

Abstract
Cantú syndrome (CS), first described in 1982, is caused by pathogenic variants in ABCC9 and KCNJ8, which encode the regulatory and pore forming subunits of ATP-sensitive potassium (KATP) channels, respectively. Multiple case reports of affected individuals have described the various clinical features of CS, but systematic studies are lacking. To define the effects of genetic variants on CS phenotypes and clinical outcomes, we have developed a standardized REDCap-based registry for CS. We report phenotypic features and associated genotypes on 74 CS subjects, with confirmed ABCC9 variants in 72 of the individuals. Hypertrichosis and a characteristic facial appearance are present in all individuals. Polyhydramnios during fetal life, hyperflexibility, edema, patent ductus arteriosus (PDA), cardiomegaly, dilated aortic root, vascular tortuosity of cerebral arteries, and migraine headaches are common features, although even with this large group of subjects, there is incomplete penetrance of CS-associated features, without clear correlation to genotype.
Keywords: ABCC9, Cantú, cardiomegaly, hypertrichosis, PDA, polyhydramnios
1 |. INTRODUCTION
Cantú syndrome (CS) (OMIM #239850) is an autosomal dominant condition caused by gain-of-function (GoF) pathogenic variants in ABCC9 and, less commonly, in KCNJ8, which encode the regulatory (SUR2) and pore-forming (Kir6.1) subunits, respectively, of ATP-sensitive potassium (KATP) channels (Brownstein et al., 2013; Cooper et al., 2014; Harakalova et al., 2012; McClenaghan et al., 2018; van Bon et al., 2012). A wide constellation of clinical features have been described in previous CS case reports (Grange, Lorch, Cole, & Singh, 2006; Grange, Nichols, & Singh, 2014; Scurr et al., 2011). Many pregnancies in which the fetus has CS are complicated by polyhydramnios, leading in some instances to repeated amniotic fluid reductions as well as preterm labor and delivery. Congenital hypertrichosis and coarse facial features, including a low frontal hairline, epicanthal folds, flat nasal bridge, long philtrum, macroglossia, prominent mouth, and full lips are defining features described in every affected individual and are usually evident at birth.
Generalized macrosomia, with large birth weight and length, as well as persistent macrocephaly, are also commonly reported (Cantu, Garcia-Cruz, Sanchez-Corona, Hernandez, & Nazar, 1982; Grange et al., 2014), as well as generalized edema at birth and lymphedema/peripheral edema (Grange et al., 2014). Affected individuals have extensive cardiovascular anomalies, including cardiomegaly, patent ductus arteriosus (PDA) and other congenital cardiac anomalies, pericardial effusion, pulmonary hypertension, dilated aortic root, and dilated and torturous cerebral blood vessels (Grange et al., 2006; Nichols, Singh, & Grange, 2013). Skeletal abnormalities including osteochondrodysplasia and generalized mild osteopenia are usually asymptomatic, although clinically significant scoliosis may occur (Concolino, Formicola, Camera, & Strisciuglio, 2000; Lazalde, Sanchez-Urbina, Nuno-Arana, Bitar, & de Lourdes Ramirez-Duenas, 2000; Rosser et al., 1998). Symptoms of a connective tissue abnormality with loose skin and hyperextensible joints are often present (Grange et al., 2014). Additional reported features include developmental delays, ADHD and autism spectrum features, without intellectual disability (Grange et al., 2014).
To date, ~60 individuals with CS have been reported in the literature, with approximately half being reported based on a clinical diagnosis. In the previously reported CS patients, genetic testing in ~35 individuals identified heterozygous pathogenic variants in ABCC9 (Harakalova et al., 2012; van Bon et al., 2012), and two cases thus far of heterozygous pathogenic variants in KCNJ8 (Brownstein et al., 2013; Cooper et al., 2014). The overall incidence is still unknown, but increased awareness of the clinical phenotype will undoubtedly lead to improved recognition and, hence, higher documented prevalence. There is considerable variation in the phenotypic spectrum, even within family members sharing the same ABCC9 variant (Roessler, Volker-Touw, Terhal, van Haaften, & van Haelst, 2018).
Despite the identification of ABCC9 and KCNJ8 variants as the underlying cause of CS, pathophysiologic mechanisms resulting in the clinical manifestations remain poorly understood, and there is no specific therapy. Development of effective targeted therapies will require well-characterized patient cohorts, with standardized data collection. Unlike common diseases, for which patient resources are routinely available and large patient cohorts can be built at a single site, such studies in rare diseases require multi-site collaboration to maximize cohort size. Appropriately designed and executed patient registries that collate such data can then provide a real-world view of common patient characteristics and outcomes, as well as a platform to facilitate clinical trials. Registries are especially valuable for rare disorders without appropriate treatments (Cavero-Carbonell et al., 2016) such as CS, for which data are generally only available in scattered case reports. Here, we present clinical and genetic findings on 74 CS subjects enrolled in an international REDCap-based CS registry. This represents the largest cohort of CS subjects studied thus far, permitting estimates of penetrance for disease features and providing data on inter-familial and intra-familial variability and genotype–phenotype correlations.
2 |. SUBJECTS AND METHODS
2.1 |. Study overview and patient identification
Development of the International Cantú Syndrome Registry (ICSR) was initiated in 2012, and resulted from the coordinated efforts of four CS clinics (at Washington University in St. Louis, Missouri in the United States, University Medical Center Utrecht in the Netherlands, the University of Bristol in the United Kingdom and the University of New South Wales in Sydney, Australia) that had already identified groups of CS subjects in their respective countries. The REDCap-based registry is maintained at Washington University in St. Louis, and the ICSR is now established as an international multicenter registry to collate clinical and genetic data on CS subjects. By effectively gathering data on CS subjects from all over the world, with data collected during recurring annual Cantú research clinics to provide longitudinal information, it is hoped that the ICSR will provide a full characterization of the clinical phenotype and mutational spectrum of CS, and a critical mass of well-phenotyped patients for rapid progression toward future interventional studies.
Clinical information, molecular data and blood samples from CS subjects and their families worldwide are collected through interaction with physicians treating CS subjects, by contacting physicians who have published on CS, and by receiving inquiries from physicians treating possible CS subjects. Individuals who present with key diagnostic criteria such as hypertrichosis, typical facial features, and cardiac abnormalities undergo sequencing for variants in ABCC9 and KCNJ8, applying targeted Sanger sequencing, whole exome sequencing (WES) or whole genome sequencing (WGS).
A questionnaire asking all subjects to self-report currently known clinical features observed in CS is completed either during the visit to a participating facility or is sent to the referring physician and/or subject directly. Additionally, subjects are invited to participate in special annual CS research clinics, which take place at the participating institutions to document clinical features and to gather data regarding the progression of CS over time. Seventy-one of the subjects reported here have been evaluated at least once through one of the CS research clinics, while the remaining three subjects consented to participate in the registry through the Washington University site and completed the questionnaire, but were not clinically evaluated in person. Some clinical and genetic findings for 29 of the 74 subjects have previously been published (See Table 1 and footnotes).
TABLE 1.
Cantú Syndrome ABCC9 variants and general clinical features
| Patient | Gender | Age (yr) | cDNA Variant | Protein Alteration | Polyhydramnios (treatment) | Congenital hypertrichosis | Wrinkled and/or loose skin | Skeletal Dysplasia | Osteopenia/Osteoporosis | Scoliosis | Pectus carinatum | Joint laxity | Hypotonia | Breathing difficulties | Exercise intolerance | Hernia | Intestinal dysmotility | Developmental delay |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CS0001a | M | 18 | c.3460 C > T | p.Arg1154Trp | − | + | − | − | − | − | na | + | + | + | − | U | − | + |
| CS0002a | F | 23 | c.3461 G > A | p.Arg1154Gln | − | + | − | − | − | + | − | + | − | + | + | − | + | − |
| CS0003a | F | 29 | c.3461 G > A | p.Arg1154Gln | − | + | − | + | − | − | − | + | na | − | + | − | − | − |
| CS0004a | F | 53 | c.3461 G > A | p.Arg1154Gln | − | + | − | + | − | + | − | − | − | + | + | − | − | − |
| CS0036 | F | 0.45 d | c.3461 G > A | p.Arg1154Gln | + | − | − | + | − | − | − | − | + | + | na | CN | + | + |
| CS0005a | F | 24 | c.3460 C > T | p.Arg1154Trp | − | + | + | + | − | +# | + | + | + | + | + | − | − | + |
| CS0006a | M | 18 | c.3014 A > T | p.His1005Leu | na | + | − | − | − | − | + | − | + | − | − | − | − | + |
| CS0007a | F | 37 | c.3346 C > T | p.Arg1116Cys | + | + | + | − | − | − | − | + | + | − | − | U | + | − |
| CS0008a | F | 6 | c.3346 C > T | p.Arg1116Cys | − | + | + | − | − | − | − | + | + | + | − | − | + | + |
| CS0009a | M | 9 | c.3346 C > T | p.Arg1116Cys | − | + | + | − | − | − | − | + | + | + | − | − | − | + |
| CS0010a | M | 11 | c.3346 C > T | p.Arg1116Cys | + | + | + | − | − | − | − | + | + | − | − | U | − | + |
| CS0011a | M | 13 | c.3347 G > A | p.Arg1116His | + | + | + | − | − | − | + | + | + | + | + | U/I | + | + |
| CS0012a | M | 13 | c.621 C > A | p.Asp207Glu | + | + | + | + | na | − | na | na | na | + | − | − | na | na |
| CS0013 | M | 4 | c.4469 T > A | p.Va1490Glu | + | + | − | + | + | − | + | − | + | + | − | − | − | + |
| CS0014 | M | 5 | c.3346 C > T | p.Arg1116Cys | na | + | + | − | − | − | − | + | − | + | − | − | − | + |
| CS0015 | M | 8 | c.3796 G > A | p.Va11266Met | + | + | + | + | − | − | − | + | − | − | − | U | + | + |
| CS0016 | F | 4 | c.2378 A > T | p.Asp793Val | + | + | − | − | − | − | − | − | + | + | − | U | − | + |
| CS0017 | F | 4 | c.3014 A > T | p.His1005Leu | + | + | + | − | − | − | − | − | + | − | − | − | − | + |
| CS0018 | M | 6 | c.3052 T > G | p.Trp1018Gly | + | + | − | − | − | − | + | + | + | − | − | U | − | + |
| CS0019 | M | 6 | c.3345 G > A | p.Arg1116His | + | + | + | + | − | − | + | + | + | + | + | − | − | + |
| CS0020 | M | 22 | c.3461 G > A | p.Arg1154Gln | + | + | + | − | − | + | + | + | + | + | + | 1 | + | + |
| CS0021 | F | 46 | c.881 G > A | p.Gly294Glu | − | + | + | − | − | + | − | − | − | + | + | − | − | + |
| CS0022 | F | 69 | c.881 G > A | p.Gly294Glu | na | + | na | − | − | − | − | − | − | − | + | − | − | + |
| CS0023 | F | 28 | c.621 C > A | p.Asp207Glu | + | + | − | − | + | − | − | + | + | − | + | − | + | + |
| CS0024 | M | 7 | c.4040 G > T | p.Arg1347Leu | − | + | + | − | − | − | − | − | − | + | − | U | + | − |
| CS0028 | F | 6 | c.3605 C > T | p.Thr1202Met | + | + | + | − | − | − | − | − | + | + | + | − | − | + |
