, NLR family pyrin domain containing 3.
Discussion
TBI is the consequence of indirect or direct external mechanical impact on the brain. It is widely recognized that TBI is a highly complex disease characterized by multiple cellular and molecular pathological changes. Therefore, drugs selectively targeting a single factor may not be as effective as multitarget drugs in the treatment of TBI. Microglia are the resident immune cells in the brain. It has been demonstrated that microglial activation during the early stages of TBI, may contribute to the restoration of homeostasis in the brain. However, when microglia remain chronically activated, these cells display a classically activated phenotype, resulting in further tissue damage and potentially contributing to neurodegeneration by releasing proinflammatory factors [42]. Inflammation is one of the hallmarks of the TBI-induced brain damage, while multiple inflammatory factors have been elucidated to be closely associated with the outcome of TBI patients. For instance, elevated levels of IL-1β in severe TBI have been associated with acute brain edema during the early stages of TBI. By contrast, lower concentrations of IL-1β are related to more favorable neurological outcomes, according to the Glasgow Outcome Scale [43]. Another crucial cytokine, TNF-α, mainly originates from microglia and plays a central role in initiating and regulating cytokine cascade. No direct association between TNF-α levelsα level and Glasgow Outcome Scale in TBI patients has been reported. However, when TNF-α is considered along with other inflammatory cytokines, its expression has been associated with worse clinical outcome [44]. Besides, MEK/ERK signaling pathway has been considered as a vital factor in mediating a pro-inflammatory environment in the brain. Therefore, in the present study, a MEK/ERK inhibitor, trametinib, was used to treat TBI mice and the results showed that trametinib treatment significantly improved the cognitive function of TBI mice and reduced inflammation.
Trametinib is a third-generation, orally available and highly selective allosteric ATP non-competitive inhibitor of the two isoforms MEK1 and MEK2 [45]. Trametinib has been approved by the Food and Drug Administration (FDA) in treating BRAFV600E/K mutated melanoma. However, its role in regulating TBI-induced brain inflammatory damage has not yet been studied. In the present study, trametinib increased the number of oligodendrocytes and remyelination in the brain trauma region during 3 and 7 days following TBI. Oligodendrocytes participate in the myelination of neurons of the CNS. It has been demonstrated that the proliferation of oligodendrocytes serves a crucial role in post-TBI cognitive recovery. These findings elucidated why trematinib-treated mice exhibited improved cognitive function following TBI. MAPK/ERK pathway is involved in tissue inflammation. Moreover, it has been previously reported that trametinib suppresses LPS-induced TNF-α secretion by macrophages [46]. In the present study, trametinib inhibited the expression of LPS-induced microglial pro-inflammatory cytokines, such as IL-1β and TNF-α. However, the expression levels of Arg1, VEGF and IL-10 were not affected following treatment with trametinib.
The results of the present study demonstrated that trametinib significantly attenuated microglial density and the expression of pro-inflammatory factors in the brain tissue. However, the results did not verify whether these effects were microglia-dependent. Subsequently, we investigated the trametinib-mediated effects in the phenotype of microglia and their chemotactic activity in vitro. Trametinib drastically inhibited LPS-induced activation and chemotactic activity of microglia. This result mimicked the TBI-induced microglia activation condition.
Notwithstanding the promising results of tremetinib, the suppression of microglial activation needs further investigation, since microglia has also been implicated in facilitating brain tissue wound healing during the late phase of TBI [7,9,47]. Except the induction of neuroinflammation, microglia cells are able to polarize into ‘M2’ anti-inflammatory phenotype and release anti-inflammatory and growth factors, such as tissue growth factor β (TGF-β) and VEGF [10,11]. These factors promote fibroblast and glial cells proliferation, and myelin repair, which in turn mediate tissue healing post TBI [48-51]. Further investigation will identify the potential role of trametinib in the late phase of TBI healing.
However, there are still some limitations in the present study. Firstly, we evaluated only the brain functions following trametinib treatment during 8 days post TBI, while the long-term outcomes were not explored. Secondly, as in the case with other TBI models, only a ‘single-hit’ model was used. However, some repetitive injury models have been emerged showing similar effects with repetitive injuries [52]. Finally, the results could not fully describe the complicated immune environment in the traumatic brain tissue, since in the current study we only determined the expression of few cytokines. Therefore, all these issues should be considered in future studies.
In summary, trematinib, a FDA approved drug, attenuated microglial activation and microglial-induced neuroinflammation during the early stages of TBI. Moreover, trametinib decreased microglial chemotactic activity and improved the cognitive function in a murine TBI model. Therefore, these findings may provide a potential therapy strategy for TBI patients.
Acknowledgements
This research was funded by the National Natural Science Foundation of China [grant no. 81602202(FH)]. We thank the microscope facility of the Tongji Hospital of Huazhong University of Science and Technology. Special thanks to Regina Piske and Maren Wendt from Max Delbrueck Center of Molecular Medicine (Berlin, Germany).
Disclosure of conflict of interest
None.
