

Abstract
Bacterial enteric pathogens have evolved efficient mechanisms to suppress mammalian inflammatory and immunoregulatory pathways. By exploiting the evolutionary relationship between the gut and pathogenic bacteria, we have developed a potential mucosal therapeutic. Our findings suggest that engineered preparations of the Salmonella acetyltransferase, AvrA, suppress acute inflammatory responses such as those observed in inflammatory bowel disease (IBD). We created 125 nm diameter cross-linked protein nanoparticles directly from AvrA and carrier protein to deliver AvrA in the absence of Salmonella. AvrA nanoparticles are internalized in vitro and in vivo into barrier epithelial and lamina propria monocytic cells. AvrA nanoparticles inhibit inflammatory signaling and confer cytoprotection in vitro, and in murine colitis models, we observe decreased clinical and histological indices of inflammation. Thus, we have combined naturally evolved immunomodulatory proteins with modern bioengineering to produce AvrA nanoparticles, a potential treatment for IBD.
Keywords: effector protein, nanoparticles, colitis, intracellular delivery, anti-inflammatory
Graphical Abstract
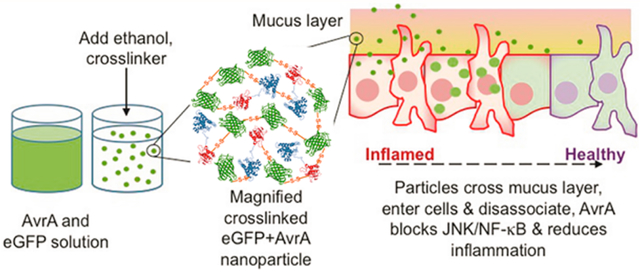
Inflammatory bowel disease (IBD) (Crohn’s disease and ulcerative colitis) are chronic relapsing autoimmune disorders of the intestinal tract that affect 1–2 of every thousand persons in developed countries, and incidence is increasing.1,2 In the intestine, they manifest with acute and chronic inflammation, tissue injury, scarring, and predisposition to adenocarcinoma and may also have systemic effects. IBD is generally recognized to represent aberrant immune recognition of the normal commensal microbiota. Current therapy involves inflammatory suppression with local 5-aminosalicylates, systemic corticosteroids, cytotoxic immunosuppressants, or biologicals, such as anti-TNF monoclonal antibodies. While effective, these are fraught with the complications of systemic immunosuppression and other toxicides.3,4 There is increasing interest in use of beneficial bacteria (probiotics) as a therapy, though to date, only modest efficacy has been reported.5
It is known that enteric bacterial pathogens have evolved mechanisms to suppress inflammatory and immunoregulatory pathways through active interference with regulators of the inflammatory response.6,7 Enteric pathogens influence eukaryotic pathways by soluble effector proteins that are translocated into the cytoplasm of target cells via a type III secretion system (TTSS) and have evolved to usurp host cellular functions for the benefit of the invading organism.8 AvrA, from Salmonella, is one such protein. It is a member of a family of acetyltransferases that covalently modify and inactivate members of the mitogen-activated protein kinase (MAPK) superfamily and thus have potent and diverse effects on a wide variety of eukaryotic growth, survival, and immune pathways.9 We have shown that AvrA overexpressed in transfected cells,10 or in a Drosophila transgenic model,11 blocked activation of nuclear factor-κB (NF-κB), JNK MAPK, and transcriptional activation of a range of inflammatory effector genes. AvrA acetylates MKK4/7, accounting for the blockade of JNK. Another laboratory demonstrated similar effects in a yeast model.12 Remarkably, in yeast, flies, human cells, and murine intestinal epithelia, AvrA-mediated signaling blockade occurs without induction of the apoptotic cell death characteristically seen during inhibition of host stress signaling pathways,9–12 thus making this activity an ideal therapeutic approach for IBD or other forms of inflammation. However, a major challenge in realizing the therapeutic potential of AvrA, or any exogenous protein effector, is the ability to deliver it locally through the harsh environment of the gastrointestinal tract and into the resident epithelial and immune cells without compromising the biological activity of the protein. Salmonella meets this challenge through use of TTSS. However, in Salmonella infection, AvrA is a SPI-1 TTSS effector protein that is co-delivered along with other virulence proteins,12,13 which can have negative effects, such as promotion of colonic tumorigenesis.14,15 Therefore, an alternative delivery approach is necessary to deliver only AvrA in the absence of Salmonella and safely access its anti-inflammatory functions.
Nanoparticles have been investigated for a variety of intraluminal gut applications including vaccination,16 diabetes,17 and IBD18–24 that target different cell types for systemic or local delivery. Previous IBD studies have encapsulated small anti-inflammatory drugs or siRNA in biodegradable polymeric micro- and nanoparticles.20 The primary benefits seen were reduced systemic side effects and reduced dosage required for the same therapeutic response. Furthermore, higher particle deposition has been seen in animals with induced colitis as compared to healthy animals, perhaps a consequence of depleted mucus, presence of phagocytic cells, or epithelial barrier disruption observed in inflamed tissue.20,21,25,26 In the case of protein drugs, however, polymeric delivery particles have limitations. Protein loading is extremely low, or the particles are too large to be internalized by cells, and harsh fabrication or degradation conditions can damage the protein.22,27–30 We have adapted a protein desolvation process31,32 to create condition-responsive cross-linked protein nanoparticles made from AvrA that can un-cross-link in the reducing environment observed inside cells, while maintaining the bioactivity of the AvrA cargo (Figure 1a). With these particles, we demonstrate the ability to suppress proinflammatory pathways in vitro and tissue inflammation in murine model colitis. This innovative approach has potential as a IBD therapeutic and establishes a drug discovery paradigm that exploits the evolution of bacterial immunoregulatory mechanisms and engineers a nanoparticle delivery strategy essential for clinical viability.
Figure 1.
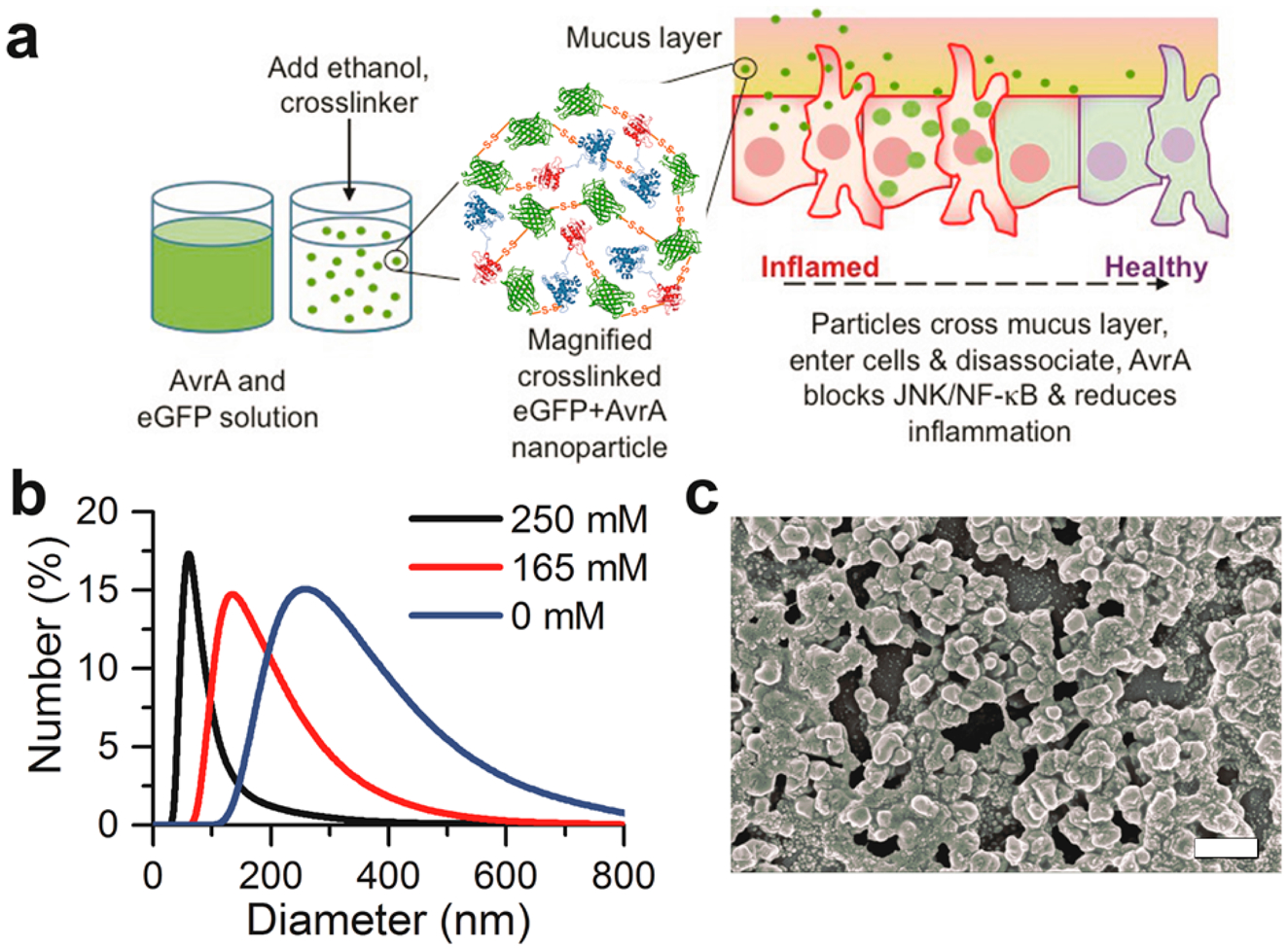
Synthesis and characterization of AvrA particles. (a) Schematic representation of eGFP-AvrA particle design, preparation, and hypothesized mode of action. Particles diffuse through the mucus and are internalized by resident cells. As the particles dissociate and release free AvrA, inflammatory function is replaced by healthy function due to AvrA activity. (b) AvrA-eGFP particle size distributions as a function of imidazole concentration during fabrication. (c) Scanning electron micrograph of 125 nm AvrA-eGFP nanoparticles used for experiments (scale bar 300 nm).
RESULTS AND DISCUSSION
Synthesis and Characterization of AvrA Nanoparticles.
The therapeutic approach described herein relies on the availability of active, soluble bacterial proteins and the ability to engineer the protein sequences for desired delivery properties. We cloned the genes of AvrA and a mutant form (mAvrA) into pGEX expression plasmids containing N-terminal glutathione S-transferase (GST) and C-terminal 6x-His tags using standard recombinant techniques to produce soluble AvrA-GST and mAvrA-GST fusion proteins. The mutant form contains a single cysteine substitution (C186A) that renders the acetyltransferase inactive and eliminates JNK inhibition and attenuates NF-κB suppressive activity.10 We expressed AvrA fusion proteins and a carrier protein, enhanced green fluorescent protein (eGFP), in E. coli and purified them (Figure S1).