| CS0029 | M | 3 | c.3460 C > T | p.Arg1154Trp | + | + | − | + | − | − | − | − | + | + | − | − | − | + |
| CS0030 | F | 41 | c.4480 G > A | p.Ala1494Thr | − | + | − | − | − | − | − | − | − | − | + | − | − | − |
| CS0031 | M | 3 | c.4480 G > A | p.Ala1494Thr | + | + | na | − | − | − | − | + | + | + | − | − | − | + |
| CS0032 | M | 9 | c.4480 G > A | p.Ala1494Thr | − | + | na | − | − | − | − | + | + | − | − | − | − | + |
| CS0033 | F | 3 | c.3461 G > A | p.Arg1154Gln | + | + | − | − | − | − | − | − | + | + | na | − | − | − |
| CS0034 | M | 34 | c.3461 G > A (mosaic) | p.Arg1154Gln | − | + | − | − | − | − | − | − | − | − | − | − | − | − |
| CS0035 | F | 59 | c.3704 C > T | p.Ser1235Phe | − | + | − | − | + | + | + | + | − | + | + | − | + | − |
| CS0037 | F | 1 | c.3056 C > A | p.Thr1019Lys | + | + | − | − | − | − | − | − | + | + | − | − | − | + |
| CS1001a | F | 25 | c.178 C > T | p.His60Tyr | + | + | + | + | − | − | − | + | − | + | − | − | − | + |
| CS1002a | F | 30 | c.3161 C > A | p.Ser1054Tyr | − | + | − | − | − | − | − | − | − | − | − | − | − | − |
| CS1003a | F | 31 | c.3461 G > A | p.Arg1154Gln | − | + | + | + | − | − | − | + | − | − | − | − | − | + |
| CS1004a | M | 11 | c.1138 G > T | p.Gly380Cys | − | + | + | − | − | − | + | − | − | − | − | − | − | + |
| CS1005a | M | 13 | c.3346 C > T | p.Arg1116Cys | − | + | + | − | − | − | − | − | − | + | − | − | − | + |
| CS1007a | M | 10 | c.3116 T > C | p.Phe1039Ser | + | + | + | − | − | − | − | − | + | − | − | U | − | + |
| CS1008 | F | 6 | c.3460 C > T | p.Arg1154Trp | + | + | − | − | − | − | − | − | − | − | − | − | − | − |
| CS1009 | M | 16 | c.3161 C > A | p.Ser1054Tyr | − | + | + | − | − | − | − | + | + | − | − | U | − | + |
| CS1010 | M | 5 | c.3460 C > T | p.Arg1154Trp | + | + | − | − | − | − | − | − | − | + | − | U | − | + |
| CS1011 | M | 15 | c.3618 C > A | p.Asn1206Lys | + | + | − | − | − | − | − | + | + | − | + | − | − | + |
| CS1012 | M | 23 | c.3460 C > T | p.Arg1154Trp | − | + | + | − | − | − | − | + | − | − | − | − | − | − |
| CS1013 | F | 3 | c.3460 C > G | p.Arg1154Gly | + | + | − | − | − | − | − | − | − | − | − | − | − | + |
| CS1014a | M | na | Not available | na | − | + | − | − | − | − | − | − | − | + | − | − | − | + |
| CS1015 | F | na | Not done | na | + | + | + | + | − | + | − | − | − | − | − | − | − | − |
| CS1016a | M | 32 | c.2444 G > C | p.Gly815Ala | + | + | − | − | − | − | − | − | − | − | − | − | − | − |
| CS1018 | F | 7 | c.3618 C > G | p.Asn1206Lys | − | + | − | − | − | + | − | + | + | + | − | U | − | − |
| CS1019 | M | 1 | c.621 C > A | p.Asp207Glu | + | + | + | − | − | − | − | + | + | + | + | I | − | + |
| CS2001a | F | 13 | c.1295 C > T | p.Pro432Leu | na | + | + | − | na | − | + | + | + | − | − | U | − | − |
| CS2002a | M | 40 | c.1433 C > T | p.Ala478Val | na | + | + | na | na | − | − | − | + | − | − | − | − | − |
| CS2003a | M | 5 | c.1433 C > T | p.Ala478Val | − | + | + | − | − | − | − | − | − | + | − | I | + | + |
| CS2004 | M | 13 | c.178 C > T | p.His60Tyr | na | + | + | na | na | na | + | + | − | + | + | U | + | + |
| CS2005 | F | 18 | c.3460 C > T | p.Arg1154Trp | na | + | + | na | + | + | + | + | + | + | + | − | − | − |
| CS2006 | F | 5 | c.3461 G > A | p.Arg1154Gln | na | + | − | na | − | − | − | − | na | + | − | U | − | − |
| CS2007a | F | 8 | c.3461 G > A | p.Arg1154Gln | na | + | na | na | na | na | na | na | na | na | na | na | na | na |
| CS2008 | F | 17 | c.3345 C > G | p.Arg.1116Gly | na | + | − | − | − | − | − | + | + | + | na | − | − | + |
| CS2009 | F | 23 | c.3461 G > A | p.Arg1154Gln | na | + | + | − | na | − | + | + | + | − | + | − | − | + |
| CS2010 | F | 18 | c.3460 C > T | p.Arg1154Trp | na | + | + | na | na | − | − | + | na | + | na | − | − | − |
| CS2011a | F | 46 | c.3345 G > A | p.Arg1116His | − | + | + | na | na | na | − | + | na | na | − | U/l | − | − |
| CS2012a | M | 12 | c.3345 G > A | p.Arg1116His | na | + | + | na | na | − | + | + | + | + | − | U/l | − | + |
| CS2013a | F | 11 | c.3460 C > T | p.Arg1154Trp | na | + | − | na | na | − | + | − | + | − | − | − | − | − |
| CS2014 | M | 4 | c.3461 G > A | p.Arg1154Gln | + | + | − | − | − | − | + | + | + | + | − | − | − | + |
| CS3001 | M | na | c.3461 G > A | p.Arg1154Gln | − | + | + | − | − | − | + | − | + | − | − | − | − | + |
| CS3002 | M | 5 | c.3460 C > T | p.Arg1154Trp | + | + | − | − | − | − | − | + | + | + | + | − | − | + |
| CS3003a | M | 1 | c.3345 G > A | p.Arg1116His | + | + | na | − | − | − | − | + | + | + | − | I | − | na |
| CS3004 | F | 37 | c.4040 G > T | p.Arg1347Leu | + | + | + | − | − | − | − | + | + | + | + | U | − | − |
| CS3005 | F | 9 | c.4040 G > T | p.Arg1347Leu | − | + | + | − | − | − | − | + | + | + | + | U | − | + |
| CS3006 | F | 7 | c.4040 G > T | p.Arg1347Leu | na | na | + | na | na | na | na | + | na | + | na | na | na | + |
| CS3009 | M | 64 | c.4040 G > T | p.Arg1347Leu | na | + | + | − | − | − | − | − | − | − | − | I | − | − |
| CS3007 | M | 5 | c.3055_3056delinsGA | p.Thr1019Glu | + | + | − | − | − | − | − | − | + | + | − | − | − | − |
| CS3008a | F | 22 | c.3058 T > G | p. Ser1020Pro | + | + | − | na | − | + | − | + | + | + | + | − | − | + |
Abbreviations: M – male; F – female; d - Died at 5 months; na – data not available; # - surgically corrected; U – Umbilical; I- Inguinal; CN – Canal of Nuck hernia.
Indicates previously reported patient.
CS0002/CS0003/CS0004: Grange et al. (2006) Am J Med Genet and van Bon et al. (2012) Am J Human Genet.
CS0012: Harakalova et al. (2012) Nat. Gen.
CS0001/CS0002/CS0004/CS0005/CS0006/CS0007/CS0008/CS0009/CS0010/CS0011: Leon-Guerrero et al (2016) Neurology.
CS1001/CS1002/CS1003/CS1004/CS1005/CS1014/CS1016: Scurr et al. (2011) Am J Med Genet.
CS1001/CS1002/CS1003/CS1004/CS1005/CS1007: Harakalova et al. (2012) Nature Genetics.
CS2002/CS2003/CS2011/CS2012: Roessler et al. (2018) Clin. Dysm.
CS2001/CS2007/CS2011/CS2012/CS2013: Harakalova et al. (2012) Nat. Gen.
CS3008: Robertson and Kirk et al., Am J Med Genet 1999;85:395–402; Scurr et al. (2011) Am J Med Genet; Harakalova et al. (2012) Nature Genetics.
CS3003: Ma et al. (2019) Am J Med Genet.
8 Kindreds are grouped together: 1) CS0002, CS0003, CS0004 and CS0036; 2) CS0007, CS0008, CS0009 and CS0010; 3) CS0021 and CS0022; 4) CS0030, CS0031 and CS0032; 5) CS0033 and CS0034; 6) CS2002 and CS2003; 7) CS2011 and CS2012; 8) CS3004, CS3005, CS3006 and CS3009.
2.2 |. Data management
Research personnel at individual institutions are responsible for obtaining and maintaining institutional review board or ethics approval, performing recruitment and enrollment procedures, and collecting and entering data into the database.
Study data are collected and managed using the Research Electronic Data Capture (REDCap) (Harris et al., 2009) database, a secure, web-based application designed to support data capture for research studies, hosted at Washington University, St. Louis. The research coordinator at each site holds the code to link anonymous patient code numbers to identifiable patient details. Pooling de-identified data from all participating institutions enables data to be analyzed with statistical power that no single clinic could achieve on its own. Data are updated or re-evaluated after follow-up visits.
2.3 |. Patient eligibility and enrollment
The ICSR is a voluntary study open to subjects of all ages who have a clinical and, in most cases, molecular diagnosis of CS. The host institution, as well as each participating health center, is granted institutional review board or ethics approval before proceeding with subject recruitment. Consent and recruitment procedures vary among participating centers, according to each clinic’s setting, available resources, and institutional policies.
2.4 |. Data collection
Data are primarily collected in questionnaire form, establishing the subject’s background and medical history, as well as by review of known phenotypic features of CS. Prior medical records, including results of molecular genetic testing, are requested after the subject or parent/guardian signs a release form. The questionnaire is revised and re-evaluated as data collection continues and new phenotypic features are discovered. Data reported in this study were collected over a period of 6 years from 2012 to 2018, most during routine visits as part of standard care for subjects with CS or during annual CS research clinics. All but three of the subjects enrolled in the ICSR have attended one of the clinics. Data on these three subjects were obtained through the questionnaire from the subject and his or her attending physician and may involve review of medical records. All data reported here were collected after consent of the subject or the parents or legal guardians for patients younger than 18 years old.
To date, 21 data modules have been developed to collect data on demographics, physicians, and specialists caring for the subject, birth and basic medical history, hair, skin, cardiovascular abnormalities, upper airway and ear, nose, and throat (ENT) concerns, pulmonary, bone and joint abnormalities, neurological and brain abnormalities, vision, hearing, gastrointestinal system, genitourinary system, reproductive system, endocrine system, immune function, learning and cognition, behavioral issues, and family history. In order to ensure standardization of information collection, checklists tracking data collection are maintained and updated for all participants.
2.5 |. Data analysis and statistics
Given the limited data available, data are presented as individual values without statistical analysis, unless otherwise noted.
3 |. RESULTS
We present results from 74 CS subjects, including 45 new subjects and 29 subjects that have been previously reported (Grange et al., 2006; Harakalova et al., 2012; Leon Guerrero et al., 2016; Ma et al., 2019; Robertson et al., 1999; Roessler et al., 2018; Scurr et al., 2011). Seventy-two of the patients have confirmed variants in ABCC9 while one has a convincing clinical diagnosis of CS but has not had molecular testing and one has had testing, but the ABCC9 variant result is not available. Information regarding genotype and general features of all 74 subjects are provided in Table 1.
3.1 |. Patient demographics
The 74 subjects (36 females [49%] and 38 males [51%]) range in age from 2 months to 67 years (Figure 1a, Table 2). Subjects were recruited by four centers, in the United States, the Netherlands, the United Kingdom, and Australia. The overwhelming majority of participants (73/74) are Caucasian and most (64/74) identify as Non-Spanish/Hispanic/Latino, reflecting the location of the participating sites. Ages at the time of diagnosis are shown in Figure 1b.
FIGURE 1.
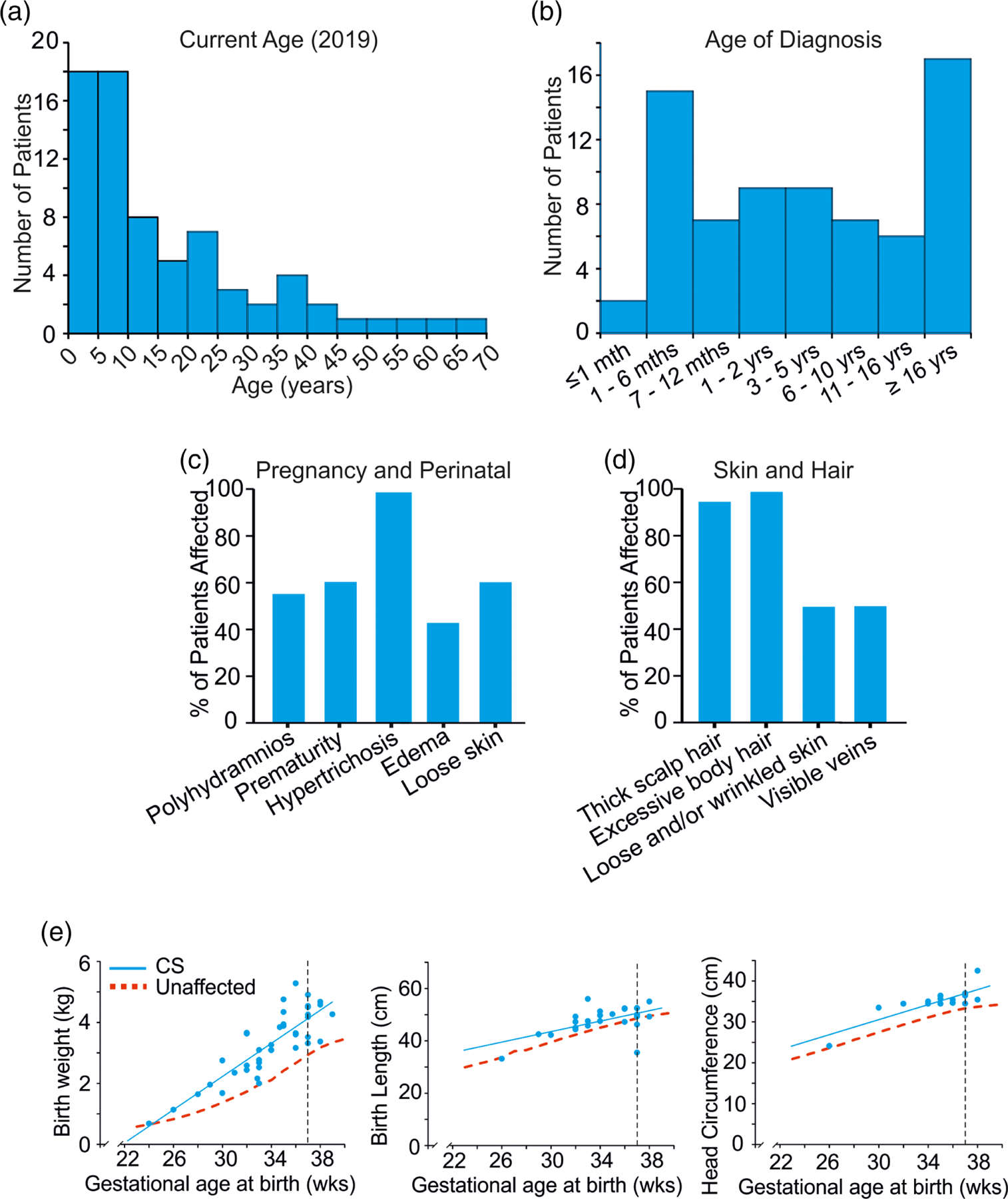
Subject characteristics (a) Current subject ages, (b) Age at diagnosis, (c) Pregnancy and perinatal features, (d) Skin and hair features, (e) Growth parameters in newborns with Cantú syndrome (Left) Birth weights of CS newborn infants plotted versus gestational age, showing elevated birth weights. (Center) Birth lengths of CS newborn infants plotted versus gestational age, showing increased birth length. (Right) Birth head circumference of CS newborn infants plotted versus gestational age, showing macrocephaly. The 50th centile for normal infants is shown in red. Vertical dashed line indicates 37 weeks. CS, Cantú syndrome
TABLE 2.
