

Abstract
Lung cancer (LC) is the major cause of death by cancer and the number of LC patients is increasing worldwide. This study investigated the therapeutic potential of gene delivery using suppressor of cytokine signaling 1 (SOCS‐1), an endogenous inhibitor of intracellular signaling pathways, for the treatment of LC. To examine the antitumor effect of SOCS‐1 overexpression on non‐small‐cell lung cancer (NSCLC) cells, NSCLC cells (A549, LU65, and PC9) were infected with adenovirus‐expressing SOCS‐1 vector. The cell proliferation assay showed that A549 and LU65, but not PC9, were sensitive to SOCS‐1 gene‐mediated suppression of cell growth. Although JAK inhibitor I could also inhibit proliferation of A549 and LU65 cells, SOCS‐1 gene delivery appeared to be more potent as SOCS‐1 could suppress focal adhesion kinase and epidermal growth factor receptor, as well as the JAK/STAT3 signaling pathway. Enhanced phosphorylation of the p53 protein was detected by means of phospho‐kinase array in SOCS‐1 overexpressed A549 cells compared with control cells, whereas no phosphorylation of p53 was observed when JAK inhibitor I was used. Furthermore, treatment with adenoviral vector AdSOCS‐1 in vivo significantly suppressed NSCLC proliferation in a xenograft model. These results suggest that the overexpression of SOCS‐1 gene is effective for antitumor therapy by suppressing the JAK/STAT, focal adhesion kinase, and epidermal growth factor receptor signaling pathways and enhancing p53‐mediated antitumor activity in NSCLC.
Lung cancer is the leading cause of cancer death in Japan and is a growing health epidemic worldwide.1 Moreover, therapies that can cure metastatic LC have not been yet established,2, 3 so there is an urgent need for the development of novel interventions to cure LC.
One of the potential therapeutic targets of NSCLC is STAT3. Constitutively activated STAT3 has been shown to promote tumor cell growth, survival, and tumor angiogenesis, and persistently activated STAT3 has been found in 50% of lung adenocarcinomas.4 It is thought that STAT3 is activated by JAK, EGFR, or Src family kinases.5 Among these TYKs, JAK family kinases play an important role in the phosphorylation of STAT3 in NSCLC.6, 7 Dysregulated activation of the JAK/STAT3 signaling pathway, the major downstream pathway of cytokines such as interleukin‐6, has been detected in various cancers including NSCLC.8 Moreover, it has been recently reported that ruxolitinib, which is a potent and selective JAK1 and JAK2 inhibitor, is associated with marked and durable clinical benefits for patients with myelofibrosis, suggesting that JAK kinases are promising therapeutic targets for cancer.9
Cytokine signaling pathways are tightly controlled by negative regulatory mechanisms under homeostatic conditions. Suppressors of cytokine signaling family proteins play a role in the negative regulation of cytokine responses by terminating the activation of the JAK/STAT and other signaling pathways.10, 11, 12
The SOCS family, characterized by a central src homology 2 domain and a conserved C‐terminus SOCS box, is composed of eight structurally related proteins.13 Of these, SOCS‐1 is known as the most potent negative regulator of pro‐inflammatory cytokine signaling.14 It interacts with phosphotyrosine residues on proteins such as JAK kinases to interfere with the activation of STAT proteins or other signaling intermediates.15, 16 Also, SOCS‐1 recruits the Elongin BC‐containing E3 ubiquitin‐ligase complex through the conserved SOCS box to promote the degradation of target proteins.17 Studies of SOCS‐1 deficient mice have indicated that SOCS‐1 is essential for the inhibition of excessive immune responses and is also involved in the suppression of tumor development.18, 19
Although it is still not clear whether SOCS shows therapeutic benefit for NSCLC, preclinical analyses of SOCS in therapies of several types of cancers have been carried out worldwide.20 Previously reported studies by us showed that SOCS‐1 or SOCS‐3 were effective when used for therapies of malignant pleural mesothelioma or gastric cancer.21, 22, 23 In addition, it has been reported that overexpression of the SOCS‐3 gene showed antitumor effects in NSCLC.24, 25, 26
In the study presented here, we used the NSCLC cell lines A549, LU65, and PC9 to investigate the possibility of the application of SOCS‐1 gene transduction to NSCLC therapies and the mechanisms of antitumor effects by SOCS‐1.