We fabricated protein nanoparticles by desolvating a solution of AvrA and eGFP protein by constant addition of ethanol while stirring (Figure 1a).32 The resulting particles were cross-linked with reducible 3,3′-dithiobis-[sulfosuccinimidylpropionate] (DTSSP) to stabilize them during delivery (Figure S2). DTSSP contains a central disulfide bond that is sensitive to intracellular reducing conditions.33 By varying imidazole concentration during synthesis, we produced spherical particles with diameters of 125 ± 25 nm and ζ-potential of −11.3 ± 0.1 mV in phosphate buffered saline (PBS) and −24.3 ± 1.1 mV in 10 mM HEPES buffer (Figure 1b,c). We selected this size in order to achieve mucosal barrier penetration and cellular internalization. Previous work has shown that the size, hydrophobicity, and charge of particles play an important role in the transport of nanoparticles in mucosa.16
In order to penetrate mucus, nanoparticles must avoid adhesion to mucin fibers and be small enough to prevent steric hindrance. AvrA-eGFP nanoparticles exhibit slightly negative ζ-potential in the presence of physiological ion concentrations, which can prevent electrostatic interactions with negatively charged mucin fibers and thus decrease adhesion to mucus. The size of AvrA-eGFP nanoparticles is within the range of the interfiber spacing of mucin to allow the particles to diffuse through the mucus.16
Though smaller particles may have better delivery properties, they also contain less AvrA than large particles. The nanoparticles contained approximately 316 AvrA molecules per particle. Real-time imaging of a Salmonella model infection has shown that a different TTSS-secreted effector, SipA, mediates biochemical functions within minutes of infection at a concentration of 1000 molecules/cell.34 Particles were fabricated from combinations of AvrA-GST or mAvrA-GST and eGFP to create fluorescent particles that could be visualized in specific experiments (Figure S3). We substituted bovine serum albumin (BSA) for eGFP in particle formulations that required nonfluorescent particles but similar properties (99.7 ± 23.1 nm in diameter, ζ-potential of −16.9 ± 0.9 mV in PBS).
Uptake and Disassociation of AvrA Nanoparticles.
We sought to take advantage of endocytic uptake mechanisms of nanoparticles containing AvrA because the Salmonella TTSS is not feasible for AvrA delivery. We confirmed uptake of AvrA-eGFP nanoparticles by cultured J774A.1 macrophages or T84 polarized epithelial cells using confocal microscopy (Figure 2a,b). T84 cells are a highly differentiated epithelial cell line that can recapitulate the barrier and uptake properties of the native epithelial monolayer.35 J774A.1 macrophages serve as a model phagocytic cell and expectedly showed more eGFP fluorescence.36,37 In the nanoparticle fabrication process, eGFP and AvrA are co-desolvated and cross-linked together. Internalized eGFP suggests that AvrA is also co-delivered and internalized. The punctate spots seen in midcell optical sections clearly show delivery of nanoparticles, whereas the lack of green fluorescence in the soluble eGFP images indicates very low uptake of soluble protein.
Figure 2.
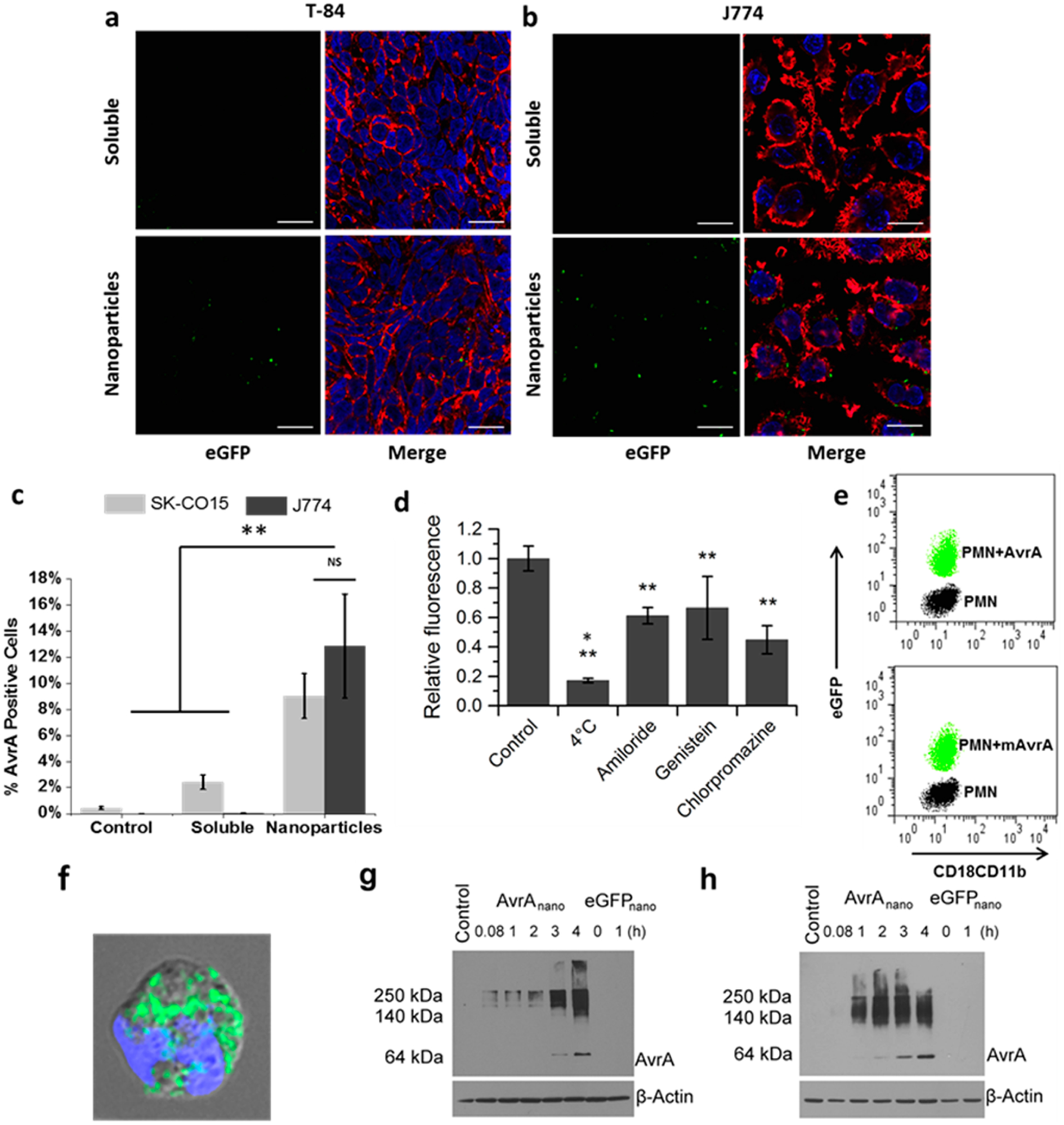
Cellular uptake of AvrA particles. Confocal images of (a) T84 and (b) J774A.1 cells incubated with mixed soluble AvrA (15 μg/mL) and eGFP (300 μg/mL), or AvrA-eGFP nanoparticles for 6 h. Images are midcell optical section overlays of eGFP fluorescence (green), nuclear Hoechst dye (blue), and actin filaments (red) labeled with rhodamine-phalloidin (scale bars 20 μm). (c) Flow cytometry quantification of soluble AvrA (15 μg/mL) and eGFP (300 μg/mL) or AvrA-eGFP nanoparticle uptake in SK-CO15 cells (light gray) and J774A.1 cells (dark gray). (d) Comparison of AvrA-eGFP nanoparticle (315 μg/mL) uptake by J774A.1 cells following pretreatment with the indicated drug. A single asterisk indicates statistical significance to all other groups; double asterisks indicate statistical significance compared only to untreated control. (e) Flow cytometry characterization of PMNs exposed to AvrA-eGFP and mAvrA-eGFP nanoparticles (500 μg/mL eGFP, 25 μg/mL (m)AvrA). eGFP represents uptake and CD18/CD11b represents activation. (f) Fluorescent/bright-field overlay of PMN uptake of AvrA-eGFP nanoparticles (scale bar 2 μm). (g) IEC-6 model intestinal epithelia and (h) IC-21 peritoneal macrophages were treated with nanoparticles for indicated times and detected by immunoblotting with anti-AvrA antibody. (*p < 0.05, **p < 0.01).
Next, we used flow cytometry with anti-AvrA antibodies to quantify uptake of specific AvrA immunoreactivity, as well as eGFP fluorescence. At 6 h, 420 times more J774A.1 cells were positive for AvrA and eGFP when treated with nanoparticle AvrA-eGFP in comparison to cells treated with soluble AvrA and eGFP (Figure 2c). SK-CO15 epithelial cells also internalized more nanoparticle AvrA-eGFP than soluble AvrA and eGFP in 6 h. Though only ~10% of J77A.1 or SK-CO15 cells labeled positive for AvrA, significantly more J774A.1 cells labeled positive for eGFP (Figure S4). This could indicate that in macrophages the nanoparticles are not completely disassociated and the antibody is not able to access all AvrA still in the nanoparticles as the eGFP signal is not dependent on particle dissociation. However, as described in the Methods, the dose used for the uptake studies was larger than the dose for in vitro activity studies in order to have sufficient signal for imaging. The low signal also necessitated the use of a quantum-dot-labeled secondary antibody to detect AvrA. This indicates AvrA is quite potent, and we can detect functional activity at lower concentrations than we can detect the “physical presence” of AvrA in cells by fluorescence. Interestingly, eGFP and AvrA-eGFP particles had the same uptake in SK-CO15 epithelial cell line, but in J774A.1 macrophages, AvrA-eGFP particles were taken up much more than eGFP only particles (Figure S4). This is surprising given that the particles with or without AvrA have the same size and ζ-potential and that AvrA is a small fraction of the total protein in a particle (4% by mass). These data indicate that AvrA may play a role in uptake of the particles, though it seems to be cell-type-specific. YopJ, a close ortholog of AvrA, is another TTSS effector from Yersinia that shares sequence similarities with AvrA.38 YopJ is an anti-inflammatory and pro-apoptotic effector that is known to be cell-selective; it induces apoptosis in macrophages and dendritic cells but not in endothelial cells or neutrophils.39,40 AvrA could similarly exhibit some manner of specificity. AvrA has been shown to be differentially expressed depending on the organ location of the Salmonella infection,41 also suggesting potential specificity. Additionally, as a pathogenic protein, AvrA could contain pathogen-associated molecular patterns (PAMPs) able to be recognized by macrophage pathogen recognition receptors (PRRs). Engagement of PRRs by any AvrA present on the surface of the nanoparticle can activate macrophages and increase endocytosis, leading to the difference in uptake seen between AvrA-eGFP and eGFP-only nanoparticle. SK-CO15 cells are epithelial cells lacking PRRs.
The route of nanoparticle uptake in J774A.1 cells was investigated by flow cytometry using the eGFP signal in the presence of endocytosis inhibitors (Figure 2d). J774A.1 cells utilize primarily energy-dependent routes, as indicated by low uptake at 4 °C. There was no strong preference toward a particular route, as inhibitors for macropinocytosis (amiloride), caveolae-mediated endocytosis (genistein), and clathrin-mediated endocytosis (chlorpromazine) all reduced nanoparticle uptake. Colocalization studies in J774A.1 cells of nanoparticles with lysosomal marker anti-Lamp1 confirmed the route of uptake to be endosomal in nature and showed that the majority of nanoparticles are found in lysosomes after 6 h (Figure S5). However, the dose of nanoparticles required for visualization was much higher than that required to detect AvrA activity, as described below. Importantly, incubation of J774A.1 cells with nanoparticles does not have any cytotoxic effect (Figure S6). Next, we exposed human polymorphonuclear leukocytes (PMNs) to AvrA-eGFP nanoparticles for 30 min ex vivo. We used flow cytometry to assess nanoparticle uptake by eGFP fluorescence and PMN activation by labeling for CD18/CD11b, which are integrins that are upregulated during activation and recruitment of leukocytes to sites of inflammation. Both AvrA-eGFP and mAvrA-eGFP nanoparticles were clearly internalized by neutrophils, and there was no increase in CD18/CD11b expression (Figure 2e,f).
