

Abstract
Clinical observations and preclinical studies both suggest that Down syndrome (DS) may be associated with significant metabolic and bioenergetic alterations. However, the relevant scientific literature has not yet been systematically reviewed. The aim of the current study was to conduct a meta-analysis of metabolites involved in bioenergetics pathways in DS to conclusively determine the difference between DS and control subjects. We discuss these findings and their potential relevance in the context of pathogenesis and experimental therapy of DS. Articles published before July 1, 2020, were identified by using the search terms “Down syndrome” and “metabolite name” or “trisomy 21” and “metabolite name”. Moreover, DS-related metabolomics studies and bioenergetics literature were also reviewed. 41 published reports and associated databases were identified, from which the descriptive information and the relevant metabolomic parameters were extracted and analyzed. Mixed effect model revealed the following changes in DS: significantly decreased ATP, CoQ10, homocysteine, serine, arginine and tyrosine; slightly decreased ADP; significantly increased uric acid, succinate, lactate and cysteine; slightly increased phosphate, pyruvate and citrate. However, the concentrations of AMP, 2,3-diphosphoglycerate, glucose, and glutamine were comparable in the DS vs. control populations. We conclude that cells of subjects with DS are in a pseudo-hypoxic state: the cellular metabolic and bio-energetic mechanisms exhibit pathophysiological alterations that resemble the cellular responses associated with hypoxia, even though the supply of the cells with oxygen is not disrupted. This fundamental alteration may be, at least in part, responsible for a variety of functional deficits associated with DS, including reduced exercise difference, impaired neurocognitive status and neurodegeneration.
Keywords: Hypoxia, Glycolysis, Krebs cycle, Oxidative phosphorylation, Meta-analysis
Down syndrome as a metabolic disease: an introduction
Down syndrome (DS; also known as Down's syndrome or trisomy 21), is the most common genetic disorder known to date. It is caused by the presence of all or part of an additional (third) copy of chromosome 21. Its incidence is estimated to be approximately 1:700–1:1000 births worldwide. In the United States, approximately 6000 babies are born each year with DS (Bull 2020; Antonarakis et al. 2020).
Three different types of DS can be distinguished. The most common form of DS (approx. 95% of cases) features a whole and separate extra copy of chromosome 21. Translocation represents a rare form of DS (approx. 3% of cases), where the extra chromosome 21 (or part of it) is attached to another chromosome. Approximately 1% of DS cases are “mosaic DS” where the subject contains a mixture of two types of cells, some containing 46 chromosomes and some containing 47 (which contain the extra chromosome 21) (Bull 2020; Antonarakis et al. 2020).
The cause of the extra full or partial chromosome remains unknown, but increased maternal age is known to markedly increase the chance of DS. For instance, DS incidence increases from 1:1300 at age 25 years to 1:25 at age 49 years. Nevertheless, approximately 80% of children with DS are born to women who are younger than 35 years of age (Bull 2020; Antonarakis et al. 2020).
Subjects with DS can exhibit diverse pathophysiological alterations and deficits, ranging from mild to severe. Developmental alterations are common (phenotypically manifesting in physical growth delays and characteristic facial features, but also congenital heart disease and neurodevelopmental problems) and a progressive intellectual disability (Bull 2020; Antonarakis et al. 2020; Lagan et al. 2020). At later stage of life, DS produces an Alzheimer-syndrome-like state; this is so common that recent classifications consider DS as a congenital form of Alzheimer’s disease. A variety of additional conditions are also more common in DS, including airway, pulmonary and hearing disorders and hematological, oncological and autoimmune diseases (Bull 2020; Antonarakis et al. 2020; Lagan et al. 2020).
The scientific literature related to DS pathogenesis is massive, with significant bodies of literature focusing on the molecular genetics of the condition as well as on the pathogenesis and the potential experimental therapy of the neurocognitive issues, which are generally considered the most significant problems limiting the life of individuals with DS (Capone et al. 2006; Rachidi and Lopes 2010; Grieco et al. 2015; Malegiannaki et al. 2019; Lukowski et al. 2019; Martínez-Cué and Rueda 2020; Urbanus et al. 2020). However, there is also a significant body of clinical observations indicating that DS is associated with fundamental metabolic alterations, which, in turn, produce phenotypical changes, as well as increased propensities to various metabolic diseases.
For instance, DS individuals have less-than-normal muscle strength (muscle hypotonia) in their hands and lower extremities (Cioni et al. 1994; Croce et al. 1996; Carmeli et al. 2002; Hawli et al. 2009; Bala et al. 2018; Dupre and Weidman-Evans 2017; Coelho-Junior et al. 2019). They also exhibit significantly reduced aerobic exercise tolerance (Aguiar et al. 2008; Pastore et al. 2000). This has been commonly attributed to potential musculoskeletal developmental issues, neuromuscular junctional problems, as well as to the fact that DS individuals have decreased manual control, perform less physical activity and may have sarcopenia (Bala et al. 2018; Dupre and Weidman-Evans 2017; Coelho-Junior et al. 2019). While the above factors can certainly play a role, skeletal muscle functional analysis conducted in DS individuals (Phillips et al. 2013; Cowley et al. 2012) and data obtained from murine DS models (Cowley et al. 2012, 2017; Cisterna et al. 2014; Brault et al. 2015; Glass and Connor 2016; Bronks and Parker 1985) point to an intrinsic bioenergetic and metabolic dysfunction in the DS skeletal muscle. Importantly, a phosphorus magnetic resonance spectroscopy study in the quadriceps muscle of DS individuals indicates that the post-exercise resynthesis kinetics of phosphocreatine is suppressed (Phillips et al. 2013), which (a) indicates that intrinsic biochemical/bioenergetic mechanisms are impaired in the skeletal muscle of DS individuals and (b) suggests that the skeletal muscle’s mitochondrial respiratory function is suppressed in DS. Moreover, comparison of the contractile properties of soleus muscle of wild-type control mice and DS mice (Ts65Dn model) indicate that (despite the fact that soleus mass, skeletal mass:body weight ratio and muscle fiber type distribution are not different between control and DS mice), skeletal muscle contractions are impaired in DS (Cowley et al. 2012). While twitch and tetanus contractile responses were not significantly impaired in DS, the skeletal muscle from DS mice developed a progressive fatigue in repeated activation/recovery protocols (Cowley et al. 2012). The contractile weakness in DS skeletal muscle is associated with the dysregulation of multiple genes and gene products regulating various metabolic processes (including glucose uptake, lipid metabolism, oxidant/antioxidant balance), and has also been linked to mitochondrial dysfunction associated with DS (Hawli et al. 2009; Cowley et al. 2012, 2017; Cisterna et al. 2014; Brault et al. 2015; Glass and Connor 2016).
It is also well documented that many DS individuals present with an increased adiposity (Cowley et al. 2017; Bronks and Parker 1985; Rubin et al. 1998; Melville et al. 2005; Fonseca et al. 2005; González-Agüero et al. 2011; Soler Marín and Xandri Graupera 2011; Adelekan et al. 2012; Hill et al. 2013). This has been commonly attributed to the fact that DS individuals perform less physical activity and perhaps to different dietary habits (Hawli et al. 2009; Bala et al. 2018; Dupre and Weidman-Evans 2017; González-Agüero et al. 2011). Moreover, various lines of biochemical and endocrinology investigations, demonstrating increased adipokine production, altered lipid handling, subchronic inflammation, subclinical or manifest insulin resistance and/or metabolic syndrome (Magni et al. 2004; Corsi et al. 2009; de Asua et al. 2014a, b) and the development of fatty liver (Seeff et al. 1967; De Matteo and Vajro 2017) point to intrinsic biochemical differences in the development of obesity in DS.
If a generalized metabolic and bioenergetic dysfunction is present in DS, this would not only have the potential to underlie some of the above-mentioned more ‘classical’ metabolic alterations, but may also contribute to organ developmental problems, or even to neurological dysfunction (the neurons being highly ATP-dependent cells), and potentially the process of neurodegeneration as well (since cellular bioenergetic deficit can cause problems with protein processing) (see in more detail in “Potential mechanisms responsible for the development of pseudohypoxia in DS. Potential therapeutic interventions.” section). The fundamental role of metabolic and bioenergetic alterations in DS have been suspected for over 70 years and has periodically re-emerged as a working hypothesis (Simon et al. 1954; Anon 1954; Breg 1977; Shapiro 1983; Blass et al. 1988; Lejeune 1990; Chango et al. 2002; Coppedè 2009; Izzo et al. 2018; Vacca et al. 2019; Antonaros et al. 2020). Jerome LeJeune, the French physician-scientist, who discovered the chromosome abnormality in humans that causes DS, constructed a “Lejeune Machine” (Lejeune 1990), to illustrate the biochemical and metabolic alterations associated with this condition. However, the relevant scientific literature has not yet been systematically reviewed and organized to delineate the various metabolic alterations associated with DS. Therefore, in the current article, we present the results of our meta-analysis of metabolites involved in bioenergetics pathways in DS. First, we overview the basic cellular bioenergetic metabolic pathways, focusing on the primary as well as alternative sources that can yield ATP, the “universal energetic currency” in mammalian cells. Next, we present the results of our meta-analysis. Finally, we discuss potential underlying pathomechanisms and implications and review possible experimental therapeutic approaches.
The principal bioenergetic pathways in mammalian cells: a brief overview
There are several metabolic pathways for the biotransformation of the fuel molecules to high energy compounds such as adenosine-5′-triphosphate (ATP), guanosine-5′-triphosphate (GTP), reduced nicotinamide adenine dinucleotide (NADH), reduced flavin adenine dinucleotide (FADH2) and reduced nicotinamide adenine dinucleotide phosphate (NADPH) (Dashty 2013). Glucose is the most important source of energy, but other monosaccharides, fatty acids, ketone bodies, amino acids, and nucleosides can also be used as an energetic source in a cell-specific manner (Berg et al. 2008).
Glycolysis
Glycolysis is an evolutionarily ancient process of releasing energy from sugar which is already present in prokaryotes. In eukaryotic cells, glycolysis takes place in the cytosol. The essence of the process is that glucose is split into two molecules of pyruvate. Besides glucose, fructose and galactose can also enter the glycolytic pathway. This multistep process yields two ATP molecules and two molecules of NADH per molecule of glucose. In aerobic glycolysis, pyruvate is converted to acetyl-CoA so it can enter the mitochondria to be involved in the Krebs cycle (also known as “citric acid cycle” or “Szent-Györgyi-Krebs cycle”). However, in an anaerobic state, pyruvate remains within the cytoplasm and converts to lactate. The two molecules of NADH that are produced in glycolysis are oxidized to NAD+ by the reduction of pyruvate to lactate. Hence, anaerobic glycolysis produces only two molecules of ATP. Unused glucose molecules are packed and stored in the form of glycogen serving as a fast energy resource in the liver and in the muscle (Melkonian and Schury 2019).
The pentose phosphate pathway
The pentose phosphate pathway is parallel to glycolysis. It occurs exclusively in the cytoplasm. The primary purpose of this alternative pathway is the generation of NADPH and the production of ribose-5-phosphate, to be used in the synthesis of nucleotides. However, this pathway is also involved in the catabolism of nucleosides, releasing energy from ribose molecules as pentose phosphate pathway is interconnected to the glycolysis through the shared used of glyceraldehyde-3-phosphate and fructose-6-phosphate (Aziz and Mohiuddin 2020).
The Krebs cycle
The acetyl-CoA molecule inside the mitochondria is fully oxidized to carbon-dioxide. The total yield of one cycle is one molecule of GTP, three molecules of NADH, and one molecule of FADH2. NADH and FADH2 are reduced electron carriers, which donate electrons to the mitochondrial electron transport chain, while GTP is readily converted to ATP by a nucleoside-diphosphate kinase enzyme (Haddad and Mohiuddin 2019).
Oxidative phosphorylation
The NADH and FADH2 donated-electrons reach the mitochondrial electron transport chain in the inner mitochondrial membrane and generate an electron flow through the mitochondrial Complexes (I, II, III and IV) thus pumping protons into the inner membrane space from the mitochondrial matrix. The resulting proton gradient is then used by the ATP synthase (also known as Complex V) to generate ATP molecules from ADP and inorganic phosphate. Oxidative phosphorylation produces 24–28 ATP molecules per glucose equivalent, more ATP than any other part of the cellular respiration (Haddad and Mohiuddin 2019).
Beta oxidation of lipids
Fats provide an efficient means for storing energy for later use. Fats store more energy than glycogen and serve as a “slow energy resource”. Fatty acids are broken down into acetyl-CoA molecules through beta oxidation inside the mitochondria. Throughout each cycle of beta-oxidation, the fatty acid is reduced by two carbon lengths, producing one molecule of acetyl-CoA, and one molecule each of NADH and FADH2. The acetyl-CoA molecule can be oxidized in the Krebs cycle, while reduced electron carriers transfer their high energy electron to the mitochondrial electron transport chain (see above). Moreover, acetyl-CoA can be converted to ketone bodies. Ketone bodies including acetoacetate, beta-hydroxybutyrate, and acetone are produced by the liver during periods of glucose depletion. Ketone bodies are transported into organs requiring energy and converted back into acetyl-CoA, which then enters the Krebs cycle (Dunn and Grider 2020).
Amino acid catabolism
Amino acids can also be used as a source of energy especially in times of starvation or high-protein content diet. The processing of amino acids results in the generation of intermediates of the citric acid cycle, including pyruvate, acetyl CoA, oxaloacetate, and α-ketoglutarate. In human blood, glutamine is the most abundant free amino acid. Glutamine catabolism, as an energy source, plays an important role in cancer cell survival (Jiang et al. 2019) and in the survival of mutant cells that harbor oxidative phosphorylation defects (Chen et al. 2018). Amino acid catabolism results in ammonia, a toxic byproduct. Ammonia can be detoxified by conversion to urea via the urea cycle, which occurs mainly in the liver. Urea produced by the liver is ultimately excreted by the kidney (Wu 2009).
Pseudohypoxia
This term “pseudohypoxia” was originally coined by Ron Tilton’s group (Williamson et al 1993), referring to hypoxia-like change in the metabolic phenotype of cells in diabetes. In general, pseudohypoxia refers to a state in which cells or tissues express hypoxia-related genes and proteins and use re-wired metabolic pathways even when there is enough oxygen present. Pseudohypoxic phenotypes have been observed in a number of pathophysiological conditions, including sepsis (Tannahill et al. 2013) and Alzheimer’s disease (Salminen et al. 2017); it is also considered a component of physiological aging (Verdin 2015). More than a century ago Warburg (Warburg et al. 1927) observed similar phenomenon in various tumor cells; even when the cells are aerobic, they tend to mobilize glycolysis (often not as an alternative, but as a supplementary process to the normal oxidative phosphorylation) to maximize ATP generation that the cancer cells require for their fast division and multiplication.
Gene expression, proteomic and metabolomic profiling of DS as it relates to cellular bioenergetics
Understanding the alterations in molecules provides important information for determining the molecular mechanisms of diseases. “Omics” technologies permit the universal detection of genes (genomics), mRNA (transcriptomics), proteins (proteomics) and metabolites (metabolomics) in biological samples (Ishihara and Akiba 2017). In DS, the genes encoded on chromosome 21 are exclusively up-regulated (and none of them are downregulated), supporting the “gene dosage theory” of DS i.e. that the extra chromosome 21 results in the upregulation of its gene products, which, in turn, directly or indirectly changes development and affects various cellular functions. However, approximately 90% of the differentially expressed genes in DS (which include both upregulated and downregulated genes) are encoded on chromosomes other than chromosome 21, indicating dysregulated signaling cascades and/or compensatory gene regulation mechanisms (Letourneau et al. 2014; Araya et al. 2019; Bally et al. 2020). Integration of different “omics” datasets (for example transcriptomics and proteomics) may provide a tool to link different networks, highlighting the cellular response to a perturbation (although, to our knowledge, in DS, several different types of omics approaches have not yet been systematically integrated). Nevertheless, “omics” approaches have many limitations. For instance, mRNA expression level is not always a good indicator for protein expression level (e.g. due to post-transcriptional regulation of protein stability), and higher protein expression level does not necessarily mean a higher rate of an enzymatic reaction (for instance, because protein function is often regulated by post-transcriptional modifications, as well as by substrate and co-factor availability). In the following section, we will focus on the meta-analysis of metabolomic alterations, which represent the various products and substrates of enzymatic biochemical reactions; analysis of these metabolites may be a more direct indication of a metabolic pathway alteration than changes in mRNA or protein expression.
Analysis methods used
Data collection and identification of studies
Journal articles published before July 1, 2020, were identified by using the search terms “Down syndrome” and “metabolite name” or “trisomy 21” and “metabolite name”. We checked reference lists and published review articles for additional potentially relevant studies. Moreover, metabolomics studies on human DS cells or individuals with DS were also collected.
Inclusion and exclusion criteria
The inclusion criteria for the present study were as follows: (1) We only included studies that measured the amount of the selected metabolites. (2) Studies had to include both a sample from DS individuals and a control comparison group (3) Studies had to provide means and standard deviations and information about sample sizes for both groups. Studies were also included, if they do not provide directly the means and standard deviations, but they can be estimated from the published data. (4) Studies providing data for the selected metabolites in different tissues or using different methods were all included. However, for measurements involving ATP or other high-energy phosphates, only direct measurements of total cellular high-energy phosphates were considered. (5) Only human samples were considered (data derived from animal models of DS were excluded). (6) Studies or experiments measuring metabolite levels after a treatment were excluded, similarly if the measurement was performed on cell lines at high (> 7) passage number. (7) Only publicly available datasets were used. In total 41 published reports and associated databases (all of which used cross-sectional study design) were identified and subjected to analysis.