Material and Methods
Cell lines
Both A549 and LU65 cell lines were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan). The PC9 cell line was kindly provided by Prof. Nishio of Kinki University of Medicine, Department of Genome Biology (Osaka, Japan). Details are described in Data S1.
Reagents
PD153035 and JAK inhibitor I were purchased from EMD Millipore (Billerica, MA, USA) and Calbiochem (La Jolla, CA, USA), respectively.
Preparation of adenoviruses
The replication‐defective recombinant adenoviral vector expressing the mouse SOCS‐1 gene was provided by Dr. Hiroyuki Mizuguchi (Osaka University, Osaka, Japan), which was constructed with an improved in vitro ligation method, as described previously.27, 28 An adenoviral vector expressing the LacZ gene was constructed using a similar method and expression of these genes was regulated by means of a CMV promoter/enhancer and intron A. Details are described in Data S1.
Phospho‐kinase array
Expression of phosphorylated proteins was detected with the Proteome Profiler Human Phospho‐Kinase Array kit (R&D Systems, Minneapolis, MN, USA). Details are described in Data S1.
Cell viability assay
The NSCLC cell lines were plated in 96‐well plates at a density of 1 × 103 cells per well and incubated in RPMI‐1640 medium containing 10% FCS. Details are described in Data S1.
SDS‐PAGE and Western blot analysis
Whole cell protein extracts were prepared from NSCLC cells and tumor tissue in RIPA buffer containing phosphatase inhibitor cocktail and a protease inhibitor cocktail (both from Nacalai Tesque, Kyoto, Japan and both at a concentration of 1×) followed by centrifugation (16 100g, 4°C, 15 min). Details are described in Data S1.
Small interfering RNA transfection
Commercial FAK siRNA was obtained from Qiagen (Hilden, Germany). Details are described in Data S1.
Mouse xenograft model
All animal experiments were carried out according to the institutional ethical guidelines for animal experimentation of the National Institute of Biomedical Innovation (Osaka, Japan). Details are described in Data S1.
Immunohistochemistry
Subcutaneously implanted tumors were harvested and paraffin embedded for immunohistochemical analysis using anti‐SOCS‐1 antibody (Abcam, Cambridge, MA, USA) and anti‐Ki‐67 antibody (Novocastra Laboratories, Newcastle, UK). A TUNEL assay (with DAPI nuclear counterstaining) for apoptosis was carried out using the ApopTag Fluorescein In Situ Apoptosis Detection Kit (Chemicon International, Temecula, CA, USA) according to the manufacturer's instructions.
Statistical analysis
Data are shown the mean ± SD (unless stated otherwise) for the number of experiments indicated. Details are described in Data S1.
Results
SOCS‐1 gene delivery shows marked antiproliferative effects in A549 and LU65 cells
We used A549, LU65, and PC9 cell lines, which have been used as NSCLC cell lines in many other experiments. Because the JAK/STAT3 pathway has recently received attention as a novel target for treatment of NSCLC, we investigated the expression levels of JAK family kinases and of STAT3, as well as tyrosine phosphorylation of STAT3 in NSCLC cells. The four known mammalian members of the JAK family are TYK2, JAK1, JAK2, and JAK3. These kinases are widely expressed in a variety of different cell types, with the exception of JAK3, which is selectively expressed in cells of hematopoietic origin.29 The expression of each JAK was at a similar level in the three cell lines (Fig. 1a). Although phosphorylated STAT3 was detectable in all cell lines, it was markedly more elevated in A549 and LU65 (Fig. 1b).