Next, we assessed AvrA-eGFP particle disassociation in vitro in cultured epithelial monolayers. Nanoparticles were incubated with IEC-6 monolayers for 5 min to 4 h, washed, lysed with SDS buffer containing DTT, and analyzed by Western blot with anti-AvrA antibodies following separation by denaturing SDS-PAGE in a 10% gel (Figure 2g). The high molecular weight AvrA immunoreactivity is indicative of nanoparticle fragments or protein aggregates as intact nanoparticles do not enter the gel.32 The signal is weak at early time points and contains only high molecular weight fragments. However, by 3 h incubation, we detected the expected MW (61 kDa) of soluble AvrA-GST, representing the intracellular dissociation of AvrA from the nanoparticles. Because nanoparticles are provided throughout the duration of the experiment, both the high molecular weight and soluble AvrA bands increase in intensity over time. We obtained similar data with uptake into IC-21 macrophages, though as expected in this phagocytic cell type, uptake and dissociation were greater and more rapid (Figure 2h). Macrophage uptake appeared to saturate, as seen by the constant high molecular weight band at later time points, but nanoparticle disassembly did not saturate and the amount of soluble AvrA continued to increase. This indicates that the reducing and denaturing conditions of lysis and SDS-PAGE were not causing nanoparticle breakup.
Though colocalization studies with anti-Lamp1 indicated the majority of AvrA-eGFP nanoparticles are endocytosed and traffic to lysosomes after 6 h (Figure S5), the detection of single AvrA-GST protein in cell lysates (Figure 2g,h) and the functional AvrA activity described below demonstrate that some AvrA protein does reach the cytosol. Though it is difficult to pinpoint endosomal escape mechanisms for most nanoparticles, there are several features of AvrA particles that could contribute to their escape. One contribution could be from osmotic pressure changes that may occur as the cross-links reduce and particles break up into soluble protein. Reducible polyarginine DNA nanocarriers have been shown to have higher transfection efficiency than those that are not reducible or when disulfide reduction was inhibited.42 Another possible mechanism is membrane destabilization due to cationic interactions and an osmotic buffering effect by protonation of the 6x-histidine tags on eGFP and AvrA-GST.43 It is also possible that AvrA itself could have endolytic properties, as there are a variety of bacterial pathogens that produce toxins or effectors that contain domains that assist in endosomal escape by different mechanisms.44 Hemolysis assays showed that pure, soluble AvrA-GST was significantly lytic with increasing concentration from 10 to 1000 μg/mL, and at higher concentrations, lysis was increased at neutral pH compared to acidic conditions (Figure S7). Considering a single particle in a 200 nm endosome, the AvrA concentration is estimated to be higher than 1000 μg/mL. This suggests AvrA may have endosomal escape properties prior to acidification. However, when the hemolytic activity of mixtures of soluble eGFP and AvrA was assessed, as would be found from a disassembled particle, the lytic activity was completely abolished (Figure S8). This suggests that soluble eGFP interferes with any lytic property of AvrA. Interestingly, intact AvrA-eGFP nanoparticles did exhibit some lytic activity, at neutral and slightly acidic pH. Nanoparticles made only of eGFP or mAvrA-eGFP showed significantly less, but nonzero, lytic activity at neutral and slightly acidic pH. These data suggest that intact AvrA-eGFP nanoparticles could contribute to destabilization of endosomal membranes at early stages of acidification. It also suggests more generally that AvrA in AvrA-eGFP nanoparticles has some interaction with cell membranes, in agreement with the observation in Figure S4 that AvrA-eGFP nanoparticles are internalized by macrophages more than eGFP nanoparticles.
To detect particle uptake in vivo, we used direct transrectal instillation of AvrA-eGFP particles into both intact and damaged/inflamed murine colons. In tissues evaluated postinstillation, eGFP was detected in the extracellular mucus layer of the epithelial cells by anti-eGFP (Figure S9). Intracellular eGFP uptake was seen within epithelial cells by 4 h in apical cells and in the base of crypts (Figure 3a,b). eGFP positive cells were seen in the lamina propria (Figure 3c) and in F4/80 positive macrophages (Figure 3d). Nanoparticle uptake in healthy tissue indicates that nanoparticles are able to penetrate healthy, fully intact mucus and could potentially be used as a protective agent to prevent the spread of inflammation. This uptake into cells was also diffusively distributed in inflamed colons when we induced injury with a seven-day pretreatment of 2% DSS in drinking water (Figure S10). We were unable to detect AvrA immunoreactivity in vivo, likely due to the small amount of AvrA taken up. Based on the colocalization of eGFP and anti-AvrA signals from in vitro flow cytometry nanoparticle uptake experiments, we conclude from the anti-eGFP fluorescence that AvrA-eGFP nanoparticles are taken up and AvrA is present with eGFP. The in vivo images indicate that these particles effectively transverse the mucosal barrier and reach the underlying epithelia, as well as lamina propria monocytes.
Figure 3.
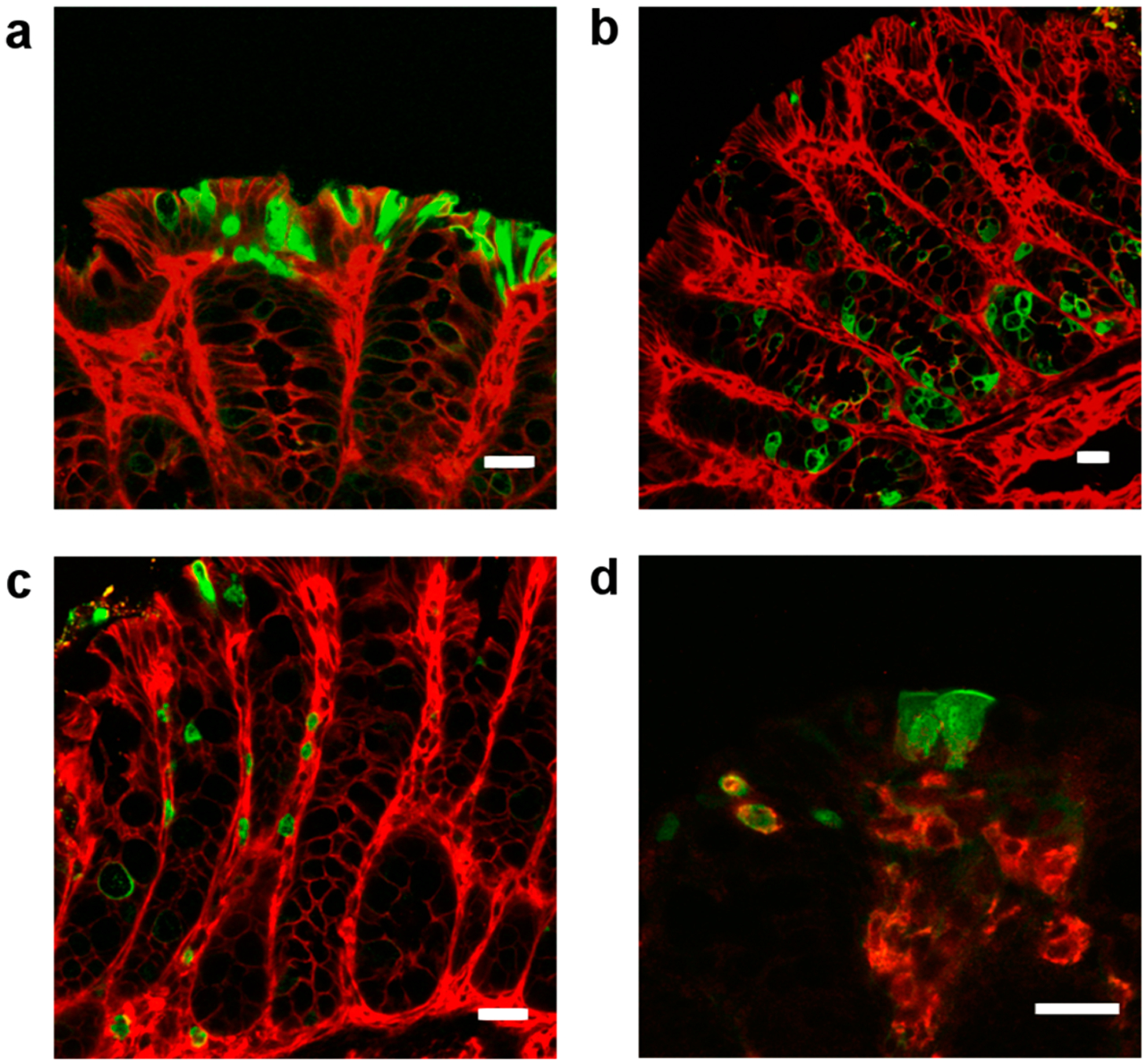
Mucosal uptake of AvrA-eGFP (12 μg eGFP) particles in healthy murine colonic surface epithelium (a), crypts (b), lamina propria (c), and macrophages (F4/80+) (d) imaged by confocal microscopy (scale bars 20 μm). Particles were instilled transrectally and imaged after 4 h. Particle uptake is marked by anti-eGFP fluorescence (green). Epithelial cells are counterstained with beta-catenin (red in (a–c)) and macrophages stained with F4/80 (red in (d), yellow indicates colocalization of eGFP and F4/80+).
Inhibition of Inflammatory Signaling by AvrA Nanoparticles.
AvrA has been shown to inhibit JNK phosphorylation and IκB degradation by transfection and transgenic approaches.10,11 Experiments with AvrA-eGFP nanoparticle preparations applied to the apical surface of polarized T84 monolayers for 3 h, to allow particle internalization and dissociation, were successful in partly suppressing both TNF-α-induced JNK activation and Iκβ degradation (Figure 4a). AvrA nanoparticles show stabilization of Ikβα after 30 min with levels reaching the prestimulation levels by 60 min. AvrA is hypothesized to act on a distal event in the NF-kB pathway downstream of Iκβα.10 Though Iκβα still is phosphorylated, this does not lead to degradation of Iκβα, thereby preventing NF-kB release and transcription of inflammatory signals. For this reason, levels of P-Iκβα increase over time in the presence of TNF-α and AvrA nanoparticles as Iκβα accumulates due to reduced degradation. JNK inhibition by AvrA nanoparticles was observed to be dose dependent (Figure 4b) and mAvrA-eGFP nanoparticles, eGFP nanoparticles, and soluble AvrA did not inhibit JNK (Figure 4c). The ability of AvrA nanoparticles to suppress NF-κB activity was confirmed by a luciferase gene reporter assay (Figure 4d). Partial suppression of NF-κB activity was observed with mAvrA nanoparticles. AvrA, has been shown to also exhibit deubiquitinase activity in the NF-κB pathway that is not entirely eliminated by the C186A mutation.45 T84 monolayers incubated with AvrA nanoparticles and stimulated with TNF-α were also assayed for IL-8 secretion at 6 h by ELISA (Figure 4e). Secreted IL-8 was markedly reduced in particle treated cells. In human neutrophils ex vivo, AvrA-eGFP nanoparticles, but not mAvrA-eGFP nanoparticles, inhibited transmigration across cultured T84 monolayers, a critical inflammatory behavior (Figure 4f). Together with the uptake experiments, it is apparent that particles internalized by cells dissociate over time and release functional AvrA. After at least 3 h is there evidence of soluble AvrA release and subsequent bioactivity. It is not yet clear what fraction of AvrA delivered is responsible for the bioactivity seen.
