

Abstract
Inherent and acquired resistance of cancer cells is increasingly recognized as a significant impediment to effective radiation cancer treatment. As important intracellular factors, aberrant tumor transmembrane signal transduction pathways, which include the prosurvival cascades (PI3K/Akt, MAPK/ERK and JAK/STAT) and the proapoptosis pathways (Wnt, p53 and TNF‐α/NF‐κB), have been proved to be crucial determinants of the probability of cell sensitivity to radiation in malignant lesions. There is increasing evidence that targeting the abnormal pathways that can regulate the activity of the DNA damage response and further influence the response of tumor cells to radiation may be suitable for improving radiation sensitization. Preclinical and clinical evidence suggest that agents targeting aberrant tumor signals can effectively improve the therapeutic effect of ionizing radiation. Therefore, in this review, we discuss the intricate interplay between tumor responses to radiation with the aberrant signal pathways, and the potential druggable targets within the pathways to sensitize tumors without significant collateral damage to normal tissues. The application of novel targeting compounds to manipulate the aberrant signal of tumor cells in clinical treatments is also addressed.
Ionizing radiation (IR) is an inalienable part of modern cancer management given the unique advantages of being non‐invasive and devoid of intense systemic toxicity. As an integral component of adjuvant treatment strategies for primary tumors and palliative treatment strategies for advanced and metastatic tumors, more than 50% of diagnosed cancer patients receive radiation therapy (RT; alone or combination with chemotherapy or surgery) worldwide. However, RT offers various degrees of success; there is still an increased recurrence and treatment failure in patients. Retrospective studies show that extracellular factors, such as location, size and hypoxia, play important roles in the lack of response to radiotherapy.1, 2 In recent 15 years, as important intracellular factors, the signaling pathways that can regulate the activity of the DNA damage response, cell growth, differentiation and development have been proved to undergo oncogenic changes far more than other molecule groups.3, 4, 5 The genetic alterations that are activated by the signal cascades in premalignant cells and further characterize neoplasm genomes have also been highlighted.6, 7, 8 Preclinical and clinical studies have indicated that the aberrant signal pathways in cancer cells are crucial determinants of the probability of the improvement of sensitivity to radiation in malignant lesions9, 10, 11, 12, 13; and a more nuanced understanding of the complex radiobiology mechanisms for the response of tumors to RT has paved the way for exploring novel or combinatorial therapeutic approaches for cancer treatment using IR. Hence, the targeted agents that can specifically inhibit the activity of the signal cascades could abrogate the radioresistance of tumors and potentiate tumor response to radiotherapy.11, 12, 14 Although some cellular signaling events triggered by clinically relevant doses of radiation and the fundamental relationship between signal pathways and tumor progression have been thoroughly reviewed elsewhere, the convergence of multiple lines of research on signaling pathways that modulate the response of tumors to RT has not occurred. Hence, the scope of this review is not only to focus on the intricate interplay between tumor responses to radiation and the aberrant signal pathways, but also to offer insight into potential druggable targets to sensitize tumors to RT without significant collateral damage to normal tissues. Moreover, recent advances in the application of novel specific targeting of chemical compounds with proven clinical efficacy in manipulating the aberrant pathways in the rational clinical treatments of tumor patients have been summarized; the toxicity and side effects of the targeting agents are also discussed.
Current Selective Agents Targeting Aberrant Signal Pathways of Tumor Cells under Preclinical and Clinical Evaluation
The benefits of IR for subsets of cancer patients derive from its ability to directly or indirectly damage DNA through interference with various DNA‐associated proteins or to disrupt normal DNA division processes, such as replication, transcription and recombination, and to induce cancer cell death.8, 15 Normal cells are proficient in reversing DNA lesions by activating appropriate repair mechanisms; however, a similar capacity to effectively recognize DNA damage and initiate DNA repair pathways leads to the phenomenon of therapeutic resistance to IR in cancer cells. Accumulating evidence suggests that these dysfunctional phenotypes elicit pro‐survival and anti‐apoptotic responses (the inducible radioresistance paradigm), and then affect the capacity of cancer cells to engage in catabolic processes, including apoptosis, postmitotic death, senescence and autophagy.16, 17 Preclinical events suggest that compared to other targeted drugs, agents targeting the aberrant signal pathways provide specific advantages to cancer patients; this is because upregulation of aberrant signal pathways often occurs in cancer cells instead of normal cells. For example, in cancer cells, proapoptotic factors have been proved to be reduced, which, combined with the overexpression of antiapoptotic proteins, enables cancer cells to be more resistant to death induced by radiation compared with normal cells.5, 6, 9, 10, 11, 12, 13, 14 Moreover, some oncogenes occur only in tumor tissues, such as proto‐oncogene fms as a receptor tyrosine kinase, which is activated only in leukemic cells.7 Thus, agents targeting these pathways in tumor cells cause little toxicity to normal tissues. Hence, a variety of targeted compounds that can selectively inhibit transferred factors in proapoptosis pathways and at the same time enhance the functions of prosurvival pathways' factors represent a promising approach to elevate the sensitization of malignant cells, and many of these agents are currently under preclinical and clinical evaluation (Table 1).
Table 1.