Figure 1.
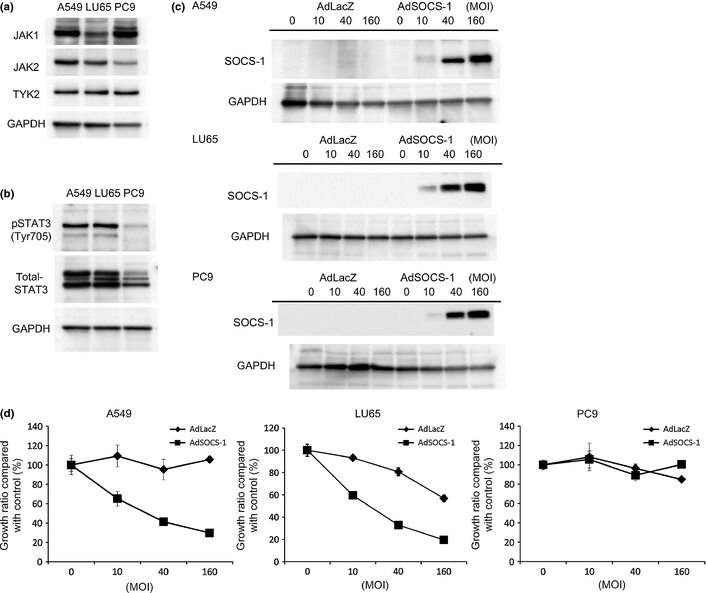
Delivery of SOCS‐1 gene shows marked antiproliferative effects in A549 and LU65 lung cancer cell lines. (a) Expression of JAK family kinases in all cell lines determined by Western blotting. Cell lysates were immunoblotted with JAK1, JAK2, or tyrosine kinase 2 (TYK2) antibodies. (b) Comparative analysis of phosphorylation and expression of signal transducer and activator of transcription 3 (STAT3) determined by Western blotting. (c) Cell lysates were prepared 48 h after treatment with AdSOCS‐1or AdLacZ adenoviral vectors at an MOI of 10–160, and immunoblotted with anti‐SOCS‐1 antibody. (d) Cells were infected with AdSOCS‐1 or AdLacZ at an MOI of 10–160. Cell proliferation was determined by MTS assay 72 h after transfection. Growth ratio of AdLacZ or AdSOCS‐1‐infected cells was calculated by a percentage of that for non‐treated cells. Each value represents the average ± SD of hexaplicate wells.
We next investigated whether SOCS‐1 could suppress proliferation of A549, LU65, and PC9. Following SOCS‐1 gene delivery, overexpression of SOCS‐1 was detected in all cell lines by Western blot analysis (Fig. 1c), but overexpression of SOCS‐1 had antiproliferative effects on A549 and LU65 but not on PC9 (Fig. 1d).
Overexpression of SOCS‐1 shows stronger antiproliferative effect than JAK inhibitor in NSCLC cells
Immunoblotting analysis showed that the expression levels of some of the JAK family kinases in all cell lines were reduced by overexpression of SOCS‐1 (Fig. 2a). Next, phosphorylation levels of STAT3 (Tyr705), a downstream molecule of the JAK kinase family, were analyzed by Western blot analysis. Phosphorylation levels of STAT3 (Tyr705) were decreased in response to overexpression of the SOCS‐1 gene in all cell lines (Fig. 2b). These results agree with previously reported findings that SOCS‐1 can act directly on JAK family kinases to suppress their kinase activity as well as to accelerate their degradation by the recruitment of an E3 ubiquitin ligase.17
Figure 2.