Demographics of CS Subjects
| Descriptive characteristics | All (n = 74) |
|---|---|
| Age | # subjectsa (%) |
| ≤5 year | 15 (21.4%) |
| >5 to 10 years | 18 (25.7%) |
| >10 to 20 | 15 (21.4%) |
| >20 years | 23 (31%) |
| Gender | |
| Female | 36/74 (48.6%) |
| Male | 38/74 (51.3%) |
| Race | |
| Caucasian | 73/74 (98.6%) |
| Black/African American | 1/74 (1.4%) |
| Ethnicity | |
| Hispanic/Latino | 10/71b (14%) |
| Not Hispanic/Latino | 61/71b (86%) |
Current ages available for only 71/74 patients.
Question not answered in three patients, but all these three indicated that they are Caucasian.
3.2 |. Pregnancy and birth
Half of the mothers of affected individuals reported prenatal abnormalities during their pregnancies, primarily abnormal ultrasound findings (Figure 1c). Macrosomia and macrocephaly were evident during third trimester ultrasounds in eight patients. Polyhydramnios was observed during gestation in 34/60 (57%) pregnancies and in multiple cases, repeated amniotic fluid reduction procedures were required (Figure 1c). Polyhydramnios is reported in both inherited and de novo CS individuals (i.e., non-CS mothers) and thus likely originates from fetal KATP channel dysfunction. Gestational age at birth ranged from 24 to 39 weeks with an average gestation period of 33.9 ± 3.4 weeks (n = 39) and 42/72 (58%) babies were born preterm (i.e., less than 37 weeks gestational age). Hence, children with CS were born significantly more prematurely than normal (Figure 1c).
Babies with CS had macrosomia with increased length and weight, as well as macrocephaly (Figure 1e). The average birth weight for all infants reported was 3.7 ± 1.0 kg (n = 68), and 4.1 ± 0.7 kg for full term infants (n = 28). 12% of CS infants had a low birth weight (i.e., <2,500 g) and 38% had a high birth weight (i.e., >4,000 g). As shown in Figure 1e, birth length and weight are consistently above average, irrespective of gestational age.
At birth, the most consistent clinical features were excessive body hair in essentially all subjects (67/68, 99%), edema in 27/63 (43%) and wrinkled and/or loose skin in 43/69 (62%) (Figure 1c). Multiple subjects reported congenital cardiac defects, as discussed below. Before or after the pregnancy with the affected CS participant, 2/41 (5%) of mothers (one of whom had CS) who responded to the question experienced a stillbirth, which is higher than the estimated risk of stillbirth in the general population (~1% of pregnancies greater than 20 weeks gestation), although the numbers are too small to be certain whether this is a genuine association or a chance observation. Of 40 women who responded to the question, miscarriages were reported by five (13%), comparable to 10–20% incidence in the general population.
3.3 |. Growth
At the most recent assessments, subjects exhibited an average BMI of 23.2 ± 8.4 (range 15.9–58.0, n = 36). A thin or even quite muscular physique was most commonly evident during childhood, while adults were more likely to be overweight due to weight gain after puberty. Mean adult height in males was at the 95th centile for the general population, although data on height measurements were available in only a limited number of men in the registry (Table 3). Females were mostly within the normal range for height for adult women (50–75th centile). However, macrocephaly persists throughout life in both men and women; mean head circumference was >98th centile (+2.5 SD) in women and at the 98th centile (+2 SD) in men (Table 3).
TABLE 3.
Growth parameters in adults with Cantú syndrome
| Characteristics | |
|---|---|
| Mean height (cm) adult female | 167.0 ± 7.8 cm (70th %ile) N = 11 |
| Normal range 151–175 cm | |
| Mean height (cm) adult male | 187.7 ± 11.4 cm (95th %ile) N = 5 |
| Normal range 163–190 cm | |
| Mean head circumference (cm) adult female | 58.7 ± 2.3 cm (>98th %ile; +2.5 SD) N = 9 |
| Normal range 52–57.5 cm | |
| Mean head circumference (cm) adult male | 57.8 ± 1.5 cm (98th %ile; +2 SD) N = 4 |
| Normal range 53–58 cm |
3.4 |. Development and behavior
Developmental delays in childhood were reported in 45/71 (63%) subjects and 29/64 (45%) reported some degree of special educational support. A majority of individuals were reported to have hypotonia (42/65, 65%) and speech delays in early childhood, requiring occupational, physical and speech therapy. Persistent intellectual impairment was not evident, however, and most individuals clearly attain normal educational levels. About 18% of the subjects in the registry currently attend first to eighth grade (or equivalent) and 12% are in high school, 21% have achieved their high school diploma or equivalent, 18% have a college degree, and 6% have a graduate degree.
With regard to behavioral abnormalities, the ICSR data are not complete for the entire cohort, but systematic information is available for the patients assessed through the Washington University site. Of these, ADHD is reported in 6/32 (19%), autism or autism spectrum disorder in 6/31 (16%), mood swings in 10/31 (32%), obsessive compulsive disorder (OCD) in 4/31 (13%), anxiety in 4/31 (13%), and depression in 6/32 (19%) of respondents. Many of the subjects self-reported more than one of these behavioral abnormalities. Four subjects had two concomitant behavioral concerns (ADHD and mood swings in one subject; depression and mood swings in one subject; ADHD and autism in two subjects), three subjects had three behavioral concerns (ADHD, mood swings and OCD in one subject; mood swings, anxiety and depression in one subject; ADHD, mood swings and anxiety in one subject) and three subjects had four behavioral concerns (ADHD, autism, OCD and mood swings in two subjects; autism, mood swings, OCD, and anxiety in one subject).
3.5 |. Craniofacial dysmorphology, hypertrichosis, and skin appearance
All affected individuals have a distinctive facial appearance with coarse features, including a low frontal hairline, epicanthal folds, puffy eyelids, flat nasal bridge with broad nasal tip, long philtrum, macroglossia, and prominent mouth with full lips (Figure 2). Hypertrichosis is a highly penetrant feature, with almost all subjects reporting thick scalp (68/72, 94%) and body hair (72/73, 99%). In 65/68 (96%) subjects, hypertrichosis was present within the first year of life, in particular, abnormal hair coverage was reported on the arms, back, cheeks, chin, feet, forehead, legs, and hands of infants (Figure 1d, 3). 21/68 (31%) subjects report variable success at managing hypertrichosis using approaches such as shaving, waxing and laser therapy.
FIGURE 2.

Facial phenotype of Cantú syndrome subjects throughout life. (a) 2 months; (b) 16 months; (c) 22 months; (d) 26 months; (e) 2.5 years; (f) 3 years; (g) 4 years; (h) 12 years; (i) 15 years; (j) 20 years; (k) 23 years; (l) 20 years; (m) 26 years; (n) 40 years; (o) 50 years: (p) 57 years. Note edema of eyelids in many of the younger subjects, epicanthal folds, widely spaced eyes, full lips, and macroglossia. Hypertrichosis of the forehead tends to decrease with age, but facial features become more prominent over time
FIGURE 3.
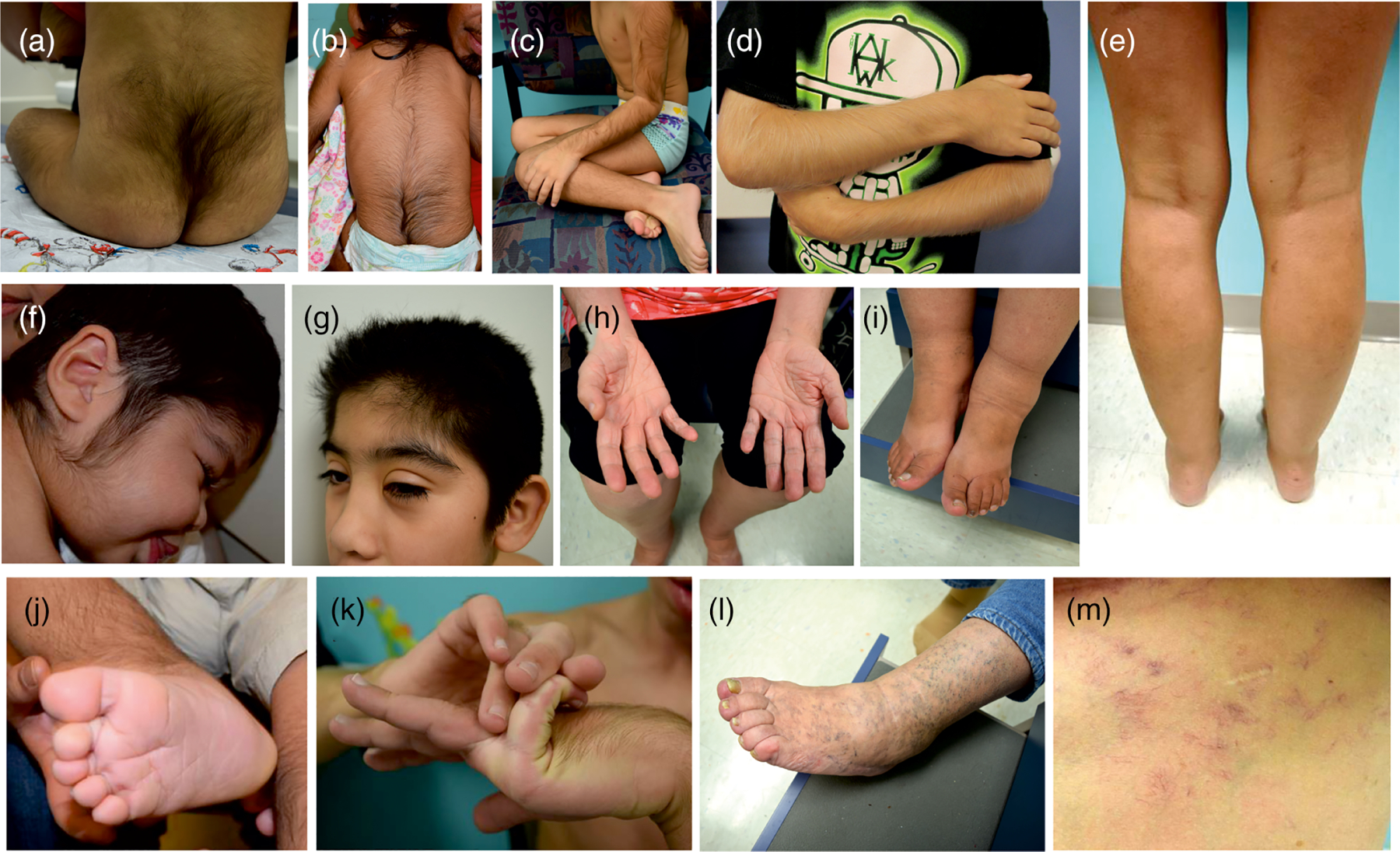
Physical characteristics (a) Hypertrichosis over the sacral area; (b) hypertrichosis of the upper and lower back; (c and d) hypertrichosis of the limbs; (e) asymmetry of lower extremities due to edema of the right leg; (f and g) hypertrichosis of the cheek and forehead; (h) wrinkled skin and deep palmar creases in an adult; (i) edema of the ankles showing indentation from socks; (j) deep plantar crease in a young child; (k) hyperflexibility of the digits; (l) prominent superficial veins and small varicosities in an older individual; (m) prominent small blood vessels and visible capillaries on the torso of an adult
Loose and/or wrinkled skin with deep palmar/plantar creases are present in a majority of infants with CS (42/69, 61%) and frequently in adults. Prominent veins or visible small blood vessels, predominantly on the lower extremities, chest and back, are reported in 33/71 (46%) cases (see Figure 3l,m). Many adults have larger varicosities on the lower extremities, occurring at a younger age than is typical in the general population.
3.6 |. Cardiovascular abnormalities
Cardiovascular features of all 74 subjects are presented in Table 4 and Figure 4a–c. Congenital heart defects are the most commonly documented CS cardiovascular abnormality, present in 56/73 (77%) subjects in our CS cohort. Persistent PDA was reported most frequently (44/73, 58%) with a variety of other defects observed including atrial septal defects (6%), ventricular septal defects (6%), and aortic coarctation (3%) (Figure 4a). Surgical closure of PDA was required in 28/52 subjects (54%). 18% of subjects reported valvular defects including mitral valve regurgitation (9%), aortic stenosis (2%), and bicuspid aortic valve (10%) (Figure 4c). Dilation of the aortic root was reported in 21/66 (32%) subjects, with 1/56 subjects reporting aortic aneurysm. Cardiac enlargement was reported by 64% of subjects, variably described as ventricular hypertrophy, hypertrophic cardiomyopathy, or dilated cardiomyopathy, based on information provided to them by their primary cardiologist or other physician (Figure 4b). However, in many of these cases, specific echocardiographic data are not available through the ICSR, and therefore cardiac features may be poorly defined; in a subset of subjects extensively characterized at the Washington University Cantú Syndrome Research Clinic between 2013 and 2018, cardiac enlargement was specifically confirmed as ventricular dilation (left ventricular end diastolic volume index >2 Z score) and increased left ventricular mass index (partially reported in (Levin et al., 2016), with increased left ventricular systolic function, in 29/32 (90%) subjects. Thus, cardiac enlargement was accompanied by a high output state in the great majority of subjects in whom cardiac output was noninvasively measured.