Drug delivery systems targeting aberrant signal pathways in cancer cells
| Inhibition sites | Agents (in clinic) | Targeting mechanisms | Application | Limitations | References |
|---|---|---|---|---|---|
| EGFR | Imatinib, panitumumab, nimotuzumab, cetuximab, Canertinib, PKI166, Lapatinib, erlotinib, Gefitinib, nilotinib, resveratrol, recombinant immunotoxin, cross linking antibodies | Inhibition of receptor‐specific ligand binding, blocking activation of receptor tyrosine kinases, blocking ATP binding to the intracellular TK domain, inhibition the phosphorylation of receptors and activation of signal transduction molecules downstream | Hepatocellular carcinoma, anaplastic thyroid cancer, head and neck squamous cell carcinoma, human colon adenocarcinoma, non‐small cell lung cancers | Neutropenia, thrombocytopenia, diarrhea, erythra | 61 |
| PI3K | XL147, PX866, GDC0941, Buparlisib, Idelalisib, NVP‐BeZ235 | Antiproliferative, bind competitively and reversibly in the ATP binding pocket of the catalytic kinase domain, inhibiting p110δ lipid kinase α and β p110 isoforms | HER2‐amplified and PIK3CA mutated cancers, lung adenocarcinomas, | Insulin resistance, limited efficacy | 9, 20 |
| Akt/PKB | Perifosine, GSK690693, Triciribine, MK2206 | Growth and invasiveness inhibition, apoptosis promotion | Cancers with AKT1 mutations and AKT1 and AKT2 amplifications | Insulin resistance, limited efficacy | 19 |
| mTOR | OSI027, AZD8055, NVP‐BeZ235, Rapamycin, Wortmannin | Anti‐proliferative activity | Breast cancer, lung cancer, prostate cancer, hamartomas | Minimal effects, limited efficacy | 23, 24 |
| Ras | Farnesyltransferase inhibitor, AFC | Inhibition of Ftase, ICMT, RCE1 | Bladder cancer, head and neck cancers, astrocytic gliomas | Low activity, less targeting, toxicity | 25 |
| Raf | Sorafenib, Vemurafenib, ZM336372, AZ628, Raf265, AAL881, LBT613 | Anti‐proliferative, anti‐angiogenic, pro‐apoptotic activities | Refractory solid tumors, renal cell carcinoma, hepatocellular carcinoma, thyroid carcinoma | Skin rush, diarrhea and hypertension | 26 |
| MEK/ERK | PD 098059, U0126, Ro 09‐2210, PD184352, PD0325901, Selumetinib, GDC‐0973, Refametinib, Trametinib, Pimasertib, Selumetinib | Inhibiting proliferation | Colorectal, pancreatic, liver, skin, and lung cancer, lymphoma | Diarrhea, asthenia, rash, nausea, and vomiting. blurred vision, acute neurotoxicity | 31 |
| MDM2 | Nutlins, RITA, quilinos, spiro‐oxidoles, HL198C, benzodiazepines | Liberating p53 and leading to growth arrest and apoptosis | Refractory solid tumours, hematologic Neoplasms | p53 toxicity to normal tissues | 28, 29 |
| JAK/STAT | AUH‐6‐96, c‐Src siRNA, cucurbitacin B, andrographolide | Inducing programmed cell death, increasing cell cycle arrest and apoptosis, enhancing chemosensitivity | Hodgkin lymphoma, head and neck squamous cell carcinoma, pancreatic cancer | Toxicity to normal tissues | 11 |
| Wnt | Dickkopf‐1, 2,4‐diamino‐quinazoline, ICG‐001‐related analogs, NSC668036 | Anti‐proliferation, immune therapy | Cancers of pancreas, stomach, liver, bile duct, breast, and cervix | Toxicity to normal tissues | 12, 43 |
| JNK | SP600125, JNK 401, semapimod, Bi‐78D3, JNKi1 | Anti‐proliferation, induction of cell death | Hepatocellular carcinoma, myelogenous leukemia | Lack specificity and selectivity for the different JNK isoforms | 10 |
| IκB | Celastrol, BMS‐345541, parthenolide, wedelolactone, PS‐1145, MLN120B, KINK‐1 | Inhibiting growth and inducing apoptosis, attenuating drug resistance | Breast cancer, lymphomas, multiple myeloma, melanoma cells | Toxicity to osteoclast and inhibiting bone resorption | 45, 46, 50 |
| NF‐κB | IκBα, parthenolide, H157I, bortezomib, MG132, PS‐34, SC‐236, DHMEQ | Chemosensitization and radiosensitization, attenuating drug resistance | Breast, lung, colorectal, pancreatic, ovarian, thyroid and prostate cancer and malignant glioma | Immune suppression and poor targeting | 47, 50 |
| p53 | Pifithrin‐α and its derivatives, MDM2 inhibitors | Reducing side effects of radiotherapy and chemotherapy and sensitizing apoptosis | Skin cancer, hematologic neoplasms, colon carcinoma | Low efficiency, toxicity to normal tissues, undefined mechanism | 14, 53 |
Strategies Inhibiting Proliferative Pathways
The plethora of proliferative signaling pathways, a feature that would be aberrantly hyperactivated or deregulated in cancer cells, has been proved to be triggered by sublethal doses of radiation within tumor cells (inducible signaling). Increased levels of the inhibitors of apoptosis proteins induced by the proliferative signaling pathways is closely associated with radio‐resistance and chemoresistance and poor clinical outcome in various types of cancer.16, 17 Hence, transfer factors within the proliferative pathways activated by radiation signals from the outside cytoplasm and the inside nucleus could serve as promising targets for novel radiosensitization strategies. Numerous promising “druggable” targets for inhibition of the anti‐apoptotic functions are being tested to enhance tumor radiosensitivity.
Agents targeting PI3K/Akt pathway
Activation of the PI3K pathway can occur in response to various extracellular signals through stimulation of membrane receptors, such as the estrogen receptor beta (erbB) receptors, insulin like growth factor (IGF)‐receptor and integrin receptors.18 Implicated in survival signaling, the PI3K/Akt cascade plays a crucial role in cancer response to current therapeutic modalities (Fig. 1). Studies show that PI3K/Akt signaling participates in angiogenesis, which is resistant to clinical low IR doses of 2–5 Gy following vascular epithelial growth factor (VEGF) stimulation and regulates capillary‐like tubule formation in tumor cells.19 After PI3K and Akt upregulated, the cell cycle is arrested, and G1 phase‐associated protein CyclinD1 and cyclin‐dependent kinase (CDK) 4 expression decrease, resistance to radiation‐induced apoptosis has been observed.20, 21, 22, 23 When PI3K or Akt activation is impaired by a PI3K inhibitor, such as LY294002, radiosensitization is detected.22 In addition, as a member of the Akt pathway, the mechanistic target of Rapamycin (mTOR) is also a promising target for cancer radiosensitivity therapy. Irradiation can activate mTOR signaling in vascular endothelium, and the mTOR inhibitors Rapamycin and RAD001 can increase apoptosis of tumor endothelial cells in response to radiation.24 Meanwhile, inhibition of mTOR could further lead to cell‐cycle arrest through upregulation of the CDK inhibitor p27kip1 and downregulation of Cyclin D1 after radiation.25 The mechanism of the radiation resistance in the laboratory and in cancer patients whose tumors harbor the activated PI3K signal pathway provides a clear clinical application for the targeted inhibitors. Inhibition of p110, a subunit of PI3K, either using specific chemical inhibitors (Wortmannin, LY294002, IC486068) or overexpression of the mutant p85, can induce PI3K to enhance apoptosis and minimize capillary tubule formation induced by radiation in cancer tissues.9 Chemical inhibition of Akt with ALX‐349 as well as overexpression of dominant‐negative mutants Akt or its downstream target glycogen synthase kinase‐3 also leads to radiosensitization of tumor cells.9 In addition, inhibitors of mTOR (RAD001, Temsirolimus [CCI‐779], AP23573) as potent radiosensitizers have been proved to lead to improved tumor‐growth delay in phase III trials.24
Figure 1.
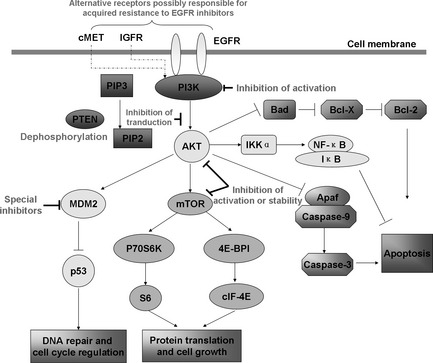
Mechanism of PI3K/Akt pathway to induce radioresistance and possible strategies to prevent acquired resistance to radiation through inhibiting the activity of relative elements. In the PI3K/Akt pathway, after GFR protein tyrosine kinases are activated, PI3K protein is recruited to the membrane by directly binding to phosphotyrosine consensus residues of growth factor receptor, leading to allosteric activation of the catalytic subunit. This activation results in production of the second messenger phosphatidylinositol‐3,4,5‐trisphosphate (PIP3). The lipid product of PI3K recruits Akt signaling protein domains to the membrane. Once activated, Akt mediates the activation of several targets, including IKKα, mTOR, MDM2 protein and Bad, Apaf/Caspase 9 proteins downregulation, and then results in cellular survival, growth and proliferation through various mechanisms. Possible strategies to prevent acquired resistance include combination therapy against alternate receptors, including cMET, IGFR and EGFR in the membrane and intracellular signal elements containing PI3K, AKT and mTOR.