Figure 4.
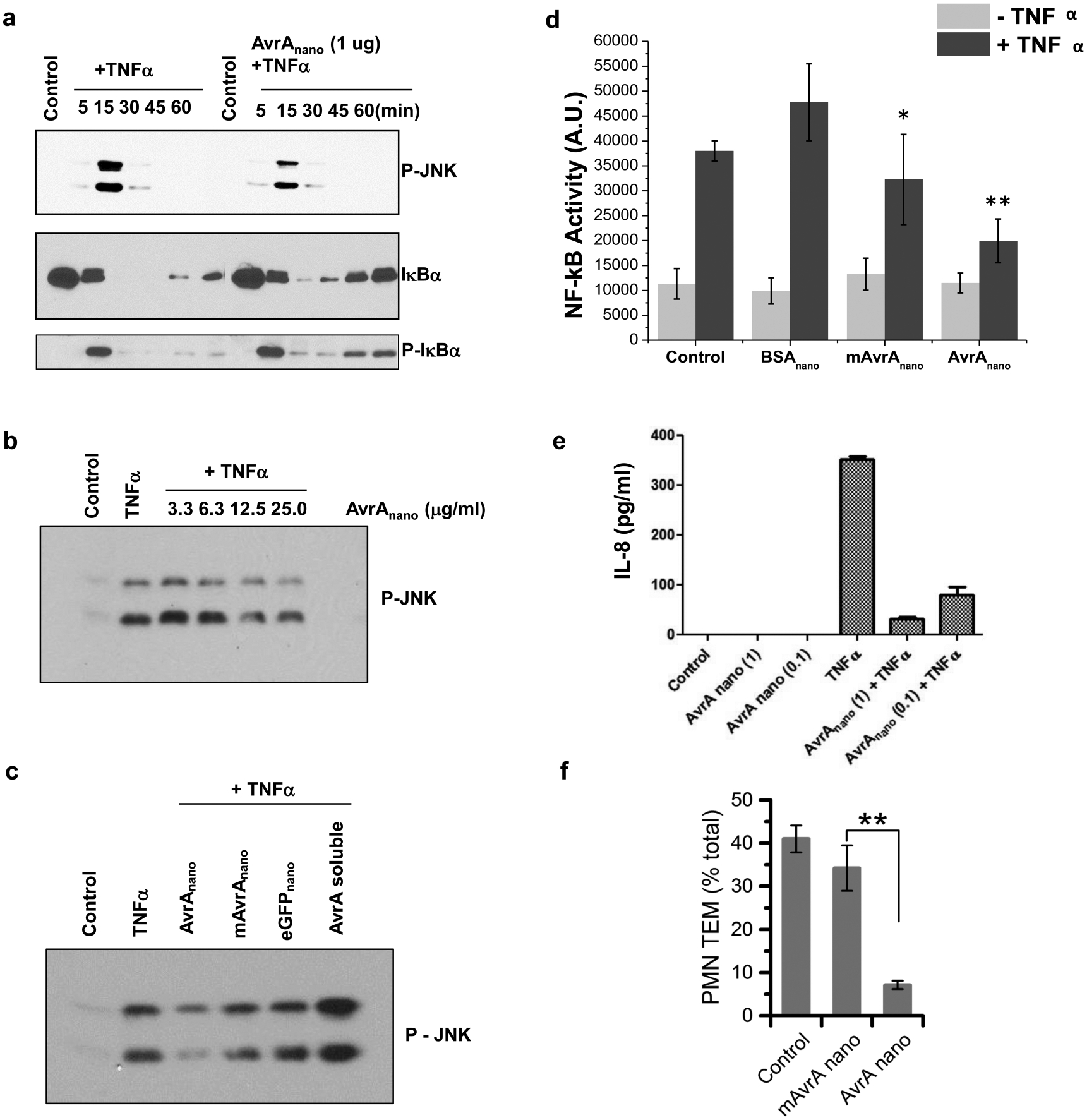
AvrA nanoparticles inhibit inflammatory signaling. AvrA nanoparticles were applied for 4 h prior to TNF- α stimulation for data shown in (a)–(d), (f). Western blots with indicated antisera showing (a) TNF- α time course; (b) AvrA nanoparticle dose response after TNF- α stimulation for 15 min; (c) AvrA nanoparticles compared to soluble AvrA, mAvrA nanoparticle and carrier nanoparticle controls. (d) Nanoparticles made from 30 μg/mL AvrA or mAvrA and 1500 μg/mL BSA were incubated with HeLa cells transfected with Luciferase NF-κB reporter gene 4 h prior to 1 h stimulation with 20 μg/mL recombinant human TNF-α (*p < 0.05, **p < 0.01 compared with + TNF- α control samples). (e) IL-8 production in polarized T84 monolayers with AvrA nanoparticles applied 3 h prior to TNF-α stimulation. (f) Transepithelial migration of PMNs following exposure to AvrA and mAvrA nanoparticles. **p < 0.01.
To demonstrate anti-inflammatory activity in vivo, we first employed a traditional murine peritonitis model of acute inflammation46 before moving into a more complex colitis model. Mice were pretreated with an intraperitoneal (IP) dose of active AvrA-BSA nanoparticles, mAvrA-BSA particles, inert BSA particles or PBS control 1 h before IP instillation of 10 mg zymosan, a fungal cell wall component commonly used as an inducer of acute inflammation. After four additional hours, mice were sacrificed and inflammatory exudates in the peritoneal cavity collected by lavage. Neutrophils were marked with anti-Ly6G antibodies and populations quantified by flow cytometry (Figure 5a). Essentially no inflammatory cells were detected in control (PBS pretreated, no zymosan) mice, while positive control (PBS pretreated, zymosan) mice showed the expected accumulation of neutrophils. Pretreatment with AvrA nanoparticles resulted in a significant reduction of neutrophil influx into the peritoneal cavity. Inert BSA particles and mutant AvrA particles showed no significant effect. The ability of AvrA nanoparticles to prevent neutrophil infiltration in the peritonitis model could transfer to prevention of neutrophil infiltration in the gastrointestinal tract. We expect this anti-inflammatory activity to be beneficial in colitis models because immune cells are the primary contributors to and perpetrators of the chronic inflammatory state seen in IBD.47
Figure 5.
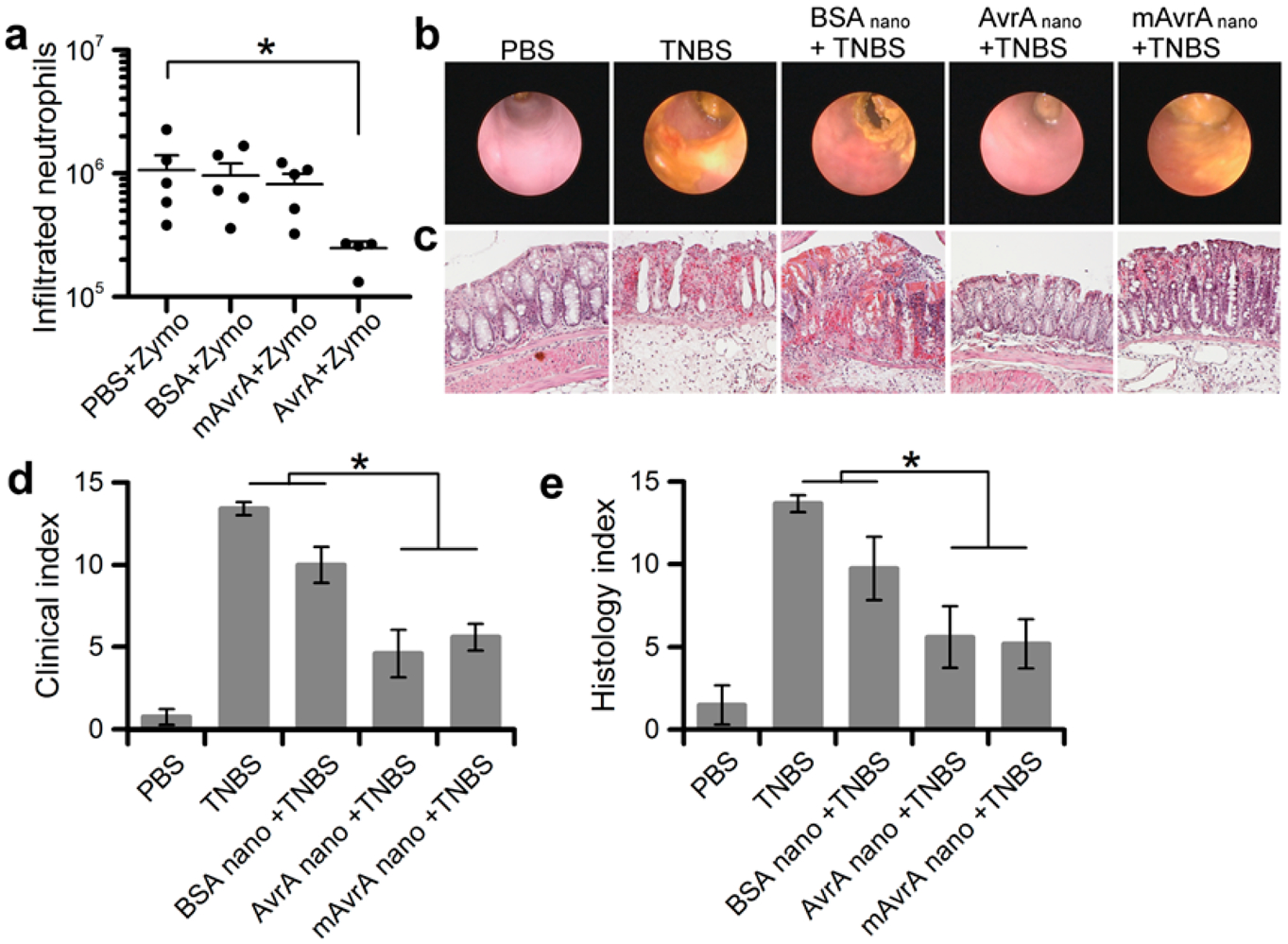
Anti-inflammatory activity of AvrA nanoparticles in murine inflammation. (a) Zymosan-induced peritonitis: quantification of neutrophils isolated by peritoneal lavage from mice (5 per indicated condition) injected peritoneally with 540 ng AvrA 1 h prior to zymosan injection. Line represents average number of neutrophils. Twenty-four hour TNBS-induced colitis models in mice: (b) representative microendoscopic images of colon and (c) representative colon histology from mice treated transrectally as indicated with 540 ng of AvrA 4 h prior to TNBS injection. (d) Quantification of clinical and endoscopic score from 5 mice per indicated condition. (e) Quantification of histological score from 5 mice per indicated condition (*p < 0.05, bars over 2 groups with no * are not statistically different).