Variables included in data analysis
The following variables were collected: (1) The first author. (2) Year of publication. (3) The chemical name of the selected metabolite. (4) Mean of metabolite content in DS group. (5) Standard deviation of metabolite content in DS group. (6) Sample size in DS group. (7) Mean of metabolite content in control group. (8) Standard deviation of metabolite content in control group. (9) Sample size in the control group.
Meta-analytic procedures
A meta-analysis was conducted with the meta R package (Balduzzi et al. 2019). Raw data from metabolomics experiments often contain unwanted technical variations between samples and data are typically right-skewed within a sample. In order to meet normality assumptions of several parametric tests, data are usually log2 transformed. Technical variations such as signal drifts or batch effects are frequently encountered in metabolic profiling thus removing these effects by normalization is now widely considered as an integral part of data processing (Li et al. 2017). Several methods are available, but still, sample normalization is sometimes ignored in metabolomics studies. We used log2 transformation if authors reported statistics on skewed data and variance stabilizing normalization (Huber et al. 2002), if the authors did not normalize, but it would have been necessary. Further details can be found in the Supplementary Material. Adjustment for potential confounders, (e.g. age or sex) was not performed, and most of the cases these data were not available on individual levels.
The following studies give directly group means (M), standard deviations (SD) and sample sizes (N): Helguera et al. (2013), Bayer and McCoy (1974), Bartels (1971, Knull et al. (1978), Puukka et al. (1982), Kedziora et al. (1972), Nelson and Benson (1972), de Asua et al. (2014a,b), Lejeune et al. (1992), Nura et al. (2008), Infantino et al. (2011), Zitnanova et al. (2004), de Sousa et al. (2015); Howell et al. (1973); Kaufman and O’Brien (1967), Campos et al. (2010), Yates et al. (1990), Zaki et al. (2017), Coppus et al. (2007), Pogribna et al. (2001) and Miles et al. (2007).
If the standard error of the mean (SEM) is reported, standard deviations can be derived by using the formula: SD = SEM*sqrt(N). This conversion was performed on the following studies: Stocchi et al. 1985, Valenti et al. 2010, Rodríguez-Sureda et al. 2015, Izzo et al. 2017, Heggarty et al. 1996, Watkins et al. 1989, Tiano et al. 2008, and Convertini et al. 2016.
In some studies, the values of data were plotted in graphs. In this situation, values were extracted using digitize R package (Poisot 2011) or Adobe Acrobat Reader measurement tools.
Mircher reported age-segmented data (Mircher et al. 1997). In this case, age-segmented data were sequentially combined according to the Cochrane textbook (Higgins and Green 2008). Being N the number of individuals in a given segment, M the mean and SD the standard deviation of segments 1 and 2, mean is calculated as (N1*M1 + N2*M2)/(N1 + N2). For SD, it is the square root of: [(N1 − 1) SD12 + (N2 − 1) SD22 + ((N1*N2/(N1 + N2)) (M12 + M22 − 2*M1*M2))]/(N1 + N2 − 1). Similarly, age and gender groups were combined for the study of Pant and colleagues (Pant et al. 1968).
Chapman and Stern (1964) and Rosner et al. (1965) reported mean values and ranges. SD from ranges was calculated using the conversion factor provided by the study of Walter and Yao (2007).
Caracausi et al. (2018) and Antonaros et al. (2020) reported raw data values. M, SD, and N were calculated for each compound both in DS and in control groups, as well as in plasma and in urine samples while the fasting condition was not considered. More details can be found in Additional file 1.
Culp-Hill et al. (2017) reported untargeted and targeted metabolomics datasets. Data were filtered for the age: 12 ≤ Age ≤ 54, in line with the original study. Normalization and log2 transformation were performed on the raw data. If the compound is present both in the targeted and the untargeted compound list, only the targeted values were considered. More details can be found in Additional file 1.
In the study of Powers et al. (2019) we used the adjusted_met_data dataset, to calculate M, SD and N for a given metabolite in DS and control plasma.
Orozco et al. (2019) reported regression coefficients from multiple linear regression performed for each metabolite to assess the association between neurodevelopmental diagnosis (independent variables) and plasma metabolites (dependent variable). Typical development (control) children were used as a reference group. Adjusting for sex, race/ethnicity, age, year of blood collection, and parental homeownership was performed. From the published parameters i.e. betas, confidence intervals (95%CImax and 95%CImin), and sample sizes (NDS for DS, and NC for control), we calculated the M and SD for meta-analysis as M (for DS) = beta, M (for control) = 0, SD (for both) = ((95%CImax − 95%CImin)/(2*t))/sqrt(1/NDS + 1/NC), when t is the t-value for the given probability (0.975) and degree of freedom. The degree of freedom for multiple regression is calculated as n − k − 1, where n = NDS + NC, and k refers to the number of regressors not including the intercept.
Obeid et al. (2012) reported median, 10% quantile, and 90% quantile. In this case, the group mean was estimated as log2(median), while SD was estimated as [log2(90%quantile)-log2(10%quantile)]/(2*z0.9), where z0.9 = 1.28.
Fuller et al. (1962) and Mertz et al. (1963) reported individual data, from which M, SD and N were calculated.
A random-effects model (Borenstein et al. 2011) was used to determine the expected high degree of heterogeneity across studies using Hedges’ g as the standardized mean difference (Hedges 1981). Besides that, the 95% confidence intervals (CI) were estimated. Positive and negative SMD values indicated higher and lower levels of metabolites in DS group, relative to the control group. Statistical significance was set at p < 0.05.
Forty-one studies met the inclusion criteria, were critically reviewed, and are summarized in Table 1.
Table 1.
Studies selected for meta-analysis
| Study | # of DS | # of CTR | Analytes measured | Biological matrix |
|---|---|---|---|---|
| Antonaros et al. (2020) | 129 | 46 | Citrate, glucose, glutamine, lactate, pyruvate, succinate, tyrosine | Plasma |
| Bartels (1971) | 15 | 15 | ATP, ADP, AMP | RBC |
| Bayer and McCoy (1974) | 8 | 19 | ATP, ADP, AMP | Platelet |
| Campos et al. (2010) | 38 | 28 | Uric acid | Urine |
| Caracausi et al. (2018) | 51 | 20 | Citrate, tyrosine | Plasma, urine |
| Chapman and Stern (1964) | 40 | 40 | Uric acid | Serum |
| Convertini et al. (2016) | 3–6 | 3–6 | Citrate | Lymphoblast, PBMC |
| Coppus et al. (2007) | 46 | 48 | Tyrosine, serine | Plasma |
| Culp-Hill et al. (2017) | 29 | 43 | Citrate, glucose, lactate, pyruvate, serine, succinate, tyrosine, uric acid, 2,3-DPG, ADP, AMP, arginine, ATP, glutamine, homocysteine, Pi | RBC |
| de Asua et al. (2014a,b) | 48 | 33 | Glucose | Plasma |
| de Sousa et al. (2015) | 30 | 30 | Uric acid | Saliva |
| Fuller et al. (1962) | 80 | 80 | Uric acid | Serum |
| Heggarty et al. (1996) | 21–22 | 18–20 | Arginine, cysteine, glutamine, serine, tyrosine | Plasma, urine |
| Helguera et al. (2013) | 10 | 10 | ATP | Neuron |
| Howell et al. (1973) | 113 | 106 | Uric acid | Serum, urine |
| Infantino et al. (2011) | 6 | 6 | Cysteine, serine | WBC |
| Izzo et al. (2017) | 5 | 4 | ATP | Fibroblast |
| Kaufman and O’Brien (1967) | 107 | 107 | Uric acid | Serum |
| Kedziora et al. (1972) | 6 | 7 | ATP, ADP, AMP, Pi, 2,3-diphosphoglycerate | RBC |
| Knull et al. (1978) | 19 | 22 | ATP, 2,3-diphosphoglycerate | RBC |
| Lejeune et al. (1992) | 79 | 206 | Serine, cysteine, tyrosine, arginine | Plasma |
| Mertz et al. (1963) | 25 | 25 | Uric acid | Serum |
| Miles et al. (2007) | 14 | 12 | CoQ10 | Plasma |
| Mircher et al. (1997) | 107 | 216 | Serine, tyrosine, arginine | Plasma, urine |
| Nelson and Benson (1972) | 20 | 20 | 2,3-diphosphoglycerate | RBC |
| Nura et al. (2008) | 48 | 37 | Serine, glutamine, arginine | Plasma |
| Obeid et al. (2012) | 35 | 47 | Cysteine, homocysteine | Plasma, ferum |
| Orozco et al. (2019) | 31 | 193 | Serine, lactate, pyruvate, tyrosine, glutamine, succinate, arginine | Plasma |
| Pant et al. (1968) | 356 | 360 | Uric acid | Plasma |
| Pogribna et al. (2001) | 42 | 36–38 | Cysteine, homocysteine | Plasma |
| Powers et al. (2019) | 72–75 | 79–90 | Citrate, glucose, arginine, cysteine, glutamine, homocysteine, serine, tyrosine, lactate, Pi, pyruvate, succinate, uric acid | Plasma |
| Puukka et al. (1982) | 10 | 10 | ATP, ADP, AMP | RBC |
| Rodríguez-Sureda et al. (2015) | 5 | 5 | ATP | Fibroblast |
| Rosner et al. (1965) | 12 | 12 | Pi, uric acid | Serum |
| Stocchi et al. (1985) | 20 | 20 | ATP, ADP, AMP | RBC |
| Tiano et al. (2008) | 30 | 30 | CoQ10, uric acid | Lymph, plasma, platelets |
| Valenti et al. (2010) | 5 | 5 | Lactate, ATP, ADP, AMP | Fibroblast |
| Watkins et al. (1989) | 15–18 | 19 | Serine, tyrosine, cysteine, arginine | Plasma |
| Yates et al. (1990) | 6 | 29–30 | Lactate | Caudate nucleus, frontal cortex |
| Zaki et al. (2017) | 43 | 43 | CoQ10, glucose | Plasma |
| Zitnanova et al. (2004) | 16 | 16 | Uric acid | Plasma |
Alterations in high energy molecule pools in DS cells and tissues
Within cells, energy is provided by oxidation of fuel molecules such as carbohydrates, lipids, and proteins. The oxidation process results in free energy production that can be stored in high-energy bonds within molecules such as nucleoside triphosphate, phosphoenolpyruvate, carbamoyl phosphate, 2,3-bisphosphoglycerate, phosphoarginine, or phosphocreatine (Bonora et al. 2012). Kedziora and his coworkers have reported decreased levels of 2,3-diphosphoglyceric acid, and increased content of AMP, GTP, NAD+, and NADP+ (Kedziora et al. 1972). Our meta-analysis performed on ATP, ADP and AMP levels revealed statistically significant changes. Importantly, we observed that cellular ATP content is significantly lower in DS individuals (Fig. 1a), while ADP is slightly reduced (Fig. 1b) and AMP is unchanged (Fig. 1c). In addition, in DS individuals, there was a tendency for an increase in inorganic phosphate (Pi) levels (Fig. 1d). Similar trends in the composition of energy molecule pool have been observed in hypoxic state. In hypoxia, there is a fall in [ATP] and an increase in [ADP], along with an increase in Pi (Taggart and Wray 1998). These alterations resulted in a lower energy charge of DS cells compared to controls (see: Additional file 1).
Fig. 1.
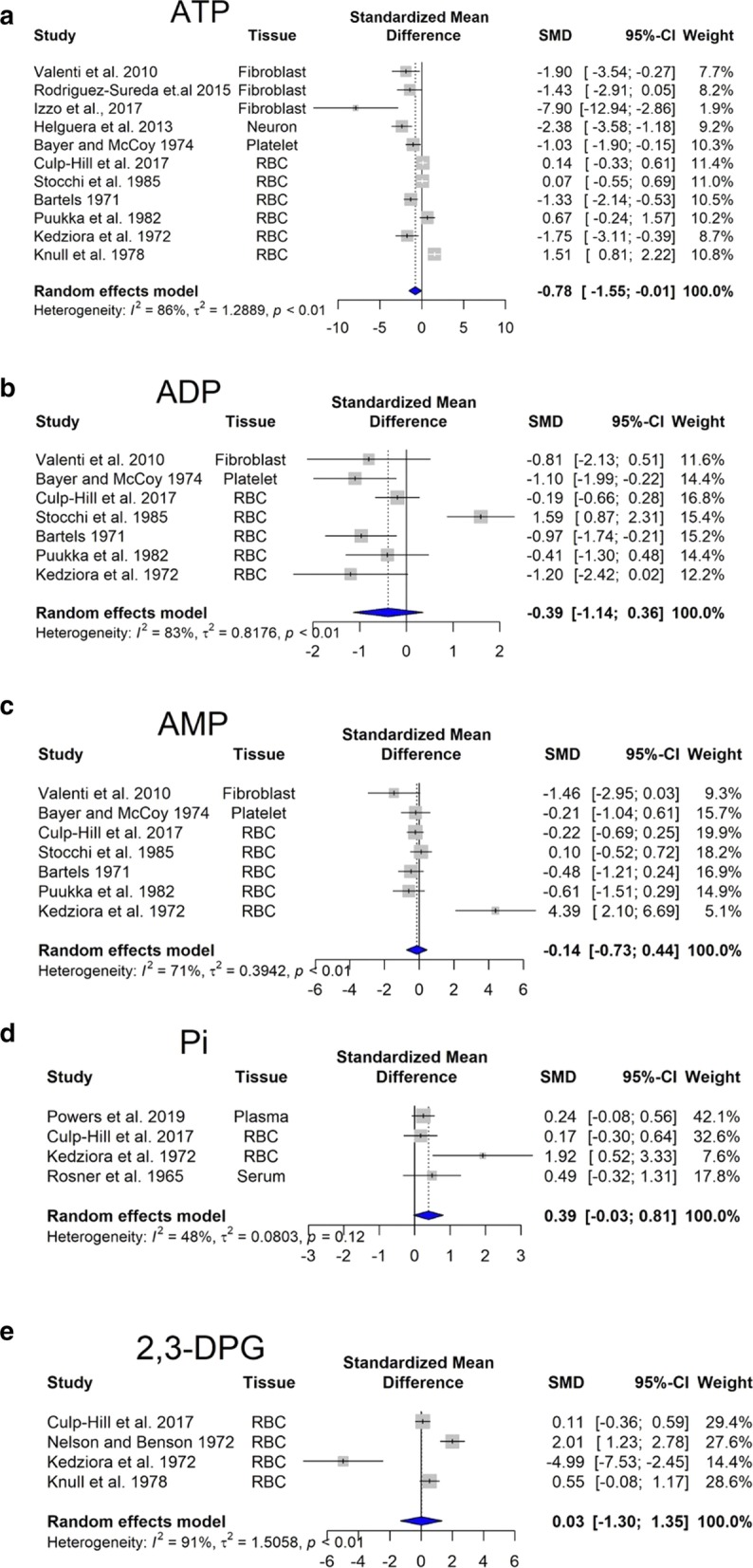
Changes in bioenergetics-related analytes in Down syndrome. Forest plot showing relative weights, standardized mean difference (Hedge’s g) with confidence intervals. Overall average effect size is displayed by filled diamond
The molecule 2,3-diphosphoglyceric acid (2,3-DPG) is very important in regulating the affinity of hemoglobin for oxygen. If the concentration of 2,3-DPG in red blood cell is low, hemoglobin affinity for oxygen is increased and tissue hypoxia may occur (MacDonald 1977). Our meta-analysis revealed no significant difference in the content of 2,3-DPG between DS and control red blood cells (Fig. 1e). This finding may indicate that oxygen delivery to DS tissues may not be impaired, but—based on the low ATP output, we hypothesize that—they may use less oxygen compared to healthy individuals (pseudo-hypoxia).
Uric acid (UA) is the end product of ATP and GTP catabolism and can reportedly act as an antioxidant. Several studies indicate elevated uric acid levels and increased prevalence of gout in DS (Igoe et al. 2017). In line with these findings, our meta-analysis showed that this elevation is statistically significant (p < 0.05) (Fig. 2). As higher urinary uric acid excretion is an indicator of severe hypoxia (Ozanturk et al. 2006), we can assume that a pseudo-hypoxic state develops in DS.
Fig. 2.

Changes in uric acid in Down syndrome. Forest plot showing relative weights, standardized mean difference (Hedges’ g) with confidence intervals. Overall average effect size is displayed by filled diamond
Further evidence for pseudohypoxia would be an increased NADH to NAD+ ratio (Gomes et al. 2013), but we have not found a study addressing this question in DS. Stocchi and colleagues reported that the total levels of nicotinamide adenine dinucleotide (NADH and NAD+) and nicotinamide adenine dinucleotide phosphate (NADPH and NADP+) were within normal ranges in DS individuals (Stocchi et al. 1985).
Changes in mitochondrial electron transport/oxidative phosphorylation in DS
As already mentioned earlier, DS is characterized by an overexpression of genes on chromosome 21, in addition to an overall dysregulation of an even larger set of genes located on the other chromosomes (Letourneau et al. 2014; Araya et al. 2019; Culp-Hill et al. 2017; Mao et al. 2005). Among these, there are genes involved in oxidative phosphorylation (OXPHOS), including all five mitochondrial complex subunits, genes implicated in mitochondrial biogenesis, and more generally in the mitochondrial function. Microarray analyses of DS samples showed a marked downregulation of genes encoding mitochondrial proteins, called nuclear-encoded mitochondrial genes (Conti et al. 2007; Chen et al. 2012; Sullivan et al. 2016; Liu et al. 2017; Sobol et al. 2019; Lee et al. 2003) (Table 2).
Table 2.