Although inhibition of PI3K/Akt pathway at various steps leads to the potentiation of radiation‐induced cell death, downregulation of phospho‐Akt does not automatically imply a decrease in tumor cell proliferation owing to PI3K/Akt survival pathway inhibition. This means overexpression of PI3K or Akt can be induced by other carcinogenic factors.9 The activation of this pathway by receptor tyrosine kinase (RTK) or by receptor‐activated Ras suggests that the PI3K/Akt pathway has an independency on growth receptors, this may be the resistant occasion to current targeted growth receptor inhibitors.9
Agents targeting MAPK/ERK pathway
In addition to signaling through PI3K, the MAPK signal cascade reaction activated by growth factor receptors (including the HGF/SF receptor [cMET], IGF receptor [IGFR] and epidermal growth factor receptor [EGFR]) has been involved in the activation of cellular proliferation pathways (Fig. 2). Studies have shown that activated mutations in Ras genes, most often in K‐Ras, are found in 30% of cancers and are generally acquired in the early process of tumorigenesis.26 Large‐scale sequencing studies have further revealed that Raf is mutated in 20% of all cancers and in more than 40% of melanomas.27 The majority of mutations is clustered in the kinase domain of Ras‐Raf and leads to the stimulation of extracellular signal‐regulated kinase (ERK)1/2 activity in cells, which is a typical survival signal and further induces cell proliferation and protects cancer cells from apoptosis. Inhibition of Ras activation can result in radiosensitization both in rodent and human tumor cell lines bearing endogenous Ras mutations.27 In addition, tumor cells with endogenous Raf activation are more resistant to radiation than their counterparts with the mutant Raf oncogene; and the intrinsic resistance can be eliminated by homologous recombination.27 Moreover, ERK activation in endothelial cells has been demonstrated to play a radioprotective role resulting in increased survival of irradiated cells.2 Another downstream factor of Ras, murine double minute 2 (MDM2), the cellular ubiquitin E3 ligase of the tumor suppressor p53, is considered to have radiosensitization activity after combination with RT in xenograft models of human cancers regardless of their p53 status.28, 29 Furthermore, the activity of the MAPK signal pathway alters several proteins expression that regulates tumor cell adhesion and motility, differentiation and proliferation, and this action makes this cascade representing significant and promising molecular targets for effective IR treatment possible.30, 31 In clinic, the initial approach to block Ras function was indirect, because targeting Ras is difficult, with more failures than successes, especially in G1 phase malignancies; however, inhibition of Farnesylation, a post‐translational modification involving the addition of a 15‐carbon group that is critical to Ras function, can decrease Ras activity.30 The farnesyl transferase inhibitors (FTI) (Tipifarnib [R115777] and Lonafarnib [SCH66336]), which have been used in phase I studies, have shown therapeutic effects on tumor growth after radiation treatment.30 Further research implies that inhibition of Farnesylation by FTI may be sufficient to abrogate Ras‐dependent cell signaling and transforming functions.30 On the basis of this, several different FTI have been tested in a large number of clinical studies as single agents as well as in combination with standard chemotherapy. As for the Raf activation, Sorafenib (BAY 43‐9006), a small‐molecule inhibitor of both A‐Raf and mutant or wild‐type B‐Raf, with additional inhibition of VEGF receptor (VEGFR)‐2, VEGFR‐3, platelet‐derived growth factor receptor (PDGFR)‐β, Flt3 and c‐KIT, has been used as single agent in a phase II trial that included 138 patients with metastatic colorectal cancer to detect the activity of anti‐tumor and radiosensitive effects.18
Figure 2.
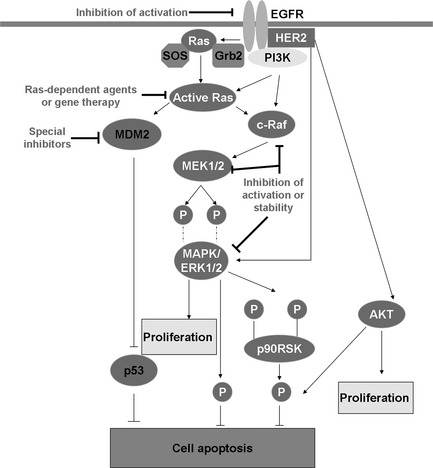
Ras‐induced MAPK prosurvival pathway: therapeutic targets and new therapies. The Ras‐Raf‐MEK‐ERK (MAPK) signaling pathway represents significant and promising molecular targets for effective treatment using radiotherapy. The dimerisation and autophosphorylation of EGFR provide docking sites for signaling molecules, including the Grb2‐SOS complex, to activate the small G‐protein Ras. This exchange elicits a conformational change in Ras, enabling it to induce Raf activation and MDM2 upregulation. Activated Raf phosphorylates and activates MEK (MAPK/ERK kinase), which, in turn, phosphorylates and activates extracellular‐signal‐regulated kinase (ERK). Activated ERK induces many substrates' activity in the cytosol to inhibit the apoptosis. ERK can also enter the nucleus to control gene expression by phosphorylating transcription factors to induce proliferation. Activated MDM2 further inhibits p53 activity and inhibits cell apoptosis. Possible strategies to prevent acquired resistance include molecular agents or gene therapy against EGFR in the membrane and intracellular signal elements containing Ras, Raf, MEK, ERK, MDM2 activation.
The activation of “classical” MAP kinases ERK1 and ERK2 by IR is dependent on the cell type, the expression of multiple growth factor receptors, and genetic alterations, so this signal pathway often shows different biological functions.32 In some cases, inhibition of MEK1/2 to regulate ERK1/2 activity enhances cell killing by radiation due to increased G2/M arrest and apoptosis, while in others, activation of ERK pathway following irradiation has been shown to promote radiosensitivity by abrogating G2/M checkpoint.32 Specifically, radiation‐induced ERK activation is closely linked to increased expression of radioprotective transcription factors and DNA repair proteins.32 The initial clinical approach to target this pathway is CI‐1040, a small‐molecule inhibitor of MEK1 and MEK2,18 which is demonstrated to decrease phospho‐MAPK (pMAPK) in tumor and surrogate tissues in phase I studies; however, there were no objective responses in 20 patients with colorectal cancer in a phase II trial.18 Two sides of biological effects of ERK pathway in tumors may lead to decrease the effects of targeting agents in some degree.