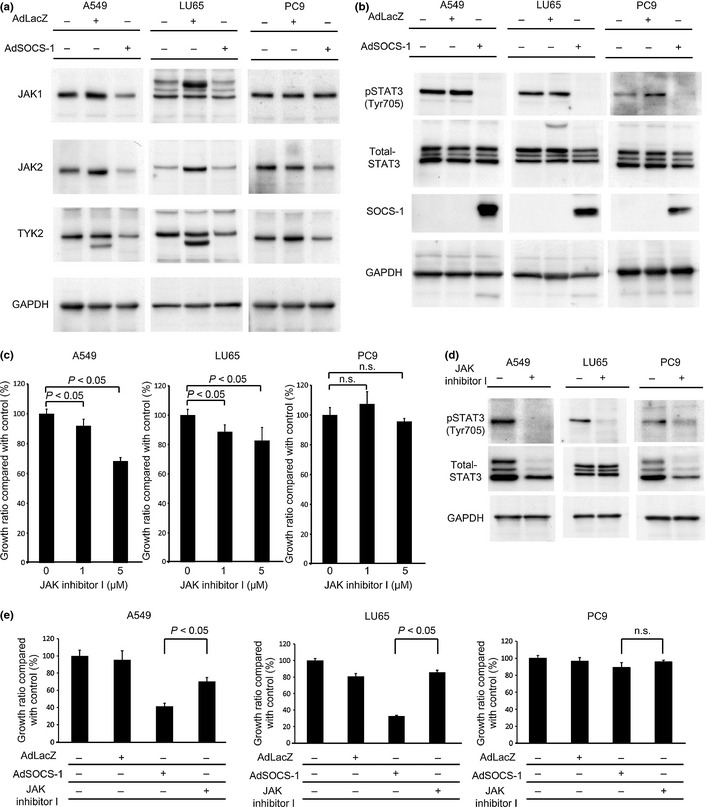
Antiproliferative effect of suppressor of cytokine signaling‐1 (SOCS‐1) correlated with JAK dependence is stronger than JAK inhibitor. (a) Cell lysates were prepared 48 h after treatment with AdSOCS‐1 or AdLacZ adenoviral vectors at an MOI of 40 and immunoblotted with JAK1, JAK2, or tyrosine kinase 2 (TYK2) antibodies. (b) SOCS‐1 suppressed activation of signal transducer and activator of transcription 3 (STAT3). Cell lysates were prepared as described above and immunoblotted with anti‐p‐STAT3 (Tyr705), anti‐STAT3, and anti‐SOCS‐1 antibodies. (c) Cells cultured in RPMI‐1640 medium containing 10% FBS were exposed to 1–5 μM JAK inhibitor I. After incubation for 72 h, viable cells were counted with the MTS assay. (d) Cells were cultured in RPMI‐1640 medium containing 0.5% FBS with 5 μM JAK inhibitor I. Cell lysates were immunoblotted with anti‐p‐STAT3 (Tyr705) and anti‐STAT3 antibodies. (e) All cell lines cultured in RPMI‐1640 medium containing 10% FBS were exposed to 5 μM JAK inhibitor I, or infected with AdLacZ or AdSOCS‐1 at an MOI of 40. After incubation for 72 h, viable cells were counted with the MTS assay.
Although the JAK/STAT3 pathway was downregulated by SOCS‐1 in all cell lines, overexpression of the SOCS‐1 gene showed an antiproliferative effect only on A549 and LU65 cells. Accordingly, we next used JAK inhibitor I, a blocker of JAK1, JAK2, and JAK3, instead of AdSOCS‐1 to examine the effects of JAK inhibition on all cell lines. Proliferation of A549 and LU65 cells was significantly suppressed by treatment with JAK inhibitor I, however, proliferation of PC9 was not significantly suppressed (Fig. 2c). Immunoblotting analysis showed that tyrosine phosphorylation of STAT3 was downregulated in all cell lines after introduction of JAK inhibitor I (Fig. 2d). These findings suggest that JAK‐mediated signals are critical for the proliferation of A549 and LU65 cells, but are not needed for that of PC9 cells. In addition, because JAK1 and JAK2 are overlapping target molecules between SOCS‐1 and JAK inhibitor I, the primary target molecules of SOCS‐1 in NSCLC cells are likely to be JAK1 and/or JAK2. Therefore, we assumed that the antiproliferative effect of SOCS‐1 was essentially determined by the JAK‐dependence of NSCLC cells.