TABLE 4.
Cardiovascular features
| Patient | PDA | ASD | VSD | PDA surgery/age | Aortic root dilation / age of diagnosis | Enlarged heart / age of diagnosis | Pericard. Eff. / treatment | Arrhythmia | Pulmonary hypertension / age of diag. | Low blood pressure | High blood pressure | Lymphedema | Abnormal blood vessels | Edema / treatment | Other cardiac defects | Cardiac medications |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CS0001 | + | − | − | +/1 m | +/7y | +/7y | − | − | − | − | − | − | + | − | − | − |
| CS0002 | − | − | − | na | − | + | +/S | + | − | − | − | + | + | + | − | − |
| CS0003 | + | − | − | − | − | + | − | − | − | + | − | + | + | + | − | − |
| CS0004 | − | − | − | na | − | + | +/S | + | − | − | − | + | + | +/sf | MV regurg. | a,ca,f,s |
| CS0005 | + | − | − | − | − | + | − | − | − | − | − | + | + | +/c | APC, MV regurg. | − |
| CS0006 | + | + | − | − | − | +/1 m | − | − | +/r | − | + | − | + | − | PFO | e |
| CS0007 | + | − | − | +/2y | +/32y | − | − | − | − | − | + | − | + | +/c | − | − |
| CS0008 | − | + | − | +/1y | − | +/2 m | − | − | +/2 m | − | − | − | na | − | − | s |
| CS0009 | − | − | − | na | − | − | − | − | − | − | − | − | − | − | − | − |
| CS0010 | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CS0011 | + | − | − | +/1 m | + | +/9 m | +/0 | − | +/9 m | − | + | − | + | − | − | − |
| CS0012 | − | − | − | na | − | + | − | − | − | − | − | − | na | − | − | a |
| CS0013 | + | − | − | +/3 m | +/4y | +/0 m | − | − | +/1 m | − | − | − | − | + | − | − |
| CS0014 | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CS0015 | + | − | − | − | − | +/0 m | − | − | − | − | − | − | − | + | PFO | − |
| CS0016 | − | − | − | na | +/3y | +/0 m | +/0 | + | − | − | − | − | + | + | − | − |
| CS0017 | + | − | − | − | +/3y | + | − | − | − | − | − | − | − | + | PFO | − |
| CS0018 | − | − | − | na | − | +/2y | +/0 | − | − | − | − | − | − | − | − | − |
| CS0019 | − | − | − | na | − | − | − | − | − | − | − | − | − | +/f | − | − |
| CS0020 | − | − | − | +/14y | +/21y | +/1 m | +/S | + | − | + | − | − | + | +/f | MV regurg. | e |
| CS0021 | + | − | − | +/1y | +/35y | + | +/P | − | − | na | na | + | + | +/f | MV regurg. | a |
| CS0022 | − | − | − | na | + | − | + | − | − | − | − | − | + | + | − | b |
| CS0023 | + | − | − | − | − | − | − | − | − | − | − | − | − | + | − | − |
| CS0024 | +/3y | +/3y | + | Enlarged left heart, bicuspid aortic valve | ||||||||||||
| CS0028 | − | − | − | +/1 m | +/1 m | +/1 m | − | − | − | + | − | − | − | − | − | − |
| CS0029 | + | − | − | +/6 m | +/2y | +/6 m | +/f | − | +/6 m | − | − | − | − | − | AV sten. | Si |
| CS0030 | − | − | − | na | +/40y | +/39y | − | − | +/r | − | + | + | − | + | MV regurg. | − |
| CS0031 | + | − | − | +/1 m | +/2y | +/0 m | +/f | − | − | − | + | − | − | − | − | − |
| CS0032 | + | − | − | − | +/8y | + | − | − | − | − | − | − | − | − | − | − |
| CS0033 | + | − | − | +/1 m | +/3 m | +/1y | + | + | +/1y | − | − | − | − | − | − | Si |
| CS0034 | − | − | − | na | +/33y | +/33y | − | − | − | − | − | − | − | − | − | − |
| CS0035 | − | − | + | − | + | + | − | + | − | − | + | − | + | +/ch | MV regurg. | − |
| CS0036 | + | − | − | +/1 m | − | +/0 m | − | − | +/1 m | − | − | − | − | +/fh | AV sten. | − |
| CS0037 | + | − | − | +15 m | − | +/5 m | +/f | − | +15 m | − | + | − | − | − | − | Si,f |
| CS1001 | + | − | − | +/1y | − | +/1 m | − | − | − | − | − | − | − | +/h | PFO | − |
| CS1002 | + | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CS1003 | − | − | − | − | − | +/7y | − | − | − | − | − | − | − | + | AV regurg. Sep. hyp. | − |
| CS1004 | + | − | − | +/3 m | − | − | − | − | − | − | − | − | − | − | − | − |
| CS1005 | + | − | − | − | − | + | − | − | − | − | − | − | − | − | − | − |
| CS1007 | − | − | − | − | − | − | − | − | + | − | − | − | − | − | − | − |
| CS1008 | − | − | + | − | − | − | − | − | − | − | − | − | − | + | − | − |
| CS1009 | − | − | − | − | − | − | − | − | − | − | + | − | − | − | − | − |
| CS1010 | + | + | − | +/2 m | − | − | − | − | − | − | − | − | − | − | − | − |
| CS1011 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CS1012 | + | − | − | +/7y | − | − | + | + | − | − | − | − | − | + | − | − |
| CS1013 | + | − | − | +/1 m | − | − | − | + | − | − | − | − | − | + | PFO | p,am |
| CS1014 | + | − | − | + | − | − | − | − | − | − | + | − | − | − | Aort. Coarct. | − |
| CS1015 | − | − | − | − | − | +/33y | + | na | +/26y | − | − | − | − | +/c | − | − |
| CS1016 | − | − | − | − | − | +/20y | + | − | − | − | + | − | − | − | − | − |
| CS1018 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| CS1019 | + | − | − | − | − | − | − | − | +/26y | − | + | − | − | − | − | − |
| CS2001 | − | − | − | na | na | +/1y | na | na | na | − | + | − | − | + | − | na |
| CS2002 | + | − | − | − | +/36y | − | − | na | − | − | − | − | na | − | − | na |
| CS2003 | + | − | − | +/1 m | − | +/1y | +/0 | − | +/r | − | − | − | − | − | − | − |
| CS2004 | + | − | − | +/1 m | − | +/0 m | +/0 | + | − | − | − | − | na | +/sf | − | m |
| CS2005 | + | − | − | +/1 m | − | +/1 m | − | − | − | − | − | − | − | − | MV regurg. | − |
| CS2006 | + | − | − | +/1 m | + | − | − | − | na | − | − | − | − | na | AV regurg. | − |
| CS2007 | + | − | − | na | na | na | na | na | na | na | na | na | na | na | − | na |
| CS2008 | − | − | − | na | na | na | na | na | na | + | − | na | − | na | − | na |
| CS2009 | + | − | − | − | na | + | na | na | na | na | na | + | na | +/c | − | na |
| CS2010 | − | − | − | − | − | na | − | − | − | − | − | na | − | +/c | Valve does not close properly | − |
| CS2011 | − | − | − | na | na | + | + | na | − | + | − | − | na | +/c | MV regurg. | f, PI, Q |
| CS2012 | + | − | − | − | na | + | − | − | na | − | − | na | na | + | − | na |
| CS2013 | − | − | − | na | na | na | na | na | − | − | − | − | na | + | − | na |
| CS2014 | + | − | − | na | − | − | − | − | − | − | − | − | − | − | − | − |
| CS3001 | + | − | − | +/1 m | − | − | − | − | − | − | + | − | − | +/fh | Aort. Coarct. | − |
| CS3002 | + | − | − | − | − | +/2y | − | − | − | − | − | − | − | − | − | − |
| CS3003 | − | − | − | +/1 m | − | + | − | − | +/1 m | − | + | − | − | + | − | − |
| CS3004 | + | − | − | − | − | − | − | − | − | − | − | + | − | +/cf | − | − |
| CS3005 | + | − | − | +/1 m | +/1 m | − | − | + | − | − | + | − | − | − | − | − |
| CS3006 | + | − | − | + | na | na | na | na | na | na | na | na | na | na | − | na |
| CS3007 | + | − | − | − | − | − | − | − | − | − | − | − | − | + | − | − |
| CS3008 | + | − | − | +/1 m | − | +/1y | − | − | +/1y | − | − | − | − | + | − | − |
| CS3009 | − | − | − | na | − | − | − | + | − | − | − | − | − | + | − | − |
Abbreviations: Am, amiodarone; Aort. Coarct., aortic coarctation; APC, aorto-pulmonary collateral; AV regurg, aortic valve regurgitation; AV sten, aortic valve stenosis; b, bisoprolol; c, compression; Ca, cardizem; a, atenolol; e, enalapril; s, spironolactone; f, furosemide; h, other diuretic; m, metoprolol; m, months of age; MV regurg., mitral valve regurgitation; na, data not available; P, pericardiocentesis; p, propranolol; PFO, patent foramen ovale; Pl, plavix; Q, quinapril; s, spironolactone; S, stripping; Sep. hyp., septal hypertrophy; Si, sildenafil; y, years of age.
FIGURE 4.
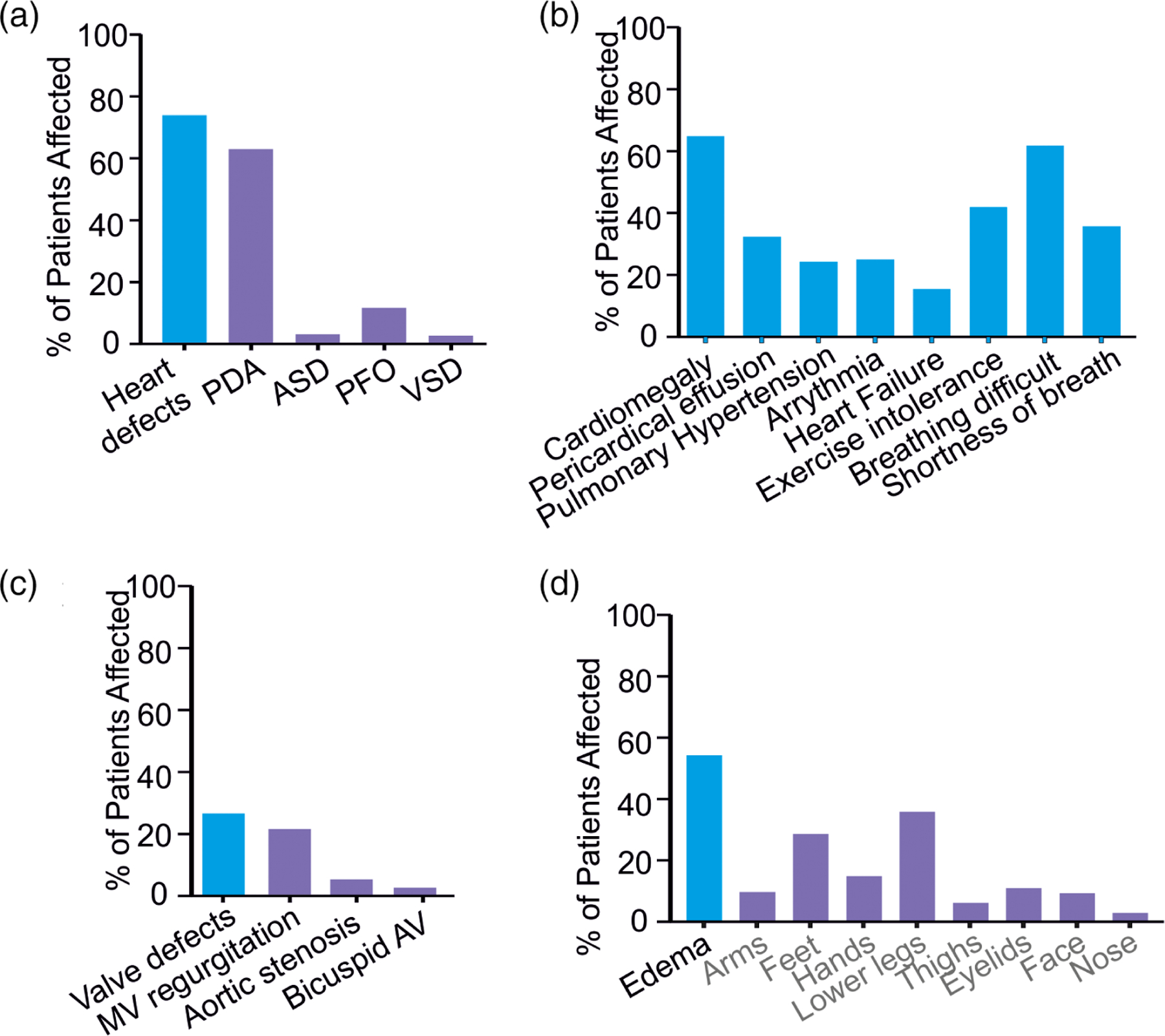
Cardiovascular and edema features (a–c) Incidence of cardiovascular abnormalities, and pulmonary and respiratory abnormalities. (d) Reported incidence of edema and location in CS subjects. CS, Cantú syndrome
Pericardial effusion was reported for 17/69 (25%) participants. In three severe cases that needed invasive treatment, pericardiocentesis was unsuccessful and pericardial stripping was required; in one individual, pericardiocentesis alone was required. In the remaining cases, medication was given, or no treatment was needed, presumably because the effusion was small and asymptomatic (Table 4). Although baseline ECG analyses typically do not reveal significant electrical defects (Levin et al., 2015), 12/65 (18%) subjects reported a history of cardiac arrhythmia. Additionally, 14% of subjects reported a history of heart failure (Figure 4b). In most cases for which information was available (8/9 patients), the heart failure was identified in infancy, and potentially related to untreated or unrepaired PDA.