Changes in genes and proteins involved in oxidative phosphorylation in DS

Genes/proteins localized on chromosome 21 are shown in green color
The first study suggesting a functional association between impaired mitochondria and DS was conducted in 1994. Prince and colleagues reported a decreased activity of mitochondrial enzymes in platelets from DS individuals compared to normal controls (Prince et al. 1994). Subsequent studies demonstrate a reduction in mitochondria energy production efficiency in DS fibroblasts due to the impairment of complex I, V, ADP/ATP translocator and adenylate kinase activities (Schapira 1999; Piccoli et al. 2013). Additionally, the expression of several different mitochondrial electron transport enzyme subunits is decreased in the brain from DS individuals (Kim et al. 2001). DS fibroblasts are characterized by increased production of reactive oxygen species (ROS), higher intra-mitochondrial Ca2+ levels, and bioenergetic deficit, with decreased basal ATP content and lower mitochondrial membrane potential (Schapira 1999; Piccoli et al. 2013; Kim et al. 2001; Panagaki et al. 2019, 2020).
Coenzyme Q10 (CoQ10), a lipid molecule is an essential component of the electron transfer chain and it is an endogenous cellular antioxidant. CoQ10 takes part in aerobic cellular respiration and generation of energy in the form of ATP. Our meta-analysis reveals that the levels of CoQ10 are significantly reduced in DS (Fig. 3).
Fig. 3.

Changes in CoQ10 in Down syndrome. Forest plot showing relative weights, standardized mean difference (Hedges’ g) with confidence intervals. Overall average effect size is displayed by filled diamond
Electron microscopy analysis of fetal DS hearts provided a deeper understanding of mitochondrial abnormalities in DS. Most of the DS mitochondria were characterized by morphological alterations, such as increased fragmentation, bigger size, irregular shape, and cristae’s patterns (Schapira 1999; Piccoli et al. 2013; Kim et al. 2001; Panagaki et al. 2019, 2020). Similar changes have been reported in hypoxic conditions, namely increased ROS production, together with a decrease in the activity in mitochondrial complexes and mitochondrial fragmentation (Lee et al. 2020a, b).
Glycolytic alterations in DS
If oxidative phosphorylation is impaired, NADH and FADH2 cannot be oxidized as rapidly in the electron transport chain and ATP cannot be generated. To maintain the energy supply of the cell, alternative pathways for producing energy are utilized.
Accordingly, there is an increased level of anaerobic glycolysis in DS. The phosphorylation of fructose 6-phosphate is irreversible, and phosphofructokinase, the enzyme that catalyzes it, is the rate-limiting enzyme in glycolysis (Mor et al. 2011). Several early reports show that there is an increase in the activity of phosphofructokinase enzyme in fibroblast and red blood cells derived from individuals with DS (Layzer and Epstein 1972; Baikie et al. 1965; Annerén et al. 1987; Conway and Layzer 1970). Nevertheless, the gene for the liver type subunit of phosphofructokinase is located on human chromosome 21 indicating a higher gene expression.
Overexpression of enzymes involved in glycolysis would predicts an increased glucose consumption of the DS cells. Our meta-analysis revealed no significant changes in glucose steady-state concentrations (Fig. 4a). However, there is an increasing trend in pyruvate concentration (Fig. 4b).
Fig. 4.

Changes in Krebs cycle-related analytes in Down syndrome. Forest plot showing relative weights, standardized mean difference (Hedges’ g) with confidence intervals. Overall average effect size is displayed by filled diamond
Glycolysis produces 2 ATP molecules and 2 NADH molecules. NADH generated during aerobic glycolysis cannot reach the electron transport chain directly. The malate-aspartate shuttle mechanism serves to move the reduced NADH across the membrane in the form of other reduced molecules (Nelson et al. 2008). However, in DS, the electron transport chain is directly inhibited {as discussed later, recent data suggest that one of the reasons is that Complex IV activity is suppressed by the increased hydrogen sulfide [H2S] production, resulting from increased cystathionine-beta-synthase [CBS] and 3-mercaptopyruvate sulfurtransferase [3-MST] expression in DS cells (Panagaki et al. 2019, 2020)}. Hence, NADH molecules remain in the cytosol and oxidized in the process of lactic fermentation to regenerate NADH molecules produced in glycolysis. Our meta-analysis revealed that lactate production, i.e. anaerobic glycolysis is significantly elevated (p < 0.05) (Fig. 4c) in Tamarkina et al. (1978) and Baikie et al. (1965) showed that lactate dehydrogenase activity in DS samples was not elevated, indicating an upstream regulation, most probably at phosphofructokinase level (Tamarkina et al. 1978). Increased blood lactate concentration is traditionally attributed to anaerobic glycolysis due to inadequate oxygen delivery (James et al. 1999).
Changes in amino acid utilization in DS
Amino acids derived from the dietary proteins serve as an energy source as they can enter the citric acid cycle, while their ammonia moiety is eliminated through the urea cycle. Glutamine is the most abundant extracellular amino acid in the blood plasma, and it is quantitatively the most important fuel among the amino acids. Nevertheless, there are no changes in its concentrations between DS and control subjects (Fig. 4a). It is immediately metabolized to glutamate by glutaminase. Later, glutamate undergoes transamination with pyruvate generating l-alanine and 2-oxoglutarate (also known as alpha-ketoglutarate). The latter metabolite is then oxidized in the Krebs cycle generating succinate (Nelson et al. 2008). Energy molecules, NADH and GTP, are generated via this pathway. GTP can be easily converted to ATP in the cell (Conway and Layzer 1970). As in DS the mitochondrial electron transport chain is inhibited, the produced NADH may be oxidized in another manner.
One possible way is the reduction of alpha-ketoglutarate to 2-hydroxyglutarate by using NADH. L-2-hydroxyglutarate is found to be produced to high levels in low oxygen conditions, including cells of the immune system and cancer cells (Tyrakis et al. 2016). In DS plasma, Powers and colleagues demonstrated an increase in the levels of 2-hydroxyglutarate (Powers et al. 2019).
Second, mitochondrial isocitrate dehydrogenase can catalyze the reverse reaction converting alpha-ketoglutarate to isocitrate and then citrate (reductive carboxylation). During this step, NADH is oxidized and the resulting citrate is then transported out of the mitochondria, to the cytosol, where the enzyme citrate lyase converts citric acid to acetyl-CoA (Corbet and Feron 2015). This is the initial step of de novo lipid biogenesis. Mitochondrial isocitrate dehydrogenase-mediated reductive carboxylation is observed when steady-state alpha-ketoglutarate levels are high, and that of citrate is low (Eales et al. 2016). In DS individuals, elevated levels of alpha-ketoglutarate have been found in plasma (Powers et al. 2019) and in red blood cells (Caracausi et al. 2018). Moreover, our meta-analysis showed a significant increase in citrate levels (Fig. 4d). In hypoxia, there is also a significant increase in the levels of molecules involved in the citric acid cycle, such as citrate, succinate, fumarate (Bénit et al. 2014; Peng et al. 2019).
Succinate produced during the citric acid cycle, is transported across the inner mitochondrial membrane by the mitochondrial dicarboxylate carrier and acts as a mediator of mitochondrial dysfunction (Tretter et al. 2016). Our meta-analysis revealed that succinate accumulates in DS (Fig. 4e). It has been shown that succinate stabilizes hypoxia-inducible factor 1-α, leading to hypoxia-like metabolic phenotypes in the absence of hypoxia, also known as pseudohypoxia. Succinate also inhibits histone and DNA demethylases changing the epigenetic profile (Tretter et al. 2016). Moreover, succinate might be involved in ROS generation, through Complex I (Chouchani et al. 2014). The presence of increased ROS levels and oxidative stress in DS individuals has been well established (Valenti et al. 2011).
Besides that, several alterations on amino acid concentrations have been described in the blood of DS in comparison with control subjects (Fig. 5). The most consistent findings are the following: increased cysteine (Fig. 5b) and decreased serine (Fig. 5c) and homocysteine (Fig. 5d) levels (most probably due to a dosage effect of the gene for CBS), as well as decreased tyrosine and arginine levels, perhaps due to the pseudo-hypoxic state of DS cells. In fact, it is known that hypoxia inhibits the synthesis of arginine from citrulline (Su and Block 1995).
Fig. 5.

Changes in selected amino acids in Down syndrome. Forest plot showing relative weights, standardized mean difference (Hedges’ g) with confidence intervals. Overall average effect size is displayed by filled diamond
Changes in lipid catabolism in DS
Fatty acid oxidation is the mitochondrial aerobic process of breaking down fatty acids into acetyl-CoA units. This process is influenced by the impaired mitochondrial oxidative phosphorylation in DS. The molecule carnitine transports long-chain fatty acids into mitochondria Carnitine exists in humans as free (unesterified) carnitine, and esterified carnitine (acyl-carnitines). Free carnitine levels were reduced in young DS children (Seven et al. 2001). Similarly, free carnitine levels were lower in the hypoxic–ischemic neonates. (Çam et al. 2005). Moreover, hypoxic–ischemic neonates had significantly increased C4, C5, C5:1, C6, C6-OH, C8 levels and decreased long-chain acylcarnitine levels (López-Suárez et al. 2019). Powers and colleagues (Powers et al. 2019) also found that C5 and C5:1 levels were significantly increased in DS.
Potential mechanisms responsible for the development of pseudohypoxia in DS. Potential therapeutic interventions
In order to conceive potential therapeutic interventions for the DS-associated pseudohypoxia, we need to understand (or at least have some reasonable working hypotheses for) the underlying pathobiochemical mechanisms. Although to identify the most relevant pathways in a system where over a thousand genes are dysregulated may seems like looking for the “needle in the haystack”, a number of potential contributing biochemical mechanisms have been identified over the last two decades.
Antioxidant strategies
ROS accumulation promotes oxidative modification of important biomolecules, including lipids, proteins, nucleic acids and mtDNA, altering the function of several organs or the whole organism. Although chromosome 21 also encodes superoxide dismutase (SOD), which would predict that DS cells may have an improved antioxidative potential, paradoxically ROS-related processes have been suggested to play a significant role in the development of various DS pathologies (Lott et al. 2006). The most likely explanation for this paradox may be that the increased SOD cannot fully cope with the massively increased ROS formation in DS. ROS can be produced by various sources in the cell; for instance, if the mitochondrial electron transport chain is blocked, electrons can back up and leak off from the mitochondria. Moreover, hypoxic cells have a number of additional sources of ROS, such as NADPH oxidase or xanthine oxidase. ROS is not only a product of mitochondria, but also a causative factor in mitochondrial dysfunction, which, in turn, further impairs the ability of the cell to perform oxidative phosphorylation and ATP generation (Vacca et al. 2019). Moreover, the accumulation of oxidative DNA damage in DS is considered as an early event promoting cell dysfunction of various types (including neurodegeneration and Alzheimer-like dementia, a distinctive and severe hallmark of DS) (Contestabile et al. 2010).
Although ROS can serve both beneficial and detrimental effects in various conditions, a generic therapeutic approach to improve mitochondrial function and to boost cellular bioenergetics would be to scavenge/neutralize excess ROS generation, with the expectation that such intervention may not only restore mitochondrial function but also improve DNA integrity. One of the practical limitations of antioxidant strategies is that the clinically available antioxidants have a relatively slow reaction rate with many oxidant species, and they are not catalytic (i.e. they are consumed in the reaction). This requires chronic administration, often at high doses. Several clinical trials based on antioxidant administration have been undertaken, although with poor or discordant outcomes. Administration of alpha-tocopherol, ascorbic acid, and alpha-lipoic acid over 2 years showed neither an improvement in cognitive functioning nor a stabilization of cognitive decline in over-50 years old DS individuals (Lott et al. 2011). A randomized study on 7 months old children, orally supplemented with a mixture of antioxidants (selenium, zinc, vitamin A, vitamin E and vitamin C) and/or folic acids for 18 months resulted in no change in oxidative stress nor in any improvement of psychomotor and language development (Ellis et al. 2008). A possible explanation to the ineffective antioxidant supplementation is that differences between normal and DS fetuses are already present at 11 weeks gestation, which implies that and at birth, any ROS-dependent damage has already occurred (Reynolds 2008). Although some promising results on learning and memory were obtained in the DS mouse model Ts65Dn after administration of antioxidants, such as vitamin E, clinical trial in DS individuals older than 50 displayed no significant efficacy of Vitamin E in slowing cognitive decline (Sano et al. 2016).
Coenzyme Q10
Since DS individuals display a reduction in cellular coenzyme CoQ10 content, another line of studies aimed to test the efficiency of CoQ10 supplementation. CoQ10 is a bioactive quinone synthetized by our organism, which is ubiquitously distributed in lipid environments, acting as a lipophilic antioxidant (Tiano and Busciglio 2011). In the bloodstream, CoQ10 is associated to lipoproteins, while at cellular level it is a component of plasma and intracellular membranes (Tiano and Busciglio 2011). It is also involved in mitochondrial bioenergetics, and CoQ10 intra-mitochondrial concentration determine the efficiency of OXPHOS chain. Due to its double role in supporting mitochondrial OXPHOS and counteracting oxidative DNA damage, several trials have been undertaken to test the efficiency of CoQ10 using the active form ubiquinol or the oxidized ubiquinone as antioxidant supplement for DS. To investigate the protective effect of ubiquinone on oxidation-dependent nucleic acid modifications, Tiano and colleagues conducted three experimental studies in DS children, testing the effect of 4 mg/kg/days for 6 month, 20 months and 4 years (Tiano et al. 2011, 2012; Larsen et al. 2018). Although some age-specific reduction of DNA oxidation was observed after 20 months (Tiano et al. 2012), no effect on DNA damage was observed in the long-term treatment study (Larsen et al. 2018).
Taken together, the above studies indicate that antioxidant supplementation alone does not improve clinical outcomes in DS individuals. The reasons may be related to a combination of factors. First, it is possible that ROS generation does not play as big a role in DS as presumed. Second, it is possible that some of the biological roles of ROS are beneficial and others are detrimental, and the interventions counteract both. Third, it is possible that the antioxidants, at the doses used in the trials, do not reach sufficiently high concentrations or do not react quickly or efficiently enough with the relevant ROS species. Fourth (and this applies to all therapeutic interventions discussed in the current section), it is possible that the pathophysiological positive feedforward events have progressed beyond the point of reversibility by the time the clinical interventions were started.
The CBS/H2S pathway
Hydrogen sulfide (H2S) is now accepted an endogenously produced gaseous transmitter and biological regulator. Three principal enzymes, cystathionine--lyase (CSE), cystathionine-β-synthase (CBS) and 3-mercaptopyruvate sulfurtransferase (3-MST) are responsible for mammalian H2S biogenesis (Szabo and Papapetropoulos 2017; Zuhra et al. 2020). CBS (encoded on chromosome 21) and 3-MST (encoded on chromosome 1) are both upregulated in DS cells (Panagaki et al. 2019; Panagaki et al. 2020; reviewed in Szabo 2020); accordingly, DS cells produce high levels of CBS. Indeed, early clinical observations have demonstrated that DS individuals have elevated plasma and urinary levels of H2S and its metabolites (Belardinelli et al. 2001; Kamoun et al. 2003). As reviewed recently (Szabo 2020), when cellular H2S levels are increased beyond an optimal concentration, H2S produce inhibitory bioenergetic effects (in principle, via inhibition of mitochondrial Complex IV, but also through other mechanisms) (Panagaki et al. 2019, 2020; Szabo 2020; Nicholls et al. 2013).1 Indeed, pharmacological inhibition or genetic silencing of CBS can restore mitochondrial function and cellular proliferation in DS fibroblasts (Panagaki et al. 2019) and it can improve neurological function in DS mice (Marechal et al. 2019). Moreover, excess H2S in normal fibroblasts, or overexpression of CBS in otherwise normal mice, can induce cellular bioenergetic deficits or neurological deficits, respectively (Panagaki et al. 2019; Marechal et al. 2019). As reviewed recently, there are several ways to pharmacologically counteract CBS activity or the excess H2S production, and some of these approaches (repurposable CBS inhibitors or H2S scavengers) are potentially translatable into the clinical arena (Szabo 2020; Kamoun 2019), although truly ‘pharmaceutical grade’ CBS inhibitors remain to be discovered and translated into clinical testing (Zuhra et al. 2020).
DYRK1
Dual-specificity tyrosine-phosphorylation regulated kinase 1A (DYRK1A) is localized in the “DS critical region” of chromosome 21 (Kimura et al. 2007; Noll et al. 2009; Guo et al. 2010; Song et al. 2015). As DYRK1A is involved in tau and amyloid precursor protein phosphorylation (Kimura et al. 2007), it is a strong candidate gene for learning defects associated with Alzheimer’s disease and DS. Among others, DYRK1A also phosphorylates enzymes related to energy pathways and the methylation cycle. DYRK1A-mediated phosphorylation at the Thr356 residue inhibits glycogen synthase kinase activity. Glycogen synthase kinase phosphorylates and inhibits glycogen synthase and leads to a decrease in glycogen synthesis in the liver and muscles, along with increased blood glucose (Song et al. 2015).
DYRK1A phosphorylates and activates SIRT1, an enzyme that deacetylates proteins. SIRT1 deacetylates and activates PGC-1α transcription factor leading to mitochondrial biogenesis (Guo et al. 2010). DYRK1A overexpression also reduces homocysteine levels independently of CBS expression. It increases the S-adenosyl-l-homocysteine hydrolase activity in the reverse direction leading to a diminution of homocysteine (Nicholls et al. 2013). Further studies are needed to clarify whether these alterations are involved in learning deficiency observed in DS or other targets of DYRK1A are more critical. Nevertheless, it was found that pharmacological inhibition of brain DYRK1A can correct recognition memory deficits in three DS mouse models, all expressing an extra copy of Dyrk1a (Nguyen et al. 2018; Kim et al. 2016). Clinical trials in adults treated with epigallocatechin-3-gallate, a natural DYRK1a inhibitor from green tea, demonstrated a modest, but detectable gain of cognition in DS individuals (de la Torre et al. 2014, 2016; Xicota et al. 2020).