Agents targeting JAK/STAT signaling pathway
Even though radioresistance in cancer cells results from a number of abnormal oncogenic signaling pathways induced by genetic or epigenetic mechanism, all the published reports seem to converge on a very limited number of nuclear transcription factors that function as final effectors and start specific gene expression patterns for a particular cancer. Among the activated proliferative signaling pathways within tumor cells, nuclear transcriptional signaling cascade JAK/STAT is unique in that active signal comes from the nucleus and the transcription factors produce effects at the convergence of the cytoplasm, while the opposite transduction cascades are observed in other pathways (Fig. 3). Major biologic effects of the JAK/STAT signal include promotion of cell survival through increased expression of anti‐apoptotic proteins such as Bcl‐2 and Bcl‐XL, mediation of tumor‐promoting inflammation and promotion of the pro‐oncogenic inflammatory pathways, including NF‐κB and IL‐6‐GP130‐JAK pathways.33 Some reports demonstrate that inhibition of STAT3 signaling can increase radiosensitivity, reduce tumorigenic properties and promote radiation‐induced apoptosis in glioblastoma‐derived tumor initiating cells.34, 35, 36 Yang et al.35 show that radiosensitivity is enhanced by the simultaneous inhibition of STAT3 and expression of erB2. Stable transfection with shRNA against STAT3 also results in enhanced radiosensitivity of human squamous cell carcinoma (A431) cells.36 Studies investigating the altered proteomic profiles of radioresistant prostate cancer cells confirm that the enhanced radioresistant phenotype of the tumor cells is accompanied by multiple mechanisms, with radiation‐induced activation of the JAK/STAT pathway playing a significant role.37 Several chemical compounds, including S3I‐201, Stattic, STA‐21 and a JAK kinase inhibitor AG490, that have been reported to successfully inhibit JAK/STAT signaling can effectively improve the sensitization to radiation of tumors without obvious toxicity.11 Moreover, other members of the STAT family have been proved to play a prominent role in tumor radioresistance; for instance, downregulation of STAT1 and STAT2 can sensitize renal cell carcinoma to radiation.38 Recent studies imply that inhibition of STAT could be one of the underlying mechanisms of radiosensitization by physiochemical compounds and plant phytochemicals, which can mediate STAT3 phosphorylation in cancer.39, 40 More importantly, studies have indicated that the abrogation of JAK/STAT signaling with these small‐molecule inhibitors is sufficient to induce growth arrest and apoptosis in various types of tumors11, 41; this means that the targeted agents can achieve radiosensitivity improvement.
Figure 3.
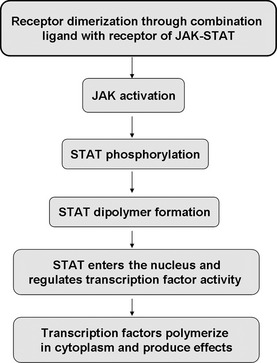
As a recently discovered nuclear signal transduction pathway, the mechanism of the JAK/STAT pathway in regulating proliferation, differentiation, apoptosis and radio‐resistance processes in numerous cancer types.
Strategies Promoting Pro‐Apoptotic Pathways
Interventional strategies that target pro‐apoptotic signaling pathways can be distinguished in those that inhibit anti‐apoptotic signaling. The first approach currently focuses on the death receptors pathway. Unlike all other apoptotic stimuli, death receptor ligands operate irrespective of the p53 status of cells, activate Caspase effector and induce apoptosis independent of the mitochondria via the extrinsic pathway. This is very attractive for cancer therapy, because the mitochondrial route for Caspase activation is frequently blocked during malignant transformation. The death receptor signal pathways are described as follows.
Agents targeting canonical Wnt signaling pathway
In normal cells, the Wnt signaling is blocked, but deregulated Wnt signaling has been reported in malignant cells, especially in more than 90% of colon cancers.12 Wnt5a expression in human melanoma biopsies has been proved to directly correlate with increasing tumor grade and inhibition of Wnt‐mediated signaling induces apoptosis in both malignant melanoma cells and non‐small‐cell lung cancer cells.12, 18 Kim et al.42 illustrate that Wnt activation is a molecular mechanism implicated in glioblastoma radioresistance and accomplishes an important radiosensitization effect through pharmacological and siRNA inhibition in U373 and 578 cells. In addition, activation of Wnt has been identified to promote infestation and metastasis of tumor cells.12 Moreover, the Wnt transcription factor T cell factor (TCF) 4 has also been reported to mediate human colorectal carcinoma resistance to chemoradiotherapy.43 Activation of the β‐catenin/TCF signaling pathway as the downstream target of Wnt not only provides a growth advantage to cancer cells, but also significantly affects the clinical outcome by inhibiting chemotherapy‐induced apoptosis.44 Therefore, targeted agents such as the β‐catenin antagonist ICG‐001, NSC668036, FJ9 and 3289–8625 and targeted gene sequences inhibiting the activation of Wnt signaling have manifested ideal effects of radiation sensitization in cancer cells.12, 44 However, the Wnt‐dependent signaling pathways conclude three different molecular pathways and the intracellular signal transduction cascades in each of these pathways are very different from each other, so, until now, the identity of the components of the Wnt signal pathway have been unclear.12 Despite the technological advances in the Wnt signal pathway interruption, the mechanisms of these compounds controlling survival gene transcription in cancer cells remain elusive. Therefore, investigating the regulation mechanisms of the Wnt pathway and targeted agents may be beneficial in future studies. Recent studies in human colorectal cancer show that targeting the β‐catenin/adenopolyposis in coli displays a novel mechanism of cyclooxygenase‐2 (COX‐2) inducing anticarcinogenic effects and COX‐2 is indicated to play important roles in the tumorigenesis and radioresistance of cancer.12
Agents targeting TNF‐α/NF‐κB signal pathway
TNF‐α is a strong anti‐tumor cytokine that could promote proliferation and differentiation of immune cells after being activated by the TNF receptor (TNFR), and further inhibit tumor angiogenesis and increase permeability of tumor vascular endothelial cells to produce anti‐tumor effects. As the prototypical death receptors, TNF‐α signal pathway has been reported to induce two downstream transductions: one can induce activation of nuclear factor kappa B (NF‐κB) to cause an anti‐apoptosis effect; the other can lead to apoptosis through regulation of Fas‐associated protein with death domain (FADD; Fig. 4). Hence, inhibition of NF‐κB results in a complex regulation with, on one hand, an increased apoptosis of irradiated cells, but on the other side, induction of lower TNF production and decreased therapeutic effects of radiation.
Figure 4.
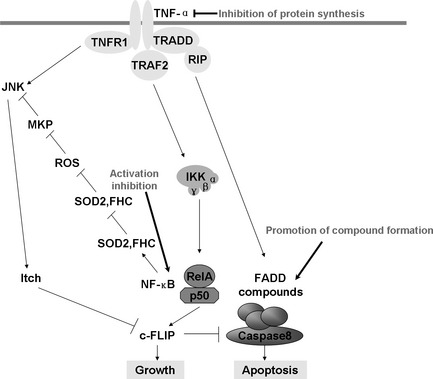
Activation of TNF‐α pathway and possible strategies to prevent resistance to radiation through inhibiting the activity of relative elements. Although TNF can bind two receptors, TNF‐R1 (TNF receptor type 1) and TNF‐R2 (TNF receptor type 2); most information regarding TNF signaling is derived from TNF‐R1. Upon contact with their ligands, TNF receptors form trimers and cause a conformational change, leading to the dissociation of the inhibitory protein SODD from the intracellular death domain and leading the adaptor protein TRADD to be binded. Following TRADD binding, three pathways can be initiated. First, TRADD recruits TRAF2 and RIP, then TRAF2, in turn, recruits the multicomponent protein kinase IKK. IKK phosphorylates an inhibitory protein, IκBα, and releases NF‐κB, which can translocate to the nucleus and mediate the transcription of a vast array of proteins involved in cell survival and proliferation, inflammatory response and anti‐apoptotic factors. Second, TNFR induces a strong activation of the stress‐related JNK group, evokes moderate response of the p38‐MAPK, and is responsible for minimal activation of the classical ERK. Of the three major TNF‐α cascades, TNFR is also involved in death signaling. TRADD binds FADD, which then recruits the cysteine protease caspase‐8. A high concentration of caspase‐8 induces its autoproteolytic activation and subsequent cleaves of effector caspases, leading to cell apoptosis. Possible strategies to prevent acquired resistance include combination therapy against TNF‐α protein synthesis in the membrance and NF‐κB activation or promoting FADD/Caspase 8 compound formation.