In this CS cohort, 16/71 (23%) subjects reported blood pressure abnormalities, with 10/16 responders reporting high blood pressure (half receiving medical treatment) and 6/16 reporting low blood pressure (Table 4). Interestingly, previous analysis demonstrated consistently low blood pressure for age (Levin et al., 2016), although this decrease did not reach clinical threshold for a hypotension diagnosis, and postural hypotension is not reported for any CS subjects.
3.7 |. Pulmonary hypertension and respiratory abnormalities
A history of pulmonary hypertension (PHN) was reported in 16/67 (24%) subjects. This was first identified in infancy in 8/10 patients for whom information is available, and was treated with sildenafil in 3 patients (Table 4). In 3/13 reporting subjects, PHN spontaneously resolved. PHN and PDA were co-incidental findings in 10/13 of these subjects. Bidirectional flow across the PDA indicated the presence of PHN. Resolution of PHN occurred with treatment (chemical or surgical) of the PDA in all 10 subjects.
Four individuals reported respiratory tract malformations. One had bronchomalacia requiring a tracheostomy. One individual developed recurrent hemoptysis at age 20 and further investigation via pulmonary angiogram revealed a large bronchial collateral artery (aorto-pulmonary collateral artery, APC) that required coiling. Another individual had hemoptysis and arterial magnetic resonance imaging (MRA) showed multiple tortuous AV communications, requiring ligation, in both lungs; this individual also had bronchiectasis on CT scan. In a third individual, bleeding from abnormal vessels in the lungs required stapling. In addition to a relatively high incidence of respiratory tract infections, exercise intolerance and difficulty breathing were reported by a large fraction of subjects (Figure 4b).
3.8 |. Edema and lymphedema
36/70 (51%) subjects report a history of edema, variously reported as edema or lymphedema, with lower legs most commonly affected (Figure 4d). The swelling in the lower legs was typically first recognized in adolescence or young adulthood, and was usually noted to spare the tops of the feet. Interestingly, edema was reported by women more frequently than men, the diagnosis being specified as lymphedema in eight female subjects, but not in any of the male subjects. Many subjects report attempting to manage edema using compression stockings as well as diuretic medications (Table 4) with varying success.
3.9 |. Skeletal and connective tissue abnormalities
Osteochondrodysplasia has previously been associated with CS (Grange et al., 2014), including thickening of the calvaria, broad ribs, platyspondyly, ovoid vertebral bodies, narrow thorax and shoulders, pectus carinatum, scoliosis, and Erlenmeyer-flask-like long bones with metaphyseal flaring. In this registry cohort, 12/64 (19%) subjects reported skeletal dysplasia or osteochondrodysplasia, 24% reported pectus carinatum, 21% reported scoliosis, 6% reported osteoporosis/osteopenia and 6% reported delayed bone age (Figure 5a). It should be noted that not all subjects have had a complete set of skeletal radiographs, so again these findings may be underreported.
FIGURE 5.
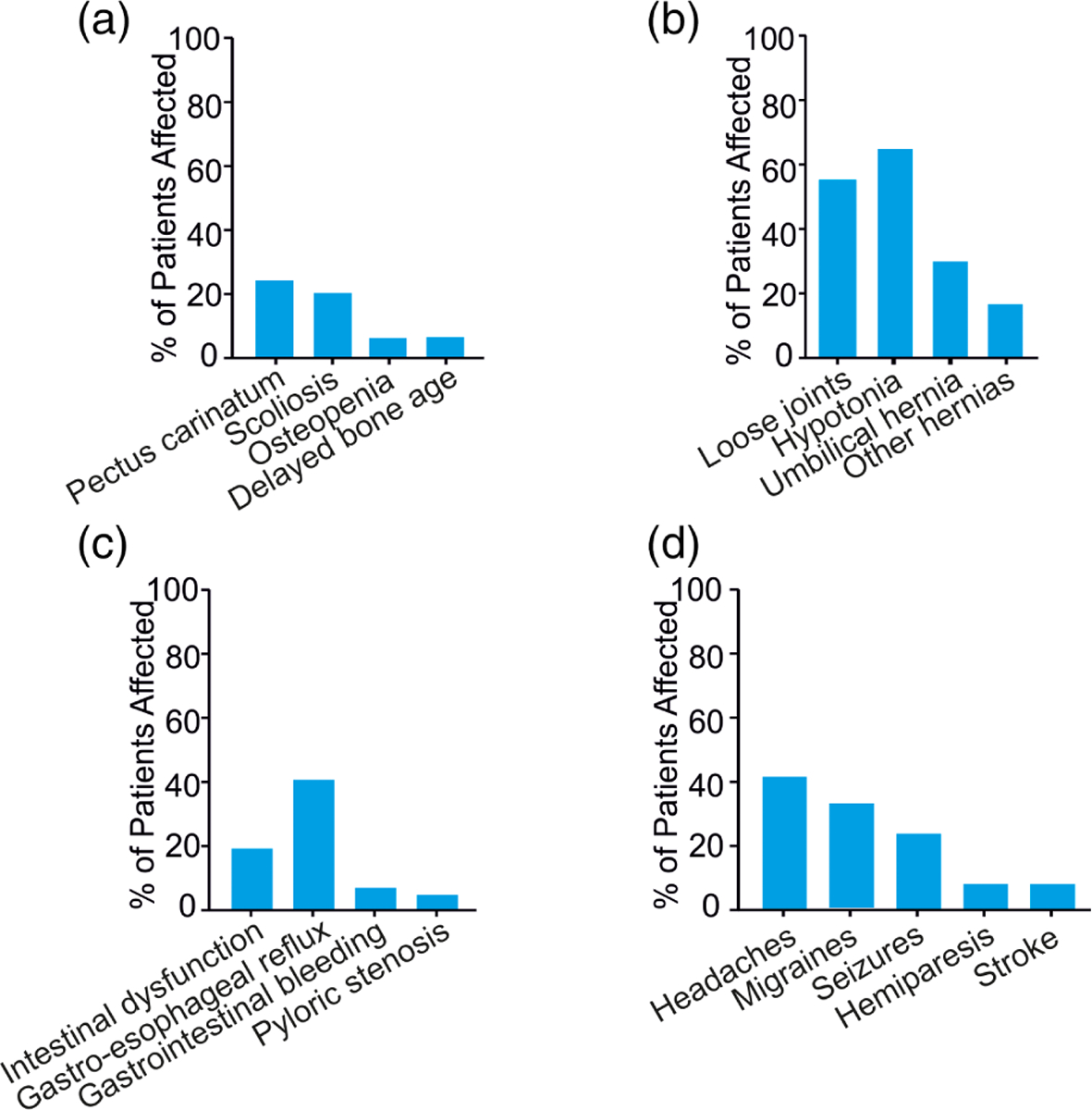
Musculoskeletal (a) and connective tissue (b) abnormalities, (c) gastrointestinal features, and (d) neurological abnormalities
Hyperflexible (“loose”) joints were reported in 40/72 (56%) of subjects (Figure 5b), in addition to loose and/or wrinkled skin with deep palmar/plantar creases, consistent with previous reports (Grange et al., 2014). Hernias were also reported, umbilical hernia (28%) being most common, followed by inguinal hernia (16.9%) (Table 1, Figure 5b).
3.10 |. Gastrointestinal features
Intestinal dysfunction was reported by 12/71 (17%) subjects, generally severe constipation or slow intestinal motility. Gastro-esophageal reflux was present in 15/36 (42%), gastrointestinal bleeding was reported in 7%, and pyloric stenosis in infancy in 4% of subjects. 22% of subjects required a feeding tube as infants or young children (Figure 5c).
3.11 |. Renal features
Renal problems were reported in 14% of subjects, and if present, consisted of recurrent kidney or bladder infections, hydronephrosis and vesico-ureteral reflux. Polycystic kidney disease was reported by one individual, and therefore it is unclear if this is related to CS, or represents a separate problem.
3.12 |. Reproductive system features
74% of adult subjects reported that they reached puberty at the expected time. Abnormal menstruation was reported in 10/22 adult females, with heavy menses frequently noted. Male reproductive system defects were reported in only 1/34 responding subjects, who required surgery for an undescended left testicle.
3.13 |. Immune function
Immune dysfunction was reported by 30/74 (41%) subjects. Recurrent respiratory infections are also cited, but specific information is not available in all cases. Low immunoglobulin levels were reported by several subjects, and five CS subjects in the cohort have been treated with IVIG. However, immunoglobulin levels checked in the CS Research Clinic at Washington University have all been essentially normal. Therefore, the relevance of this finding is unclear at this time, and further investigation is needed.
3.14 |. Neurological and neurovascular features
There were a variety of associated abnormal neurological features (Table 5) consistent with those reported previously (Leon Guerrero et al., 2016). 27/68 (40%) subjects report frequent headaches, with 16 of these 27 (60%) reporting migraine type headaches, often with associated aura, photophobia, phonophobia, and occasionally with transient hemiparesis (Figure 5d). In addition, 17/72 (24%) subjects report a history of various seizures types including febrile, tonic–clonic, and absence seizures, and temporal lobe epilepsy. 8% report hemiparesis, sometimes transient and associated with migraines, and 6% report a history of stroke (Figure 5d and Table 5). One individual had a stroke in infancy and has persistent unilateral weakness and two adults had strokes in their fourth and fifth decades. One adult had evidence of a prior stroke on brain imaging but the age at which it occurred is unknown and she has no current clinical symptoms.
TABLE 5.
Neurological features
| Patient | Headaches/migraine | Headache medication | Seizures / type of seizures | Seizure medication | Brain abnormalities | Hemiparesis | Stroke / age |
|---|---|---|---|---|---|---|---|
| CS0001 | Migraine | na | − | − | − | Transient, migraine-assoc. | − |
| CS0002 | Headache | OTC | − | − | − | − | − |
| CS0003 | Headache | OTC | − | − | − | − | − |
| CS0004 | Migraine | t,f,z | − | − | − | During migraine, transient left hemiplegia | − |
| CS0005 | Migraine | OTC | Febrile | na | White matter lesions | − | − |
| CS0006 | Migraine | OTC | Grand mal | I | Calcifications | − | − |
| CS0007 | Migraine/aura | OTC | Febrile/grand mal | na | Blocked blood vessel | − | MRI |
| CS0008 | − | − | − | − | − | − | − |
| CS0009 | − | − | − | − | − | − | − |
| CS0010 | − | − | − | − | − | − | − |
| CS0011 | Migraine/cluster headaches | na | − | Decreased white matter | − | − | |
| CS0012 | na | na | − | na | na | na | |
| CS0013 | − | − | Seizure/1 m | p | External hydrocephalus | − | − |
| CS0014 | − | − | − | − | “Extra ventricle” | na | na |
| CS0015 | Headache/aura | OTC | − | − | − | − | − |
| CS0016 | − | − | Convulsions during sleep | k, t,d,c | Dysgenesis of corpus callosum, diffuse leptomeningeal enhancement | − | − |
| CS0017 | − | − | − | − | − | − | − |
| CS0018 | Migraine | a | Febrile seizures | na | Brain lesions in pons and other areas | − | − |
| CS0019 | − | − | Seizures after stroke | p, k | Damage from stroke, right side weaker | na | +/2 mo |
| CS0020 | Migraine | na | − | − | Periventricular leukomalacia | − | − |
| CS0021 | − | − | − | − | − | na | na |
| CS0022 | − | − | − | − | − | na | na |
| CS0023 | Headache | na | − | − | − | − | − |
| CS0024 | Headache | na | Febrile seizures | na | − | − | − |
| CS0028 | − | − | Febrile seizures | na | − | − | − |
| CS0029 | − | − | − | − | − | − | − |
| CS0030 | Migraine (photo/phonophobia) | na | − | − | − | − | − |
| CS0031 | − | − | − | − | − | − | − |
| CS0032 | Migraine (photo/phonophobia) | OTC | − | − | − | − | − |
| CS0033 | − | − | − | − | − | − | − |
| CS0034 | − | − | − | − | − | − | − |
| CS0035 | Migraine (numbness/tingling in extremities) | na | Seizure during stroke | − | Calcium deposits; panhypopituitarism | + | +/51 yr, 53 yr |
| CS0036 | na | na | − | − | Intraventricular hemorrhage, mild lateral ventricular dilatation without third/fourth ventricular dilatation, bilateral thalamostriate vasculopathy, blood products in the atrium of the lateral ventricles, extra-axial fluid with debris | − | − |
| CS0037 | − | − | − | − | − | − | − |
| CS1001 | − | − | − | − | − | − | − |
| CS1002 | − | − | − | − | − | − | − |
| CS1003 | Headache | na | − | − | Enlarged pituitary | − | − |
| CS1004 | − | − | − | − | − | − | − |
| CS1005 | − | − | − | − | − | − | − |
| CS1007 | − | − | − | − | − | − | − |
| CS1008 | − | − | − | − | − | na | na |
| CS1009 | Headache | na | − | − | − | − | − |
| CS1010 | − | − | − | − | − | − | − |
| CS1011 | Headache | OTC | − | − | − | − | − |
| CS1012 | − | − | − | − | − | − | − |
| CS1013 | − | − | Neonatal seizure after diuretic intake | na | − | − | − |
| CS1014 | − | − | − | − | − | na | na |
| CS1015 | − | − | − | − | − | na | na |
| CS1016 | − | − | − | − | − | na | na |
| CS1018 | Headache | na | Grand mal | na | − | − | − |
| CS1019 | − | − | − | − | − | − | − |
| CS2001 | − | − | − | − | − | − | − |
| CS2002 | − | − | − | − | − | na | na |
| CS2003 | − | na | − | − | − | − | − |
| CS2004 | Migraine (photo/phonophobia, nausea, visual aura) | na | + | na | na | ||
| CS2005 | − | − | − | − | − | − | − |
| CS2006 | na | na | − | − | − | na | na |
| CS2007 | na | na | na | na | na | na | na |
| CS2008 | Migraine | na | Minor seizure in infancy | na | − | − | − |
| CS2009 | Migraine (photo/phonophobia, nausea, aphasia, paresthesia) | na | − | − | Abnormally small pituitary | − | − |
| CS2010 | − | − | − | − | − | − | − |
| CS2011 | Migraine (photo/phonophobia, nausea, visual aura, hemiparesis) | na | + | na | Calcifications | Possible TIA | +/44 yr |
| CS2012 | − | − | − | − | − | − | − |
| CS2013 | Headache | na | + | na | na | − | − |
| CS2014 | − | − | − | − | − | − | − |
| CS3001 | − | − | − | − | − | na | na |
| CS3002 | − | − | − | − | − | na | na |
| CS3003 | na | na | − | − | − | − | − |
| CS3004 | − | − | Temporal lobe epilepsy | na | − | − | − |
| CS3005 | − | − | − | − | − | − | − |
| CS3006 | na | na | na | na | na | na | na |
| CS3007 | Headache | na | − | − | − | − | − |
| CS3008 | Migraine, VP shunt, right hemiparesis | na | − | − | − | Right side weak | + |
| CS3009 | Headache | na | − | − | − | − | − |
Abbreviations: a, amitriptyline; c, CBD oil; d, diazepam; f, fioricet; k, keppra; l, lamictal; MRI, evidence from MR imaging; na, data not available; OTC, over the counter drugs; p, phenobarbital; t, topamax; z, zofran.