References
- 1.Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- 2.Wells AJ, Hutchinson PJ. The management of traumatic brain injury. Surg (United Kingdom) 2018;36:613–620. [Google Scholar]
- 3.Mckee AC, Daneshvar DH. The neuropathology of traumatic brain injury. Handb Clin Neurol. 2015;127:45–66. doi: 10.1016/B978-0-444-52892-6.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76:886–899. doi: 10.1016/j.neuron.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 6.Patterson ZR, Holahan MR. Understanding the neuroinflammatory response following concussion to develop treatment strategies. Front Cell Neurosci. 2012;6:58. doi: 10.3389/fncel.2012.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron. 2017;95:1246–1265. doi: 10.1016/j.neuron.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Loane DJ, Kumar A. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol. 2016;275:316–327. doi: 10.1016/j.expneurol.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 11.Helmut K, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 12.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 13.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. [Google Scholar]
- 14.Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 15.Peyssonnaux C, Eychène A. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell. 2001;93:53–62. doi: 10.1016/s0248-4900(01)01125-x. [DOI] [PubMed] [Google Scholar]
- 16.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 17.Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283:125–134. doi: 10.1016/j.canlet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 18.Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17:1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Bergami P, Huang C, Goydos JS, Yip D, Bar-Eli M, Herlyn M, Smalley KS, Mahale A, Eroshkin A, Aaronson S, Ronai Z. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell. 2007;11:447–460. doi: 10.1016/j.ccr.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, Van Belle P, Elder DE, Herlyn M. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–759. [PubMed] [Google Scholar]
- 21.Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 22.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, Algazi A, Lewis K, Long GV, Puzanov I, Lebowitz P, Singh A, Little S, Sun P, Allred A, Ouellet D, Kim KB, Patel K, Weber J. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandalà M, Millward M, Arance A, Bondarenko I, Haanen JB, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, DeMarini DJ, Irani JG, Casey M, Ouellet D, Martin AM, Le N, Patel K, Flaherty K. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 24.Minelli A, Lyons S, Nolte C, Verkhratsky A, Kettenmann H. Ammonium triggers calcium elevation in cultured mouse microglial cells by initiating Ca2+ release from thapsigargin-sensitive intracellular stores. Pflugers Arch Eur J Physiol. 2000;439:370–377. doi: 10.1007/s004249900188. [DOI] [PubMed] [Google Scholar]
- 25.Yao X, Liu S, Ding W, Yue P, Jiang Q, Zhao M, Hu F, Zhang H. TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. J Neuroimmunol. 2017;310:38–45. doi: 10.1016/j.jneuroim.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 26.Kreipke CW, Morgan R, Kallakuri S, Rafols JA. Behavioral pre-conditioning enhances angiogenesis and cognitive outcome after brain trauma. Neurol Res. 2007;29:388–394. doi: 10.1179/016164107X204710. [DOI] [PubMed] [Google Scholar]
- 27.Hu F, Dzaye O, Hahn A, Yu Y, Scavetta RJ, Dittmar G, Kaczmarek AK, Dunning KR, Ricciardelli C, Rinnenthal JL, Heppner FL, Lehnardt S, Synowitz M, Wolf SA, Kettenmann H. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro Oncol. 2015;17:200–210. doi: 10.1093/neuonc/nou324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiggins HL, Rappoport JZ. An agarose spot assay for chemotactic invasion. Biotechniques. 2010;48:121–124. doi: 10.2144/000113353. [DOI] [PubMed] [Google Scholar]
- 29.Ifuku M, Buonfiglioli A, Jordan P, Lehnardt S, Kettenmann H. TLR2 controls random motility, while TLR7 regulates chemotaxis of microglial cells via distinct pathways. Brain Behav Immun. 2016;58:338–347. doi: 10.1016/j.bbi.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Roozenbeek B, Maas AI, Menon DK. Changing patterns in the epidemiology of traumatic brain injury. Nat Rev Neurol. 2013;9:231–236. doi: 10.1038/nrneurol.2013.22. [DOI] [PubMed] [Google Scholar]
- 31.D’Hooge R, De Deyn PP. Applications of the Morris water maze in the study of learning and memory. Brain Res Rev. 2001;36:60–90. doi: 10.1016/s0165-0173(01)00067-4. [DOI] [PubMed] [Google Scholar]
- 32.Scheller A, Bai X, Kirchhoff F. The role of the oligodendrocyte lineage in acute brain trauma. Neurochem Res. 2017;42:2479–2489. doi: 10.1007/s11064-017-2343-4. [DOI] [PubMed] [Google Scholar]
