

Abstract
We present a genome assembly from an individual male Sciurus carolinensis (the eastern grey squirrel; Vertebrata; Mammalia; Eutheria; Rodentia; Sciuridae). The genome sequence is 2.82 gigabases in span. The majority of the assembly (92.3%) is scaffolded into 21 chromosomal-level scaffolds, with both X and Y sex chromosomes assembled.
Keywords: Sciurus carolinensis, grey squirrel, genome sequence, chromosomal
Species taxonomy
Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Euarchontoglires; Glires; Rodentia; Sciuromorpha; Sciuridae; Sciurinae; Sciurini; Sciurus; Sciurus carolinensis Gmelin, 1788 (NCBI txid 30640).
Background
The eastern grey squirrel, Sciurus carolinensis, is native to eastern North America, where it plays important roles in forest regeneration through its habit of caching food nuts and seeds ( Corbet & Hill, 1991) 1. In North America, S. carolinensis has been introduced outside its native range such that it is now found from the Canadian Pacific northwest to Florida. S. carolinensis was introduced to Britain (in 1876), Ireland (in 1911), Italy (in 1948), South Africa (before 1900), Australia (in 1880s, extirpated in 1973) and Pitcairn island (in 1987) (see https://www.cabi.org/isc/datasheet/49075). S. carolinensis, which thrives in urban parklands and gardens, is classed as invasive in Europe and on Pitcairn island. In Britain and Ireland the expansion of S. carolinensis populations has driven decline in populations of the native red squirrel, Sciurus vulgaris, which we have also assembled ( Mead et al., 2020). The negative impact of S. carolinensis is through interspecific competition, leading to competitive exclusion of S. vulgaris, and by their carriage of squirrelpox virus, to which they are resistant but S. vulgaris are not ( Chantrey et al., 2014) ( Darby et al., 2014). The S. carolinensis genome will aid analyses of resistance and susceptibility to squirrelpox, as well as to the genomics of invasiveness.
Genome sequence report
The genome was sequenced from DNA extracted from a naturally deceased male S. carolinensis collected as part of a squirrel monitoring project run by the Wildlife Trust for Lancashire, Manchester and North Merseyside. A total of 74-fold coverage in Pacific Biosciences single-molecule long reads (N50 28 kb) and 40-fold coverage in 10X Genomics read clouds (from molecules with an estimated N50 of 19 kb) were generated. Primary assembly contigs were scaffolded with chromosome conformation HiC data (42-fold coverage). A contamination check identified a small number of low-coverage contigs that were likely to have derived from an apicomplexan parasite infecting the squirrel ( Léveillé et al., 2020); these were removed. Subsequent manual assembly curation corrected 272 missing/misjoins and removed three haplotypic duplications, reducing the scaffold number by 19% and increasing the scaffold N50 by 242% The final assembly has a total length of 2.82 Gb in 752 sequence scaffolds with a scaffold N50 of 148.2 Mb ( Table 1). The majority, 92.3%, of the assembly sequence was assigned to 21 chromosomal-level scaffolds representing 19 autosomes (numbered by sequence length), and the X and Y sex chromosomes ( Figure 1– Figure 5; Table 2) plus 13 unlocalised scaffolds (assigned to chromosomes but with ambiguous placement). The assembly has a BUSCO ( Simão et al., 2015) completeness of 93.7% using the mammalia_odb9 reference set. The primary assembly is a large-scale mosaic of both haplotypes (i.e. is not fully phased) and we have therefore also deposited the contigs corresponding to the alternate haplotype. The S. carolinensis mSciCar1 genome sequence is largely collinear with that of S. vulgaris mSciVul1 ( Figure 4).
Table 1. Genome data for Sciurus carolinensis mSciCar1.