Neuroimaging of the brain was available for 61% of subjects and revealed a variety of abnormal findings (Figure 6). Head and neck MR angiography revealed dilated and tortuous arteries, including internal carotids and vessels of the circle of Willis, in 10/10 (100%) patients assessed at the Washington University site (Leon Guerrero et al., 2016). Abnormal or tortuous blood vessels were also observed in the retina of 34% of participants. Various additional brain abnormalities were reported in 11/49 (22%) subjects, including hydrocephalus, “extra ventricle,” dysgenesis of the corpus callosum, diffuse leptomeningeal enhancement, FLAIR hyperintensities in the subcortical white matter, intraventricular hemorrhage, mild lateral ventricular dilatation, bilateral thalamostriate vasculopathy, calcifications, and pituitary abnormalities.
FIGURE 6.
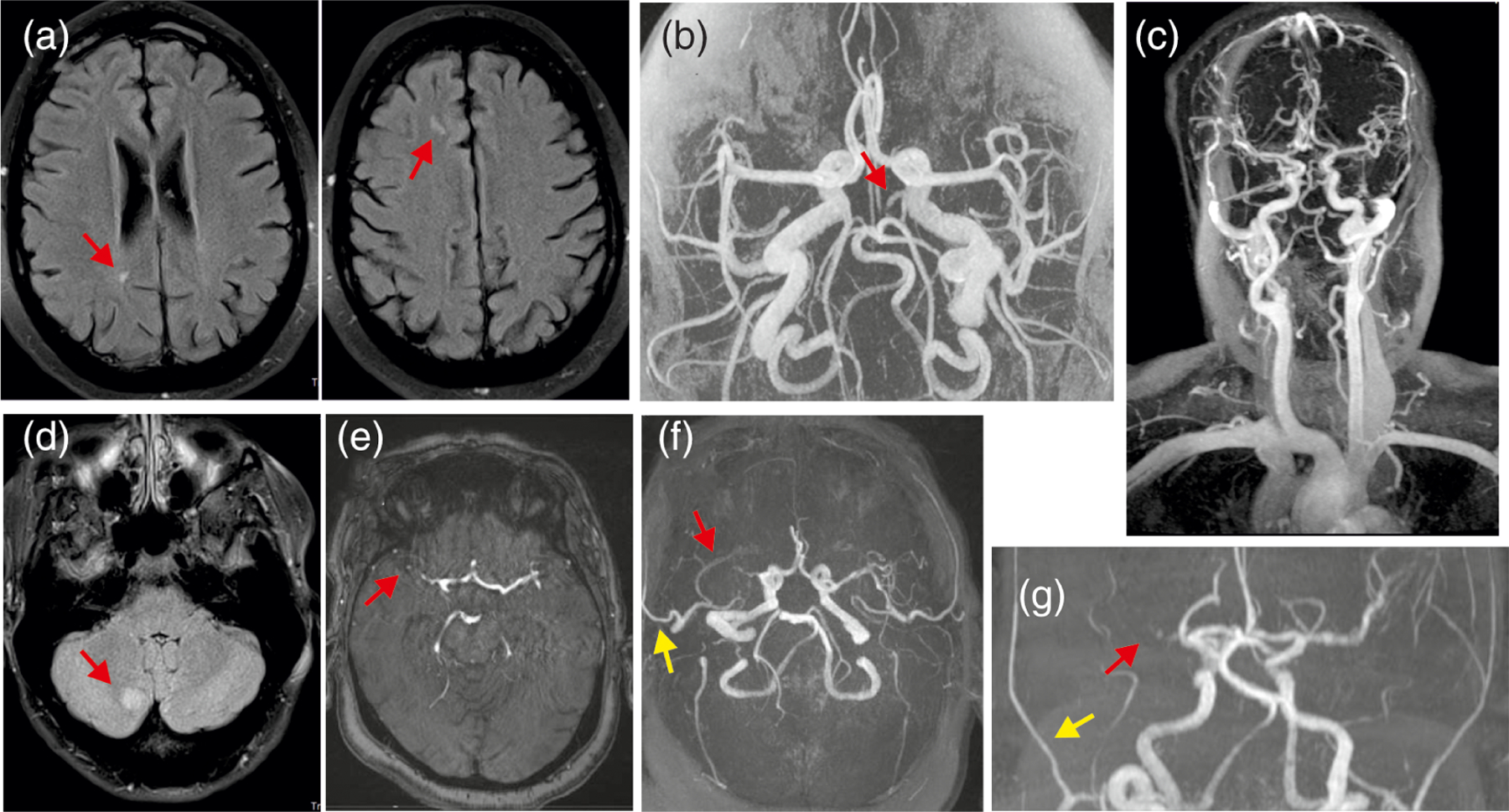
Neuroimaging features (a) MR images from a 49 year old female with CS. Nonspecific white matter lesions are frequently seen on MR imaging in CS and may be due to ischemia or demyelination, but etiology is unclear; (b and c) Diffusely dilated and tortuous cerebral arteries have been identified in essentially all CS subjects undergoing imaging at Washington University; the red arrow indicates a persistent trigeminal artery in a 20 year old female with CS; (d) right cerebellar lesion without enhancement, mass effect or diffusion restriction, possibly due to ischemia or demyelination in a 33 year old female with CS; (e–g) MR images from a 57 year old female with CS showing mildly dilated cerebral arteries, occlusion of right MCA consistent with old stroke (red arrows), as well as right temporal artery collateral (yellow arrows). CS, Cantú syndrome
3.15 |. Vision
Myopia and hyperopia are the most frequent vision disturbances in CS subjects; 36% of patients report problems with eyesight and 32% wear glasses. Infrequently, nystagmus, strabismus, retinal, or optic nerve anomalies were reported.
3.16 |. Hearing and ENT
Five of 15 subjects reporting hearing loss (33%) were diagnosed with conductive deficits, 1/14 (7%) was diagnosed with sensorineural deficits, and 9/15 (60%) were of unknown origin. Thirty-four subjects at the Washington University site responded to questions about ENT concerns and prior surgery; 8/34 (24%) subjects reported laryngomalacia. Three of 31 (8.8%) subjects seen in the Washington University clinic had a tracheostomy, which was eventually removed in one case. Five of 32 (14%) reported sleep apnea, and either adenoidectomy alone or both tonsillectomy and adenoidectomy was required.
3.17 |. Endocrine problems
A single individual reported panhypopituitarism, with multiple pituitary hormone deficiencies, including ACTH, TSH, FSH, and LH. One individual has diabetes insipidus. One individual has been treated for hypothyroidism since infancy. One individual was diagnosed with growth hormone deficiency and was on replacement therapy.
3.18 |. Medications
Eleven of 64 (17%) affected subjects report taking cardiac medications, including ACE inhibitors, such as Quinapril and Enalapril, and beta-blockers such as Atenolol and Bisoprolol (Table 4), either currently or in the past. Of the subjects reporting hypertension, 63% are currently being treated with ACE inhibitors, and 60% with edema report current or previous medication treatment with diuretics, including furosemide, spironolactone and amiloride. Three subjects reported previous treatment for pulmonary hypertension with sildenafil (See cardiovascular section).
3.19 |. Genetic testing and CS genotype
ABCC9 variants were identified in 72/74 (97%) participants in the ICSR cohort (Table 1, Table 6) and none harbor a variant in KCNJ8. Most subjects (60/74; 81%) were initially given a clinical diagnosis of CS, subsequently confirmed by ABCC9 sequencing or research or targeted exome sequencing in 58/60 cases. However, in 12 subjects (12/72; 16.6%), the diagnosis was arrived at through exome sequencing, without CS being clinically suspected. In 16/72 (22%) subjects with a molecular diagnosis, the ABCC9 variant was inherited from an affected parent (16% mother, 6% father). Of 54 reporting participants, 11 have one or more affected sibling(s). This cohort includes eight families with two or three generations of affected individuals; five of these families have been evaluated at the Washington University CS clinic, two families are from the Netherlands and one is from Australia (See Table 1). In one of the families, a father was found to have mosaicism for the ABCC9 variant in blood, consistent with somatic and germline mosaicism in him. Clinically, he was only mildly affected in segments of his body, predominantly hypertrichosis, without evidence of systemic features, and he was only identified after diagnosis of a severely affected daughter. In the remaining individuals with negative family history, the ABCC9 variant was presumed to be de novo.
TABLE 6.
ABCC9 variant distribution in 72 subjects with Cantú syndrome
| cDNA variant | Protein alteration | # of subjects |
|---|---|---|
| (NM_005691.3) | (# of kindreds) | |
| c.178 C > T | p.His60Tyr | 2 (2) |
| c.621 C > A | p.Asp207Glu | 3 (3) |
| c.881 G > A | p.Gly294Glu | 2 (1) |
| c.1138 G > T | p.Gly380Cys | 1 (1) |
| c.1295 C > T | p.Pro432Leu | 1 (1) |
| c.1433 C > T | p.Ala478Val | 2 (1) |
| c.2378 A > T | p.Asp793Val | 1 (1) |
| c.2444 G > C | p.Gly815Ala | 1 (1) |
| c.3014 A > T | p.His1005Leu | 2 (2) |
| c.3052 T > G | p.Trp1018Gly | 1 (1) |
| c.3055_3,056 del ins GA | p.Thr1019Glu | 1 (1) |
| c.3056 C > A | p.Thr1019Lys | 1 (1) |
| c.3058 T > G | p.Ser1020Pro | 1 (1) |
| c.3116 T > C | p.Phe1039Ser | 1 (1) |
| c.3161 C > A | p.Ser1054Tyr | 2 (2) |
| c.3346 C > T | p.Arg1116Cys | 6 (3) |
| c.3345 C > G | p.Arg1116Gly | 1 (1) |
| c.3347 G > A | p.Arg1116His | 5 (4) |
| c.3461 G > A | p.Arg1154Gln | 13 (9) |
| c.3460 C > G | p.Arg1154Gly | 1 (1) |
| c.3460 C > T | p.Arg1154Trp | 10 (10) |
| c.3605 C > T | p.Thr1202Met | 1 (1) |
| c.3618 C > A | p.Asn1206Lys | 2 (2) |
| c.3704 C > T | p.Ser1235Phe | 1 (1) |
| c.3796 G > A | p.Val1266Met | 1 (1) |
| c.4040 G > T | p.Arg1347Leu | 5 (3) |
| c.4469 T > A | p.Val1490Glu | 1 (1) |
| c.4480 G > A | p.Ala1494Thr | 3 (1) |
| Unknown | Unknown | 2 |
In every case, the diagnosis of CS was made after birth, with no instances of prenatal diagnosis. Since the two genes associated with CS have only recently been identified, older individuals often lack a molecular diagnosis. Hence, a majority of confirmed subjects were diagnosed either within the first year of life or after age 17, and some parents participating in this study were only diagnosed with CS after their affected children received the diagnosis. Interestingly, 41% of participants initially received other diagnoses, including lysosomal storage disorders such as a mucopolysaccharidosis, or Beckwith-Wiedemann syndrome, based on coarse facial features, or Pompe disease based on neonatal cardiomegaly.