Caloric restriction
The prevalence of obesity among individuals with DS is relatively high compared to the general population (Bronks and Parker 1985; Rubin et al. 1998; Melville et al. 2005; Fonseca et al. 2005; González-Agüero et al. 2011; Soler and Xandri 2011; Bertapelli et al. 2016). To explain is phenomenon, several hypotheses have been proposed, but the underlying mechanisms have not yet been elucidated. The following factors have been proposed to increase the risk of obesity in DS: (1) Leptin is a hormone secreted by adipocytes, acting in the hypothalamus to suppress appetite. (2) High leptin levels in obese subjects, generally thought to be the outcome of increased leptin resistance. (3) Children with DS had increased leptin for percent body fat than their unaffected siblings. This difference may contribute to the increased risk of obesity in children with DS (Magge et al. 2008). Resting energy expenditure represents the amounts of calories required for a 24-h period by the body during a non-active period. Lower resting energy expenditure could partially explain the increased risk of obesity in youth with DS. Hill and colleagues found that children with DS had lower resting energy expenditure compared to siblings without DS (Hill et al. 2013; Mendonca et al. 2010). Whether traditional dietary interventions (e.g. caloric restriction) have the potential to improve metabolic status (beyond improving BMI, for instance), remains to be seen.
Low sulfur diet
Generally, a low sulfur diet involves the reduction of meats, dairy products, eggs, onions, peas and cruciferous vegetables. The primary aim of a potential low sulfur diet would be to reduce the generation of H2S which is the result of increased CBS and 3-MST activity (see above). Amino acids, methionine and cysteine are the primary sources of sulfur. A mathematical model (Orozco et al. 2019) suggests that a low-methionine diet might offer a perspective for reversing the metabolic imbalance in DS, but clinical investigations remain to be conducted to test this theory.
Vitamin supplementation (folate and cobalamine)
Folate is an important vitamin that contributes to cell division and growth. Studies on the effect of vitamin supplements on metabolic or clinical outcomes in DS showed limited, if any, evidence of improvement (Lumley et al. 2001; Blehaut et al. 2010).
Choline supplementation
Supplementing the maternal diet of Ts65Dn mice with choline during pregnancy and lactation substantially improved attention (Strupp et al. 2016). Therefore, choline metabolism might be an important target in DS. However, there are no clinical studies on choline supplementation in DS.
Amino acid supplementation
Lejeune et al. has found differences in serine concentrations between control children and DS individuals and recommended a dietary supplementation (Lejeune et al. 1992). Many of the currently marketed food supplement formulas for DS include amino acids.
Zinc and selenium
Several studies have shown zinc and selenium serum levels are decreased in DS (Barañano and Hartman 2008). These trace elements are involved in the synthesis and regulation of the activity of proteins and enzymes that are necessary for our endogenous antioxidant response (Saghazadeh et al. 2017; Parisotto et al. 2015). It has been suggested that antioxidant therapies might have some benefit for DS individuals, although the clinical trials have not been particularly encouraging (see above).
Ketogenic diet
The ketogenic diet contains high fat, moderate protein, and very low carbohydrate, which induce the hepatic production of ketone bodies. Ketone bodies cross the blood–brain barrier and are converted to acetyl coenzyme A, enters the citric acid cycle to produce energy for the brain. The ketogenic diet traditionally has been used in cases of intractable epilepsy (Barañano and Hartman 2008). As epilepsy is a common comorbidity in DS and it is also thought to contribute to cognitive dysfunction for cognitive decline in DS, it was proposed that the ketogenic diet may be a therapeutic option for cognitive decline in DS (Kaneko et al. 2018).
Physical exercise
Lower physical activity due to overall weakness has been well documented in DS (Cioni et al. 1994; Croce et al. 1996; Carmeli et al. 2002; Hawli et al. 2009; Bala et al. 2018; Dupre and Weidman-Evans 2017; Coelho-Junior et al. 2019; Aguiar et al. 2008; Pastore et al. 2000; Phillips et al. 2013). Consumption of high carbohydrate and nutrient-poor food products is more frequent in DS children as these products are easy to chew. The increased expression and activity of CBS in children with DS results in an increased plasma level of cysteine (Pogribna et al. 2001). Plasma concentrations of cysteine correlate strongly with fat mass, but the mechanism of how increased cysteine levels induce fat accumulation is unclear (Elshorbagy et al. 2012). One can also postulate that beta-oxidation of fatty acids in DS is impaired as the mitochondrial electron transport chain is not functioning properly, producing increased acyl-carnitine levels.
Physical exercise may improve DS outcomes through effects on the mitochondria. It is known that electron transport chain activity i.e. mitochondrial function, and mitochondrial numbers can improve with exercise training (Menshikova et al. 2006). Mitochondrial function is regulated by mitochondrial matrix free Ca2+ concentration. Stimuli such as action potentials or hormone-receptor bindings can trigger a signal transduction process leading to Ca2+ oscillations between intracellular organelles (Pecze et al. 2015). This leads to the activation of mitochondrial dehydrogenases (Denton 2009). Moreover, mitochondria undergo several structural changes in response to the energetic challenge. High energy demand induces mitochondrial fusion allowing efficient mixing of mitochondrial content within an extended mitochondrial network (Westermann 2012). Activation of the PGC-1α transcription factor, the “master regulator” of mitochondrial biogenesis is also triggered by physical exercise (Drake et al. 2016). PGC-1α regulates the expression of nuclear genes encoding mitochondrial proteins, thereby regulating mitochondrial content and function (Hood et al. 2016). All of the abovementioned changes would be expected to exert beneficial effects in DS individuals. Functional fitness in DS can be meaningfully improved through physical exercise (Dodd and Shields 2005). For instance, a non-randomized controlled trial has found that the functional fitness of adults with DS improved with an aquatic training but was insufficient to improve balance and upper body strength (Boer and de Beer 2019).
Implications, conclusions and outlook
Since mitochondrial function is the crucial contributor to ATP generation in mammalian cells, and ATP is essential to the maintenance of all fundamental cellular function in resting cells, (and even more so in dividing ones), one would expect that a significant impairment of cellular ATP synthesis would result in significant suppression of all fundamental cellular functions, including cellular division cell proliferation, maintenance of membrane potential, and—for neurons—cellular excitability ability to form synapses, release neurotransmitters and induce post-synaptic currents. Indeed, multiple studies demonstrate that DS cells have an impairment in all of the above-mentioned processes, and there is evidence that various interventions aimed at restoring mitochondrial activity can improve these functional parameters (Izzo et al. 2017; Panagaki et al. 2019; Park et al. 2000; Jablonska et al. 2006; Contestabile et al. 2007; Trazzi et al. 2011; Valenti et al. 2013; Weick et al. 2013; Bhattacharyya et al. 2009; Stern et al. 2015; Coskun et al. 2017; Huo et al. 2018; Mizuno et al. 2018; Mollo et al. 2019).
A generalized energetic deficit would also predict disruptions in the cell cycle and impairment of nuclear and mitochondrial DNA repair (all extremely energetically demanding processes), contributing to genomic instability. Indeed, there is direct experimental evidence showing significant impairment of DNA repair and cell cycle in human DS cells and in animal models of DS (e.g. Contestabile et al. 2007; Pincheira et al. 1994; Druzhyna et al. 1998; Maluf and Erdtmann 2001; Rosner et al. 2003; Necchi et al. 2015; Wang et al. 2016). While various compensatory bioenergetic processes (e.g. an upregulation of glycolysis) may be able to ‘make up for’ some of the ‘missing’ ATP), the consequences of the metabolic pseudohypoxia may be more severe when the cells experience sudden increases in energetic demand. For instance, it is conceivable that such fundamental bioenergetic mechanisms may contribute to the well-documented impairment of exercise tolerance in DS individuals (see: Introduction). An impaired mitochondrial function in DS may also predict that DS tissues have a reduced O2 utilization; for example, measured as whole-body-O2 consumption, or as tissue O2 extraction (i.e. quantification of arteriovenous oxygen difference). Such measurements are commonplace in the field of metabolic disease and in the field of sepsis (where, in many pathophysiological conditions, the tissues’ ability to extract O2 is impaired; for instance, in sepsis, due to a metabolic ‘poisoning’ of the mitochondria) (Carré and Singer 2008); indeed, the available body of clinical data in DS indicates that a similar reduced O2 utilization is characteristic of DS individuals (Seron and Greguol 2014; Mendonca et al. 2018). The same process (impaired mitochondrial function, and the resulting compensatory upregulation of glycolysis) would also produce increases in systemic lactate production (again, a common observation in septic shock, where, in fact, elevated lactate levels are excellent predictors of mortality); indeed, our meta-analysis has clearly demonstrated that DS cells produce increased levels of lactate (Fig. 4c) and clinical studies have also reported elevated plasma lactate levels in DS individuals (Caracausi et al. 2018; Gross et al. 2019).
It is plain to see how a generalized cellular bioenergetic deficit (pseudohypoxia) would predispose to various embryonic developmental problems and to various fundamental functional disturbances in later life. These problems would be expected even on the background of a (otherwise unperturbed) organism (i.e. in the absence of any additional gene dysregulation); it is obvious that a globalized bioenergetic deficit would be even more deleterious in cells where hundreds of genes and gene products are dysregulated due to the gene dosage effect of chromosome 21, as well as the even larger number of perturbances of genes encoded on all other chromosomes. Let’s consider one single bioenergetic disturbance, the inhibition of mitochondrial Complex IV activity (which is reversibly inhibited by H2S and irreversibly inhibited by cyanide). It is well documented in the environmental toxicology literature that even a partial inhibition of this enzyme (caused by H2S or cyanide administration) can cause various developmental and neurodevelopmental defects in developing embryos (Singh 1981; Tewe and Maner 1981; Frakes et al. 1986; Hannah and Roth 1991; Skrajny et al. 1992; U.S. EPA 2010); Complex IV inhibition also produces significant bioenergetic and functional (including neurocognitive) defects in juvenile and adult organisms (Egekeze and Oehme 1980; Persson et al. 1985; Nicholson et al. 1998; Schneider et al. 1998; Nam et al. 2004; Fiedler et al. 2008; Reed et al. 2014; Tshala-Katumbay et al. 2016). In conditions with insufficient energy production in the brain, convulsions also frequently occur (Folbergrovà et al. 1981; Nakanishi et al. 1991; Shukla et al. 1989). Indeed, there is a significant increase in the incidence of convulsive events in DS individuals (Bull et al. 2011; Barca et al. 2014).
A global pseudohypoxia, of course, would be also expected to be a proximate cause of neurological dysfunction (since neurons are highly ATP-dependent cells, highly susceptible to disturbances in cellular bioenergetic processes) (Hoyer 1990; Zeiger et al. 2011; Funes et al. 2014; Nicholls et al. 2015; Hebert-Chatelain et al. 2016; Plotegher et al. 2020). We hypothesize that neuronal pseudohypoxia may also potentially contribute to the process of delayed neurodegeneration (Alzheimer-like symptoms) in DS. Indeed, DS cells exhibit significant problems with protein processing (Aivazidis et al. 2017), and it is well documented that cellular bioenergetic deficit, on its own, can directly causes problems with the processing of various cellular proteins, including amyloid beta (Hoyer 1993; Gabuzda et al. 1994; Meier-Ruge et al. 1994; Busciglio et al. 2002; Ferreira et al. 2010; Wilkins and Swerdlow 2017). It is conceivable that the chronic cellular bioenergetic deficit in DS contributes to dysfunction amyloid processing, which, in turn, may culminate in an Alzheimer’s-like neurodegenerative process. This hypothesis, nevertheless, remain to be further investigated in future studies.
In the current analysis, we have primarily focused on studies utilizing human DS cells or on clinical studies on DS individuals (as opposed to animal studies). There are, in fact, multiple DS mouse models available (reviewed in Gupta et al. 2016; Herault et al. 2017; Muñiz Moreno et al. 2020), and these models do mimic some of the features of the human DS. However, in mice, the genes that are on human chromosome 21 are localized on 3 different chromosomes, and in most published studies utilizing various DS mice strains use animal models that do not overexpress all of the human-DS genes. Therefore, in some case these models are useful and potentially clinically translatable, while in others they may not be. For instance, one of the most commonly used mouse model (Ts65Dn) overexpresses DYRK1, and therefore it may be considered an adequate model of the human DS (at least as far as DYRK1-related processes are concerned). However, the same mouse not overexpress the mouse-equivalent of the human cbs gene; given the emerging evidence for the potential role of CBS and H2S in DS, the utility of this particular mouse strain to study H2S-related processes is questionable. Clearly, future animal studies of DS should utilize mice where the set of ‘gene dosage’ closely approximates the human condition. While such animals (containing segments of all relevant 3 extra mouse chromosomes) have been created, they are fragile and have significant fertility problems, limiting their utility for experimentation (Yu et al. 2010). There are also mouse models overexpressing the human chromosome 21, or part of it (O’Doherty et al. 2005; Miyamoto et al. 2014; Kazuki et al. 2020); while these animals have the ‘correct’ extra genes, these are human genes encoding human proteins: the interaction of these human proteins with the mouse proteins is likely to be different than human–human or mouse-mouse protein–protein interactions, which introduces a significant complicating factor. Clearly, the field of DS research requires better animal tools, and undoubtedly, such tools will be forthcoming in the future.
The radical therapy of DS, i.e. complete switching off the extra chromosome in all somatic cells is, of course, the logical “definitive therapeutic solution”. (Although, even in this case, one would have to consider what would be the ideal timing of such intervention, perhaps intrauterine, because by the time of birth, significant, perhaps irreversible developmental problems can occur). Such studies have been pursued by several laboratories and turning off chromosome 21 has been successful in cell-based models (Jiang et al. 2013; Mentis 2016; Chiang et al. 2018). However, it remains unclear if/when these technical advances (sometimes known as ‘chromosome therapy’) would be clinically translatable. In the meantime, continuing experimental studies, both omics-based and hypothesis-driven, are expected to further elucidate already identified targets, identify potentially translatable pharmacological or nutritional tools to target them, and to identify novel targets for pharmacological interventions in DS.
The (generally) disappointing experience with clinical trials in DS, so far, can be attributed to many different factors (Gardiner 2014; Lee et al. 2020a, b), some of these issues relate to clinical trial design (duration, dosage), while others may relate to the fact that possibly in the trials so far, the pathways/targets were not the most relevant for the pathogenesis of DS. Other reasons may include the relative ‘bluntness’ of the pharmacological tools tested so far (for instance, insufficient potency/specificity and/or suboptimal dose of the antioxidants used in the trials). From the pathomechanisms and associated therapeutic interventions discussed in “Changes in lipid catabolism in DS” section, in our opinion, for the therapeutic improvement of the pseudohypoxic state in DS, ROS scavengers/antioxidants (provided that agents of sufficient activity, perhaps catalytic antioxidants) may be worth for renewed attention. Moreover, future generations of DYRK1 inhibitors and/or CBS inhibitors may be promising approaches as they appear to target important checkpoints in the pathophysiology of DS. It is also conceivable that a complex condition such as DS requires simultaneous pharmacological intervention at multiple ‘levels’, similar to what is considered common practice, for instance, in cancer therapy. These issues remain to be tackled both at the level of future experimental and preclinical studies as well as during the next wave of clinical trials.
Conclusion
The metabolomic alterations, as shown in the above sections, as well as in the Additional file 1/Supplement, as summarized in Fig. 6, suggest that DS cells are in a pseudo-hypoxic state: they show pathophysiological alterations that resemble cellular responses to hypoxia, even though their oxygen supply is not disrupted. This alteration may importantly contribute a variety of functional deficits associated with DS. Several potential therapeutic approaches exist to counteract this DS-associated pseudohypoxia.
Fig. 6.

Metabolic and bioenergetic alterations in DS. Upregulated pathways and metabolites are highlighted in yellow. As mitochondrial oxidative phosphorylation is suppressed in DS, cells downregulate various proteins of the electron transport chain and re-wire their energetic pathways. Glycolysis is upregulated, producing ATP and as a byproduct, lactate. Because the mitochondrial NADH consumption is inhibited, NADH does not enter the mitochondria through the malate-aspartate cycle, but, rather, it is converted back to NAD+ by lactic fermentation. As pyruvate is transformed into lactate, it does not enter the cyclic acid cycle. Similarly, lipids are also inhibited to enter the Krebs cycle and they are accumulated in the form of acyl-carnitines. As another compensatory pathway, glutamine, glutamate and other amino acids enter the Krebs cycle as 2-oxoglutarate and generate GTP, which can immediately be transformed to ATP and NADH. To oxidize the NADH, the Krebs cycle can run in reverse direction, thus producing citrate. As a result, various metabolites and enzymes involved in cyclic acid cycle are upregulated. As cells use amino acids as an alternative energy source, excess ammonia is generated (this can, in turn, be eliminated via the urea cycle)
Supplementary information
Additional file 1. Supplementary material. Supplementary Fig. 1. Log2 transformation and normalization on raw datasets. The boxplot shows the distribution of plasma samples after log2 transformation. Data are from Caracausi et al. 2018. Supplementary Fig. 2. Log2 transformation and normalization on raw datasets. The boxplot shows the distribution of plasma samples after log2 transformation. Data are from Antonaros et al. 2020. Supplementary Fig. 3. Boxplot showing the distribution of plasma samples after log2 transformation. The authors (Caracausi et al. 2018) used Probabilistic Quotient Normalization (PQN). Supplementary Fig. 4. Boxplot showing the distribution of plasma samples after log2 transformation. Normalized and log2 transformed samples from the study of Powers et al. 2019. Supplementary Fig. 5. Targeted metabolomic result from Culp-Hill study (Culp-Hill et al. 2017). Supplementary Fig. 6. Untargeted metabolomic result from Culp-Hill study (Culp-Hill et al. 2017) showing significant technical variations. Supplementary Fig. 7. Untargeted metabolomic result from Culp-Hill study Culp-Hill et al. 2017). We applied variance stabilization normalization to reduce technical variations. Supplementary Table 1. Significant regression coefficients from a multiple linear regression model applied on each metabolite found in plasma. The raw data are from Caracausi’s study (Caracausi et al. 2018). Supplementary Table 2. Significant regression coefficients from a multiple linear regression model applied on each metabolite found in urine. The raw data are from Caracausi’s study (Caracausi et al. 2018). Supplementary Fig. 8. Energy charge values in DS cells vs. control. Supplementary Fig. 9. Meta-analysis of amino acids not included in the Main Text.