| Project accession data | |
|---|---|
| Assembly identifier | mSciCar1 |
| Species | Sciurus carolinensis |
| Specimen | NHMUK ZD 2019.214 |
| NCBI taxonomy ID | 30640 |
| BioProject | PRJEB35386 |
| Biosample ID | SAMEA994726 |
| Isolate information | Wild isolate; male |
| Raw data accessions | |
| PacificBiosciences
SEQUEL I |
ERR3313242-ERR3313245,
ERR3313247-ERR3313255, ERR3313329, ERR3313331, ERR3313332, ERR3313342- ERR3313348 |
| 10X Genomics Illumina | ERR3316153-ERR3316156,
ERR3316173-ERR3316176 |
| Hi-C Illumina | ERR3312499-ERR3312500,
ERR3850937 |
| Genome assembly | |
| Assembly accession | GCA_902686445.1 |
| Accession of alternate
haplotype |
GCA_902685475.1 |
| Span (Mb) | 2,815,397,268 |
| Number of contigs | 2576 |
| Contig N50 length (Mb) | 13.98 |
| Number of scaffolds | 752 |
| Scaffold N50 length (Mb) | 148.23 |
| Longest scaffold (Mb) | 208.99 |
| BUSCO * genome score | C:93.7%[S:92.3%,D:1.4%],F:2.8%,M
:3.5%,n:4104 |
* BUSCO scores based on the mammalia_odb9 BUSCO set using v3.0.2. C= complete [S= single copy, D=duplicated], F=fragmented, M=missing, n=number of orthologues in comparison. A full set of BUSCO scores is available at https://blobtoolkit.genomehubs.org/view/mSciCar1_1/dataset/mSciCar1_1/busco.
Figure 1. Genome assembly of Sciurus carolinensis mSciCar1: Metrics.
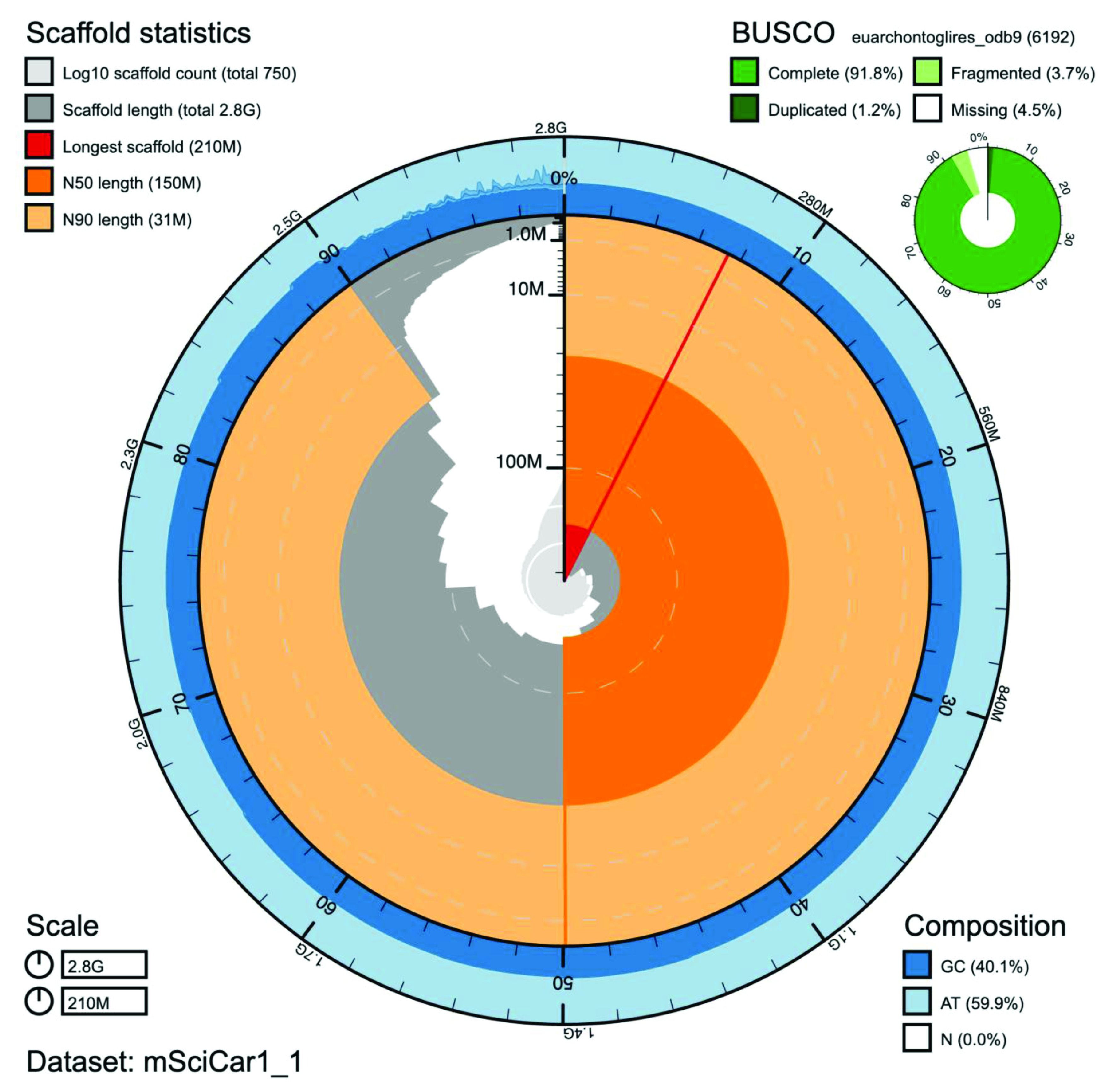
BlobToolKit Snailplot showing N50 metrics for S. carolinensis assembly mSciCar1 and BUSCO scores for the Euarchontoglires set of orthologues. The interactive version is available here.