We identified 28 different ABCC9 variants in 72 participants (Table 1, Table 6), applying targeted Sanger sequencing of the ABCC9 gene, or exome sequencing (ES) or genome sequencing (GS). Fifteen variants have not previously been reported. The majority of reported subjects show simple missense variants in ABCC9, and only 1 individual harbors a deletion–insertion variant involving two nucleotides. Figure 7 shows the protein architecture of the ABCC9-encoded KATP-channel SUR2 subunit, including all documented variants and their distribution across different protein domains. A majority of variants (68%) cluster around transmembrane domain 2 (TMD2). Variants at p.Arg1154 (identified in 24 subjects, 33%) and p.Arg1116 (identified in 11 subjects, 15%), represent the most commonly observed (Table 6).
FIGURE 7.
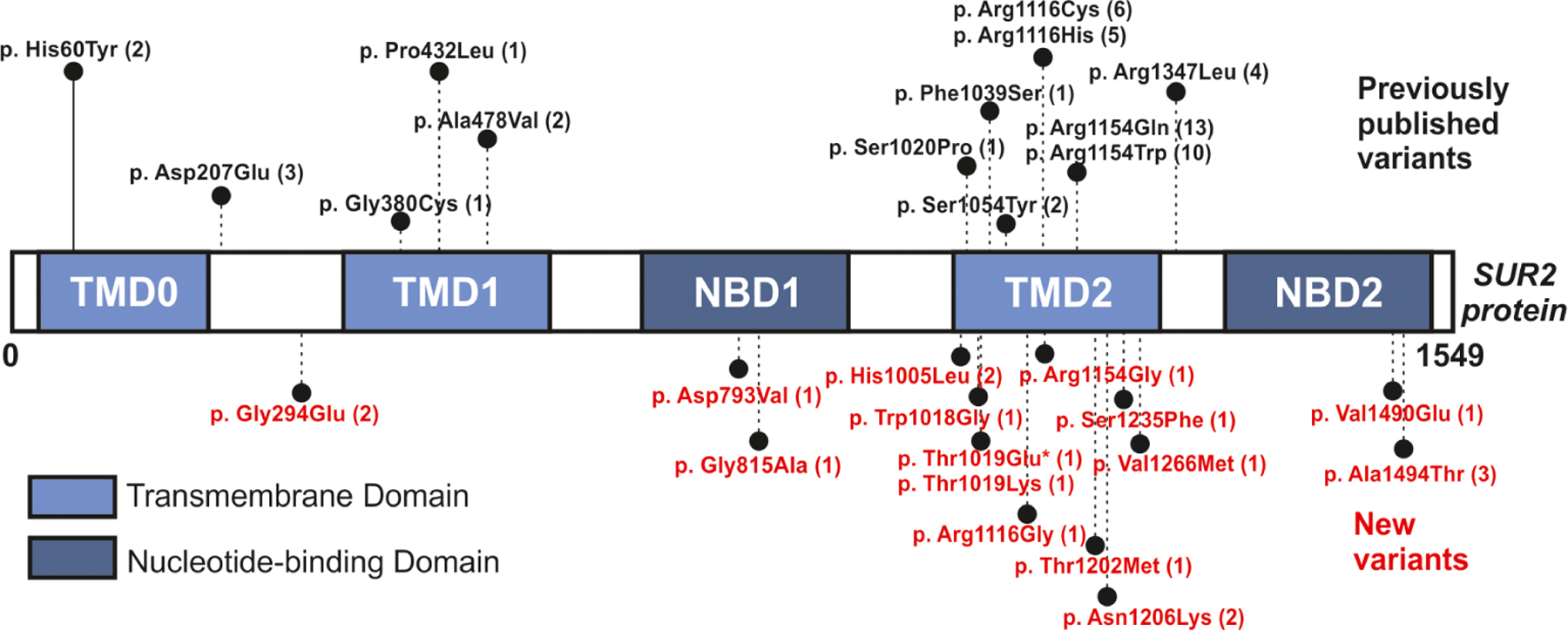
Location of CS variants in the SUR2 protein Schematic of SUR2 protein domain architecture: TMD0, transmembrane domain 0, with five predicted transmembrane helices; TMD1, with six transmembrane helices; NBD1, nucleotide-binding domain 1; TMD2, with six transmembrane helices; and NBD2. Amino acid changes identified in CS subjects are indicated. The number of subjects harboring each variant is indicated in brackets. Previously published variants are shown in black and new variants in orange. Transmembrane domains (TMDs) are highlighted in light blue, nucleotide-binding domains (NBDs) in dark blue. Deletion–insertion variants are indicated by an asterisk. CS, Cantú syndrome
3.20 |. Comparison of phenotypes by genetic variants
We evaluated the correlation of ABCC9 variants with syndromic features. We have not identified any obvious difference in frequency or degree of craniofacial dysmorphology, hypertrichosis or skin appearance depending on the site of the gene variant, and cardiovascular and neurological anomalies were similar across the various variant sites (Tables 4 and 5). Even within families having multiple affected members, there is highly variable expression of CS features, in common with other autosomal dominant conditions. In one family (Figure 8), in which the mother was diagnosed only after her children, the 8-year-old boy (Figure 8b) has autism spectrum disorder and developmental delays, the 6-year-old boy (Figure 8c) has typical features of CS, without evidence of autism, while the 3.5 year old girl (Figure 8d) had esophageal atresia and a PDA that required surgery, as well as pulmonary hypertension and a requirement for oxygen supplementation and gastrostomy tube feedings for the first 2 years of life.
FIGURE 8.
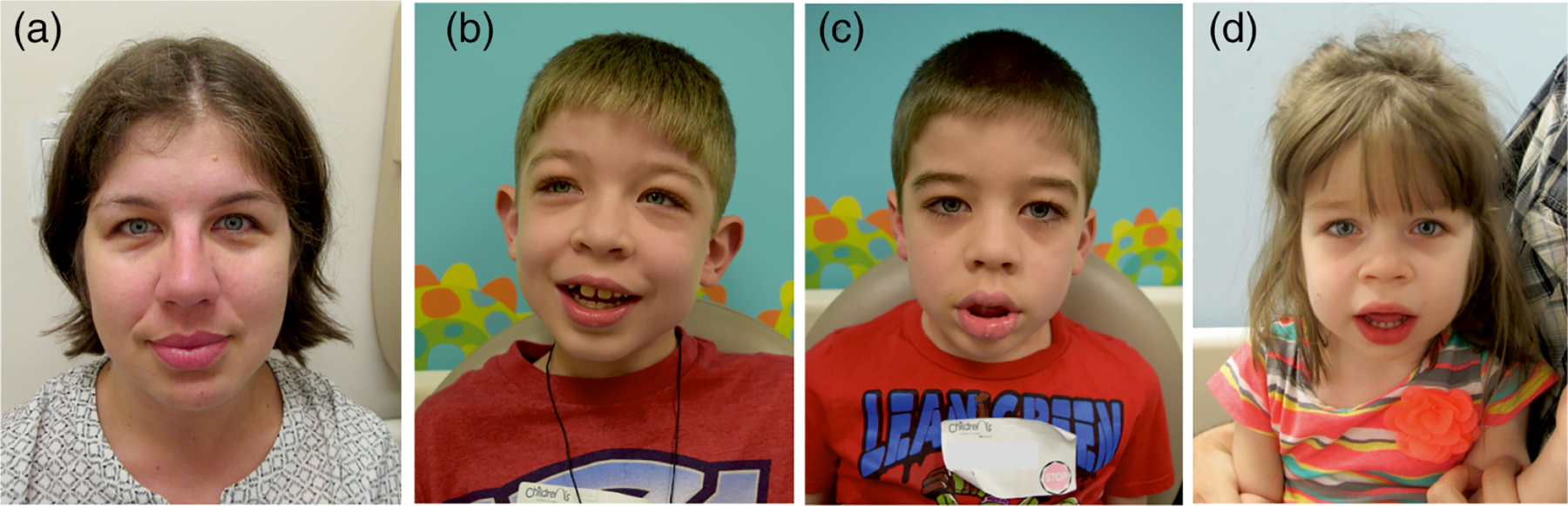
Intra-family presentation of Cantú syndrome mildly affected mother (a) and her three children who are variably affected (see text)
4 |. DISCUSSION
The identification of pathogenic variants in ABCC9 and KCNJ8 as the molecular genetic basis of CS has led to the diagnosis of numerous additional patients, as well as increased awareness of the syndrome in the field of clinical genetics. This study reports multiple novel CS-associated ABCC9 variants, further defines the genetic basis of the disease, and provides quantitative assessment of the penetrance of disease features.
Notably, the majority of subjects in our cohort are children or young adults, with 59% below the age of 15 at the most recent examinations (Table 2, Figure 1), consistent with a previous analysis of all reported cases of CS in which 61% of patients were children (Roessler et al., 2018). Since the genes associated with CS have only recently been identified, we suspect there may be many affected individuals, particularly older individuals, who have not received genetic testing and therefore remain undiagnosed.
4.1 |. The spectrum of CS features
Abnormal growth is common in CS, and may be identified both prenatally and postnatally. Birth length and weight are consistently above average, irrespective of gestational age, which suggests the possibility that the increased incidence of prematurity may be related to excessive in utero growth. Polyhydramnios is a known risk factor for premature delivery, but we did not observe a correlation between presence of polyhydramnios and early delivery. Our analysis shows that macrocephaly is typically present at birth and persists throughout life (Figure 1, Table 3). Both birth weight and length are increased, with some adult males continuing to show elevated height (Table 3). A thin or even quite muscular appearance during childhood changes due to weight gain that occurs after puberty.
Previous reports have noted developmental delay in CS patients (Grange et al., 2014). It is clear that developmental milestones are delayed in a majority of CS patients, and motor development may be impaired due to hypotonia or joint laxity. There is also an increased frequency of reported behavioral problems, including ADHD and autism spectrum features in young patients. However, despite early developmental delays, most patients with CS attain normal cognition and intellectual function by adulthood; speech develops normally in most patients and assessed intelligence tends to be normal, with most children attending regular schools. Cerebrovascular abnormalities have been recognized in CS patients, including dilated and tortuous cerebral vessels, as well as white matter changes and persistent fetal circulation (Figure 6) (Brownstein et al., 2013; Leon Guerrero et al., 2016). Additional neuroimaging studies in our larger cohort reveal these features to be common. Although further studies are required, increased blood flow, associated with vascular dilation in the brain, may be causally associated with the increased incidence of headaches and migraines in CS patients (Table 5). There appears to be an increased frequency of stroke in this CS cohort, but the long term risk is unclear at this time. Brain imaging, including evaluation of the cerebral vasculature, should be considered for all patients with CS, especially those with headaches and other neurological symptoms.
Previous analyses of CS patients have revealed multiple cardiovascular abnormalities, both developmental and pathophysiological (Grange et al., 2014). PDA was reported most frequently and required surgical closure in almost half of affected individuals. Cardiomegaly, due to ventricular enlargement, with ventricular systolic function either normal or increased, is a common finding. The resultant increased cardiac output (high output state) is distinct from dilated cardiomyopathy, in which the ventricles are dilated and have decreased systolic function. It is also distinct from hypertrophic cardiomyopathy in which ventricular walls are thickened but cardiac chamber cavity is either normal or smaller than normal. There is no evidence of diastolic dysfunction in the majority of patients. The majority of subjects that have been followed long term (>5 years) have not developed heart failure, but they are still relatively young. Two older individuals have developed high output heart failure, potentially exacerbated by inappropriate treatment with ACE-inhibitors. Thus, while the “high output state” may ultimately lead to cardiac dysfunction, long-term natural history, and cardiac outcomes are yet to be determined.
Dilated and tortuous large arteries, as well as persistent fetal arteries, are common findings in CS (Hiraki et al., 2014; Leon Guerrero et al., 2016). PDA is disproportionately associated with pulmonary hypertension, suggesting a common origin due to increased blood volume. The consistent finding of cardiomegaly with preserved cardiac function (Levin et al., 2016) is replicated in mice (Huang et al., 2018) and zebrafish (Tessadori et al., 2018) carrying CRISPR/Cas9-engineered CS variants in ABCC9. These animal studies suggest that cardiac enlargement and enhanced cardiac output is a secondary response to decreased vascular resistance and lowered blood pressure (Huang et al., 2018). In this case, as with persistent PDA, increased pulmonary blood flow may contribute to the development of pulmonary hypertension in the long-term due to risk of the development of pulmonary vascular occlusive disease, and therefore, treatment with phosphodiesterase type 5 inhibitors such as sildenafil might not be appropriate.
Edema has been reported previously in CS subjects (Grange et al., 2014), and is a common finding in the present cohort; generalized edema is often present at birth and resolves, but reappears later in life, usually in adolescence or young adulthood. In some subjects, this is reported specifically as lymphedema, and it is unclear at this juncture whether lymphatic dysfunction is the underlying basis more broadly. As discussed below, it is possible that both vasodilation and lymphatic dysfunction may share a common origin in CS, related to decreased smooth muscle activity. A high frequency of pericardial effusion, clearly detected in the present cohort, may also be related to increased blood volume or perhaps to reduced cardiac lymphatic drainage. Treatment is often needed, including pericardial stripping in extreme cases.
4.2 |. Genotype–phenotype correlation and CS
Since the initial reports of an association of CS with variants in ABCC9 (Harakalova et al., 2012; van Bon et al., 2012), and KCNJ8 (Brownstein et al., 2013; Cooper et al., 2014), it has become clear that pathogenic variants in one or the other of these two genes is the primary cause of CS, but the expression of disease features is clearly quite variable. A sub-set of patients with ABCC9 variants, but without evidence of cardiovascular or other features beyond hypertrichosis and characteristic facial features, have previously been defined as distinct syndromes—hypertrichosis with acromegaloid facial features (HAFF) or acromegaloid facial appearance (AFA) (Afifi, Abdel-Hamid, Eid, Mostafa, & Abdel-Salam, 2016; Harakalova et al., 2012). The present study further emphasizes the variability of clinical features, even with the same gene variant, and in multiple individuals within a single family. Patients reported as having HAFF or AFA clearly fall within the milder end of the CS spectrum, and therefore, we strongly suggest that such alternative terms should no longer be used.