Acknowledgements
The editorial assistance of Dr. A. Marton is appreciated.
Abbreviations
- ATP
Adenosine-5′-triphosphate
- ADP
Adenosine-5′-diphosphate
- AMP
Adenosine-5′-monophosphate
- 2,3-DPG
2,3-Diphosphoglyceric acid
- GTP
Guanosine-5′-triphosphate
- NADH
Reduced nicotinamide adenine dinucleotide
- FADH2
Reduced flavin adenine dinucleotide
- NADPH
Reduced nicotinamide adenine dinucleotide phosphate
- CoA
Coenzyme A
- DS
Down syndrome
- DYRK1A
Dual-specificity tyrosine-phosphorylation regulated kinase 1A
- M
Mean
- Pi
Inorganic phosphate
- SD
Standard deviation
- SEM
Standard error of mean N sample size
- sqrt
Square root of
- ROS
Reactive oxygen species
- CBS
Cystathionine beta-synthase
Authors’ contributions
All authors performed literature review and writing. LP also conducted the meta-analysis. All authors read and approved the final manuscript.
Funding
Jerome LeJeune Foundation (Paris).
Availability of supporting data
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
None.
Footnotes
Technical footnote: with a few recent exceptions (Panagaki et al. 2019; Panagaki et al. 2020), most of the studies investigating mitochondrial function, or specific function of various mitochondrial complexes in DS utilized isolated mitochondrial preparations (in many cases also using high saturating concentrations of electron donors). These experiments would ‘miss’ the inhibitory effect of a gaseous mediator, such as H2S, which would no longer be present in the mitochondria after subcellular isolation and in vitro processing. Moreover, none of the studies focusing on the expression of mitochondrial electron complexes (e.g. Table 2) would “see” a H2S-mediated inhibition of Complex IV, because this occurs on the enzymatic activity level, and not on the level of protein expression. And yet, such an inhibitory effect can markedly suppress the activity of the entire mitochondrial electron transport, and, in our opinion, is therefore more functionally relevant than the expression (omics) type surveys.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s10020-020-00225-8.
References
- Adelekan T, Magge S, Shults J, Stallings V, Stettler N. Lipid profiles of children with Down syndrome compared with their siblings. Pediatrics. 2012;129(6):e1382–e1387. doi: 10.1542/peds.2011-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguiar AS, Jr, Tuon T, Albuquerque MM, Rocha GS, Speck AE, Araújo JC, et al. The exercise redox paradigm in the Down's syndrome: improvements in motor function and increases in blood oxidative status in young adults. J Neural Transm (Vienna) 2008;115(12):1643–1650. doi: 10.1007/s00702-008-0120-x. [DOI] [PubMed] [Google Scholar]
- Aivazidis S, Coughlan CM, Rauniyar AK, Jiang H, Liggett LA, Maclean KN, et al. The burden of trisomy 21 disrupts the proteostasis network in Down syndrome. PLoS ONE. 2017;12(4):e0176307. doi: 10.1371/journal.pone.0176307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annerén KG, Korenberg JR, Epstein CJ. Phosphofructokinase activity in fibroblasts aneuploid for chromosome 21. Hum Genet. 1987;76(1):63–65. doi: 10.1007/BF00283052. [DOI] [PubMed] [Google Scholar]
- Anon XX. Is mongolism a metabolic error? Br Med J. 1954;2(4891):802–803. [PubMed] [Google Scholar]
- Antonarakis SE, Skotko BG, Rafii MS, Strydo A, Pape SE, Bianchi DW, et al. Down syndrome. Nat Rev Dis Primers. 2020;6(1):9. doi: 10.1038/s41572-019-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonaros F, Ghini V, Pulina F, Ramacieri G, Cicchini E, Mannini E, et al. Plasma metabolome and cognitive skills in Down syndrome. Sci Rep. 2020;10(1):10491. doi: 10.1038/s41598-020-67195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya P, Waugh KA, Sullivan KD, Núñez NG, Roselli E, Smith KP, et al. Trisomy 21 dysregulates T cell lineages toward an autoimmunity-prone state associated with interferon hyperactivity. Proc Natl Acad Sci USA. 2019;116(48):24231–24241. doi: 10.1073/pnas.1908129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz H, Mohiuddin SS. Biochemistry. Hexose Monophosphate Pathway: StatPearls Publishing, Treasure Island, FL; 2020. [PubMed] [Google Scholar]
- Baikie A, Loder PB, De Gruchy G, Pitt D. Phosphohexokinase activity of erythrocytes in mongolism—another possible marker for chromosome 21. Lancet. 1965;1(7382):412–414. doi: 10.1016/S0140-6736(65)90007-3. [DOI] [PubMed] [Google Scholar]
- Bala U, Leong MP, Lim CL, Shahar HK, Othman F, Lai MI, et al. Defects in nerve conduction velocity and different muscle fibre-type specificity contribute to muscle weakness in Ts1Cje Down syndrome mouse model. PLoS ONE. 2018;13(5):e0197711. doi: 10.1371/journal.pone.0197711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balduzzi S, Rücker G, Schwarzer G. How to perform a meta-analysis with R: a practical tutorial. Evid Based Ment Health. 2019;22(4):153–160. doi: 10.1136/ebmental-2019-300117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bally BP, Farmer WT, Jones EV, Jessa S, Kacerovsky JB, Mayran A, et al. Human iPSC-derived Down syndrome astrocytes display genome-wide perturbations in gene expression, an altered adhesion profile, and increased cellular dynamics. Hum Mol Genet. 2020;29(5):785–802. doi: 10.1093/hmg/ddaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barañano KW, Hartman AL. The ketogenic diet: uses in epilepsy and other neurologic illnesses. Curr Treat Options Neurol. 2008;10(6):410–419. doi: 10.1007/s11940-008-0043-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barca D, Tarta-Arsene O, Dica A, Iliescu C, Budisteanu M, Motoescu C, et al. Intellectual disability and epilepsy in Down syndrome. Maedica (Buchar) 2014;9(4):344–350. [PMC free article] [PubMed] [Google Scholar]
- Bartels H. Blood adenine nucleotides in Down's syndrome. Blut. 1971;22(4):216–217. doi: 10.1007/BF01633617. [DOI] [PubMed] [Google Scholar]
- Bayer SM, McCoy EE. A comparison of the serotonin and ATP content in platelets from subjects with Down's syndrome. Biochem Med. 1974;9(3):225–232. doi: 10.1016/0006-2944(74)90056-8. [DOI] [PubMed] [Google Scholar]
- Belardinelli MC, Chabli A, Chadefaux-Vekemans B, Kamoun P. Urinary sulfur compounds in Down syndrome. Clin Chem. 2001;47(8):1500–1501. doi: 10.1093/clinchem/47.8.1500. [DOI] [PubMed] [Google Scholar]
- Bénit P, Letouzé E, Rak M, Aubry L, Burnichon N, Favier J, et al. Unsuspected task for an old team: succinate, fumarate and other Krebs cycle acids in metabolic remodeling. Biochim Biophys Acta. 2014;1837(8):1330–1337. doi: 10.1016/j.bbabio.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. Berlin: Springer; 2008. [Google Scholar]
- Bertapelli F, Pitetti K, Agiovlasitis S, Guerra-Junior G. Overweight and obesity in children and adolescents with Down syndrome—prevalence, determinants, consequences, and interventions: a literature review. Res Dev Disabil. 2016;57:181–192. doi: 10.1016/j.ridd.2016.06.018. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A, McMillan E, Chen SI, Wallace K, Svendsen CN. A critical period in cortical interneuron neurogenesis in down syndrome revealed by human neural progenitor cells. Dev Neurosci. 2009;31(6):497–510. doi: 10.1159/000236899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass JP, Sheu RK, Cedarbaum JM. Energy metabolism in disorders of the nervous system. Rev Neurol (Paris) 1988;144(10):543–563. [PubMed] [Google Scholar]
- Blehaut H, Mircher C, Ravel A, Conte M, De Portzamparc V, Poret G, et al. Effect of leucovorin (folinic acid) on the developmental quotient of children with Down's syndrome (trisomy 21) and influence of thyroid status. PLoS ONE. 2010;5(1):e8394. doi: 10.1371/journal.pone.0008394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer PH, de Beer Z. The effect of aquatic exercises on the physical and functional fitness of adults with Down syndrome: a non-randomised controlled trial. J Intellect Disabil Res. 2019;63(12):1453–1463. doi: 10.1111/jir.12687. [DOI] [PubMed] [Google Scholar]
- Bonora M, Patergnani S, Rimessi A, De Marchi E, Suski JM, Bononi A, et al. ATP synthesis and storage. Purinergic Signal. 2012;8(3):343–357. doi: 10.1007/s11302-012-9305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borenstein M, Hedges LV, Higgins JP, Rothstein HR. Introduction to meta-analysis. Hoboken: Wiley; 2011. [Google Scholar]
- Brault V, Duchon A, Romestaing C, Sahun I, Pothion S, Karout M, et al. Opposite phenotypes of muscle strength and locomotor function in mouse models of partial trisomy and monosomy 21 for the proximal Hspa13-App region. PLoS Genet. 2015;11(3):e1005062. doi: 10.1371/journal.pgen.1005062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breg WR. Down syndrome: a review of recent progress in research. Pathobiol Annu. 1977;7:257–303. [PubMed] [Google Scholar]
- Bronks R, Parker AW. Anthropometric observation of adults with Down syndrome. Am J Ment Defic. 1985;90(1):110–113. [PubMed] [Google Scholar]
- Bull MJ. Down syndrome. N Engl J Med. 2020;382(24):2344–2352. doi: 10.1056/NEJMra1706537. [DOI] [PubMed] [Google Scholar]
- Bull MJ. Committee on Genetics. Clinical report - Health supervision for children with Down syndrome. Pediatrics. 2011;128(2):393–406. doi: 10.1542/peds.2011-1605. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, et al. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron. 2002;33(5):677–688. doi: 10.1016/S0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- Çam H, Yildirim B, Aydin A, Say A. Carnitine levels in neonatal hypoxia. J Trop Pediatr. 2005;51(2):106–108. doi: 10.1093/tropej/fmh089. [DOI] [PubMed] [Google Scholar]
- Campos C, Guzmán R, López-Fernández E, Casado Á. Urinary uric acid and antioxidant capacity in children and adults with Down syndrome. Clin Biochem. 2010;43(3):228–233. doi: 10.1016/j.clinbiochem.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Capone G, Goyal P, Ares W, Lannigan E. Neurobehavioral disorders in children, adolescents, and young adults with Down syndrome. Am J Med Genet C Semin Med Genet. 2006;142C(3):158–172. doi: 10.1002/ajmg.c.30097. [DOI] [PubMed] [Google Scholar]
- Caracausi M, Ghini V, Locatelli C, Mericio M, Piovesan A, Antonaros F, et al. Plasma and urinary metabolomic profiles of Down syndrome correlate with alteration of mitochondrial metabolism. Sci Rep. 2018;8(1):2977. doi: 10.1038/s41598-018-20834-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeli E, Kessel S, Coleman R, Ayalon M. Effects of a treadmill walking program on muscle strength and balance in elderly people with Down syndrome. J Gerontol A Biol Sci Med Sci. 2002;57(2):M106–M110. doi: 10.1093/gerona/57.2.M106. [DOI] [PubMed] [Google Scholar]
- Carré JE, Singer M. Cellular energetic metabolism in sepsis: the need for a systems approach. Biochim Biophys Acta. 2008;1777(7–8):763–771. doi: 10.1016/j.bbabio.2008.04.024. [DOI] [PubMed] [Google Scholar]
- Chango A, Mircher C, James SJ, Réthoré MO, Nicolas JP. One carbon metabolism and trisomy 21: analysis of the genetic polymorphism. Ann Biol Clin (Paris) 2002;60(6):647–653. [PubMed] [Google Scholar]
- Chapman M, Stern J. (1964) Uric acid in Downs disease. J Ment Defic Res. 1964;8(Suppl):119–124. doi: 10.1111/j.1365-2788.1964.tb00806.x. [DOI] [PubMed] [Google Scholar]
- Chen C-P, Chen Y-H, Chern S-R, Chang S-J, Tsai T-L, Li S-H, et al. Placenta proteome analysis from Down syndrome pregnancies for biomarker discovery. Mol BioSyst. 2012;8(9):2360–2372. doi: 10.1039/c2mb25081k. [DOI] [PubMed] [Google Scholar]
- Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, et al. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018;27(5):1007–1025.e5. doi: 10.1016/j.cmet.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang JC, Jiang J, Newburger PE, Lawrence JB. Trisomy silencing by XIST normalizes Down syndrome cell pathogenesis demonstrated for hematopoietic defects in vitro. Nat Commun. 2018;9(1):5180. doi: 10.1038/s41467-018-07630-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515(7527):431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioni M, Cocilovo A, Di Pasquale F, Araujo MB, Siqueira CR, Bianco M. Strength deficit of knee extensor muscles of individuals with Down syndrome from childhood to adolescence. Am J Ment Retard. 1994;99(2):166–174. [PubMed] [Google Scholar]
- Cisterna B, Costanzo M, Scherini E, Zancanaro C, Malatesta M. Ultrastructural features of skeletal muscle in adult and aging Ts65Dn mice, a murine model of Down syndrome. Muscles Ligaments Tendons J. 2014;3(4):287–294. [PMC free article] [PubMed] [Google Scholar]
- Coelho-Junior HJ, Villani ER, Calvani R, Carfì A, Picca A, Landi F, et al. Sarcopenia-related parameters in adults with Down syndrome: a cross-sectional exploratory study. Exp Gerontol. 2019;119:93–99. doi: 10.1016/j.exger.2019.01.028. [DOI] [PubMed] [Google Scholar]
- Contestabile A, Benfenati F, Gasparini L. Communication breaks-Down: from neurodevelopment defects to cognitive disabilities in Down syndrome. Prog Neurobiol. 2010;91(1):1–22. doi: 10.1016/j.pneurobio.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Contestabile A, Fila T, Ceccarelli C, Bonasoni P, Bonapace L, Santini D, et al. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus. 2007;17(8):665–678. doi: 10.1002/hipo.20308. [DOI] [PubMed] [Google Scholar]
- Conti A, Fabbrini F, D'Agostino P, Negri R, Greco D, Genesio R, et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genomics. 2007;8:268. doi: 10.1186/1471-2164-8-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertini P, Menga A, Andria G, Scala I, Santarsiero A, Castiglione Morelli MA, et al. The contribution of the citrate pathway to oxidative stress in Down syndrome. Immunology. 2016;149(4):423–431. doi: 10.1111/imm.12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway MM, Layzer RB. Blood cell phosphofructokinase in Down's syndrome. Humangenetik. 1970;9(2):185–190. doi: 10.1007/BF00278934. [DOI] [PubMed] [Google Scholar]
- Coppedè F. The complex relationship between folate/homocysteine metabolism and risk of Down syndrome. Mutat Res. 2009;682(1):54–70. doi: 10.1016/j.mrrev.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Coppus AW, Fekkes D, Verhoeven WM, Tuinier S, Egger JI, van Duijn CM. Plasma amino acids and neopterin in healthy persons with Down's syndrome. J Neural Transm (Vienna) 2007;114(8):1041–1045. doi: 10.1007/s00702-007-0656-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet C, Feron O. (2015) Metabolic and mind shifts: from glucose to glutamine and acetate addictions in cancer. Curr Opin Clin Nutr Metab Care. 2015;18(4):346–353. doi: 10.1097/MCO.0000000000000178. [DOI] [PubMed] [Google Scholar]
- Corsi MM, Dogliotti G, Pedroni F, Galliera E, Malavazos AE, Villa R, et al. Adipocytokines in Down's syndrome, an atheroma-free model: Role of adiponectin. Arch Gerontol Geriatr. 2009;48(1):106–109. doi: 10.1016/j.archger.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Coskun P, Helguera P, Nemati Z, Bohannan RC, Thomas J, Samuel SE, et al. Metabolic and growth rate alterations in lymphoblastic cell lines discriminate between Down syndrome and Alzheimer's disease. J Alzheimers Dis. 2017;55(2):737–748. doi: 10.3233/JAD-160278. [DOI] [PubMed] [Google Scholar]
- Cowley PM, Keslacy S, Middleton FA, DeRuisseau LR, Fernhall B, Kanaley JA, et al. Functional and biochemical characterization of soleus muscle in Down syndrome mice: insight into the muscle dysfunction seen in the human condition. Am J Physiol Regul Integr Comp Physiol. 2012;303(12):R1251–R1260. doi: 10.1152/ajpregu.00312.2012. [DOI] [PubMed] [Google Scholar]
- Cowley PM, Nair DR, DeRuisseau LR, Keslacy S, Atalay M, DeRuisseau KC. Oxidant production and SOD1 protein expression in single skeletal myofibers from Down syndrome mice. Redox Biol. 2017;13:421–425. doi: 10.1016/j.redox.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce RV, Pitetti KH, Horvat M, Miller J. Peak torque, average power, and hamstrings/quadriceps ratios in nondisabled adults and adults with mental retardation. Arch Phys Med Rehabil. 1996;77(4):369–372. doi: 10.1016/S0003-9993(96)90086-6. [DOI] [PubMed] [Google Scholar]