Figure 2. Genome assembly of Sciurus carolinensis mSciCar1: GC-coverage plot.
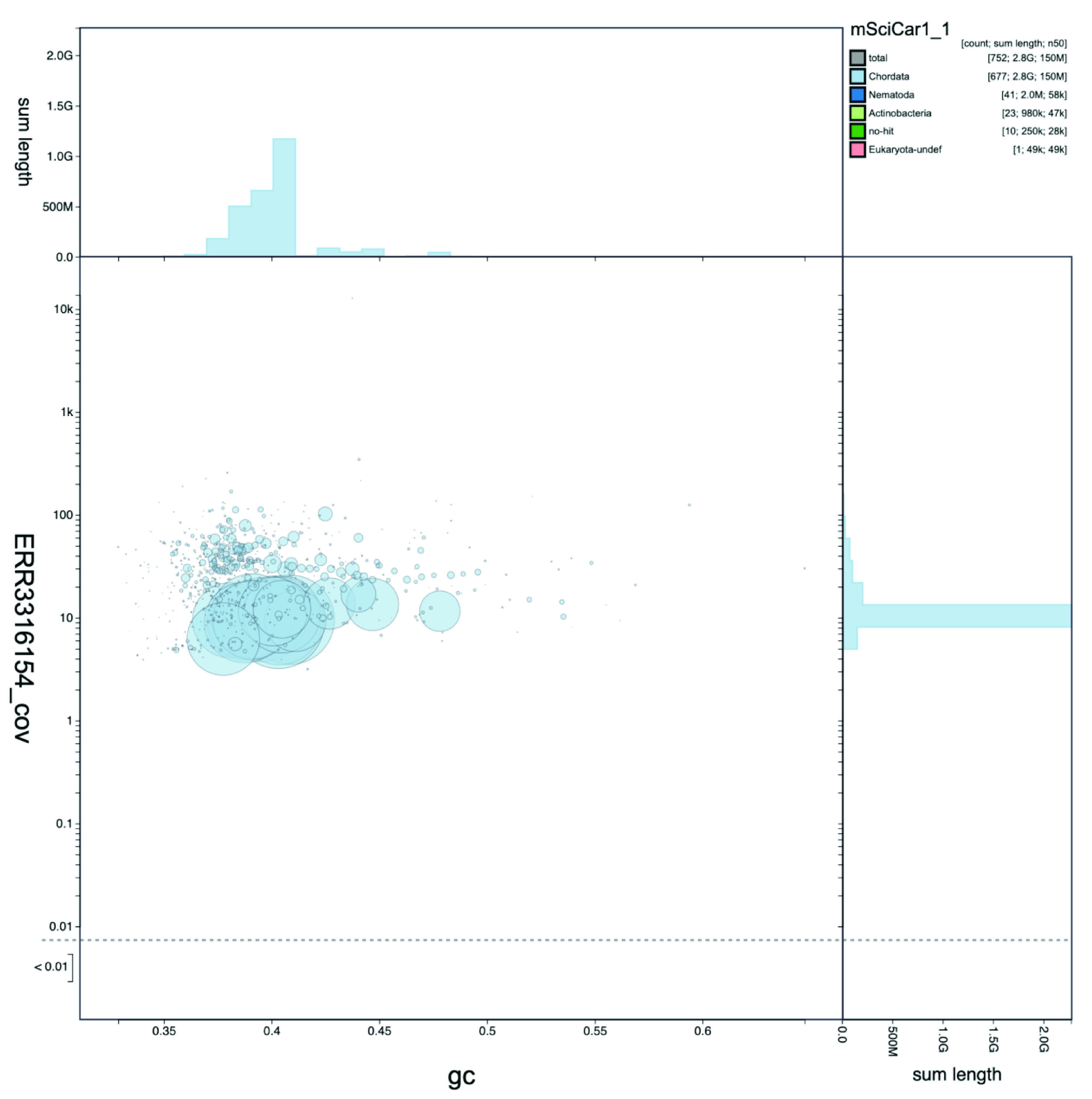
BlobToolKit GC-coverage plot of S. carolinensis mSciCar1 from long read data submission ERR3316154. The interactive version is available here.