The impact of NF‐κB inhibition on radiosensitivity has been explored in various models. Expression of the NF‐κB inhibitor (IKB) can result in increased apoptosis in irradiated head and neck squamous carcinomas cells13 and overexpression of wild‐type IKB can sensitize human glioblastomas to radiation.45 Moreover, the specific knockout of the I kappa B kinase (IKK2) gene in intestinal epithelial cells demonstrates a radioprotective role of NF‐κB.13 Using genetic approaches, it has been shown that expression of the repressors of NF‐κB enhances radiation‐induced apoptosis in cancer cells.13, 46
Evaluation of all these studies clearly demonstrates novel strategies that can target proapoptosis signal in TNF‐α related pathways and depress NF‐κB activation. Because a number of different steps are involved in NF‐κB activation, there are several potential methods to downregulate NF‐κB in target tissues using a wide range of agents (corticosteroids, phytochemicals, proteasome inhibitors and synthetic peptides).47 These approaches can be roughly categorized as: (i) inhibition of the key steps in NF‐κB pathway; (ii) targeting the upstream components of the NF‐κB pathway; or (iii) pharmacological inhibition of the key components of the effector. Parthenolide of the sesquiterpene lactone can sensitize human hybrid CGL1 cells to radiation by inhibiting NF‐κB activity and enhancing apoptosis.48 In addition, in prostate cancer cells, Parthenolide can mediate radiosensitization by inhibiting radiation‐induced NF‐κB activation and the expression of its downstream target superoxide dismutase 2 (SOD2), the gene coding for an important anti‐apoptotic and antioxidant enzyme (manganese superoxide dismutase).49 Cepharanthin, the anti‐inflammatory biscoclaurine alkaloid extracted from the roots of Stephania cepharantha hayata, enhances the radiosensitivity of oral squamous carcinoma cells by inhibiting the activation of radiation‐induced NF‐κB and the production of NF‐κB‐mediated downstream pro‐inflammatory cytokines IL‐6 and IL‐8.47 Apart from plant phytochemicals, a few other synthetic compounds have also shown to exert their radiosensitizing effects through NF‐κB suppression, such as docosahexaenoic acid and pitavastatin.50 Indomethacin, which can suppress NF‐κB activation, has rendered HeLa cells more susceptible to apoptosis after irradiation.48 Similarly, the radiosensitizing properties of genistein, a soy isoflavone, are mediated by suppression of radiation‐induced NF‐κB, leading to altered expression of regulatory cell cycle proteins such as Cyclin B or p21WAF1/Cip1, and, thus, promoting G2/M arrest.47 As for the natural agents, the proteasome inhibitor Velcade1 (Bortezomib or PS‐341) has been shown to increase radiation‐induced apoptosis and to augment radiosensitivity in colorectal cancer cells in vitro and in vivo through NF‐κB inhibition.51 In addition, the radiation sensitization phenomenon of bortezomib has been observed through NF‐κB activity and the variability in Bortezomib‐induced radiosensitization of cervical cancer cell lines has been attributed to differential NF‐κB signaling patterns among cell lines.51 All the evidence implies that NF‐κB activated in the tumorigenic process has become a promising target to enhance radiation sensitization. However, during treatment, the “double‐face” of NF‐κB and the downstream factor‐reactive oxygen species might require some consideration and the effects of drugs may be degraded in some sides.13
Agents targeting p53 signal pathway
p53 transcription factor, known as a DNA damage‐inducible molecule, has been reported to suppress cancer progression through induction of cell‐cycle arrest, apoptosis, or senescence in response to a variety of cellular stimuli. Its activity is regulated by murine double minute 2 (MDM2) and then feedbacks to block MDM2 activities (Fig. 1). In addition to losing wild‐type p53(wtp53) activity, a high percentage of human tumors are characterized by mutations (Fig. 5) that convert the tumor suppressor function into a negative action or into an oncogenic signaling coordinator with the ability to induce gene expression distinct from the wild‐type counterpart. These acquired functions enable mutant p53 to promote a large spectrum of cancer phenotypes and lead to unchecked proliferation, tumor growth and therapeutic resistance. In breast and colorectal tumors, p53 mutations are reported to show resistance to many chemical anticancer agents and radiation.52 Another study shows that mutant p53 can initiate a feedback loop that involves ERK‐mediated transcription of early growth response‐1 (Egr‐1), which, in turn, increases the secretion of epithelial growth factor reactor (EGFR) ligands through stimulation of EGFR signaling.53 Although previous studies show that radiation inhibits the upregulation of mutant p53 instead of wt p53, recent reports indicate that the expression of wtp53 is required for the radiation stimuli.14 Additional data show that radiation resistance is associated with decreased wtp53 and activated ERK in leukemia.14 Currently, p53 targeting technologies containing antisense technology, RNAi technology, tranfection exogenous genes technology and microRNA technology have proved to upregulate the sensitivity of radiation in most cancer models.14, 52, 53, 54 Recombinant human p53 adenovirus parenteral solution has been approved for use by the State Food and Drug Administration. It is the first gene therapy drug to be approved and has demonstrated good efficacy in terms of radiation sensitivity in the treatment of head and neck and gastric cancer.14
Figure 5.
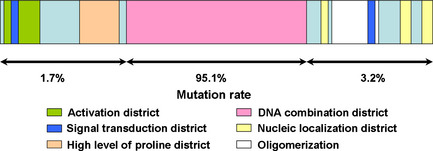
Mutation location and mutation ratio of p53.
Interaction between the Anti‐Apoptotic Pathways and the Pro‐Apoptotic Pathways
Although multiple biological effects at the cell and tissue level induced by radiation can be divided into pro‐apoptosis and anti‐apoptosis response, the tumor cell signal transduction contains multi‐components and multi‐linked pathways and even now some signal transduction pathways and middle factors' functions are not clear. In fact, after activation, many signal pathways show multi‐interaction and cross function. For example, p53 and the Bcl‐2 family are involved in the apoptosis signals; however, as important downstream cascade elements in the proliferative signals, they also play important functions in the pro‐survival tumor cells (Figs 1, 2). Using the antisense oligonucleotide to inhibit the function of the antiapoptotic protein Bcl‐2 in colorectal and other cancers reveals no significant cytocidal activity.55 Hence, attempts in the clinic to block the function of single pathway have been met with limited success. Instead of trying to interrupt anti‐apoptotic or pro‐apoptotic pathways, which belong to traditional membrane‐based and intracellular signaling pathways, a novel approach has emerged attempting direct stimulation of survival or apoptosis via engagement of a family of membrane‐bound anti‐apoptotic or pro‐apoptotic receptors. Because many of the pathways can be activated by the EGFR and TNF‐related apoptosis‐inducing ligand (TRAIL), their interruption promotes amplified effects of target agents.18 Clinical data provide proof that inhibiting the EGFR signal transduction pathway can result in antitumor activity,18, 55 such as using the IgG1 monoclonal antibody Cetuximab, which is typical used as an inhibitor of EGFR and approved by the FDA for the treatment of patients with irinotecan‐refractory disease.55 However, the presence of EGFR does not imply that EGFR signaling is a dominant pathway for the given cancer.56, 57 Indeed, recent data suggest that some EGFR‐negative tumors may respond to EGFR inhibition.56, 57 Thus, despite modest clinical benefit, many patients (EGFR positive or negative) do not respond to Cetuximab.56, 57 Although there are a number of plausible explanations for the resistance of cancers to EGFR inhibitors, the main hypothesis is there is significant cross‐talk between the PI3K/AKT and other pathways, such as the apoptosis pathways and the NF‐κB and, possibly, the Wnt pathway.