Immunoblotting analysis showed that introduction of both the SOCS‐1 gene (at 40 MOI) and of JAK inhibitor I (5 μM) could effectively suppress STAT3 phosphorylation (Fig. 2b,d). However, SOCS‐1 was more effective than JAK inhibitor I for inhibiting proliferation of A549 and LU65 cells (Fig. 2e). Because SOCS‐1 is known as an adaptor of several molecules other than JAK family kinases,19 it was expected that AdSOCS‐1 could also exert JAK‐independent action in A549 and LU65. To investigate in detail the mechanisms of antitumor effects induced by overexpression of SOCS‐1, we used A549 cells for further analyses.
Focal adhesion kinase is downregulated by overexpression of SOCS‐1 in A549
Focal adhesion kinase is known as an adhesion molecule and previous investigations have indicated that FAK contributes to tumor cell proliferation, survival, and metastasis.30 SOCS‐1 was found to inhibit FAK‐dependent signaling events by suppressing FAK‐associated kinase activity and tyrosine phosphorylation of FAK,31 and also by promoting polyubiquitination and degradation of FAK in a SOCS box‐dependent manner.17 For this reason, we next focused on the FAK pathway and examined the levels of FAK expression and of FAK tyrosine phosphorylation after overexpression of the SOCS‐1 gene.
Focal adhesion kinase expression was upregulated by introduction of the LacZ gene in A549 cells (Fig. 3a), probably reflecting the previously reported non‐specific stimulation by adenovirus vectors.32 Compared to the cells introduced by LacZ, FAK phosphorylation (Tyr397) was downregulated after the introduction of the SOCS‐1 gene in A549 (Fig. 3a). We also investigated whether JAK inhibitor I, instead of SOCS‐1, can suppress activation of FAK in A549 cells, and found that JAK inhibitor could not (Fig. 3b).
Figure 3.
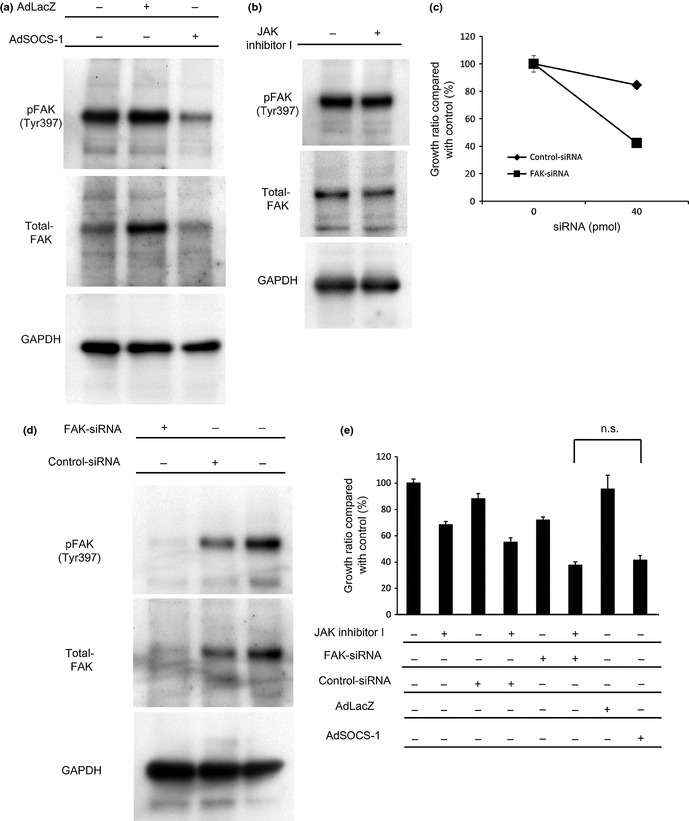
Focal adhesion kinase (FAK) is downregulated by adenovirus vector containing suppressor of cytokine signaling‐1 (AdSOCS‐1) in A549 lung cancer cells. (a) Cell lysates were prepared 48 h after treatment with AdSOCS‐1 or AdLacZ at an MOI of 40, and immunoblotted with p‐FAK (Tyr397) and FAK antibodies. (b) Cell lysates were prepared 48 h after exposure to 5 μM JAK inhibitor I and