Next, we tested AvrA particles in in vivo models of colitis. TNBS-colitis is a rapid local transmural colitis model induced by rectal administration of the hapten reagent 2,4,6-rinitrobenzenesulfonic acid (TNBS) and is associated with a Th1 T cell response.48 Inflammatory responses can be observed within 24 h. Mice were instilled luminally (transrectally) with particles 4 h prior to TNBS administration to assess if the particles could modify this colitis model. At 48 h, TNBS results in marked colitis as measured by clinical parameters of vascular pattern, size of the ulceration, stool consistency, and rectum stenosis using a small animal veterinary endoscope and standard histological indices. Remarkably, these parameters were significantly improved in mice pretreated with AvrA particles, while control BSA particles had no effect (Figure 5b–e). Interestingly, mutant AvrA particles also showed significant activity, suggesting the therapeutic enzymatic activity of AvrA in this model is not abolished with a single mutation in the acetyltransferase active site. Mutant AvrA partial activity is reported in literature and seen in the in vitro activity data (Figure 4d), possibly because the deubiquitinase activity in the NF-κB pathway is not entirely eliminated by the C186A mutation.45 Indeed, many, if not most TTSS effectors are bi- or multi-modular proteins with multiple catalytic functions,49,50 and other activities in AvrA may account for partial activity seen in vivo. A second active site, an SH2-like domain, is also present in AvrA and Yersinia ortholog YopJ.49 Mutation of this D/E-X-E active site reduces YopJ anti-inflammatory function, and is expected to have a similar function in AvrA. This domain should be unaffected by the C186A mutant used in these studies and so its function would remain intact. We have also seen partial function of nanoparticles made from the same mutant of YopJ, in agreement with multiple domains responsible for bioactivity.51
Dextran sodium sulfate (DSS) colitis is another in vivo model where the chemical irritant, supplied in drinking water, results in epithelial erosions and subsequent inflammation in the colon within 4–5 days. With this model, we tested nanoparticles both as prophylaxis, administered at 1 day prior and 1 day post initiation of DSS (Figures 6a–d), and as a therapeutic design, administered 7 days post initiation of DSS after clinical symptoms have become manifest (Figures 6e,f). In both experimental designs, suppression of clinical indices and scores of colonic inflammation were observed. From these experiments we conclude that sufficient quantities of bioactive AvrA are delivered to colonic tissue to suppress acute inflammatory events in distinct colitis models.
Figure 6.
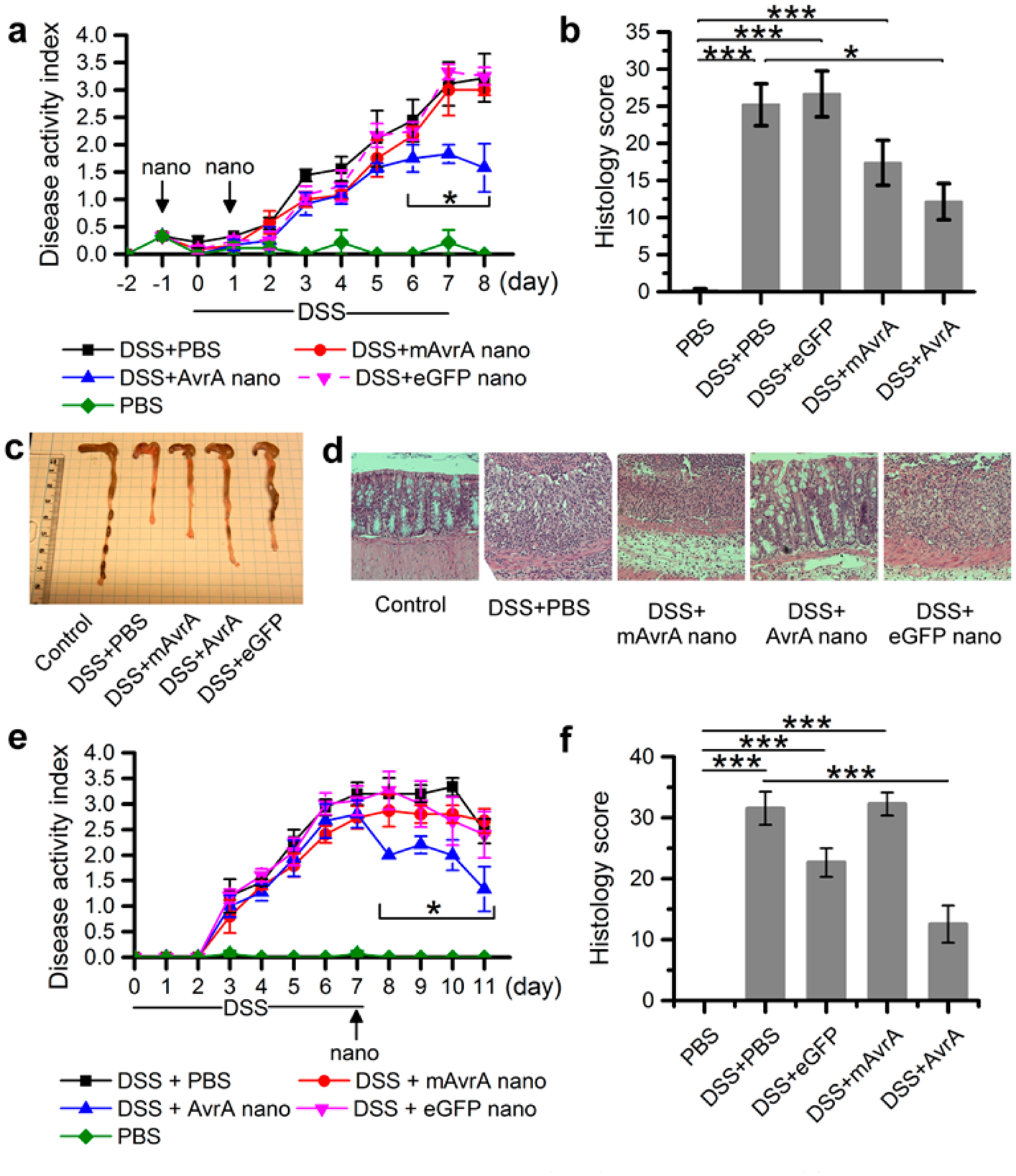
Anti-inflammatory effect of AvrA on dextran sodium sulfate (DSS)-induced colitis. (a) Clinical disease activity index and (b) histological scoring of 5 mice per indicated condition receiving two injections (intrarectal) of nanoparticles (900 ng AvrA per injection) while subjected to DSS challenge for 7 days. (c) Representative colon gross pathology from mice treated as indicated in (a). (d) Representative colon histology from mouse treated as indicated in (a). (e) Clinical disease activity index and (f) histological scoring of 5 mice per indicated condition treated once with nanoparticles (900 ng AvrA) after DSS-induced colitis (*p < 0.05).
IBD results from aberrant mucosal immune activation, often in the context of genetic susceptibility, resulting in the influx of acute and chronic inflammatory cells into the mucosa. Both the overlying epithelial monolayer and the underlying immune cells of the lamina propria possess the ability to initiate inflammatory reactions during injury.52 In our murine experiments, restricted to transrectal delivery of nanoparticles into the distal colon, both surface epithelial and monocytic cells in the lamina propria took up nanoparticle formulations of AvrA under control and colitic conditions. AvrA particles potently suppressed histologic inflammation and clinical injury in several commonly used in vivo models of colitis. It is widely known that there is not a perfect model of IBD. In our studies, we utilized chemical models of acute/self-limited inflammation with DSS and TNBS that occur even in the absence of a functional adaptive immune system. Thus, our data demonstrates an inflammatory suppressive role outside of adaptive regulatory mechanisms. We speculate that use of AvrA nanoparticles in the treatment of a chronic model of IBD, such as IL-10 knockout53 or T-bet and RAG2 double knockout,54 would lead to similar results of suppression of inflammation markers and disease activity scores because the JNK and NF-κB targets of AvrA are highly conserved inflammatory signaling molecules. Though complications arising from tissue restructuring, such as neoplasia and fibrosis, would likely not be reversed by AvrA nanoparticles, further damage could be prevented and a reversal of symptoms may be observed. To make these nanoparticles clinically relevant, oral delivery is necessary. Future work will entail encapsulation of nanoparticles and oral delivery that will allow AvrA-eGFP nanoparticles access to Peyer’s patches and other components of the gut associated lymphoid tissue (GALT) resident in the distal ileum. It will be interesting to determine if such therapeutic strategies affect adaptive immunity.
The protein nanoparticle platform is highly adaptable to a variety of biological molecules and chemistries. Nanoparticle formation, stabilization, and targeting ligands can be independently modified to suit the biological delivery requirements of different diseased tissues and routes of administration. For example, AvrA nanoparticles could also be used in other models of inflammation that do not require systemic access, such as in the joints, airways, skin, or eyes. In this work both BSA and eGFP were used as carriers for AvrA and in either case, AvrA was successfully delivered and retained its function. The versatility of the protein nanoparticle would allow use of recombinant human serum albumin (HSA) as a carrier in future human clinical applications as it is more physiologically benign and FDA approved. While AvrA was chosen as a model immunosuppressive protein for construction into nanoparticles, this approach could be applied to other proteins. Orthologous acetyltransferases (YopJ, VopA, AopP) have been detected in a variety of bacteria that associate intimately with eukaryotic hosts, and these effectors exhibit extended activity against a wider spectrum of MAPKs. Our lab and others have been studying these related effectors that have variable effects on MAPK and are more potently immune suppressive, but can also be potently pro-apoptotic.55,56 The many other classes of bacterial effectors demonstrate a potentially vast repository of biochemical activities relevant to manipulation of eukaryotic inflammatory signaling, and exploitation of bacterial virulence proteins in yeast and mammalian immune cells has been explored as a synthetic biology approach.57 However, this method is not limited to bacterial proteins or single types of proteins. Combination therapies of bacterial proteins and human cytokines or small molecule anti-inflammatory agents, for example, may have value in future immunological therapeutics.
CONCLUSIONS
Enteric bacteria have coevolved with humans to develop specific effector proteins capable of immunomodulation. We engineered one such protein, AvrA, to form protein nanoparticles, enabling intracellular delivery of AvrA in the absence of Salmonella. AvrA nanoparticles inhibited inflammatory pathways in vitro and reduced inflammation in murine colitis models, indicating their potential as a treatment for IBD. Future work could expand the protein nanoparticle platform to other bacterial proteins for development of effective therapeutics to combat chronic inflammatory disease.
METHODS
Cells.
J774A1 cells were purchased from American Type Culture Collection and cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% (v/v) fetal bovine serum (FBS). SK-CO15 epithelial cells were cultured in DMEM with 10% FBS and 1% nonessential amino acids. IEC 6 cells and T84 cells were cultured as described previously.58 Briefly, primary rat intestinal IEC 6 epithelial cells were maintained in DMEM (4.5 g of glucose/L) supplemented with 10% FBS and 4 μg/mL insulin (Invitrogen). T84 model human intestinal epithelial cells were prepared on 0.33 cm2 permeable filters and cultured in DMEM/F12 supplemented with 5% FBS. T84 cells were used after they had achieved a stable transepithelial resistance of >1000 Ω·cm2. Mouse macrophage IC21 cells were maintained in RPMI-1640 medium with 10% FBS. All media were supplemented with 1% penicillin/streptomycin, and cells were incubated in a 5% CO2 humidified air atmosphere.