Multifunctional Agents for Radiosensitivity Through Inhibiting Various Signal Transduction Pathways in Tumor Cells
Although aberrant signal pathways of tumors are emerging as promising radiation therapeutic targets, there are potential concerns and challenges related to the clinical use of current targeted drugs owing to their low cancer selectivity and specificity. For example, some targeted agents might bind specifically to normal cellular components and this special combination is a hindrance for in vivo application. A further concern is the possible intrinsic drug‐resistant mechanisms, such as the intervention of multidrug resistance pumps, which might exclude these potential drugs from cancer cells.58 Delivering these agents selectively and maintaining sufficient concentrations at the tumor sites is important to improve the therapeutic efficacy of these targeted agents and to decrease side effects. Based on the above analysis, the application of radiosensitivity targeting strategies through development of multi‐targeting agents or direct conjugation of multifarious therapeutic molecular agents may accomplish promising efficacy and the novel strategies are described in the following.
Epithelial Growth Factor Receptor‐targeting Agents for Radiosensitization in Aberrant Signal Pathways
The EGFR (HER1) is a transmembrane glycoprotein member of the ErbB receptor family. The main function of EGFR is to transfer the extracellular signal into cells and then activate downstream pathways to obtain the biological effects. PI3K/Akt and MAPK pathways are mainly dependent on EGFR (Figs 1, 2). Hence, EGFR targeting agents as broad‐spectrum radiosensitizers have shown clinical promise; the representative drug is cetuximab, which has shown target specificity in clinic.18 Recent reports illustrate that this agent can effectively increase the radiation sensitivity of tumor cells and it exhibits better targeting and therapeutic effects.59 However, unlike HER‐2 expression, EGFR is not a robust predictor of response to EGFR‐targeted therapy; patients with a low degree of EGFR expression still respond to EGFR antagonists.59, 60 In a human glioblastoma cell model, IGFR mediated resistance to anti‐EGFR therapy partly through PI3K/AKT, providing a particularly vivid example of the redundancy of downstream signaling.61 The alternate receptors are possibly responsible for acquired resistance to EGFR inhibitors in tumor therapy.61 For example, the PI3K/AKT and Ras/MAPK pathways can also be activated by cMET or IGFR.61 The development of acquired resistance to EGFR inhibitors results in downstream mutations or activation of alternative survival pathways, and is emerging as a potential treatment barrier for the optimization of EGFR‐targeted therapy. Hence, the broad‐spectrum RTK inhibitors, which have the advantage of targeting multiple RTK sites, may be attractive. One such small molecule agent is PTK787, which targets VEGFR2, PDGF and cMET.61 Another drug of SU11248 that is a selective inhibitor of VEGFR2 and PDGFR as well as Kit and VEGFR1 and is designed to inhibit kinase activity of fibroblast growth factor, PDGF and VEGF receptors has demonstrated significant radiation sensitization effects in preclinical models.61 Nilotinib (Tasigna), inhibiting PDGFR, Bcr2Abl and c2kit receptors and protein kinase C (PKC) 412 (N‐benzoylstaurosporine), which is a staurosporine derivative with a broad therapeutic index in early phase I–II clinical trials, was initially thought to be exclusively a competitive inhibitor of adenosine triphosphate binding to three PKC isoforms.61 Additional mechanism studies have revealed that PKC 412 has an antiangiogenic effect through inhibition of autophosphorylation of the VEGFR tyrosine kinase and further inhibition of the activity of Flt‐3 receptor tyrosine kinases.61 However, these agents offering effective treatment are also associated with side effects in addition to radiosensitivity, such as toxicity to normal cells for large treatment dosage and short half‐life periods.61 Therefore, target specificity and the broad therapeutic window are still convincing arguments for exploring the role of novel molecular‐defined anticancer agents.
Therapeutic Alliance of Various Molecular Agents
The mechanism of intrinsic resistance of radiotherapy is complex and contains various cellular and molecular alterations, not solely the transduction pathway modifications. Traditional therapeutic alliance of molecular agents targeting signaling cascades is accomplished through a combination of two or three signal targeting agents. This strategy has been proved to produce effective outcomes in improving sensitivity to IR of tumor cells. However, DNA damage restoration, activation of oncogenes and angiogenesis factors have also been linked to multiple aspects of tumorigenesis and radiation resistance, and these targeted agent combinations may also provide far more effective sensitization.62 For example, binding the DNA repair enzymes to cell signaling proteins can improve the lethal effects of radiation effectively.62 Utilizing hypoxia sensitizers that can increase the sensitivity of hypoxic cells has also been demonstrated to kill radioresistant hypoxic cells effectively during a radiotherapy course.4, 63 Moreover, application of vascular targeting agents is another common strategy of radiation sensitization, so exploring mechanisms of these agents with signal inhibitors may provide novel combination modality.2 Morever, radiation therapy is often given in combination with a course of chemotherapy, especially in distant tumor tissues; therapeutic gain is sought by exploiting synergy between radiation and drug effects. Exploring chemotherapy agents as novel radiosensitizers could effectively prevent normal tissue damage induced by profuse reduplicate drug application.
Conclusions
Despite the technological advances in the field of radiation oncology, overcoming intrinsic and inducible tumor radioresistance remains a major conceptual and therapeutic challenge. This is particularly the case when the resistance mechanism is similar in tumor cells and normal cells. Understanding the cellular and molecular basis for innate and acquired resistance of cancer cells to radiotherapy is a prerequisite for overcoming this difficulty. Over the past decades, great strides have been made in discovering and analyzing the aberrant signaling pathways in tumor cells. Research suggests that targeting the special cascades in tumor cells offers the promise of a means of enhancing the intrinsic sensitivity of tumors to radiation and a means of decreasing the normal tissue side effects of radiation. Rationally designed targeted agents could upregulate cancer cell radiation sensitivity effectively. As described above, the developing small molecular agents targeting tumor signal pathways exhibit obvious advantages over conventional cytotoxic radiation sensitizers. The development of multifunctional agents targeting multiple components of signal pathways in tumor cells further supplies advantages over the side effects induced by conventional radiotherapy. This inspires the development of multifunctional, multi‐targeting radiosensitizers utilizing artificial structures drugs and targeting gene sequences. Therefore, in this article, the relationships between aberrant signal pathways and radioresistance and various targeting radiation sensitivity agents have been reviewed; clinical and preclinical data also show significant promise. The use of these strategies provides not only a potential avenue to explore new pathways responsible for tumor radioresistance but also a potential framework to incorporate two or more strategies to provide multifunctional capabilities.