We attempted to correlate severity of clinical features with genotype for the most common variants (p.Arg1116 and p.Arg1154), but it is clear that there is essentially as much variability of features among patients carrying these variants as between genotypes. While it remains important to draw quantitative phenotype-to-genotype comparisons where possible, a bigger cohort may be required. In the present study, there are several families with multiple affected family members in whom variability is demonstrated (Figure 8). Variability is present in multiple organ systems, and especially notable in cardiac-related abnormalities. In some family members, recognition of CS would have been difficult if the proband had not been diagnosed and available for comparison. These observations confirm findings of intrafamilial variability previously described in the literature (Czeschik et al., 2013; Hiraki et al., 2014; Roessler et al., 2018).
It is important to note that even in this extended cohort no individual has been recognized to carry a pathogenic missense variant in ABCC9 without showing a clinically relevant phenotype. One individual in this cohort who was only mildly affected was found to have somatic and germline mosaicism for the ABCC9 variant, suggesting that mosaicism might account for milder clinical features in some cases. Since not all relatives of individuals with CS have been evaluated for the presence of the variant found in the proband, we cannot rule out nonpenetrance; however, the present data do not suggest this. So far, only two individuals harboring variants in KCNJ8 have been identified (Brownstein et al., 2013; Cooper et al., 2014). In human neonatal diabetes resulting from equivalent mutations in the paralogous KCNJ11 (Kir6.2) and ABCC8 (SUR1) genes, heterozygous activating variants in KCNJ11 account for approximately three times as many cases as variants in ABCC8 (Naylor, Greeley, Bell, & Philipson, 2011). Both of the identified individuals with a KCNJ8 variant have key clinical features of CS including congenital hypertrichosis, macrosomia at birth, macrocephaly, coarse facial appearance, cardiomegaly, skeletal abnormalities, and developmental delay, with reported severity equal to or perhaps greater than that seen in ABCC9-related CS. Mouse models of CS in which human CS variant equivalents have been introduced to the endogenous ABCC9 and KCNJ8 loci (Huang et al., 2018) indicate that, at least for the Kir6.1 (p. V65M) variant, the features are more severe than for the introduced ABCC9 (SUR2 [p.A478V]) variant. It is therefore unclear why the incidence of KCNJ8-related CS is so low, and raises the possibility that such mutations might frequently be lethal. Regulation of Kir6.2/SUR1 and Kir6.1/SUR2–dependent KATP channel activity by cytoplasmic nucleotides is qualitatively similar (in both cases, ATP inhibits the channel by binding to the Kir6 subunit, Mg-nucleotides activate by interaction with the SUR subunit), but there are important quantitative differences (Nichols, 2006), and tissue-dependent modulation by second messengers may be very different (Flagg, Enkvetchakul, Koster, & Nichols, 2010).
5 |. CONCLUSIONS AND PROSPECTUS
The current study represents the largest survey of individuals (n = 74) with CS published to date. The most notable diagnostic features are hypertrichosis and facial dysmorphology, which are typically evident at birth. Additionally, we confirm a high incidence of PDA, cardiomegaly, edema and migraines or severe headache in CS patients. More than half of the mothers of CS babies exhibit polyhydramnios, and babies with CS are found to be at a high risk for preterm delivery (<37 weeks). Macrocephaly is typically observed at birth and persists throughout life. Adult females fall within the normal range for height, but adult males may be taller than average, although data are limited in this cohort. Developmental delay is commonly reported in young children, but intellectual disability is not common, and adult intelligence is typically normal. The complex clinical presentation of CS may be attributed to the presence of SUR2/Kir6.1 channels in multiple tissues throughout the body (Nichols et al., 2013), as well as the variable severity of features resulting from any given gene variant, even the same variant within the same family. This variability points to yet unidentified genetic or other modifiers that influence the phenotypic outcome.
The ICSR has been built not only in order to generate a critical mass of affected individuals to identify new clinical characteristics of the disorder, but also to ensure rapid progression toward clinical application of current genetic and scientific study results. Moving forward, the hope is that the registry will continue to expand to include as many individuals as possible from around the world and to generate longitudinal data, essential to understanding the long-term effects and implications of CS pathophysiology, as well as to provide essential data and a large well-characterized patient cohort for development of a directed pharmacological treatment for CS. Currently, there are no targeted therapies for CS available and it is not known to what extent associated features can be reversed once manifest. Hence, clinical management at this time involves symptomatic treatments to address secondary complications.
There are undoubtedly still numerous undiagnosed or misdiagnosed cases of CS, especially individuals of older ages. While commercial gene sequencing is available for clinical genetic diagnostic purposes in most advanced countries, such facilities are not available to all. The authors welcome contact from patients/caregivers, both regarding inclusion in the ICSR, and for potential inclusion in annual clinics.1
ACKNOWLEDGMENTS
We thank the patients and families for their participation and consent to publish this work. We acknowledge the technical assistance of Mr. Michael Lugo (Washington University School of Medicine) in setting up the initial registry scripts.
Funding information
American Heart Association, Grant/Award Number: 19POST34380407; Children’s Discovery Institute, Grant/Award Number: CH-MD-II-2015-488; CIMED Pilot and Feasibility Program, Grant/Award Numbers: CIMED-14-03, CIMED-17-01; E-Rare Joint Transnational Cantu Treat Program, Grant/Award Number: I-2101-B26; National Institutes of Health, Grant/Award Number: HL140024
Footnotes
For inquiries regarding inclusion in the registry, cantu-syndrome@wustl.edu; for research-based genetic sequencing where not otherwise available, g.vanhaaften@umcutrecht.nl
REFERENCES
- Afifi HH, Abdel-Hamid MS, Eid MM, Mostafa IS, & Abdel-Salam GM (2016). De novo mutation in ABCC9 causes hypertrichosis acromegaloid facial features disorder. Pediatric Dermatology, 33(2), e109–e113. [DOI] [PubMed] [Google Scholar]
- Brownstein CA, Towne MC, Luquette LJ, Harris DJ, Marinakis NS, Meinecke P, … Beggs AH (2013). Mutation of KCNJ8 in a patient with Cantu syndrome with unique vascular abnormalities - support for the role of K(ATP) channels in this condition. European Journal of Medical Genetics, 56(12), 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu JM, Garcia-Cruz D, Sanchez-Corona J, Hernandez A, & Nazar Z (1982). A distinct osteochondrodysplasia with hypertrichosis?Individualization of a probable autosomal recessive entity. Human Genetics, 60(1), 36–41. [DOI] [PubMed] [Google Scholar]
- Cavero-Carbonell C, Gras-Colomer E, Guaita-Calatrava R, Lopez-Briones C, Amoros R, Abaitua I, … Zurriaga O (2016). Consensus on the criteria needed for creating a rare-disease patient registry. A Delphi study. Journal of Public Health, 38(2), e178–e186. [DOI] [PubMed] [Google Scholar]
- Concolino D, Formicola S, Camera G, & Strisciuglio P (2000). Congenital hypertrichosis, cardiomegaly, and osteochondrodysplasia (cant√Ð syndrome): A new case with unusual radiological findings. American Journal of Medical Genetics, 92(3), 191–194. [PubMed] [Google Scholar]
- Cooper PE, Reutter H, Woelfle J, Engels H, Grange DK, van Haaften G, … Nichols CG (2014). Cantu syndrome resulting from activating mutation in the KCNJ8 gene. Human Mutation, 35(7), 809–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeschik JC, Voigt C, Goecke TO, Ludecke HJ, Wagner N, Kuechler A, & Wieczorek D (2013). Wide clinical variability in conditions with coarse facial features and hypertrichosis caused by mutations in ABCC9. American Journal of Medical Genetics Part A, 161A(2), 295–300. [DOI] [PubMed] [Google Scholar]
- Flagg TP, Enkvetchakul D, Koster JC, & Nichols CG (2010). Muscle KATP channels: Recent insights to energy sensing and Myoprotection. Physiological Reviews, 90(3), 799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grange DK, Lorch SM, Cole PL, & Singh GK (2006). Cantu syndrome in a woman and her two daughters: Further confirmation of autosomal dominant inheritance and review of the cardiac manifestations. American Journal of Medical Genetics Part A, 140(15), 1673–1680. [DOI] [PubMed] [Google Scholar]
- Grange DK, Nichols CG, & Singh GK (2014). Cantu syndrome and related disorders. In Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Stephens K, & Bean LJH (Eds.). Seattle (WA): GeneReviews(R). [Google Scholar]
- Harakalova M, van Harssel JJT, Terhal PA, van Lieshout S, Duran K, Renkens I, … Cuppen E (2012). Dominant missense mutations in ABCC9 cause Cantú syndrome. Nature Genetics, 44(7), 793–796. [DOI] [PubMed] [Google Scholar]
- Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, & Conde JG (2009). Research electronic data capture (REDCap)–A metadata-driven methodology and workflow process for providing translational research informatics support. Journal of Biomedical Informatics, 42(2), 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraki Y, Miyatake S, Hayashidani M, Nishimura Y, Matsuura H, Kamada M, … Matsumoto N (2014). Aortic aneurysm and craniosynostosis in a family with Cantu syndrome. American Journal of Medical Genetics Part A, 164A(1), 231–236. [DOI] [PubMed] [Google Scholar]
- Huang Y, McClenaghan C, Harter TM, Hinman K, Halabi CM, Matkovich SJ, … Nichols CG (2018). Cardiovascular consequences of KATP overactivity in Cantu syndrome. JCI Insight, 3(15) pii: 121153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazalde B, Sanchez-Urbina R, Nuno-Arana I, Bitar WE, & de Lourdes Ramirez-Duenas M (2000). Autosomal dominant inheritance in Cantu syndrome (congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly). American Journal of Medical Genetics, 94(5), 421–427. [DOI] [PubMed] [Google Scholar]
- Leon Guerrero CR, Pathak S, Grange DK, Singh GK, Nichols CG, Lee JM, & Vo KD (2016). Neurologic and neuroimaging manifestations of Cantu syndrome: A case series. Neurology, 87, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin MD, Singh GK, Zhang HX, Uchida K, Kozel BA, Stein PK, et al. (2016). KATP channel gain-of-function leads to increased myocardial L-type Ca2+ current and contractility in Cantu syndrome. Proceedings of the National Academy of Sciences of the United States of America, 113(24), 6773–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin MD, Zhang H, Uchida K, Grange DK, Singh GK, & Nichols CG (2015). Electrophysiologic consequences of KATP gain of function in the heart: Conduction abnormalities in Cantu syndrome. Heart Rhythm: The Official Journal of the Heart Rhythm Society, 12(11), 2316–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Gurnasinghani S, Kirk EP, McClenaghan C, Singh GK, Grange DK, … Nichols CG (2019). Glibenclamide treatment in a Cantu syndrome patient with a pathogenic ABCC9 gain-of-function variant: Initial experience. American Journal of Medical Genetics Part A, 179(8), 1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClenaghan C, Hanson A, Sala-Rabanal M, Roessler HI, Josifova D, Grange DK, … Nichols CG (2018). Cantu syndrome-associated SUR2 (ABCC9) mutations in distinct structural domains result in KATP channel gain-of-function by differential mechanisms. The Journal of Biological Chemistry, 293(6), 2041–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor RN, Greeley SA, Bell GI, & Philipson LH (2011). Genetics and pathophysiology of neonatal diabetes mellitus. Journal of Diabetes Investigation, 2(3), 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG (2006). KATP channels as molecular sensors of cellular metabolism. Nature, 440, 471–476. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Singh GK, & Grange DK (2013). KATP channels and cardiovascular disease: Suddenly a syndrome. Circulation Research, 112(7), 1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson SP, Kirk E, Bernier F, Brereton J, Turner A, & Bankier A (1999). Congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly: Cantú syndrome. American Journal of Medical Genetics, 85(4), 395–402. [PubMed] [Google Scholar]
- Roessler HI, Volker-Touw CML, Terhal PA, van Haaften G, & van Haelst MM (2018). Cantu syndrome, the changing phenotype: A report of the two oldest Dutch patients. Clinical Dysmorphology, 27(3), 78–83. [DOI] [PubMed] [Google Scholar]
- Rosser EM, Kaariainen H, Hurst JA, Baraitser M, Hall CM, Clayton P, & Leonard JV (1998). Three patients with the osteochondrodysplasia and hypertrichosis syndrome–Cantu syndrome. Clinical Dysmorphology, 7(2), 79–85. [DOI] [PubMed] [Google Scholar]
- Scurr I, Wilson L, Lees M, Robertson S, Kirk E, Turner A, … Smithson S (2011). Cantu syndrome: Report of nine new cases and expansion of the clinical phenotype. American Journal of Medical Genetics Part A, 155A(3), 508–518. [DOI] [PubMed] [Google Scholar]
- Tessadori F, Roessler HI, Savelberg SMC, Chocron S, Kamel SM, Duran KJ, … Bakkers J (2018). Effective CRISPR/Cas9-based nucleotide editing in zebrafish to model human genetic cardiovascular disorders. Disease Models & Mechanisms, 11(10), dmm035469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bon BW, Gilissen C, Grange DK, Hennekam RC, Kayserili H, Engels H, … Hoischen A (2012). Cantu syndrome is caused by mutations in ABCC9. American Journal of Human Genetics, 90(6), 1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]