Figure 3. Genome assembly of Sciurus carolinensis mSciCar1: Cumulative sequence plot.
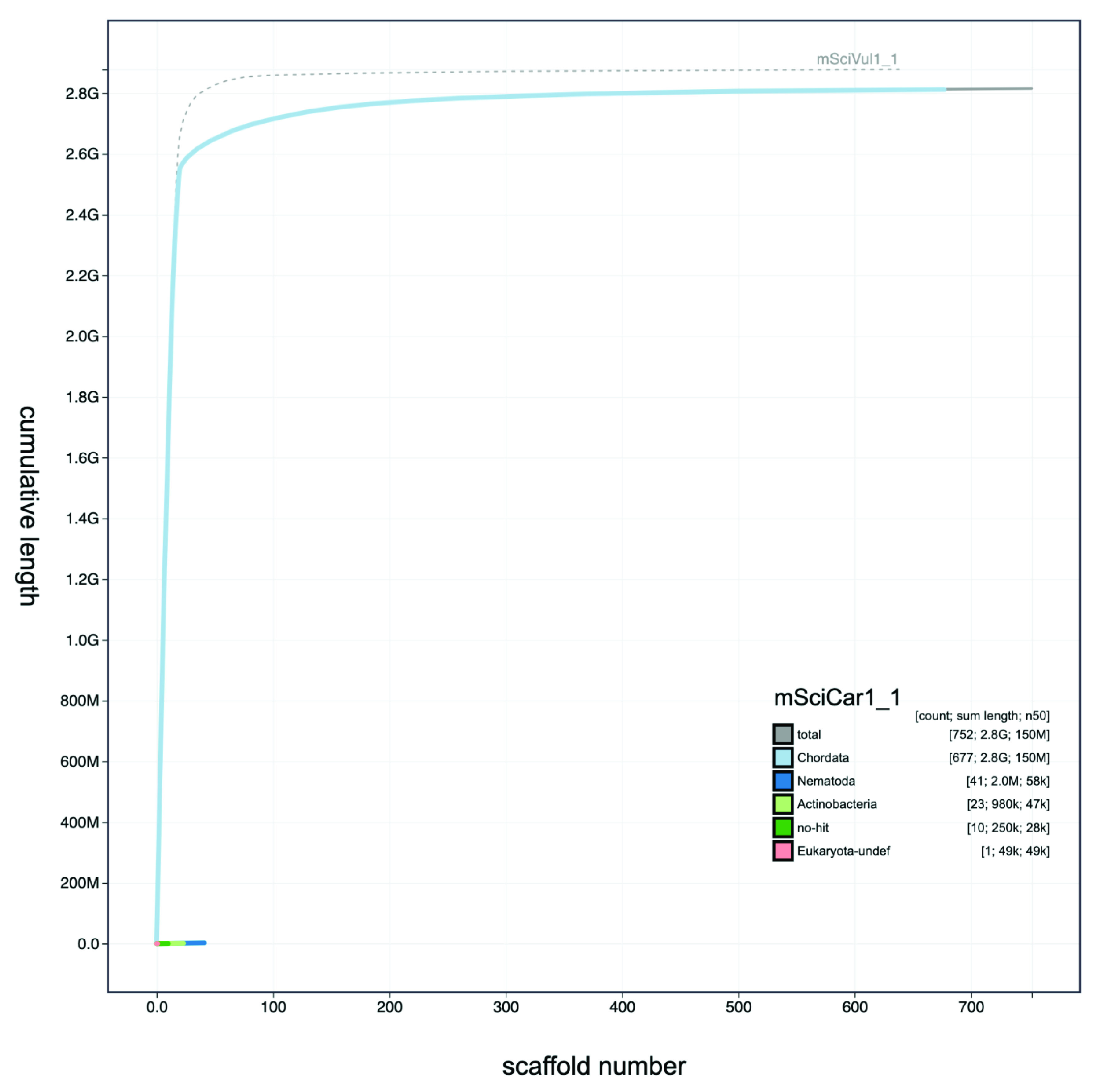
The blue line in the main plot shows the cumulative sequence plot for mSciCar. The sashed line shows the cumulative sequence plot of S. vulgaris mSciVul1 for comparison. The interactive version is available here.