immunoblotted with p‐FAK (Tyr397) and FAK antibodies. (c) Cells were exposed to either FAK siRNA or non‐specific siRNA as control. Cells were cultured in RPMI‐1640 medium containing 10% FBS. After a 3‐day culture, viable cells were counted with the MTS assay. (d) Protein extracts prepared at 48 h after treatment with FAK siRNA or non‐specific siRNA were blotted with anti‐p‐FAK (Tyr397) and anti‐FAK antibodies. (e) A549 cells cultured in RPMI‐1640 medium containing 10% FBS were exposed to 5 μM JAK inhibitor I or 40 pmol FAK siRNA; 40 pmol non‐specific siRNA was used as control. Infections of AdLacZ or AdSOCS‐1 were carried out at an MOI of 40. Cell proliferation activity was assessed by MTS assay 72 h after treatment. n.s., no significant change detected by Tukey's post‐hoc comparisons.
We assumed that downregulation of FAK by SOCS‐1 could contribute to the inhibition of A549 cell proliferation. Therefore, examined whether FAK siRNA indeed had an antiproliferative effect on A549, and found that proliferation of A549 cells was suppressed by FAK knockdown, indicating that these cells are dependent on FAK (Fig. 3c,d). We also investigated the combined effect of JAK inhibitor I and FAK siRNA on A549 cell proliferation. The results showed that the combined effect was stronger than that of JAK inhibitor I alone (Fig. 3e). These findings suggest that FAK has an important role in A549 cell proliferation independent of JAK and that SOCS‐1‐mediated FAK inhibition might contribute to the suppression of proliferation of A549 cells treated with AdSOCS‐1.
Epidermal growth factor receptor is downregulated by overexpression of SOCS‐1 in A549
Expression of SOCS‐1 reportedly interacts with the cytoplasmic domain of EGFR and is likely to induce ubiquitination and degradation of ligand‐bound EGFR.33 This notion was confirmed by means of immunoblotting analysis in our study, in that EGFR expression was downregulated 48 h after introduction of the SOCS‐1 gene in A549 cells. The EGFR phosphorylation (Tyr1068) was also downregulated after overexpression of the SOCS‐1 gene had occurred in A549 cells (Fig. 4a). In contrast, JAK inhibitor I did not suppress EGFR activation in A549 cells (Fig. 4b).
Figure 4.