- 33.Takase H, Washida K, Hayakawa K, Arai K, Wang X, Lo EH, Lok J. Oligodendrogenesis after traumatic brain injury. Behav Brain Res. 2018;340:205–211. doi: 10.1016/j.bbr.2016.10.042. [DOI] [PubMed] [Google Scholar]
- 34.Marion CM, Radomski KL, Cramer NP, Galdzicki Z, Armstrong RC. Experimental traumatic brain injury identifies distinct early and late phase axonal conduction deficits of white matter pathophysiology, and reveals intervening recovery. J Neurosci. 2018;38:8723–8736. doi: 10.1523/JNEUROSCI.0819-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang S, Ballerini P, Buccella S, Giuliani P, Jiang C, Huang X, Rathbone MP. Remyelination after chronic spinal cord injury is associated with proliferation of endogenous adult progenitor cells after systemic administration of guanosine. Purinergic Signal. 2008;4:61–71. doi: 10.1007/s11302-007-9093-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helmy A, Carpenter KL, Menon DK, Pickard JD, Hutchinson PJ. The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J Cereb Blood Flow Metab. 2011;31:658–670. doi: 10.1038/jcbfm.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feuerstein GZ, Liu T, Barone FC. Cytokines, inflammation, and brain injury: role of tumor necrosis factor-alpha. Cerebrovasc Brain Metab Rev. 1994;6:341–360. [PubMed] [Google Scholar]
- 38.Qu WS, Tian DS, Guo ZB, Fang J, Zhang Q, Yu ZY, Xie MJ, Zhang HQ, Lü JG, Wang W. Inhibition of EGFR/MAPK signaling reduces microglial inflammatory response and the associated secondary damage in rats after spinal cord injury. J Neuroinflammation. 2012;9:178. doi: 10.1186/1742-2094-9-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taetzsch T, Levesque S, McGraw C, Block ML. NFkB P50 in neurotoxic microglial activation. Free Radic Biol Med. 2012;53:S64. [Google Scholar]
- 40.Mattson MP. NF-κB in the survival and plasticity of neurons. Neurochem Res. 2005;30:883–893. doi: 10.1007/s11064-005-6961-x. [DOI] [PubMed] [Google Scholar]
- 41.Nonaka M, Chen XH, Pierce JE, Leoni MJ, McIntosh TK, Wolf JA, Smith DH. Prolonged activation of NF-κB following traumatic brain injury in rats. J Neurotrauma. 1999;16:1023–1034. doi: 10.1089/neu.1999.16.1023. [DOI] [PubMed] [Google Scholar]
- 42.Donat CK, Scott G, Gentleman SM, Sastre M. Microglial activation in traumatic brain injury. Front Aging Neurosci. 2017;9:208. doi: 10.3389/fnagi.2017.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nwachuku EL, Puccio AM, Adeboye A, Chang YF, Kim J, Okonkwo DO. Time course of cerebrospinal fluid inflammatory biomarkers and relationship to 6-month neurologic outcome in adult severe traumatic brain injury. Clin Neurol Neurosurg. 2016;149:1–5. doi: 10.1016/j.clineuro.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 44.Santarsieri M, Kumar RG, Kochanek PM, Berga S, Wagner AK. Variable neuroendocrine-immune dysfunction in individuals with unfavorable outcome after severe traumatic brain injury. Brain Behav Immun. 2015;45:15–27. doi: 10.1016/j.bbi.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lugowska I, Koseła-Paterczyk H, Kozak K, Rutkowski P. Trametinib: a MEK inhibitor for management of metastatic melanoma. Onco Targets Ther. 2015;8:2251–2259. doi: 10.2147/OTT.S72951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi-Lin D, Yuan X, Zhan S, Luo-Jia T, Chao-Yang T. Trametinib, a novel MEK kinase inhibitor, suppresses lipopolysaccharide-induced tumor necrosis factor (TNF)-α production and endotoxin shock. Biochem Biophys Res Commun. 2015;458:667–673. doi: 10.1016/j.bbrc.2015.01.160. [DOI] [PubMed] [Google Scholar]
- 47.Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173:692–702. doi: 10.1111/bph.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobolyi A, Vincze C, Pál G, Lovas G. The neuroprotective functions of transforming growth factor beta proteins. Int J Mol Sci. 2012;13:8219–8258. doi: 10.3390/ijms13078219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mattson MP, Barger SW, Furukawa K, Bruce AJ, Wyss-Coray T, Mark RJ, Mucke L. Cellular signaling roles of TGF beta, TNF alpha and beta APP in brain injury responses and Alzheimer’s disease. Brain Res Brain Res Rev. 1997;23:47–61. doi: 10.1016/s0165-0173(96)00014-8. [DOI] [PubMed] [Google Scholar]
- 50.Merrill MJ, Oldfield EH. A reassessment of vascular endothelial growth factor in central nervous system pathology. J Neurosurg. 2005;103:853–868. doi: 10.3171/jns.2005.103.5.0853. [DOI] [PubMed] [Google Scholar]
- 51.Ju S, Xu C, Wang G, Zhang L. VEGF-C induces alternative activation of microglia to promote recovery from traumatic brain injury. J Alzheimer’s Dis. 2019;68:1687–1697. doi: 10.3233/JAD-190063. [DOI] [PubMed] [Google Scholar]
- 52.O’Connor WT, Smyth A, Gilchrist MD. Animal models of traumatic brain injury: a critical evaluation. Pharmacol Ther. 2011;130:106–113. doi: 10.1016/j.pharmthera.2011.01.001. [DOI] [PubMed] [Google Scholar]