Figure 4. Genome assembly of Sciurus carolinensis mSciCar1: Whole genome alignment with Sciurus vulgaris mSciVul1.
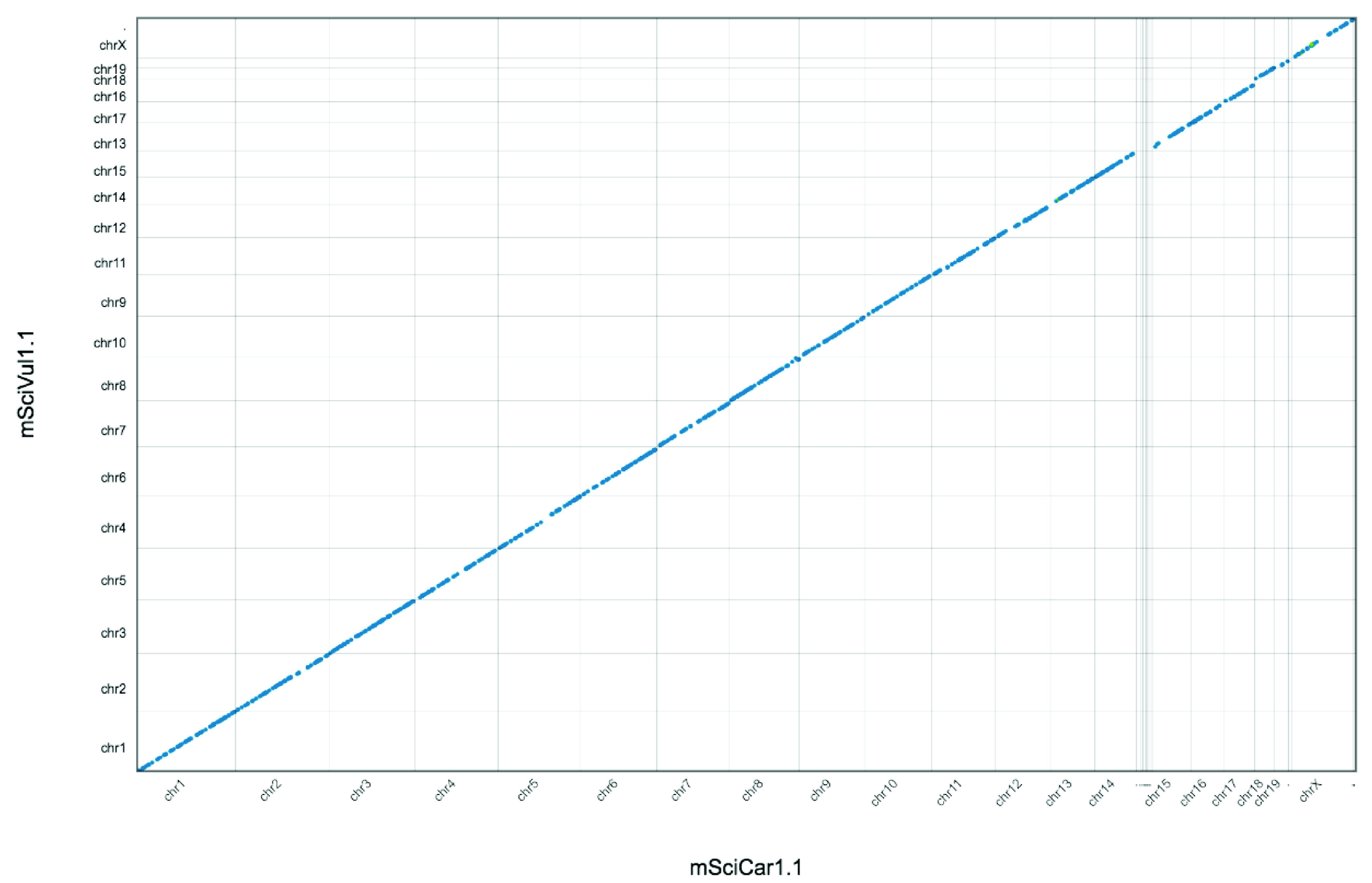
A nucmer ( Kurtz et al., 2004) pairwise alignment of mSciCar1 (x-axis) with mSciVul1 (Y axis).