- Culp-Hill R, Zheng C, Reisz JA, Smith K, Rachubinski A, Nemkov T, et al. Red blood cell metabolism in Down syndrome: hints on metabolic derangements in aging. Blood Adv. 2017;1(27):2776–2780. doi: 10.1182/bloodadvances.2017011957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashty M. (2013) A quick look at biochemistry: carbohydrate metabolism. Clin Biochem. 2013;46(15):1339–1352. doi: 10.1016/j.clinbiochem.2013.04.027. [DOI] [PubMed] [Google Scholar]
- de Asua DR, Parra P, Costa R, Moldenhauer F, Suarez C. Evaluation of the impact of abdominal obesity on glucose and lipid metabolism disorders in adults with Down syndrome. Res Dev Disabil. 2014;35(11):2942–2949. doi: 10.1016/j.ridd.2014.07.038. [DOI] [PubMed] [Google Scholar]
- de Asua DR, Parra P, Costa R, Moldenhauer F, Suarez C. A cross-sectional study of the phenotypes of obesity and insulin resistance in adults with down syndrome. Diabetes Metab J. 2014;38(6):464–471. doi: 10.4093/dmj.2014.38.6.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre R, de Sola S, Hernandez G, Farré M, Pujol J, Rodriguez J, et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down's syndrome (TESDAD): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(8):801–810. doi: 10.1016/S1474-4422(16)30034-5. [DOI] [PubMed] [Google Scholar]
- de la Torre R, de Sola S, Pons M, Duchon A, de Lagran MM, Farré M, et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol Nutr Food Res. 2014;58(2):278–288. doi: 10.1002/mnfr.201300325. [DOI] [PubMed] [Google Scholar]
- De Matteo A, Vajro P. Down syndrome and pediatric nonalcoholic fatty liver disease: a causal or casual relationship? J Pediatr. 2017;189:11–13. doi: 10.1016/j.jpeds.2017.07.011. [DOI] [PubMed] [Google Scholar]
- de Sousa MC, Vieira RB, dos Santos DS, Carvalho CAT, Camargo SEA, Mancini MNG, et al. Antioxidants and biomarkers of oxidative damage in the saliva of patients with Down's syndrome. Arch Oral Biol. 2015;60(4):600–605. doi: 10.1016/j.archoralbio.2014.09.013. [DOI] [PubMed] [Google Scholar]
- Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787(11):1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Dodd KJ, Shields N. A systematic review of the outcomes of cardiovascular exercise programs for people with Down syndrome. Arch Phys Med Rehab. 2005;86(10):2051–2058. doi: 10.1016/j.apmr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Drake JC, Wilson RJ, Yan Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. 2016;30(1):13–22. doi: 10.1096/fj.15-276337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druzhyna N, Nair RG, LeDoux SP, Wilson GL. Defective repair of oxidative damage in mitochondrial DNA in Down's syndrome. Mutat Res. 1998;409(2):81–89. doi: 10.1016/S0921-8777(98)00042-1. [DOI] [PubMed] [Google Scholar]
- Dunn J, Grider MH. Physiology, Adenosine Triphosphate (ATP) Treasure Island: StatPearls Publishing; 2020. [PubMed] [Google Scholar]
- Dupre C, Weidman-Evans E. Musculoskeletal development in patients with Down syndrome. JAAPA. 2017;30(12):38–40. doi: 10.1097/01.JAA.0000526779.77230.79. [DOI] [PubMed] [Google Scholar]
- Eales K, Hollinshead K, Tennant D. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis. 2016;5(1):e190. doi: 10.1038/oncsis.2015.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egekeze JO, Oehme FW. Cyanides and their toxicity: a literature review. Vet Q. 1980;2(2):104–114. doi: 10.1080/01652176.1980.9693766. [DOI] [PubMed] [Google Scholar]
- Ellis JM, Tan HK, Gilbert RE, Muller DP, Henley W, Moy R, et al. Supplementation with antioxidants and folinic acid for children with Down’s syndrome: randomised controlled trial. BMJ. 2008;336(7644):594–597. doi: 10.1136/bmj.39465.544028.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshorbagy AK, Smith AD, Kozichm V, Refsum H. Cysteine and obesity. Obesity (Silver Spring) 2012;20(3):473–481. doi: 10.1038/oby.2011.93. [DOI] [PubMed] [Google Scholar]
- Ferreira IL, Resende R, Ferreiro E, Rego AC, Pereira CF. Multiple defects in energy metabolism in Alzheimer's disease. Curr Drug Targets. 2010;11(10):1193–1206. doi: 10.2174/1389450111007011193. [DOI] [PubMed] [Google Scholar]
- Fiedler N, Kipen H, Ohman-Strickland P, Zhang J, Weisel C, Laumbach R, et al. Sensory and cognitive effects of acute exposure to hydrogen sulfide. Environ Health Perspect. 2008;116(1):78–85. doi: 10.1289/ehp.10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folbergrovà J, Nilsson B, Sakabe T, Siesjö BK. The influence of hypoxia on the concentrations of cyclic nucleotides in the rat brain. J Neurochem. 1981;36(5):1670–1674. doi: 10.1111/j.1471-4159.1981.tb00417.x. [DOI] [PubMed] [Google Scholar]
- Fonseca CT, Amaral DM, Ribeiro MG, Beserra IC, Guimarães MM. Insulin resistance in adolescents with Down syndrome: a cross-sectional study. BMC Endocr Disord. 2005;5:6. doi: 10.1186/1472-6823-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frakes RA, Sharma RP, Willhite CC, Gomez G. Effect of cyanogenic glycosides and protein content in cassava diets on hamster prenatal development. Fundam Appl Toxicol. 1986;7(2):191–198. doi: 10.1016/0272-0590(86)90147-8. [DOI] [PubMed] [Google Scholar]
- Fuller RW, Luce MW, Mertz ET. Serum uric acid in mongolism. Science. 1962;137(3533):868–869. doi: 10.1126/science.137.3533.868. [DOI] [PubMed] [Google Scholar]
- Funes HA, Apostolova N, Alegre F, Blas-Garcia A, Alvarez A, Marti-Cabrera M, et al. Neuronal bioenergetics and acute mitochondrial dysfunction: a clue to understanding the central nervous system side effects of efavirenz. J Infect Dis. 2014;210(9):1385–1395. doi: 10.1093/infdis/jiu273. [DOI] [PubMed] [Google Scholar]
- Gabuzda D, Busciglio J, Chen LB, Matsudaira P, Yankner BA. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem. 1994;269(18):13623–13628. [PubMed] [Google Scholar]
- Gardiner KJ. Pharmacological approaches to improving cognitive function in Down syndrome: current status and considerations. Drug Des Devel Ther. 2014;9:103–125. doi: 10.2147/DDDT.S51476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass TJ, Connor NP. Digastric muscle phenotypes of the Ts65Dn mouse model of Down syndrome. PLoS ONE. 2016;11(6):e0158008. doi: 10.1371/journal.pone.0158008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Agüero A, Ara I, Moreno LA, Vicente-Rodríguez G, Casajús JA. Fat and lean masses in youths with Down syndrome: gender differences. Res Dev Disabil. 2011;32(5):1685–1693. doi: 10.1016/j.ridd.2011.02.023. [DOI] [PubMed] [Google Scholar]
- Grieco J, Pulsifer M, Seligsohn K, Skotko B, Schwartz A. Down syndrome: cognitive and behavioral functioning across the lifespan. Am J Med Genet C Semin Med Genet. 2015;169(2):135–149. doi: 10.1002/ajmg.c.31439. [DOI] [PubMed] [Google Scholar]
- Gross TJ, Doran E, Cheema AK, Head E, Lott IT, Mapstone M. Plasma metabolites related to cellular energy metabolism are altered in adults with Down syndrome and Alzheimer's disease. Dev Neurobiol. 2019;79(7):622–638. doi: 10.1002/dneu.22716. [DOI] [PubMed] [Google Scholar]
- Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285(17):13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Dhanasekaran AR, Gardiner KJ. Mouse models of Down syndrome: gene content and consequences. Mamm Genome. 2016;27(11–12):538–555. doi: 10.1007/s00335-016-9661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad A, Mohiuddin SS. Biochemistry Citric Acid Cycle. Treasure Island: StatPearls Publishing; 2019. [PubMed] [Google Scholar]
- Hannah RS, Roth SH. Chronic exposure to low concentrations of hydrogen sulfide produces abnormal growth in developing cerebellar Purkinje cells. Neurosci Lett. 1991;122(2):225–228. doi: 10.1016/0304-3940(91)90864-P. [DOI] [PubMed] [Google Scholar]
- Hawli Y, Nasrallah M, El-Hajj FG. Endocrine and musculoskeletal abnormalities in patients with Down syndrome. Nat Rev Endocrinol. 2009;5(6):327–334. doi: 10.1038/nrendo.2009.80. [DOI] [PubMed] [Google Scholar]
- Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, et al. A cannabinoid link between mitochondria and memory. Nature. 2016;539(7630):555–559. doi: 10.1038/nature20127. [DOI] [PubMed] [Google Scholar]
- Hedges LV. Distribution theory for Glass's estimator of effect size and related estimators. J Stat Educ. 1981;6(2):107–128. doi: 10.3102/10769986006002107. [DOI] [Google Scholar]
- Heggarty H, Ball R, Smith M, Henderson M. Amino acid profile in Down's syndrome. Arch Dis Child. 1996;74(4):347–349. doi: 10.1136/adc.74.4.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helguera P, Seiglie J, Rodriguez J, Hanna M, Helguera G, Busciglio J. Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab. 2013;17(1):132–140. doi: 10.1016/j.cmet.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herault Y, Delabar JM, Fisher EMC, Tybulewicz VLJ, Yu E, Brault V. Rodent models in Down syndrome research: impact and future opportunities. Dis Model Mech. 2017;10(10):1165–1186. doi: 10.1242/dmm.029728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JPT, Green S. Cochrane handbook for systematic reviews of interventions. Chichester: Wiley; 2008. [Google Scholar]
- Hill DL, Parks EP, Zemel BS, Shults J, Stallings VA, Stettler N. Resting energy expenditure and adiposity accretion among children with Down syndrome: a 3-year prospective study. Eur J Clin Nutr. 2013;67(10):1087–1091. doi: 10.1038/ejcn.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood DA, Tryon LD, Carter HN, Kim Y, Chen CC. Unravelling the mechanisms regulating muscle mitochondrial biogenesis. Biochem J. 2016;473(15):2295–2314. doi: 10.1042/BCJ20160009. [DOI] [PubMed] [Google Scholar]
- Howell A, Mason A, Brown E, Watts R, Chanarin I, McPherson K, et al. Red cell size and uric acid in Down's syndrome. Scand J Haematol. 1973;11(2):140–147. doi: 10.1111/j.1600-0609.1973.tb00108.x. [DOI] [PubMed] [Google Scholar]
- Hoyer S. Brain glucose and energy metabolism during normal aging. Aging (Milano) 1990;2(3):245–258. doi: 10.1007/BF03323925. [DOI] [PubMed] [Google Scholar]
- Hoyer S. Abnormalities in brain glucose utilization and its impact on cellular and molecular mechanisms in sporadic dementia of Alzheimer type. Ann NY Acad Sci. 1993;695:77–80. doi: 10.1111/j.1749-6632.1993.tb23032.x. [DOI] [PubMed] [Google Scholar]
- Huber W, Von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18(S1):S96–104. doi: 10.1093/bioinformatics/18.suppl_1.S96. [DOI] [PubMed] [Google Scholar]
- Huo HQ, Qu ZY, Yuan F, Ma L, Yao L, Xu M, et al. Modeling Down syndrome with patient iPSCs reveals cellular and migration deficits of GABAergic neurons. Stem Cell Reports. 2018;10(4):1251–1266. doi: 10.1016/j.stemcr.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoe A, Roller BA, Elangovan A, Kaelber KL, Kaelber D. A case series of gout and Downs Syndrome—a new paradigm for detecting disease associations using big data. In: Arthritis and Rheumatology, Vol 69. Hoboken: Wiley. 2017.
- Infantino V, Castegna A, Iacobazzi F, Spera I, Scala I, Andria G, et al. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down's syndrome. Mol Genet Metab. 2011;102(3):378–382. doi: 10.1016/j.ymgme.2010.11.166. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Akiba S. A comprehensive diverse ‘-omics’ approach to better understanding the molecular pathomechanisms of down syndrome. Brain Sci. 2017;7(4):44. doi: 10.3390/brainsci7040044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo A, Mollo N, Nitti M, Paladino S, Calì G, Genesio R, et al. Mitochondrial dysfunction in down syndrome: molecular mechanisms and therapeutic targets. Mol Med. 2018;24(1):2. doi: 10.1186/s10020-018-0004-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo A, Nitti M, Mollo N, Paladino S, Procaccini C, Faicchia D, et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum Mol Genet. 2017;26(6):1056–1069. doi: 10.1093/hmg/ddx016. [DOI] [PubMed] [Google Scholar]
- Jablonska B, Ford D, Trisler D, Pessac B. The growth capacity of bone marrow CD34 positive cells in culture is drastically reduced in a murine model of Down syndrome. C R Biol. 2006;329(9):726–732. doi: 10.1016/j.crvi.2006.06.004. [DOI] [PubMed] [Google Scholar]
- James JH, Luchette FA, McCarter FD, Fischer JE. Lactate is an unreliable indicator of tissue hypoxia in injury or sepsis. Lancet. 1999;354(9177):505–508. doi: 10.1016/S0140-6736(98)91132-1. [DOI] [PubMed] [Google Scholar]
- Jiang J, Jing Y, Cost GJ, Chiang J-C, Kolpa HJ, Cotton AM, et al. Translating dosage compensation to trisomy 21. Nature. 2013;500(7462):296–300. doi: 10.1038/nature12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Srivastava S, Zhang J. Starve cancer cells of glutamine: Break the spell or make a hungry monster? Cancers (Basel) 2019;11(6):804. doi: 10.3390/cancers11060804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamoun PP. Mental retardation in Down syndrome: two ways to treat. Med Hypotheses. 2019;131:109289. [DOI] [PubMed]
- Kamoun P, Belardinelli MC, Chabli A, Lallouchi K, Chadefaux-Vekemans B. Endogenous hydrogen sulfide overproduction in Down syndrome. Am J Med Genet A. 2003;116A(3):310–311. doi: 10.1002/ajmg.a.10847. [DOI] [PubMed] [Google Scholar]
- Kaneko KN, Wong M, Corley MJ, Lee RWY. The ketogenic diet as a potential therapy in Down syndrome. J Pediatr Pediatr Med. 2018;2(2):11–17. doi: 10.29245/2578-2940/2018/2.1121. [DOI] [Google Scholar]
- Kaufman JM, O'Brien WM. Hyperuricemia in mongolism. N Engl J Med. 1967;276(17):953–956. doi: 10.1056/NEJM196704272761704. [DOI] [PubMed] [Google Scholar]
- Kazuki Y, Gao FJ, Li Y, Moyer AJ, Devenney B, Hiramatsu K, et al. A non-mosaic transchromosomic mouse model of Down syndrome carrying the long arm of human chromosome 21. Elife. 2020;9:e56223. doi: 10.7554/eLife.56223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedziora J, Hübner H, Kanski M, Jeske J, Leyko W. Efficiency of the glycolytic pathway in erythrocytes of children with Down's syndrome. Pediatr Res. 1972;6(1):10–17. doi: 10.1203/00006450-197201000-00002. [DOI] [PubMed] [Google Scholar]
- Kim H, Lee K-S, Kim A-K, Choi M, Choi K, Kang M, et al. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Dis Models Mech. 2016;9(8):839–848. doi: 10.1242/dmm.025668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G. The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer's disease. Life Sci. 2001;68(24):2741–2750. doi: 10.1016/S0024-3205(01)01074-8. [DOI] [PubMed] [Google Scholar]
- Kimura R, Kamino K, Yamamoto M, Nuripa A, Kida T, Kazui H, et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum Mol Genet. 2007;16(1):15–23. doi: 10.1093/hmg/ddl437. [DOI] [PubMed] [Google Scholar]
- Knull HR, Bronstein WW, Porter PJ. Adenosine triphosphate and diphosphoglycerate levels in red blood cells from patients with Down's syndrome. Experientia. 1978;34(9):1133–1134. doi: 10.1007/BF01922912. [DOI] [PubMed] [Google Scholar]
- Lagan N, Huggard D, Mc Grane F, Leahy TR, Franklin O, Roche E, et al. Multiorgan involvement and management in children with Down syndrome. Acta Paediatr. 2020;109(6):1096–1111. doi: 10.1111/apa.15153. [DOI] [PubMed] [Google Scholar]
- Larsen EL, Padella L, Bergholdt HKM, Henriksen T, Santoro L, Gabrielli O, et al. The effect of long-term treatment with coenzyme Q10 on nucleic acid modifications by oxidation in children with Down syndrome. Neurobiol Aging. 2018;67:159–161. doi: 10.1016/j.neurobiolaging.2018.03.001. [DOI] [PubMed] [Google Scholar]
- Layzer RB, Epstein CJ. Phosphofructokinase and chromosome 21. Am J Hum Genet. 1972;24(5):533–543. [PMC free article] [PubMed] [Google Scholar]
- Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. 2020;21(5):268–283. doi: 10.1038/s41580-020-0227-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Duran-Martinez M, Khantsis S, Bianchi DW, Guedj F. Challenges and opportunities for translation of therapies to improve cognition in Down syndrome. Trends Mol Med. 2020;26(2):150–169. doi: 10.1016/j.molmed.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Lee S, Jun HS, Jeong HJ, Cha WT, Cho YS, et al. Expression of the mitochondrial ATPase6 gene and Tfam in Down syndrome. Mol Cells. 2003;15(2):181–185. [PubMed] [Google Scholar]