Animals.
All experimental protocols used C57BL/6 (WT) mice (The Jackson Laboratory). All procedures using animals were reviewed and approved by the Emory University Institutional Animal Care and Use Committee and were performed according to National Institutes of Health criteria.
Genes.
AvrA and mAvrA genes were amplified by PCR using the primers 5′-TCATGAATTCCCATGATATTTTCGGTGCAGGAGCTATCATG-3′ and 5′-ATGAGTCGACTTAATGATGATGATGATGATGCGGTTTAAGTAAAGACTTATATTCAGCTATCCT-3′ and inserted into the bacterial expression plasmid pGEX-4T-2 (GE Lifesciences) between SalI and EcoRI sites. Since pGEX-4T-2 does not encode a 6xHis tag, the reverse primer was designed to introduce it. Constructs were confirmed by DNA sequencing. eGFP gene in a pPROTet plasmid (Clontech Laboratories) was a kind gift of Dr. Andreas Bommarius.
Production of Recombinant Proteins.
Protein expression was performed in BL21 Escherichia coli. Bacterial cultures were grown to o.d. 0.7 at 37 °C and induced with 0.4 mM isopropyl β-d-thiogalactoside (IPTG) (only for AvrA fusions) at 25 °C for 4 h. AvrA-GST, mAvrA-GST, and eGFP proteins were purified on Ni-NTA agarose (Qiagen) following manufacturer’s native purification protocols in imidazole-containing buffers. Purified proteins were buffer exchanged using 10k MWCO centrifugal ultrafiltration devices (Millipore) into PBS buffer (10 mM NaH2PO4, 137 mM NaCl, 2.7 mM KCl, 2 mM KH2PO4; pH 7.4).
Nanoparticle Production.
Protein particles were prepared by the desolvation technique as previously described.28 In brief, 600 μg of eGFP and ~5 μg of AvrA-GST or mAvrA-GST in 100 μL imidazole solution (250 mM imidazole, 300 mM NaCl, 50 mM Na2PO4 pH 8) were placed in a glass vial. The protein solution was desolvated by continuous, drop-by-drop addition of 400 μL ethanol at a rate of 1 mL/min. After desolvation, particles were cross-linked with 2 mg/mL dithiobis(sulfosuccinimidylpropionate) (DTSSP; Pierce) at a ratio of cross-linker to lysines of 1:2.2. After being stirred for 2 h, the cross-linking reaction was stopped by centrifugation at 1000g for 1 min and supernatant removed. Particles were resuspended in PBS and sonicated on ice for 1 s every 15 s at 30% amplitude for a total of 5 min.
Nanoparticle Characterization.
Particle size distribution was measured by dynamic light scattering using a Zetasizer Nano ZS90 (Malvern Instruments Ltd.). All samples were measured at 25 °C and a scattering angle of 90°. Average particle size was calculated as the arithmetic mean of the distribution of at least 3 batches of particles and the standard deviation was calculated as the variance between average diameters of the batches. The ζ-potential was determined by measuring the electrophoretic mobility of the nanoparticles in PBS and 10 mM Hepes buffer using the same instrument.
Particles were prepared for scanning electron microscopy (SEM) imaging by further cross-linking the particles with glutaraldehyde after fabrication. A 5 μL droplet of these nanoparticles suspended in water was placed on an SEM specimen stub. The droplet was frozen in liquid nitrogen vapor and lyophilized for 24 h. Samples were coated with gold/palladium and imaged at 3 kV using a Zeiss Ultra60 FE-SEM.
Confocal Microscopy.
J774.A1 cells were seeded at a density of 104 cells per well in an 8-well chamber slide system (Nunc LabTek II, Thermo Scientific) with growth medium. After 14 h, cells were incubated for 6 h with fresh cell media containing 300 μg/mL soluble AvrA-GST and eGFP or AvrA-eGFP nanoparticles. Cells were washed twice with ice cold PBS and fixed with 3.7% paraformaldehyde for 15 min at room temperature. Cells were permeabilized with 1% Triton X-100 in PBS for 15 min at room temperature and rinsed three times in PBS. Cells were incubated with 2 μM Hoechst 33342 (AnaSpec Inc.) and 0.165 μM rhodamine phalloidin (Biotium) in blocking buffer for 20 min at room temperature. Cells were washed three times with PBS and mounted for imaging in a Zeiss LSM 700 confocal microscope.
T84 cells were cultured in transwell chambers in a 24 well plate. Soluble AvrA-GST and eGFP or AvrA-eGFP nanoparticles were administered to cells and cells were prepared as described for J774A1 cells. Transwell membranes were cut using a razor blade and mounted onto a glass slide for imaging.
Detection of AvrA Nanoparticles in Cells.
J774A1 macrophages or SK-CO15 epithelial cells were plated at a density of 2 × 105 cells per well in a 24-well dish. After 14–16 h of incubation, cell medium was replaced with fresh media containing 300 μg/mL AvrA-eGFP nanoparticles or soluble AvrA-GST an eGFP and incubated for 6 h. Control cells were incubated with AvrA-eGFP nanoparticles or soluble AvrA-GST and eGFP at 4 °C. Cells were washed twice with ice cold PBS, scraped or trypsinized, and fixed with 3.7% paraformaldehyde for 15 min at room temperature. Fixed cells were washed 3 times by centrifugation with ice cold PBS and permeabilized with 0.25% Triton X-100 in PBS for 15 min at room temperature. Cells were rinsed three times in PBS and incubated with purified anti-AvrA antibody in 6% BSA and 10% FBS in PBS for 1 h. After this, cells were washed 3 times with PBS and incubated with 20 nM Qdot 655 VIVID secondary antibody conjugate (Invitrogen) in 6% BSA in PBS for 1 h. Cells were washed 3 times with PBS and analyzed in an LSR II flow cytometer (Becton Dickinson and Company). Positive events were identified as cells with simultaneous increase in green fluorescence and quantum dot fluorescence.
Endocytosis Inhibition Assay.
J774.A1 macrophages were plated at a density of 2 × 104 cells per well in a 24-well dish. After 14–16 h of incubation, cell media was replaced with fresh media containing endocytosis inhibitors. Macropinocytosis was inhibited using 2 mM amiloride (MP Biomedicals, LLC). Caveolae-mediated endocytosis was inhibited using 300 μM genistein (TCI America). Clathrinmediated endocytosis was inhibited using 20 μg/mL chlorpromazine (Alfa Aesar). Energy-dependent endocytosis was inhibited by incubation at 4 °C (cells were also incubated at 4 °C for 1 h prior to nanoparticle introduction). Cell viability in the presence of all inhibitors was measured according to the methods described in the Supporting Information and was found to be at least 80% of untreated cell control.
After 1 h pretreatment with inhibitors, cell media was replaced with fresh inhibitor and AvrA-eGFP nanoparticles or positive controls (clathrin-mediated: transferrin-CF640R, caveolae-mediated: BSA-CF640R, macropinocytosis: 10000 MW Dextran-CF640R, all purchased from Biotium) suspended in PBS (50% v/v). Twenty-five μg/mL of positive control or 300 μg/mL nanoparticles were used. Cells were incubated for an additional 3 h, then washed twice with ice cold PBS, scraped, and resuspended in PBS. Cells were analyzed in an Accuri C6 flowcytometer and relative endocytosis was quantified as the ratio of mean fluorescence of the sample population to fluorescence of the control population (no particles given).
Intracellular Particle Degradation.
IEC6 epithelial cells and IC-21 peritoneal macrophages were seeded at a density of 8 × 104 cells per well in a 48-well plate and incubated overnight. Cells were treated with 40 μg/mL AvrA-eGFP or eGFP nanoparticles for 5 min to 4 h. Next, cell medium was removed, and cells were washed once with PBS. Cells were lysed with 1× SDS lysis buffer (62.5 mM Tris-Cl, pH 6.8, 10% glycerol, 2% SDS, 50 mM DTT, 0.1% bromophenol blue), and cell lysates were collected and immediately stored at −80 °C until analysis by Western blot. Lysates were loaded onto 10% SDS-PAGE gels and transferred onto nitrocellulose membranes and immunostained with anti-AvrA antibodies.
Enzyme-Linked Immunosorbent Assay (ELISA) for Cytokine IL-8.
Polarized T84 cells were pretreated with AvrA-eGFP nanoparticles for 3 h and culture medium was collected after recombinant human TNFα (R&D Systems) stimulation (20 ng/mL) for 5 h. IL-8 levels released by T84 cells were quantified using ELISA according to the manufacturer’s instructions (R&D Systems).
JNK and IκBα Western Blot.
For cell signal transduction inhibition experiments, HeLa cells were seeded in 24-well plates at 80% confluency and incubated overnight. AvrA-eGFP nanoparticles (0.835 μg/mL) or PBS were added to cells for 4 h followed by 20 ng/mL rhTNFα for 5, 15, 30, 45, and 60 min. For immunoblotting, cells were harvested at different time points, washed in PBS, and lysed in RIPA buffer (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) with a cocktail of complete protease inhibitors (Roche Applied Science). Sample protein concentration was determined by Bio-Rad protein assay. Lysates with equal protein concentration were separated by 10% SDS-PAGE at 100 V for 60 min. Proteins were transferred to nitrocellulose membranes and immunostained with anti-p-JNK or IκBα antibodies.
NF-kB Luciferase Assay.
HeLa cells were seeded at 6 × 106 cells/well in a 6-well plate and incubated overnight at 37 °C. To prepare the transfection agent, 2 μg of pGL4 plasmid (Promega) and 15 μL of lipofectamine (Life Technologies) were added to 300 μL of serum-free media (DMEM, ATCC) and incubated at room temperature for 5 min. The cell medium was aspirated, and the transfection solution was added directly on top of confluent cells for 1 min. Six milliliters of 1% FBS in DMEM was then added to the well. The transfection agent was incubated with the cells overnight at 37 °C. Cells were then trypsinized and plated at 50000 cells/well in a 96-well plate overnight in 1% FBS in DMEM. Cells were pretreated with nanoparticles (300 μg/mL) for 4 h and then stimulated with 20 ng/mL of recombinant human TNF-α (R&D Systems) for 1 h. Afterward, the medium was aspirated and 50 μL BrightGlo reagent (Promega) and 50 μL of serum-free medium was added to each well. Luminescence was measured in a BioTek Synergy 2 plate reader.
Ex Vivo PMN Activity Assays.
For PMN isolation, human blood was drawn and handled according to protocols for the protection of human subjects, as approved by the Emory University Hospital Institutional Review Board. PMN were isolated by density gradient centrifugation and were used in experiments within 2 h of isolation. Freshly isolated PMN were incubated with either AvrA-eGFP or mAvrA-eGFP nanoparticles for 30 min (37 °C). The uptake of nanoparticles tagged with eGFP fluorescence was examined by flow cytometry, using FACS Calibur and FlowJo software. To examine the activation state of neutrophils after nanoparticle uptake, neutrophils were stained for the activation marker CD11b/CD18. For microscopy studies, PMN incubated with nanoparticles were PFA fixed, permeabilized with 1% Triton 100 and incubated with nuclear stain, TO-PRO-3 iodide (Molecular Probes). All images were acquired on a LSM510 confocal microscope (Carl Zeiss, Thornwood, NY) with Plan-Neofluor 60× objective.