Figure 5. Genome assembly of Sciurus carolinensis mSciCar1: Hi-C contact map.
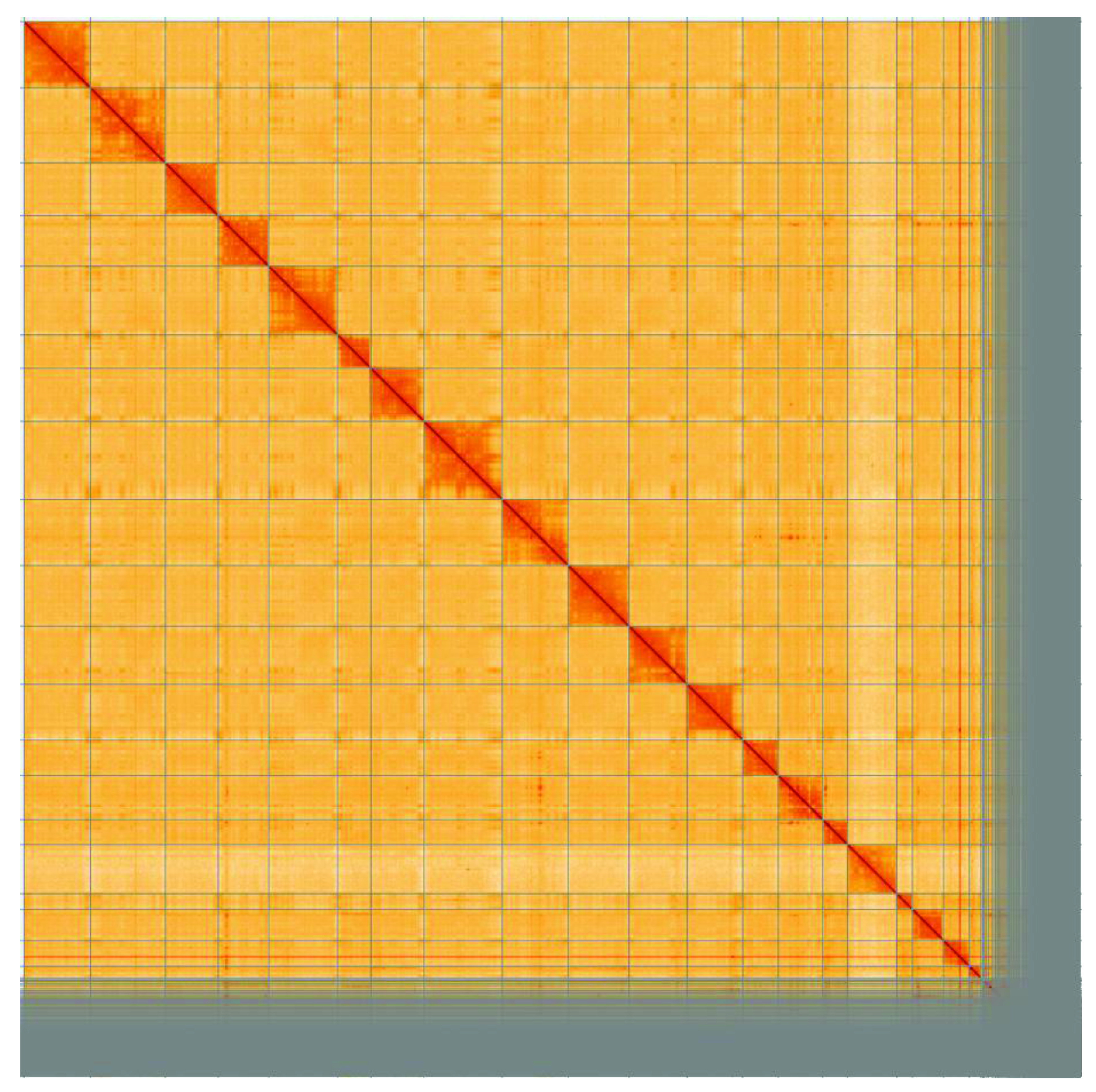
Hi-C scaffolding of the S. carolinensis mSciCar1 assembly visualised in HiGlass ( Kerpedjiev et al., 2018).