- Lejeune J. Pathogenesis of mental deficiency in trisomy 21. Am J Med Genet Suppl. 1990;7:20–30. doi: 10.1002/ajmg.1320370705. [DOI] [PubMed] [Google Scholar]
- Lejeune J, Rethoré MO, de Blois MC, Peeters M, Naffah J, Megarbane A, et al. Amino acids and trisomy 21. Ann Genet. 1992;35(1):8–13. [PubMed] [Google Scholar]
- Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, et al. Domains of genome-wide gene expression dysregulation in Down's syndrome. Nature. 2014;508(7496):345–350. doi: 10.1038/nature13200. [DOI] [PubMed] [Google Scholar]
- Li B, Tang J, Yang Q, Li S, Cui X, Li Y, et al. NOREVA: normalization and evaluation of MS-based metabolomics data. Nucleic Acids Res. 2017;45(W1):W162–W170. doi: 10.1093/nar/gkx449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Borel C, Li L, Müller T, Williams EG, Germain P-L, et al. Systematic proteome and proteostasis profiling in human Trisomy 21 fibroblast cells. Nat Commun. 2017;8(1):1212. doi: 10.1038/s41467-017-01422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Suárez O, Concheiro-Guisán A, Sánchez-Pintos P, Cocho JA, Lorenzo JRF, Couce ML. Acylcarnitine profile in neonatal hypoxic-ischemic encephalopathy: the value of butyrylcarnitine as a prognostic marker. Medicine (Baltimore) 2019;98(15):e15221. doi: 10.1097/MD.0000000000015221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT, Doran E, Nguyen VQ, Tournay A, Head E, Gillen DL. Down syndrome and dementia: a randomized, controlled trial of antioxidant supplementation. Am J Med Genet A. 2011;155A(8):1939–1948. doi: 10.1002/ajmg.a.34114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT, Head E, Doran E, Busciglio J. Beta-amyloid, oxidative stress and Down syndrome. Curr Alzheimer Res. 2006;3(5):521–528. doi: 10.2174/156720506779025305. [DOI] [PubMed] [Google Scholar]
- Lukowski AF, Milojevich HM, Eales L. Cognitive functioning in children with Down syndrome: current knowledge and future directions. Adv Child Dev Behav. 2019;56:257–289. doi: 10.1016/bs.acdb.2019.01.002. [DOI] [PubMed] [Google Scholar]
- Lumley J, Watson L, Watson M, Bower C. Periconceptional supplementation with folate and/or multivitamins for preventing neural tube defects. Cochrane Database Syst Rev. 2001;1:001056. doi: 10.1002/14651858.CD001056. [DOI] [PubMed] [Google Scholar]
- MacDonald R. Red cell 2,3-diphosphoglycerate and oxygen affinity. Anaesthesia. 1977;32(6):544–553. doi: 10.1111/j.1365-2044.1977.tb10002.x. [DOI] [PubMed] [Google Scholar]
- Magge SN, O’Neill KL, Shults J, Stallings VA, Stettler N. Leptin levels among prepubertal children with Down syndrome compared with their siblings. J Pediatr. 2008;152(3):321–326. doi: 10.1016/j.jpeds.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni P, Ruscica M, Dozio E, Roti E, Licastro F, Motta M, et al. Free and bound leptin in prepubertal children with Down's syndrome and different degrees of adiposity. Eur J Clin Nutr. 2004;58(11):1547–1549. doi: 10.1038/sj.ejcn.1602000. [DOI] [PubMed] [Google Scholar]
- Malegiannaki AC, Katsarou D, Liolios A, Zisi V. Ageing and Down syndrome: neurocognitive characteristics and pharmacological treatment. Hell J Nucl Med. 2019;22(Suppl):123–132. [PubMed] [Google Scholar]
- Maluf SW, Erdtmann B. Genomic instability in Down syndrome and Fanconi anemia assessed by micronucleus analysis and single-cell gel electrophoresis. Cancer Genet Cytogenet. 2001;124(1):71–75. doi: 10.1016/S0165-4608(00)00322-8. [DOI] [PubMed] [Google Scholar]
- Mao R, Wang X, Spitznagel EL, Jr, Frelin LP, Ting JC, Ding H, et al. Primary and secondary transcriptional effects in the developing human Down syndrome brain and heart. Genome Biol. 2005;6(13):R107. doi: 10.1186/gb-2005-6-13-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal D, Brault V, Leon A, Martin D, Lopes Pereira P, Loaëc N, et al. CBS overdosage is necessary and sufficient to induce cognitive phenotypes in mouse models of Down syndrome and interacts genetically with Dyrk1a. Hum Mol Genet. 2019;28(9):1561–1577. doi: 10.1093/hmg/ddy447. [DOI] [PubMed] [Google Scholar]
- Martínez-Cué C, Rueda N. Cellular senescence in neurodegenerative diseases. Front Cell Neurosci. 2020;14:16. doi: 10.3389/fncel.2020.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Ruge W, Bertoni-Freddari C, Iwangoff P. Changes in brain glucose metabolism as a key to the pathogenesis of Alzheimer's disease. Gerontology. 1994;40(5):246–252. doi: 10.1159/000213592. [DOI] [PubMed] [Google Scholar]
- Melkonian EA, Schury MP. Biochemistry, Anaerobic Glycolysis. Treasure Island: StatPearls Publishing; 2019. [PubMed] [Google Scholar]
- Melville CA, Cooper SA, McGrother CW, Thorp CF, Collacott R. Obesity in adults with Down syndrome: a case-control study. J Intellect Disabil Res. 2005;49(Pt 2):125–133. doi: 10.1111/j.1365-2788.2004.00616.x. [DOI] [PubMed] [Google Scholar]
- Mendonca GV, Borges A, Wee SO, Fernhall B. Oxygen uptake efficiency slope during exercise in adults with Down syndrome. J Appl Res Intellect Disabil. 2018;31(5):897–904. doi: 10.1111/jar.12449. [DOI] [PubMed] [Google Scholar]
- Mendonca GV, Pereira FD, Fernhall B. Reduced exercise capacity in persons with Down syndrome: cause, effect, and management. Ther Clin Risk Manag. 2010;6:601–610. doi: 10.2147/TCRM.S10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006;61(6):534–540. doi: 10.1093/gerona/61.6.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis AF. Epigenomic engineering for Down syndrome. Neurosci Biobehav Rev. 2016;71:323–327. doi: 10.1016/j.neubiorev.2016.09.012. [DOI] [PubMed] [Google Scholar]
- Mertz ET, Fuller RW, Concon JM. Serum uric acid in young mongoloids. Science. 1963;141(3580):535. doi: 10.1126/science.141.3580.535. [DOI] [PubMed] [Google Scholar]
- Miles MV, Patterson BJ, Chalfonte-Evans ML, Horn PS, Hickey FJ, Schapiro MB, et al. Coenzyme Q10 (ubiquinol-10) supplementation improves oxidative imbalance in children with trisomy 21. Pediatr Neurol. 2007;37(6):398–403. doi: 10.1016/j.pediatrneurol.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Mircher C, Salabelle A, Peeters MA, Rabier D, Parvy P, Kamoun P, et al. Variation of amino acids in relation to age in Down syndrome subjects. Arch Pediatr. 1997;4(11):1093–1099. doi: 10.1016/S0929-693X(97)88974-9. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Suzuki N, Sakai K, Asakawa S, Okazaki T, Kudoh J, et al. A novel mouse model for Down syndrome that harbor a single copy of human artificial chromosome (HAC) carrying a limited number of genes from human chromosome 21. Transgenic Res. 2014;23(2):317–329. doi: 10.1007/s11248-013-9772-x. [DOI] [PubMed] [Google Scholar]
- Mizuno GO, Wang Y, Shi G, Wang Y, Sun J, Papadopoulos S, et al. Aberrant calcium signaling in astrocytes inhibits neuronal excitability in a human Down syndrome stem cell model. Cell Rep. 2018;24(2):355–365. doi: 10.1016/j.celrep.2018.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollo N, Nitti M, Zerillo L, Faicchia D, Micillo T, Accarino R, et al. Pioglitazone improves mitochondrial organization and bioenergetics in Down syndrome cells. Front Genet. 2019;10:606. doi: 10.3389/fgene.2019.00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor I, Cheung E, Vousden K. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harbor Laboratory Press, Woodbury, NY. 2011;76:211–216. doi: 10.1101/sqb.2011.76.010868. [DOI] [PubMed] [Google Scholar]
- Muñiz Moreno MDM, Brault V, Birling MC, Pavlovic G, Herault Y. Modeling Down syndrome in animals from the early stage to the 4.0 models and next. Prog Brain Res. 2020;251:91–143. doi: 10.1016/bs.pbr.2019.08.001. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Kamata O, Ukai K, Yamamoto K. Inhibitory effects of NMDA receptor antagonists on hypoxia-induced seizures in dietary Mg^+- deficient mice. Eur J Pharmacol. 1991;204(1):29–34. doi: 10.1016/0014-2999(91)90831-A. [DOI] [PubMed] [Google Scholar]
- Nam B, Kim H, Choi Y, Lee H, Hong E-S, Park J-K, et al. Neurologic sequela of hydrogen sulfide poisoning. Ind Health. 2004;42(1):83–87. doi: 10.2486/indhealth.42.83. [DOI] [PubMed] [Google Scholar]
- Necchi D, Pinto A, Tillhon M, Dutto I, Serafini MM, Lanni C, et al. Defective DNA repair and increased chromatin binding of DNA repair factors in Down syndrome fibroblasts. Mutat Res. 2015;780:15–23. doi: 10.1016/j.mrfmmm.2015.07.009. [DOI] [PubMed] [Google Scholar]
- Nelson DL, Cox MM, Lehninger AL. Principles of Biochemistry. New York: W.H Freeman; 2008. [Google Scholar]
- Nelson E, Benson PF. Elevated erythrocyte 2,3-diphosphoglycerate concentration in primary trisomic Down's syndrome. Arch Dis Child. 1972;47(254):672. doi: 10.1136/adc.47.254.672-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TL, Duchon A, Manousopoulou A, Loaëc N, Villiers B, Pani G, et al. Correction of cognitive deficits in mouse models of Down syndrome by a pharmacological inhibitor of DYRK1A. Dis Model Mech. 2018;11(9):035634. doi: 10.1242/dmm.035634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Brand MD, Gerencser AA. Mitochondrial bioenergetics and neuronal survival modelled in primary neuronal culture and isolated nerve terminals. J Bioenerg Biomembr. 2015;47(1–2):63–74. doi: 10.1007/s10863-014-9573-9. [DOI] [PubMed] [Google Scholar]
- Nicholls P, Marshall DC, Cooper CE, Wilson MT. Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem Soc Trans. 2013;41(5):1312–1316. doi: 10.1042/BST20130070. [DOI] [PubMed] [Google Scholar]
- Nicholson RA, Roth SH, Zhang A, Zheng J, Brookes J, Skrajny B, et al. Inhibition of respiratory and bioenergetic mechanisms by hydrogen sulfide in mammalian brain. J Toxicol Environ Health. 1998;54(6):491–507. doi: 10.1080/009841098158773. [DOI] [PubMed] [Google Scholar]
- Noll C, Planque C, Ripoll C, Guedj F, Diez A, Ducros V, et al. DYRK1A, a novel determinant of the methionine-homocysteine cycle in different mouse models overexpressing this Down-syndrome-associated kinase. PLoS ONE. 2009;4(10):e7540. doi: 10.1371/journal.pone.0007540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nura NM, Nuri MSh, Zienat Kh. Amino acids profile in mentally retarded Libyan children. Bull Egypt Soc Physiol Sci. 2008;28:1–10. [Google Scholar]
- O'Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, et al. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309(5743):2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid R, Hartmuth K, Herrmann W, Gortner L, Rohrer TR, Geisel J, et al. Blood biomarkers of methylation in Down syndrome and metabolic simulations using a mathematical model. Mol Nutr Food Res. 2012;56(10):1582–1589. doi: 10.1002/mnfr.201200162. [DOI] [PubMed] [Google Scholar]
- Orozco JS, Hertz-Picciotto I, Abbeduto L, Slupsky CM. Metabolomics analysis of children with autism, idiopathic-developmental delays, and Down syndrome. Transl Psychiatry. 2019;9(1):243. doi: 10.1038/s41398-019-0578-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozanturk E, Ucar ZZ, Varol Y, Koca H, Demir AU, Kalenci D, et al. Urinary uric acid excretion as an indicator of severe hypoxia and mortality in patients with obstructive sleep apnea and chronic obstructive pulmonary disease. Rev Port Pneumol. 2016;22(1):18–26. doi: 10.1016/j.rppnen.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Panagaki T, Randi EB, Szabo C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation and cellular bioenergetics in human Down syndrome fibroblasts. Biomolecules. 2020;10(4):653. doi: 10.3390/biom10040653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagaki T, Randi EB, Augsburger F, Szabo C. Overproduction of H2S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc Natl Acad Sci USA. 2019;116(38):18769–18771. doi: 10.1073/pnas.1911895116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant SS, Moser HW, Krane SM. Hyperuricemia in Down's syndrome. J Clin Endocrinol Metab. 1968;28(4):472–478. doi: 10.1210/jcem-28-4-472. [DOI] [PubMed] [Google Scholar]
- Parisotto EB, Giaretta AG, Zamoner A, Moreira EAM, Fröde TS, Pedrosa RC, et al. Persistence of the benefit of an antioxidant therapy in children and teenagers with Down syndrome. Res Dev Disabil. 2015;45–46:14–20. doi: 10.1016/j.ridd.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Park E, Alberti J, Mehta P, Dalton A, Sersen E, Schuller-Levis G. Partial impairment of immune functions in peripheral blood leukocytes from aged men with Down's syndrome. Clin Immunol. 2000;95(1 Pt 1):62–69. doi: 10.1006/clim.2000.4834. [DOI] [PubMed] [Google Scholar]
- Pastore E, Marino B, Calzolari A, Digilio MC, Giannotti A, Turchetta A. Clinical and cardiorespiratory assessment in children with Down syndrome without congenital heart disease. Arch Pediatr Adolesc Med. 2000;154(4):408–410. doi: 10.1001/archpedi.154.4.408. [DOI] [PubMed] [Google Scholar]
- Pecze L, Blum W, Schwaller B. (2015) Routes of Ca2+ shuttling during Ca2+ oscillations focus on the role of mitochondrial Ca2+ handling and cytosolic Ca2+ buffers. J Biol Chem. 2015;290(47):28214–28230. doi: 10.1074/jbc.M115.663179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng M, Yang D, Hou Y, Liu S, Zhao M, Qin Y, et al. Intracellular citrate accumulation by oxidized ATM-mediated metabolism reprogramming via PFKP and CS enhances hypoxic breast cancer cell invasion and metastasis. Cell Death Dis. 2019;10(3):228. doi: 10.1038/s41419-019-1475-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Persson SA, Cassel G, Sellström A. Acute cyanide intoxication and central transmitter systems. Fundam Appl Toxicol. 1985;5(6 Pt 2):S150–S159. doi: 10.1016/0272-0590(85)90124-1. [DOI] [PubMed] [Google Scholar]
- Phillips AC, Sleigh A, McAllister CJ, Brage S, Carpenter TA, Kemp GJ, et al. Defective mitochondrial function in vivo in skeletal muscle in adults with Down's syndrome: a 31P-MRS study. PLoS ONE. 2013;8(12):e84031. doi: 10.1371/journal.pone.0084031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli C, Izzo A, Scrima R, Bonfiglio F, Manco R, Negri R, et al. Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Hum Mol Genet. 2013;22(6):1218–1232. doi: 10.1093/hmg/dds529. [DOI] [PubMed] [Google Scholar]
- Pincheira J, Rodriguez M, Bravo M, Navarrete MH, Lopez-Saez JF. Defective G2 repair in Down syndrome: effect of caffeine, adenosine and niacinamide in control and X-ray irradiated lymphocytes. Clin Genet. 1994;45(1):25–31. doi: 10.1111/j.1399-0004.1994.tb03985.x. [DOI] [PubMed] [Google Scholar]
- Plotegher N, Perocheau D, Ferrazza R, Massaro G, Bhosale G, Zambon F, et al. Impaired cellular bioenergetics caused by GBA1 depletion sensitizes neurons to calcium overload. Cell Death Differ. 2020;27(5):1588–1603. doi: 10.1038/s41418-019-0442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogribna M, Melnyk S, Pogribny I, Chango A, Yi P, James SJ. Homocysteine metabolism in children with Down syndrome: in vitro modulation. Am J Hum Genet. 2001;69(1):88–95. doi: 10.1086/321262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poisot T. The digitize package: extracting numerical data from scatterplots. R J. 2011;3(1):25–26. doi: 10.32614/RJ-2011-004. [DOI] [Google Scholar]
- Powers RK, Culp-Hill R, Ludwig MP, Smith KP, Waugh KA, Minter R, et al. Trisomy 21 activates the kynurenine pathway via increased dosage of interferon receptors. Nat Commun. 2019;10(1):4766. doi: 10.1038/s41467-019-12739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince J, Jia S, Båve U, Anneren G, Oreland L. Mitochondrial enzyme deficiencies in Down's syndrome. J Neural Transm Park Dis Dement Sect. 1994;8(3):171–181. doi: 10.1007/BF02260938. [DOI] [PubMed] [Google Scholar]
- Puukka R, Puukka M, Leppilampi M, Linna SL, Kouvalainen K. Erythrocyte adenosine deaminase, purine nucleoside phosphorylase and phosphoribosyltransferase activity in patients with Down's syndrome. Clin Chim Acta. 1982;126(3):275–281. doi: 10.1016/0009-8981(82)90301-1. [DOI] [PubMed] [Google Scholar]
- Rachidi M, Lopes C. Molecular and cellular mechanisms elucidating neurocognitive basis of functional impairments associated with intellectual disability in Down syndrome. Am J Intellect Dev Disabil. 2010;115(2):83–112. doi: 10.1352/1944-7558-115.2.83. [DOI] [PubMed] [Google Scholar]