PMN transepithelial migration across human intestinal epithelial cells (T84) was examined in the presence of either AvrA-eGFP or mAvrA-eGFP nanoparticles (25 μg/mL) using a transwell setup. In these assays, nanoparticles were added to the upper chambers of transwells (basolateral side of the epithelium) immediately prior to the addition of PMNs. PMN migration in the physiologically relevant, basolateral-to-apical direction was induced with addition of N-formyl-methionine-leucine-phenylalanine (fMLF) (100 nM) to the bottom chamber and quantified by assaying for the PMN azurophilic granule protein MPO as previously described.59 Data are shown as percent migrated out of total PMN applied (1 × 106/well).
In Vivo Peritonitis and Colitis Models.
Care of experimental animals was performed in accordance with Emory University IACUC institutional guidelines. Zymosan-induced peritonitis was used to study acute polymorphonuclear leukocyte (PMN) response and infiltration in vivo.46 C57BL/6J female mice were treated intraperitoneally (IP) with nanoparticles containing 540 ng AvrA or vehicle (PBS). After 1 h, mice were injected IP with 10 μg zymosan (Sigma-Aldrich) to induce peritonitis. Four hours after zymosan administration, the animals were sacrificed by CO2 asphyxiation. The peritoneal lavage fluid was collected with 3 mL of cold PBS/EDTA solution. The total number of PMN in the lavage was labeled with antimouse Ly-6G (Gr-l) Ab (eBioscience) and assessed by flow cytometry in a LSR II flow cytometer (Beckman Coulter) using a known concentration of fluorescent beads (Molecular Probes) as a control.
Trinitrobenzenesulfonic acid (TNBS)-induced colitis was induced by exposure to TNBS.60 Briefly, mice were presensitized with 1% TNBS on a shaved 1.5 cm × 1.5 cm area on the back of the mouse between the shoulders. After 7 days, mice were fasted and nanoparticles containing 540 ng AvrA or vehicles (PBS) were injected intrarectally. Four hours later, mice were anesthetized with ketamine and xylazine and a 100 μL enema containing 2.5% TNBS was administered. After 24 h, colon images were taken by a high-resolution miniaturized colonoscopy system (Karl Stortz). Clinical analysis of TNBS-induced colitis was conducted in a blind experiment scoring from 0 to 3 the thickening of the colon, changes of the vascular pattern, size of the ulceration, stool consistency, and rectum stenosis. A clinical index was calculated as the sum of the individual scores.61 At the end of experiment, the mice were sacrificed and the colon was dissected out, cut open, and solid feces carefully removed. A swiss roll was made, put in a cassette, soaked in 10% formalin overnight, and sent to a pathological core facility for processing. Histological examination was carried out with hematoxylin–eosin staining in paraffin sections. Histology index was determined as the sum of the scores for inflammatory cell infiltration in colons, edema, and erosion; each ranging from 0 to 3.60
Dextran sulfate sodium (DSS) colitis was induced by giving 3% (wt/vol) DSS (36000–50000 MW; MP Biomedicals) in autoclaved Milli-Qwater as drinking water and allowing mice to drink ad libidum for 7 days. Nine hundred nanograms (AvrA dose) of eGFP+AvrA or eGFP+mAvrA nanoparticles or vehicles (PBS) was intrarectally injected at indicated times. Disease activity was monitored daily. Disease activity index was calculated as the sum of the scores of stool consistency (0: hard, 2: soft, 4: diarrhea), fecal occult blood using Hemoccult Sensa (Beckman Coulter) (0: negative, 2: positive, 4: macroscopic) and weight loss (0: <1%, 1: 1–5%, 2: 5–10%, 3: 10–20%, 4: >20%). Disease score was calculated as the average of these three parameters. At the end of experiment, the mice were sacrificed and the colon was dissected out, cut open, and solid feces carefully removed. A swiss roll was made, put in a cassette, soaked in 10% formalin overnight, and sent to a pathological core facility for processing. Histological examination was performed by two independent observers on hematoxylin–eosin slides of paraffin colon sections. Histology score was assessed for severity of inflammation (0: none, 1: slight, 2: moderate, 3: severe), PMN infiltration/HPF (0: <5, 1: 5–20, 2: 21–60, 3: 61 − 100, 4: >100), depth of injury (0: none, 1: mucosa, 2: mucosa and submucosa, 3: transmural), crypt damage (0: none, 1: basal 1/3, 2: basal 2/3, 3: only surface epithelium intact, 4: entire crypt lost), and adjusted to tissue involvement by multiplication of percentage factor (x1: 0–25%, x2: 26–50%, x3: 51–75%, x4: 76–100%).62
Mucosal Uptake of AvrA Particles in Murine Colonic Epithelia.
C57BL/6J female mice were fasted overnight and AvrA-eGFP nanoparticles (12 μg eGFP) were injected intrarectally the following day. After 5 h, mice were sacrificed and colon tissue was removed. Distal colon was fixed in 4% paraformaldehyde and embedded in Tissue-Tek O.C.T. Compound. Twenty micrometer frozen sections were immunostained with anti-β-catenin and anti-GFP antibodies, and mucosal uptake of eGFP particles were imaged using a Zeiss LSM 510 confocal laser scanning system.
Statistical Analysis.
One-way ANOVA was performed, followed by post hoc comparisons using Tukey’s multiple comparison test.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (1R56 DK095074-01A1), the Kenneth Rainin Foundation (10H2), and the Crohn’s and Colitis Foundation of America (274694).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b03239.
Further characterization of protein purity, nanoparticle cross-linker and contents, in vitro nanoparticle uptake and cytotoxicity, and in vivo nanoparticle uptake (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Cosnes J; Gower-Rousseau C; Seksik P; Cortot A Epidemiology and Natural History of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1785–1794. [DOI] [PubMed] [Google Scholar]
- (2).Molodecky NA; Soon IS; Rabi DM; Ghali WA; Ferris M; Chernoff G; Benchimol EI; Panaccione R; Ghosh S; Barkema HW; Kaplan GG Increasing Incidence and Prevalence of the Inflammatory Bowel Diseases with Time, Based on Systemic Review. Gastroenterology 2012, 142, 46–54. [DOI] [PubMed] [Google Scholar]
- (3).Burger D; Travis S Conventional Medical Management of Inflammatory Bowel Disease. Gastroenterology 2011, 140, 1827–1837. [DOI] [PubMed] [Google Scholar]
- (4).Plevy SE; Targan SR Future Therapeutic Approaches for Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Floch MH; Walker WA Advances in Clinical Use of Probiotics. J. Clin. Gastroenterol 2008, 42, S45. [DOI] [PubMed] [Google Scholar]
- (6).Donnenberg MS Pathogenic Strategies of Enteric Bacteria. Nature 2000, 406, 768–774. [DOI] [PubMed] [Google Scholar]
- (7).Ruter C; Hardwidge PR “Drug from Bugs’: Bacterial Effector Protein as Promising Biological (Immune-)Therapeutics. FEMS Microbiol. Lett 2014, 351, 126–132. [DOI] [PubMed] [Google Scholar]
- (8).Galan JE Common Themes in the Design and Function of Bacterial Effectors. Cell Host Microbe 2009, 5, 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hao YH; Wang Y; Burdette D; Mukherjee S; Keitany G; Goldsmith E; Orth K Structural Requirements for Yersinia Yopj Inhibition of Map Kinase Pathways. PLoS One 2008, 3, e1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Collier-Hyams LS; Zeng H; Sun J; Tomlinson AD; Bao ZQ; Chen H; Madara JL; Orth K; Neish AS Cutting Edge: Salmonella Avra Effector Inhibits the Key Proinflammatory, Anti-Apoptotic Nf-Kappa B Pathway. J. Immunol 2002, 169, 2846–2850. [DOI] [PubMed] [Google Scholar]
- (11).Jones RM; Wu H; Wentworth C; Luo L; Collier-Hyams L; Neish AS Salmonella Avra Coordinates Suppression of Host Immune and Apoptotic Defenses Via Jnk Pathway Blockade. Cell Host Microbe 2008, 3, 233–244. [DOI] [PubMed] [Google Scholar]
- (12).Du F; Galan JE Selective Inhibition of Type Iii Secretion Activated Signaling by the Salmonella Effector Avra. PLoS Pathog. 2009, 5, e1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kaur J; Jain SK Role of Antigens and Virulence Factors of Salmonella Enterica Serovar Typhi in Its Pathogenesis. Microbiol. Res 2012, 167, 199–210. [DOI] [PubMed] [Google Scholar]
- (14).Lu R; Wu S; Zhang YG; Xia Y; Zhou Z; Kato I; Dong H; Bissonnette M; Sun J Salmonella Protein Avra Activates the Stat3 Signaling Pathway in Colon Cancer. Neoplasia 2016, 18, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lu R; Wu S; Zhang Y. g.; Xia Y; Liu X; Zheng Y; Chen H; Schaefer KL; Zhou Z; Bissonnette M; Li L; Sun J Enteric Bacterial Protein Avra Promotes Colonic Tumorigenesis and Activates Colonic Beta-Catenin Signaling Pathway. Oncogenesis 2014, 3, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lai SK; Wang YY; Hanes J Mucus-Penetrating Nanoparticles for Drug and Gene Delivery to Mucosal Tissues. Adv. Drug Delivery Rev 2009, 61, 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li MG; Lu WL; Wang HC; Zhang X; Wang XQ; Zheng AP; Zhang Q Distribution, Transition, Adhesion and Release of Insulin Loaded Nanoparticles in the Gut of Rats. Int. J. Pharm 2007, 329, 182–191. [DOI] [PubMed] [Google Scholar]
- (18).Lamprecht A; Ubrich N; Yamamoto H; Schafer U; Takeuchi H; Maincent P; Kawashima Y; Lehr CM Biodegradable Nanoparticles for Targeted Drug Delivery in Treatment of Inflammatory Bowel Disease. J. Pharmacol. Exp. Ther 2001, 299, 775–781. [PubMed] [Google Scholar]
- (19).Makhlof A; Tozuka Y; Takeuchi H Ph-Sensitive Nanospheres for Colon-Specific Drug Delivery in Experimentally Induced Colitis Rat Model. Eur. J. Pharm. Biopharm 2009, 72, 1–8. [DOI] [PubMed] [Google Scholar]
- (20).Meissner Y; Lamprecht A Alternative Drug Delivery Approaches for the Therapy of Inflammatory Bowel Disease. J. Pharm. Sci 2008, 97, 2878–2891. [DOI] [PubMed] [Google Scholar]