Table 2. Chromosomal pseudomolecules in the genome assembly of Sciurus carolinensis mSciCar1.
| ENA accession | Chromosome | Size (Mb) | GC% |
|---|---|---|---|
| LR738590.1 | 1 | 208.99 | 40.3 |
| LR738591.1 | 2 | 199.83 | 40.8 |
| LR738592.1 | 3 | 183.55 | 40.3 |
| LR738593.1 | 4 | 177.11 | 39.5 |
| LR738594.1 | 5 | 175.91 | 39.1 |
| LR738595.1 | 6 | 162.27 | 38.7 |
| LR738596.1 | 7 | 154.99 | 39.1 |
| LR738597.1 | 8 | 148.23 | 40.5 |
| LR738598.1 | 9 | 141.42 | 38.8 |
| LR738599.1 | 10 | 140.98 | 38.1 |
| LR738600.1 | 11 | 135.23 | 40.1 |
| LR738602.1 | 12 | 118.65 | 40.1 |
| LR738603.1 | 13 | 94.68 | 41.1 |
| LR738604.1 | 14 | 88.65 | 40.2 |
| LR738605.1 | 15 | 83.14 | 40.5 |
| LR738606.1 | 16 | 68.57 | 44.7 |
| LR738607.1 | 17 | 66.05 | 42.7 |
| LR738608.1 | 18 | 41.56 | 47.8 |
| LR738609.1 | 19 | 30.99 | 44 |
| LR738601.1 | X | 131.72 | 37.8 |
| LR738610.1 | Y | 4.81 | 38.3 |
| - | Unplaced | 258.08 | 40 |
Methods
The eastern grey squirrel specimen was collected by the Wildlife Trust for Lancashire, Manchester and North Merseyside as part of an ongoing programme of recovery of dead squirrels. A full tissue dissection and preservation in 80% ethanol was undertaken and the specimen accessioned by the Natural History Museum, London.
DNA was extracted using an agarose plug extraction from spleen tissue following the Bionano Prep Animal Tissue DNA Isolation Soft Tissue Protocol 2. Pacific Biosciences CLR long read and 10X Genomics read cloud sequencing libraries were constructed according to the manufacturers’ instructions. Sequencing was performed by the Scientific Operations core at the Wellcome Sanger Institute on Pacific Biosciences SEQUEL I (single molecule long read) and Illumina HiSeq X (10X Genomics Chromium). HiC data were generated using the Dovetail v1.0 kit and sequenced on HiSeq X.
See Table 3 for software versions and sources. Assembly was carried out using Falcon-unzip ( Chin et al., 2016), haplotypic duplication was identified and removed with purge_dups ( Guan et al., 2020) and a first round of scaffolding carried out with 10X Genomics read clouds using scaff10x. Scaffolding with Hi-C data was carried out using SALSA2. The Hi-C scaffolded assembly was polished with arrow using the PacBio data, then polished with the 10X Genomics Illumina data by aligning to the assembly with longranger align, calling variants with freebayes ( Garrison & Marth, 2012) and applying homozygous non-reference edits using bcftools consensus. Two rounds of the Illumina polishing were applied. The assembly was checked for contamination and corrected using the gEVAL system ( Chow et al., 2016). Since Hi-C data were sparse, curation was aided by synteny with the assembly for Sciurus vulgaris simultaneously being curated by the Wellcome Sanger Institute. The genome was analysed within the BlobToolKit environment ( Challis et al., 2019).
Table 3. Software tools used.
| Software
tool |
Version | Source |
|---|---|---|
| Falcon-unzip | falcon-kit 1.2.2 | ( Chin et al., 2016) |
| purge_dups | 1.0.0 | ( Guan et al., 2020) |
| SALSA2 | 2.2 | ( Ghurye et al., 2018) |
| scaff10x | 4.2 |
https://github.com/wtsi-
hpag/Scaff10X |
| arrow | GenomicConsensus
2.3.3 |
https://github.com/
PacificBiosciences/ GenomicConsensus |
| longranger align | 2.2.2 |
https://
support.10xgenomics. com/genome-exome/ software/pipelines/latest/ advanced/other-pipelines |
| freebayes | v1.1.0-3-g961e5f3 | ( Garrison & Marth, 2012) |
| bcftools
consensus |
1.9 |
http://samtools.github.
io/bcftools/bcftools.html |
| gEVAL | 2016 | ( Chow et al., 2016) |
| BlobToolKit | 1 | ( Challis et al., 2019) |
| nucmer from
MUMmer 3 |
3.0 | ( Kurtz et al., 2004) |
Data availability
Underlying data
European Nucleotide Archive: Sciurus carolinensis (grey squirrel) genome assembly, mSciCar1. BioProject accession number PRJEB35386; https://identifiers.org/ena.embl:PRJEB35386.