Identifying novel signal elements that are connected to radioresistance can bring to novel agents exploitation for radiosensitization. However, there are still challenges to improve the transfer of these therapeutic agents to clinical use, such as short half‐life periods, side effects impacting normal organs, targeting and penetrating tumors and long‐term toxicity. Hence, a rigid examination of these agents must be undertaken in future, practical and validated pharmacodynamic biomarkers connected with the agents need to be developed. Drug delivery systems also provide a novel radiosensitization strategy, such as nanomaterials that have been demonstrated to be excellent tools for molecular imaging, diagnosis and targeted delivery. In addition, modification of the radiosensitivity of cancer stem‐like cells that are key contributors to radioresistance and are responsible for tumor progression and recurrence may be another approach to consider in radiation therapy.
In conclusion, the identification of aberrant signal pathways broaden the current concept of radiation sensitivity, exploring multi‐targeting molecular agents and new targeting sensitization technologies which can achieve personalized radiotherapy will be under intensive development in future.
Funding Sources
This work was supported by National Natural Science Foundation Program (NSFC81072523) and State Education Ministry Program (FANEDD200777) grants from China.
Disclosure Statement
The authors have no conflict of interest.
(Cancer Sci 2013; 104: 1401–1410)
References
- 1. Yorke E, Gelblum D, Ford E. Patient safety in external beam radiation therapy. Am J Roentgenol 2011; 196: 768–72. [DOI] [PubMed] [Google Scholar]
- 2. Milosevic MF. Hypoxia, anerobic metabolism, and interstitial hypertension. In: Siemann DW, ed. Tumor Microenvironment, 1st edn. Chichester: John Wiley & Sons Ltd, 2010; 183–205. [Google Scholar]
- 3. Wachsberger P, Burd R, Dicker AP. Tumor response to ionizing radiation combined with antiangiogenesis or vascular targeting agents: exploring mechanisms of interaction. Clin Cancer Res 2003; 9: 1957–71. [PubMed] [Google Scholar]
- 4. Kasten‐Pisula U, Menegakis A, Brammer I et al The extreme radiosensitivity of the squamous cell carcinoma SKX is due to a defect in double‐strand break repair. Radiother Oncol 2009; 90: 257–64. [DOI] [PubMed] [Google Scholar]
- 5. Zhivotovsky B, Joseph B, Orrenius S. Tumor radiosensitivity and apoptosis. Exp Cell Res 1999; 248: 10–7. [DOI] [PubMed] [Google Scholar]
- 6. Nielsen KV, Müller S, Møller S et al Aberrations of ERBB2 and TOP2A genes in breast cancer. Mol Oncol 2010; 4: 161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Toffalini F, Demoulin JB. New insights into the mechanisms of hematopoietic cell transformation by activated receptor tyrosine kinases. Blood 2010; 116: 2429–37. [DOI] [PubMed] [Google Scholar]
- 8. Squatrito M, Holland EC. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res 2011; 71: 5945–9. [DOI] [PubMed] [Google Scholar]
- 9. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev 2010; 20: 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9: 537–49. [DOI] [PubMed] [Google Scholar]
- 11. Kim BH, Yi CH, Guo Q et al A small‐molecule compound identified through a cell‐based screening inhibits JAK/STAT pathway signaling in human cancer cells. Mol Cancer Ther 2008; 7: 2672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sato N, Yamabuki T, Takano A et al Wnt inhibitor Dickkopf‐1 as a target for passive cancer immunotherapy. Cancer Res 2010; 70: 5326–36. [DOI] [PubMed] [Google Scholar]
- 13. Baud V, Karin M. Is NF‐[kappa]B a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov 2009; 8: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Galina S. Therapeutic targeting of p53 by small molecules. Semi Cancer Biol 2010; 20: 46–56. [DOI] [PubMed] [Google Scholar]
- 15. Zhu Y, Hu J, Hu Y et al Targeting DNA repair pathways: a novel approach to reduce cancer therapeutic resistance. Cancer Treat Rev 2009; 35: 590–6. [DOI] [PubMed] [Google Scholar]
- 16. Valerie K, Yacoub A, Hagan MP et al Radiation‐induced cell signaling: inside‐out and outside‐in. Mol Cancer Ther 2007; 6: 789–801. [DOI] [PubMed] [Google Scholar]
- 17. Dent P, Yacoub A, Contessa J et al Stress and radiation‐induced activation of multiple intracellular signaling pathways. Radiat Res 2003a; 159: 283–300. [DOI] [PubMed] [Google Scholar]
- 18. Viloria‐Petit A, Crombet T, Jothy S et al Inhibition of vascular endothelial growth factor receptor signaling leads to reversal of tumor resistance to radiotherapy. Cancer Res 2001a; 61: 2413–9. [PubMed] [Google Scholar]
- 19. Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res 2002; 90: 1243–50. [DOI] [PubMed] [Google Scholar]
- 20. Jeong SJ, Pise‐Masison CA, Radonovich MF et al Activated AKT regulates NF‐[kappa]B activation, p53 inhibition and cell survival in HTLV‐1‐transformed cells. Oncogene 2005; 24: 6719–28. [DOI] [PubMed] [Google Scholar]
- 21. Soderlund K, Perez‐Tenorio G, Stal O. Activation of the phosphatidylinositol 3‐kinase/Akt pathway prevents radiation‐induced apoptosis in breast cancer cells. Int J Oncol 2005; 26: 25–32. [PubMed] [Google Scholar]
- 22. Nakamura JL, Karlsson A, Arvold ND et al PKB/Akt mediates radiosensitization by the signaling inhibitor LY294002 in human malignant gliomas. J Neurooncol 2005; 71: 215–22. [DOI] [PubMed] [Google Scholar]
- 23. Shinohara ET, Cao C, Niermann K et al Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene 2005; 24: 5414–22. [DOI] [PubMed] [Google Scholar]
- 24. Gera JF, Mellinghoff IK, Shi Y et al AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating Cyclin D1 and c‐myc expression. J Biol Chem 2004; 279: 2737–46. [DOI] [PubMed] [Google Scholar]
- 25. Gupta AK, Bakanauskas VJ, Cerniglia GJ et al The Ras radiation resistance pathway. Cancer Res 2001; 61: 4278–82. [PubMed] [Google Scholar]
- 26. Khazak V, Astsaturov I, Serebriiskii IG, Golemis EA. Selective Raf inhibition in cancer therapy. Expert Opin Ther Targets 2007; 11: 1587–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones HA, Hahn SM, Bernhard E, McKenna WG. Ras inhibitors and radiation therapy. Semin Radiat Oncol 2001; 11: 328–37. [DOI] [PubMed] [Google Scholar]
- 28. Michael PD, Ross F, Peter MF. Small‐molecule inhibitors of MDM2 as new anticancer therapeutics. Semin Cancer Biol 2010; 20: 10–8. [DOI] [PubMed] [Google Scholar]