- (21).Moulari B; Pertuit D; Pellequer Y; Lamprecht A The Targeting of Surface Modified Silica Nanoparticles to Inflamed Tissue in Experimental Colitis. Biomaterials 2008, 29, 4554–4560. [DOI] [PubMed] [Google Scholar]
- (22).Pertuit D; Moulari B; Betz T; Nadaradjane A; Neumann D; Ismaili L; Refouvelet B; Pellequer Y; Lamprecht A 5-Amino Salicylic Acid Bound Nanoparticles for the Therapy of Inflammatory Bowel Disease. J. Controlled Release 2007, 123, 211–218. [DOI] [PubMed] [Google Scholar]
- (23).Theiss AL; Laroui H; Obertone TS; Chowdhury I; Thompson WE; Merlin D; Sitaraman SV Nanoparticle-Based Therapeutic Delivery of Prohibitin to the Colonic Epithelial Cells Ameliorates Acute Murine Colitis. Inflammatory Bowel Dis. 2011, 17, 1163–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wilson DS; Dalmasso G; Wang LX; Sitaraman SV; Merlin D; Murthy N Orally Delivered Thioketal Nanoparticles Loaded with Tnf-Alpha-Sirna Target Inflammation and Inhibit Gene Expression in the Intestines. Nat. Mater 2010, 9, 923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lamprecht A; Schafer U; Lehr CM Size-Dependent Bioadhesion of Micro- and Nanoparticulate Carriers to the Inflamed Colonic Mucosa. Pharm. Res 2001, 18, 788–793. [DOI] [PubMed] [Google Scholar]
- (26).Swidsinski A; Loening-Baucke V; Theissig F; Engelhardt H; Bengmark S; Koch S; Lochs H; Dorffel Y Comparative Study of the Intestinal Mucus Barrier in Normal and Inflamed Colon. Gut 2007, 56, 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cheng X; Liu R; He Y A Simple Method for the Preparation of Monodisperse Protein-Loaded Microspheres with High Encapsulation Efficiencies. Eur. J. Pharm. Biopharm 2010, 76, 336–341. [DOI] [PubMed] [Google Scholar]
- (28).Jin T; Zhu J; Wu F; Yuan W; Geng LL; Zhu H Preparing Polymer-Based Sustained-Release Systems without Exposing Proteins to Water-Oil or Water-Air Interfaces and Cross-Linking Reagents. J. Controlled Release 2008, 128, 50–59. [DOI] [PubMed] [Google Scholar]
- (29).Park TG; Lu W; Crotts G Importance of in Vitro Experimental Conditions on Protein Release Kinetics, Stability and Polymer Degradation in Protein Encapsulated Poly (-Lactic Acid-CoGlycolic Acid) Microspheres. J. Controlled Release 1995, 33, 211–222. [Google Scholar]
- (30).Vila A; Sanchez A; TobIo M; Calvo P; Alonso MJ Design of Biodegradable Particles for Protein Delivery. J. Controlled Release 2002, 78, 15–24. [DOI] [PubMed] [Google Scholar]
- (31).Langer K; Anhorn MG; Steinhauser I; Dreis S; Celebi D; Schrickel N; Faust S; Vogel V Human Serum Albumin (Hsa) Nanoparticles: Reproducibility of Preparation Process and Kinetics of Enzymatic Degradation. Int. J. Pharm 2008, 347, 109–117. [DOI] [PubMed] [Google Scholar]
- (32).Estrada LH; Chu S; Champion JA Protein Nanoparticles for Intracellular Delivery of Therapeutic Enzymes. J. Pharm. Sci 2014, 103, 1863–1871. [DOI] [PubMed] [Google Scholar]
- (33).Cheng R; Feng F; Meng F; Deng C; Feijen J; Zhong Z Glutathione-Responsive Nano-Vehicles as a Promising Platform for Targeted Intracellular Drug and Gene Delivery. J. Controlled Release 2011, 152, 2–12. [DOI] [PubMed] [Google Scholar]
- (34).Schlumberger MC; Muller AJ; Ehrbar K; Winnen B; Duss I; Stecher B; Hardt WD Real-Time Imaging of Type Iii Secretion: Salmonella Sipa Injection into Host Cells. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 12548–12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Madara JL; Stafford J; Dharmsathaphorn K; Carlson S Structural Analysisof a Human Intestinal Epithelial Cell Line. Gastroenterology 1987, 92, 1133–1145. [DOI] [PubMed] [Google Scholar]
- (36).Yen HJ; Hsu SH; Tsai CL Cytotoxicity and Immunological Response of Gold and Silver Nanoparticles of Different Sizes. Small 2009, 5, 1553–1561. [DOI] [PubMed] [Google Scholar]
- (37).Prabhu A; Shelburne CE; Gibbons DF Cellular Proliferation and Cytokine Responses of Murine Macrophage Cell Line J774a.1to Polymethylmethacrylate and Cobalt-Chrome Alloy Particles. J. Biomed. Mater. Res 1998, 42, 655–663. [DOI] [PubMed] [Google Scholar]
- (38).Hardt WD; Galan JE A Secreted Salmonella Protein with Homology to an Avirulence Determinant of Plant Pathogenic Bacteria. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 9887–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Spinner JL; Seo KS; O’Loughlin JL; Cundiff JA; Minnich SA; Bohach GA; Kobayashi SD Neutrophils Are Resistant to Yersinia Yopj/P-Induced Apoptosis and Are Protected from Ros-Mediated Cell Death by the Type Iii Secretion System. PLoS One 2010, 5, e9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Monack DM; Mecsas J; Ghori N; Falkow S Yersinia Signals Macrophages to Undergo Apoptosis and Yopj Is Necessary for This Cell Death. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 10385–10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Giacomodonato MN; Noto Llana M; Aya Castaneda R; Buzzola FR; Sarnacki SH; Cerquetti MC Avra Effector Protein of Salmonella Enterica Serovar Enteritidis Is Expressed and Translocated in Mesenteric Lymph Nodes at Late Stages of Infection in Mice. Microbiology 2014, 160, 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yao C; Tai Z; Wang X; Liu J; Zhu Q; Wu X; Zhang L; Zhang W; Tian J; Gao Y; et al. Reduction-Responsive Cross-Linked Stearyl Peptide for Effective Delivery of Plasmid DNA Int. J. Nanomed 2015, 10, 3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Martens TF; Remaut K; Demeester J; De Smedt SC; Braeckmans K Intracellular Delivery of Nanomaterials: How to Catch Endosomal Escape in the Act. Nano Today 2014, 9, 344–364. [Google Scholar]
- (44).Varkouhi AK; Scholte M; Storm G; Haisma HJ Endosomal Escape Pathways for Delivery of Biologicals. J. Controlled Release 2011, 151, 220–228. [DOI] [PubMed] [Google Scholar]
- (45).Ye Z; Petrof EO; Boone D; Claud EC; Sun J Salmonella Effector Avra Regulation of Colonic Epithelial Cell Inflammation by Deubiquitination. Am. J. Pathol 2007, 171, 882–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Cash JL; White GE; Greaves DR Zymosan-Induced Peritonitis as a Simple Experimental System for the Study of Inflammation. Methods Enzymol 2009, 461, 379–396. [DOI] [PubMed] [Google Scholar]
- (47).Xavier RJ; Podolsky DK Unravelling the Pathogenesis of Inflammatory Bowel Disease. Nature 2007, 448, 427–434. [DOI] [PubMed] [Google Scholar]
- (48).Boismenu R; Chen Y Insights from Mouse Models of Colitis. J. Leukocyte Biol 2000, 67, 267–278. [DOI] [PubMed] [Google Scholar]
- (49).Schesser K; Spiik AK; Dukuzumuremyi JM; Neurath MF; Pettersson S; Wolf-Watz H The Yopj Locus Is Required for Yersinia-Mediated Inhibition of Nf-Kb Activation and Cytokine Expression: Yopj Contains a Eukaryotic Sh2-Like Domain That Is Essential for Its Repressive Activity. Mol. Microbiol 1998, 28, 10671079. [DOI] [PubMed] [Google Scholar]
- (50).Srikanth C; Wall DM; Maldonado-Contreras A; Shi HN; Zhou D; Demma Z; Mumy KL; McCormick BA Salmonella Pathogenesis and Processing of Secreted Effectors by Caspase-3. Science 2010, 330, 390–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Herrera Estrada L; Padmore TJ; Champion JA Bacterial Effector Nanoparticles as Breast Cancer Therapeutics. Mol. Pharmaceutics 2016, 13, 710–719. [DOI] [PubMed] [Google Scholar]
- (52).Roda G; Sartini A; Zambon E; Calaflore A; Marocchi M; Caponi A; Belluzi A; Roda E Intestinal Epithelial Cells in Inflammatory Bowel Disease. World J. Gastroenterol 2010, 16, 4264–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Mizoguchi A Animal Models of Inflammatory Bowel Disease. Prog. Mol. Biol. Transl Sci 2012, 105, 263–320. [DOI] [PubMed] [Google Scholar]
- (54).Garrett WS; Lord GM; Punit S; Lugo-Villarino G; Mazmanian SK; Ito S; Glickman JN; Glimcher LH Communicable Ulcerative Colitis Induced by T-Bet Deficiency in the Innate Immune System. Cell 2007, 131, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Mukherjee S; Keitany G; Li Y; Wang Y; Ball HL; Goldsmith EJ; Orth K Yersinia Yopj Acetylates and Inhibits Kinase Activation by Blocking Phosphorylation. Science 2006, 312, 1211–1214 [DOI] [PubMed] [Google Scholar]
- (56).Trosky JE; Li Y; Mukherjee S; Keitany G; Ball H; Orth K Vopa Inhibits Atp Binding by Acetylating the Catalytic Loop of Mapk Kinases. J. Biol. Chem 2007, 282, 34299–34305. [DOI] [PubMed] [Google Scholar]
- (57).Wei P; Wong WW; Park JS; Corcoran EE; Peisajovich SG; Onuffer JJ; Weiss A; Lim WA Bacterial Virulence Proteins as Tools to Rewire Kinase Pathways in Yeast and Immune Cells. Nature 2012, 488, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zeng H; Wu H; Sloane V; Jones R; Yu Y; Lin P; Gewirtz AT; Neish AS Flagellin/Tlr5 Responses in Epithelia Reveal Intertwined Activation of Inflammatory and Apoptotic Pathways. Am. J. Physiol.: Gastrointest. Liver Physiol 2005, 290, G96–G108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Sumagin R; Robin AZ; Nusrat A; Parkos CA Transmigrated Neutrophils in the Intestinal Lumen Engage Icam-1 to Regulate the Epithelial Barrier and Neutrophil Recruitment. Mucosal Immunol. 2014, 7, 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wirtz S; Popp V; Kindermann M; Gerlach K; Weigmann B; Fichtner-Feigl S; Neurath MF Chemically Induced Mouse Models of Acute and Chronic Intestinal Inflammation. Nat. Protoc 2017, 12, 1295–1309. [DOI] [PubMed] [Google Scholar]
- (61).Alam A; Leoni G; Wentworth CC; Kwal JM; Wu H; Ardita CS; Swanson PA; Lambeth JD; Jones RM; Nusrat A; Neish AS Redox Signaling Regulates Commensal-Mediated Mucosal Homeostasis and Restitution and Requires Formyl Peptide Receptor 1. Mucosal Immunol. 2014, 7, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Nava P; Koch S; Laukoetter MG; Lee WY; Kolegraff K; Capaldo CT; Beeman N; Addis C; Gerner-Smidt K; Neumaier I; Skerra A; Li L; Parkos CA; Nusrat A Interferon-Gamma Regulates Intestinal Epithelial Homeostasis through Converging Beta-Catenin Signaling Pathways. Immunity 2010, 32, 392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.