- Reed BR, Crane J, Garrett N, Woods DL, Bates MN. Chronic ambient hydrogen sulfide exposure and cognitive function. Neurotoxicol Teratol. 2014;42:68–76. doi: 10.1016/j.ntt.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds T. Antioxidants do not improve early childhood development in children with Down's syndrome. J Pediatr. 2008;153(3):441. doi: 10.1016/j.jpeds.2008.05.055. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Sureda V, Vilches Á, Sánchez O, Audí L, Domínguez C. Intracellular oxidant activity, antioxidant enzyme defense system, and cell senescence in fibroblasts with trisomy 21. Oxid Med Cell Longev. 2015;2015:509241. doi: 10.1155/2015/509241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner F, Ong BH, Paine RS, Mahanand D. Biochemical differentiation of trisomic Down's syndrome (mongolism) from that due to translocation. N Engl J Med. 1965;273(25):1356–1361. doi: 10.1056/NEJM196512162732503. [DOI] [PubMed] [Google Scholar]
- Rosner M, Kowalska A, Freilinger A, Prusa AR, Marton E, Hengstschläger M. Cell cycle and cell size regulation in Down syndrome cells. J Neural Transm Suppl. 2003;67:51–58. doi: 10.1007/978-3-7091-6721-2_4. [DOI] [PubMed] [Google Scholar]
- Rubin SS, Rimmer JH, Chicoine B, Braddock D, McGuire DE. Overweight prevalence in persons with Down syndrome. Ment Retard. 1998;36(3):175–181. doi: 10.1352/0047-6765(1998)036<0175:OPIPWD>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Saghazadeh A, Mahmoudi M, Ashkezari AD, Rezaie NO, Rezaei N. Systematic review and meta-analysis shows a specific micronutrient profile in people with Down Syndrome: lower blood calcium, selenium and zinc, higher red blood cell copper and zinc, and higher salivary calcium and sodium. PLoS ONE. 2017;12(4):e0175437. doi: 10.1371/journal.pone.0175437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kauppinen A, Kaarniranta K. Hypoxia/ischemia activate processing of Amyloid Precursor Protein: impact of vascular dysfunction in the pathogenesis of Alzheimer's disease. J Neurochem. 2017;140(4):536–549. doi: 10.1111/jnc.13932. [DOI] [PubMed] [Google Scholar]
- Sano M, Aisen PS, Andrews HF, Tsai W-Y, Lai F, Dalton AJ. Vitamin E in aging persons with Down syndrome: a randomized, placebo-controlled clinical trial. Neurology. 2016;86(22):2071–2076. doi: 10.1212/WNL.0000000000002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial involvement in Parkinson's disease, Huntington's disease, hereditary spastic paraplegia and Friedreich's ataxia. Biochim Biophys Acta. 1999;1410(2):159–170. doi: 10.1016/S0005-2728(98)00164-9. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Tobe EH, Mozley PD, Jr, Barniskis L, Lidsky TI. Persistent cognitive and motor deficits following acute hydrogen sulphide poisoning. Occup Med (Lond) 1998;48(4):255–260. doi: 10.1093/occmed/48.4.255. [DOI] [PubMed] [Google Scholar]
- Seeff LB, Levitsky J, Tillman PW, Perou ML, Zimmerman HJ. Histopathology of the liver in Down's syndrome. Am J Dig Dis. 1967;12(11):1102–1113. doi: 10.1007/BF02233876. [DOI] [PubMed] [Google Scholar]
- Seron BB, Greguol M. Assessment protocols of maximum oxygen consumption in young people with Down syndrome–a review. Res Dev Disabil. 2014;35(3):676–685. doi: 10.1016/j.ridd.2013.12.008. [DOI] [PubMed] [Google Scholar]
- Seven M, Cengiz M, Tüzgen S, İscan MY. Plasma carnitine levels in children with Down syndrome. Am J Hum Biol. 2001;13(6):721–725. doi: 10.1002/ajhb.1117. [DOI] [PubMed] [Google Scholar]
- Shapiro BL. Down syndrome—a disruption of homeostasis. Am J Med Genet. 1983;14(2):241–269. doi: 10.1002/ajmg.1320140206. [DOI] [PubMed] [Google Scholar]
- Shukla VK, Sethi AK, Garg SK, Ganguly NK, Kulkami SK. Effect of venoruton on hypoxic stress-induced neurotoxicity in mice and oxygen free radical generation by human neutrophils. Arch Int Pharmacodyn Ther. 1989;299:127–133. [PubMed] [Google Scholar]
- Simon A, Ludwig C, Gofman JW, Crook GH. Metabolic studies in mongolism: serum protein-bound iodine, cholesterol, and lipoprotein. Am J Psychiatry. 1954;111(2):139–144. doi: 10.1176/ajp.111.2.139. [DOI] [PubMed] [Google Scholar]
- Singh JD. The teratogenic effects of dietary Cassava on the pregnant albino rat: a preliminary report. Teratology. 1981;24(3):289–291. doi: 10.1002/tera.1420240307. [DOI] [PubMed] [Google Scholar]
- Skrajny B, Hannah RS, Roth SH. Low concentrations of hydrogen sulphide alter monoamine levels in the developing rat central nervous system. Can J Physiol Pharmacol. 1992;70(11):1515–1518. doi: 10.1139/y92-215. [DOI] [PubMed] [Google Scholar]
- Sobol M, Klar J, Laan L, Shahsavani M, Schuster J, Annerén G, et al. Transcriptome and proteome profiling of neural induced pluripotent stem cells from individuals with Down syndrome disclose dynamic dysregulations of key pathways and cellular functions. Mol Neurobiol. 2019;56(10):7113–7127. doi: 10.1007/s12035-019-1585-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler Marín A, Xandri Graupera JM. Nutritional status of intellectual disabled persons with Down syndrome. Nutr Hosp. 2011;26(5):1059–1066. doi: 10.1590/S0212-16112011000500021. [DOI] [PubMed] [Google Scholar]
- Song WJ, Song EAC, Jung MS, Choi SH, Baik HH, Jin BK, et al. Phosphorylation and inactivation of glycogen synthase kinase 3β (GSK3β) by dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) J Biol Chem. 2015;290(4):2321–2333. doi: 10.1074/jbc.M114.594952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Segal M, Moses E. Involvement of potassium and cation channels in hippocampal abnormalities of embryonic Ts65Dn and Tc1 trisomic mice. EBioMedicine. 2015;2(9):1048–1062. doi: 10.1016/j.ebiom.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocchi V, Magnani M, Cucchiarini L, Novelli G, Dallapiccola B. Red blood cell adenine nucleotides abnormalities in Down syndrome. Am J Med Genet. 1985;20(1):131–135. doi: 10.1002/ajmg.1320200116. [DOI] [PubMed] [Google Scholar]
- Strupp BJ, Powers BE, Velazquez R, Ash JA, Kelley CM, Alldred MJ, et al. Maternal choline supplementation: A potential prenatal treatment for Down syndrome and Alzheimer’s disease. Curr Alzheimer Res. 2016;13(1):97–106. doi: 10.2174/1567205012666150921100311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Block ER. Hypoxia inhibits l-arginine synthesis from l-citrulline in porcine pulmonary artery endothelial cells. Am J Physiol. 1995;269(5 Pt 1):L581–L587. doi: 10.1152/ajplung.1995.269.5.L581. [DOI] [PubMed] [Google Scholar]
- Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, et al. Trisomy 21 consistently activates the interferon response. Elife. 2016;5:e16220. doi: 10.7554/eLife.16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. The re-emerging pathophysiological role of the cystathionine-β-synthase—hydrogen sulfide system in Down syndrome. FEBS J. 2020;287(15):3150–3160. doi: 10.1111/febs.15214. [DOI] [PubMed] [Google Scholar]
- Szabo C, Papapetropoulos A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol Rev. 2017;69(4):497–564. doi: 10.1124/pr.117.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart MJ, Wray S. Hypoxia and smooth muscle function: key regulatory events during metabolic stress. J Physiol. 1998;509(Pt 2):315–325. doi: 10.1111/j.1469-7793.1998.315bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamarkina A, Filippov I, Annenkov G, Beniashvili G. Changes in the mole fraction ratio of lactate dehydrogenase subunits in the lymphocytes of Down's syndrome patients. Genetika. 1978;14(2):354–358. [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewe OO, Maner JH. Long-term and carry-over effect of dietary inorganic cyanide (KCN) in the life cycle performance and metabolism of rats. Toxicol Appl Pharmacol. 1981;58(1):1–7. doi: 10.1016/0041-008X(81)90109-5. [DOI] [PubMed] [Google Scholar]
- Tiano L, Busciglio J. Mitochondrial dysfunction and Down's syndrome: is there a role for coenzyme Q(10)? BioFactors. 2011;37(5):386–392. doi: 10.1002/biof.184. [DOI] [PubMed] [Google Scholar]
- Tiano L, Carnevali P, Padella L, Santoro L, Principi F, Brugè F, et al. Effect of Coenzyme Q10 in mitigating oxidative DNA damage in Down syndrome patients, a double blind randomized controlled trial. Neurobiol Aging. 2011;32(11):2103–2105. doi: 10.1016/j.neurobiolaging.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Tiano L, Padella L, Carnevali P, Gabrielli O, Bruge F, Principi F, et al. Coenzyme Q10 and oxidative imbalance in Down syndrome: biochemical and clinical aspects. BioFactors. 2008;32(1–4):161–167. doi: 10.1002/biof.5520320119. [DOI] [PubMed] [Google Scholar]
- Tiano L, Padella L, Santoro L, Carnevali P, Principi F, Brugè F, et al. Prolonged coenzyme Q10 treatment in Down syndrome patients, effect on DNA oxidation. Neurobiol Aging. 2012;33(3):626.e1–8. doi: 10.1016/j.neurobiolaging.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Trazzi S, Mitrugno VM, Valli E, Fuchs C, Rizzi S, Guidi S, et al. APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum Mol Genet. 2011;20(8):1560–1573. doi: 10.1093/hmg/ddr033. [DOI] [PubMed] [Google Scholar]
- Tretter L, Patocs A, Chinopoulos C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta. 2016;1857(8):1086–1101. doi: 10.1016/j.bbabio.2016.03.012. [DOI] [PubMed] [Google Scholar]
- Tshala-Katumbay DD, Ngombe NN, Okitundu D, David L, Westaway SK, Boivin MJ, et al. Cyanide and the human brain: perspectives from a model of food (cassava) poisoning. Ann NY Acad Sci. 2016;1378(1):50–57. doi: 10.1111/nyas.13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature. 2016;540(7632):236–241. doi: 10.1038/nature20165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. EPA. IRIS Toxicological Review of Hydrogen Cyanide and Cyanide Salts (Final Report). U.S. Environmental Protection Agency, Washington, DC. 2010;EPA/635/R-08/016F.
- Urbanus E, van Rijn S, Swaab HA. Review of neurocognitive functioning of children with sex chromosome trisomies: Identifying targets for early intervention. Clin Genet. 2020;97(1):156–167. doi: 10.1111/cge.13586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacca RA, Bawari S, Valenti D, Tewari D, Nabavi SF, Shirooie S, et al. Down syndrome: neurobiological alterations and therapeutic targets. Neurosci Biobehav Rev. 2019;98:234–255. doi: 10.1016/j.neubiorev.2019.01.001. [DOI] [PubMed] [Google Scholar]
- Valenti D, De Rasmo D, Signorile A, Rossi L, de Bari L, Scala I, et al. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down's syndrome. Biochim Biophys Acta. 2013;1832(4):542–552. doi: 10.1016/j.bbadis.2012.12.011. [DOI] [PubMed] [Google Scholar]
- Valenti D, Manente GA, Moro L, Marra E, Vacca RA. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: involvement of the cAMP/PKA signalling pathway. Biochem J. 2011;435(3):679–688. doi: 10.1042/BJ20101908. [DOI] [PubMed] [Google Scholar]
- Valenti D, Tullo A, Caratozzolo MF, Merafina RS, Scartezzini P, Marra E, et al. Impairment of F1F0-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem J. 2010;431(2):299–310. doi: 10.1042/BJ20100581. [DOI] [PubMed] [Google Scholar]
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350(6265):1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Walter SD, Yao X. Effect sizes can be calculated for studies reporting ranges for outcome variables in systematic reviews. J Clin Epidemiol. 2007;60(8):849–852. doi: 10.1016/j.jclinepi.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chang J, Shao L, Feng W, Luo Y, Chow M, et al. Hematopoietic stem cells from Ts65Dn mice are deficient in the repair of DNA double-strand breaks. Radiat Res. 2016;185(6):630–637. doi: 10.1667/RR14407.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SE, Thomas DE, Clifford M, Tidmarsh SF, Sweeney AE, Ah-Sing E, et al. Plasma amino acids in patients with senile dementia and in subjects with Down's syndrome at an age vulnerable to Alzheimer changes. J Ment Defic Res. 1989;33(Pt 2):159–166. doi: 10.1111/j.1365-2788.1989.tb01462.x. [DOI] [PubMed] [Google Scholar]
- Weick JP, Held DL, Bonadurer GF, 3rd, Doers ME, Liu Y, Maguire C, et al. Deficits in human trisomy 21 iPSCs and neurons. Proc Natl Acad Sci USA. 2013;110(24):9962–9967. doi: 10.1073/pnas.1216575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochim Biophys Acta. 2012;1817(10):1833–1838. doi: 10.1016/j.bbabio.2012.02.033. [DOI] [PubMed] [Google Scholar]
- Wilkins HM, Swerdlow RH. Amyloid precursor protein processing and bioenergetics. Brain Res Bull. 2017;133:71–79. doi: 10.1016/j.brainresbull.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42(6):801–813. doi: 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids. 2009;37(1):1–17. doi: 10.1007/s00726-009-0269-0. [DOI] [PubMed] [Google Scholar]
- Xicota L, Rodríguez J, Langohr K, Fitó M, Dierssen M, de la Torre R, et al. Effect of epigallocatechin gallate on the body composition and lipid profile of down syndrome individuals: implications for clinical management. Clin Nutr. 2020;39(4):1292–1300. doi: 10.1016/j.clnu.2019.05.028. [DOI] [PubMed] [Google Scholar]
- Yates CM, Butterworth J, Tennant MC, Gordon A. Enzyme activities in relation to pH and lactate in postmortem brain in Alzheimer-type and other dementias. J Neurochem. 1990;55(5):1624–1630. doi: 10.1111/j.1471-4159.1990.tb04948.x. [DOI] [PubMed] [Google Scholar]
- Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet. 2010;19(14):2780–2791. doi: 10.1093/hmg/ddq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki ME, El-Bassyouni HT, Tosson A, Youness E, Hussein J. Coenzyme Q10 and pro- inflammatory markers in children with Down syndrome: clinical and biochemical aspects. J Pediatr (Rio J) 2017;93(1):100–104. doi: 10.1016/j.jped.2016.04.012. [DOI] [PubMed] [Google Scholar]
- Zeiger SL, Stankowski JN, McLaughlin B. Assessing neuronal bioenergetic status. Methods Mol Biol. 2011;758:215–235. doi: 10.1007/978-1-61779-170-3_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitnanová I, Korytár P, Aruoma OI, Sustrová M, Garaiová I, Muchová J, et al. Uric acid and allantoin levels in Down syndrome: antioxidant and oxidative stress mechanisms? Clin Chim Acta. 2004;341(1–2):139–146. doi: 10.1016/j.cccn.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Zuhra K, Augsburger F, Majtan T, Szabo C. Cystathionine-β-synthase: molecular regulation and pharmacological inhibition. Biomolecules. 2020;10(5):697. doi: 10.3390/biom10050697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplementary material. Supplementary Fig. 1. Log2 transformation and normalization on raw datasets. The boxplot shows the distribution of plasma samples after log2 transformation. Data are from Caracausi et al. 2018. Supplementary Fig. 2. Log2 transformation and normalization on raw datasets. The boxplot shows the distribution of plasma samples after log2 transformation. Data are from Antonaros et al. 2020. Supplementary Fig. 3. Boxplot showing the distribution of plasma samples after log2 transformation. The authors (Caracausi et al. 2018) used Probabilistic Quotient Normalization (PQN). Supplementary Fig. 4. Boxplot showing the distribution of plasma samples after log2 transformation. Normalized and log2 transformed samples from the study of Powers et al. 2019. Supplementary Fig. 5. Targeted metabolomic result from Culp-Hill study (Culp-Hill et al. 2017). Supplementary Fig. 6. Untargeted metabolomic result from Culp-Hill study (Culp-Hill et al. 2017) showing significant technical variations. Supplementary Fig. 7. Untargeted metabolomic result from Culp-Hill study Culp-Hill et al. 2017). We applied variance stabilization normalization to reduce technical variations. Supplementary Table 1. Significant regression coefficients from a multiple linear regression model applied on each metabolite found in plasma. The raw data are from Caracausi’s study (Caracausi et al. 2018). Supplementary Table 2. Significant regression coefficients from a multiple linear regression model applied on each metabolite found in urine. The raw data are from Caracausi’s study (Caracausi et al. 2018). Supplementary Fig. 8. Energy charge values in DS cells vs. control. Supplementary Fig. 9. Meta-analysis of amino acids not included in the Main Text.
Data Availability Statement
Not applicable.