- 29. Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med 2007; 13: 23–31. [DOI] [PubMed] [Google Scholar]
- 30. Chinnaiyan P, Allen GW, Harari PM. Radiation and new molecular agents, Part II: targeting HDAC, HSP90, IGF‐1R, PI3K, and Ras. Semin Radiat Oncol 2006; 16: 59–64. [DOI] [PubMed] [Google Scholar]
- 31. Fremin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol 2010; 3: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene 2003b; 22: 5885–96. [DOI] [PubMed] [Google Scholar]
- 33. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9: 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang Z, Cheng L, Guryanova O, Wu Q, Bao S. Cancer stem cells in glioblastoma‐molecular signaling and therapeutic targeting. Protein Cell 2010; 1: 638–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang YP, Chang YL, Huang PI et al Resveratrol suppresses tumorigenicity and enhances radiosensitivity in primary glioblastoma tumor initiating cells by inhibiting the STAT3 axis. J Cell Physiol 2012; 227: 976–93. [DOI] [PubMed] [Google Scholar]
- 36. Bonner JA, Trummell HQ, Willey CD, Plants BA, Raisch KP. Inhibition of STAT3 results in radiosensitization of human squamous cell carcinoma. Radiother Oncol 2009; 92: 339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skvortsova I, Skvortsov S, Stasyk T et al Intracellular signaling pathways regulating radioresistance of human prostate carcinoma cells. Proteomics 2008; 8: 4521–33. [DOI] [PubMed] [Google Scholar]
- 38. Kim KW, Mutter RW, Cao C et al Inhibition of signal transducer and activator of transcription 3 activity results in down‐regulation of Survivin following irradiation. Mol Cancer Ther 2006; 5: 2659–65. [DOI] [PubMed] [Google Scholar]
- 39. Hui Z, Tretiakova M, Zhang Z et al Radiosensitization by inhibiting STAT1 in renal cell carcinoma. Int J Radiat Oncol 2009; 73: 288–95. [DOI] [PubMed] [Google Scholar]
- 40. Deorukhkar A, Krishnan S, Sethi G, Aggarwal BB. Back to basics: how natural products can provide the basis for new therapeutics. Expert Opin Investig Drugs 2007; 16: 1753–73. [DOI] [PubMed] [Google Scholar]
- 41. Dorai T, Aggarwal BB. Role of chemopreventive agents in cancer therapy. Cancer Lett 2004; 215: 129–40. [DOI] [PubMed] [Google Scholar]
- 42. Kim Y, Kim KH, Lee J et al Wnt activation is implicated in glioblastoma radioresistance. Lab Invest 2012; 92: 466–73. [DOI] [PubMed] [Google Scholar]
- 43. Kendziorra E, Ahlborn K, Spitzner M et al Silencing of the Wnt transcription factor TCF4 sensitizes colorectal cancer cells to (chemo‐) radiotherapy. Carcinogenesis 2011; 32: 1824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi‐Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res 2010; 16: 3153–62. [DOI] [PubMed] [Google Scholar]
- 45. Idris AI, Libouban H, Nyangoga H, Landao‐Bassonga E, Chappard D, Ralston SH. Pharmacologic inhibitors of IκB kinase suppress growth and migration of mammary carcinosarcoma cells in vitro and prevent osteolytic bone metastasis in vivo. Mol Cancer Ther 2009; 8: 2339–47. [DOI] [PubMed] [Google Scholar]
- 46. Pajonk F, Pajonk K, McBride WH. Inhibition of NF‐κB, clonogenicity, and radiosensitivity of human cancer cells. J Natl Cancer Inst 1999; 91: 1956–60. [DOI] [PubMed] [Google Scholar]
- 47. Deorukhkar A, Krishnan S. Targeting inflammatory pathways for tumor radiosensitization. Biochem Pharmacol 2010; 80: 1904–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mendonca MS, Chin‐Sinex H, Gomez‐Millan J et al Parthenolide sensitizes cells to X‐ray‐induced cell killing through inhibition of NF‐κB and split‐dose repair. Radiat Res 2007; 168: 689–97. [DOI] [PubMed] [Google Scholar]
- 49. Sun Y, Clair DK, Fang F et al The radiosensitization effect of parthenolide in prostate cancer cells is mediated by nuclear factor‐κB inhibition and enhanced by the presence of PTEN. Mol Cancer Ther 2007; 6: 2477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Magné N, Toillon RA, Bottero V et al NF‐κB modulation and ionizing radiation: mechanisms and future directions for cancer treatment. Cancer Lett 2006; 231: 158–68. [DOI] [PubMed] [Google Scholar]
- 51. Russo SM, Tepper JE, Baldwin AS Jr et al Enhancement of radiosensitivity by proteasome inhibition: implications for a role of NF‐κB. Int J Radiat Oncol 2001; 50: 183–93. [DOI] [PubMed] [Google Scholar]
- 52. Kirsch DG, Kastan MB. Tumor‐suppressor p53: implications for tumor development and prognosis. J Clin Oncol 1998; 16: 3158–68. [DOI] [PubMed] [Google Scholar]
- 53. Vassilev LT. p53 activation by small molecules: application in oncology. J Med Chem 2005; 48: 4491–9. [DOI] [PubMed] [Google Scholar]
- 54. Nielsen LL, Maneval DC. P53 tumor suppressor gene therapy for cancer. Cancer Gene Ther 1998; 5: 52–63. [PubMed] [Google Scholar]
- 55. Cohen SJ, Cohen RB, Meropol NJ. Meropol targeting signal transduction pathways in colorectal cancer‐more than skin deep. J Clin Oncol 2005; 23: 5374–85. [DOI] [PubMed] [Google Scholar]
- 56. Chung KY, Shia J, Kemeny NE et al Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 2005; 23: 1803–10. [DOI] [PubMed] [Google Scholar]
- 57. Lenz H, Mayer R, Gold P et al Activity of cetuximab in patients with colorectal cancer refractory to both irinotecan and oxaliplatin. Proc Am Soc Clin Oncol 2004; 22: 247s. [Google Scholar]
- 58. Zhang E, Zhang C, Su Y, Cheng T, Shi C. Newly developed strategies for multifunctional mitochondria‐targeted agents in cancer therapy. Drug Discov Today 2011; 16: 140–6. [DOI] [PubMed] [Google Scholar]
- 59. Cappuzzo1 F, Finocchiaro G, Rossi E et al EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol 2008; 19: 717–23. [DOI] [PubMed] [Google Scholar]
- 60. Viloria‐Petit A, Crombet T, Jothy S et al Acquired resistance to the antitumor effect of epidermal growth factor receptor‐blocking antibodies in vivo. Cancer Res 2001b; 61: 5090–101. [PubMed] [Google Scholar]
- 61. Weickhardt AJ, Tebbutt NC, Mariadason JM. Strategies for overcoming inherent and acquired resistance to EGFR inhibitors by targeting downstream effectors in the Ras/PI3K pathway. Curr Cancer Drug Targets 2010; 10: 824–33. [DOI] [PubMed] [Google Scholar]
- 62. Roti Roti JL, Vander Waal RP, Laszlo A. Thermal modulation of radiation‐induced DNA damage responses. Curr Cancer Res 2011; 2: 227–49. [Google Scholar]
- 63. Wardman P, Rothkamm K, Folkes LK, Woodcock M, Johnston PJ. Radiosensitization by nitric oxide at low radiation doses. Radiat Res 2007; 167: 475–84. [DOI] [PubMed] [Google Scholar]