The genome sequence is released openly for reuse. The S. carolinensis genome sequencing initiative is part of the Wellcome Sanger Institute’s “25 genomes for 25 years” project 3. It is also part of the Vertebrate Genome Project (VGP) 4 and the Darwin Tree of Life (DToL) project 5. The specimen has been preserved in ethanol and deposited with the Natural History Museum, London under registration number NHMUK ZD 2019.214, where it will remain accessible to the research community for posterity. All raw data and the assembly have been deposited in the ENA. The genome will be annotated and presented through the Ensembl pipeline at the European Bioinformatics Institute. Raw data and assembly accession identifiers are reported in Table 1.
Acknowledgements
We thank Mike Stratton and Julia Wilson for their continuing support for the 25 genomes for 25 years project. The Wildlife Trust for Lancashire, Manchester and North Merseyside thank many members of the public for support.
Funding Statement
This work was supported by the Wellcome Trust through core funding to the Wellcome Sanger Institute (WT206194). SMcC and RD were supported by Wellcome grant WT207492. MB was supported through Wellcome grant WT218328.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
[version 1; peer review: 2 approved]
Footnotes
References
- Challis R, Richards E, Rajan J, et al. : BlobToolKit – Interactive Quality Assessment of Genome Assemblies. bioRxiv. 2019. 10.1101/844852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantrey J, Dale TD, Read JM, et al. : European red squirrel population dynamics driven by squirrelpox at a gray squirrel invasion interface. Ecol Evol. 2014;4(19):3788–99. 10.1002/ece3.1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Peluso P, Sedlazeck FJ, et al. : Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods. 2016;13(12):1050–54. 10.1038/nmeth.4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow W, Brugger K, Caccamo M, et al. : gEVAL - a web-based browser for evaluating genome assemblies. Bioinformatics. 2016;32(16):2508–10. 10.1093/bioinformatics/btw159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet GB, Hill JE: A World List of Mammalian Species (3rd Edition).Natural History Museum Publications/Oxford University Press, 1991, 243 Pp., HB £30.00. Oryx.Cambridge University Press,1991;25(3):174 10.1017/S0030605300034268 [DOI] [Google Scholar]
- Darby AC, McInnes CJ, Kjær KH, et al. : Novel host-related virulence factors are encoded by squirrelpox virus, the main causative agent of epidemic disease in red squirrels in the UK. PLoS One. 2014;9(7):e96439. 10.1371/journal.pone.0096439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, Marth G: Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv [q-bio.GN].arXiv,2012. Reference Source [Google Scholar]
- Ghurye J, Rhie A, Walenz BP, et al. : Integrating Hi-C Links with Assembly Graphs for Chromosome-Scale Assembly. bioRxiv. 2018. 10.1101/261149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye J, Rhie A, Walenz BP, et al. : Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol. 2019;15(8):e1007273. 10.1371/journal.pcbi.1007273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D, McCarthy SA, Wood J, et al. : Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020; pii: btaa025. 10.1093/bioinformatics/btaa025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerpedjiev P, Abdennur N, Lekschas F, et al. : HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018;19(1):125. 10.1186/s13059-018-1486-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, et al. : Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léveillé, AN, El Skhawy N, Barta JR: Multilocus sequencing of Hepatozoon cf. griseisciuri infections in Ontario eastern gray squirrels ( Sciurus carolinensis) uncovers two genotypically distinct sympatric parasite species. Parasitol Res. 2020;119(2):713–724. 10.1007/s00436-019-06583-5 [DOI] [PubMed] [Google Scholar]
- Mead D, Fingland K, Cripps R, et al. : The genome sequence of the Eurasian red squirrel, Sciurus vulgaris Linnaeus 1758 [version 1; peer review: awaiting peer review]. Wellcome Open Res. 2020;5:18 10.12688/wellcomeopenres.15679.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, et al. : BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–12. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]