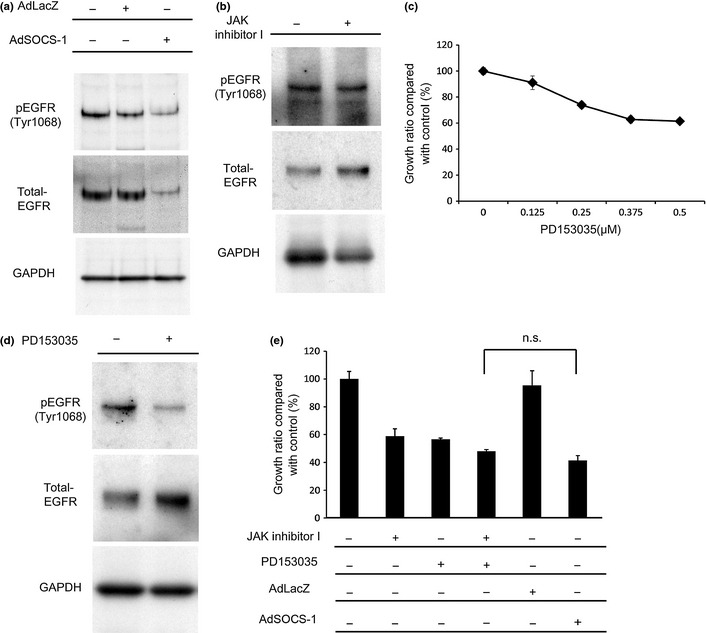
Epidermal growth factor receptor (EGFR) is downregulated by adenovirus vector containing suppressor of cytokine signaling‐1 (AdSOCS‐1) in A549 lung cancer cells. (a) Cells were infected with AdLacZ or AdSOCS‐1 at an MOI of 40 for 24 h. Cell lysates were immunoblotted with anti‐p‐EGFR (Tyr1068) and anti‐EGFR antibodies. (b) Cell lysates were prepared 48 h after exposure to 5 μM JAK inhibitor I and immunoblotted with anti‐p‐EGFR (Tyr1068) and anti‐EGFR antibodies. (c) Cells cultured in RPMI‐1640 medium containing 10% FBS were exposed to 0.125–0.5 μM PD153035. Cell proliferation activity was assessed by MTS assay 72 h after exposure. (d) A549 cells were cultured in RPMI‐1640 medium containing 0.5% FBS with 0.5 μM PD153035. Cell lysates were prepared 48 h after treatment and immunoblotted with anti‐p‐EGFR (Tyr1068) and anti‐EGFR antibodies. (e) A549 cells cultured in RPMI‐1640 medium containing 10% FBS were exposed to 5 μM JAK inhibitor I or 0.5 μM PD153035. Infection of AdLacZ or AdSOCS‐1 was carried out at an MOI of 40. Cell proliferation activity was assessed by MTS assay 72 h after treatment. n.s., no significant change detected by Tukey's post‐hoc comparisons.
We next examined the antiproliferative effects of the EGFR inhibitor PD153035 on A549 cells. Expression of EGFR in A549 cells was much lower than that in PC9 cells (data not shown), which harbor a deletion of an EGFR mutation.34 Nevertheless, our findings show that proliferation of A549 cells with wild‐type EGFR were also somewhat dependent on EGFR (Fig. 4c,d). We also investigated the combined effect of JAK inhibitor I and PD153035 on proliferation of A549 cells and found that they have an additive effect (Fig. 4e). This finding suggests that EGFR is also involved in A549 cell proliferation independent of JAK and that its downregulation by SOCS‐1 may have an inhibitory effect on proliferation of A549 cells.
Accordingly, JAK1, JAK2, FAK, and EGFR should be considered critical for the proliferation of A549 cells, so that simultaneous inhibition of these molecules by SOCS‐1 may have a potent antiproliferative effect on A549.
Upregulation of p53 by AdSOCS‐1 in A549 cells
We also used a phospho‐kinase array to determine the expression profile of phosphorylated proteins in A549 in order to identify other target molecules for SOCS‐1 in addition to JAK, FAK, and EGFR (Fig. 5a). Phosphorylation of p53 was upregulated in A549 cells after the introduction of the SOCS‐1 gene. This was confirmed by immunoblotting analysis, which showed that p53 phosphorylation was upregulated in A549 cells by overexpression of the SOCS‐1 gene (Fig. 5b). We also investigated whether JAK inhibitor I, instead of SOCS‐1, could activate p53 in A549 cells, but found that it did not (Fig. 5b). Given the well‐established antitumor effect of p53 phoshorylation,35, 36, 37, 38 SOCS‐1‐induced p53 activation may thus also contribute to the suppression of A549 cell proliferation.
Figure 5.
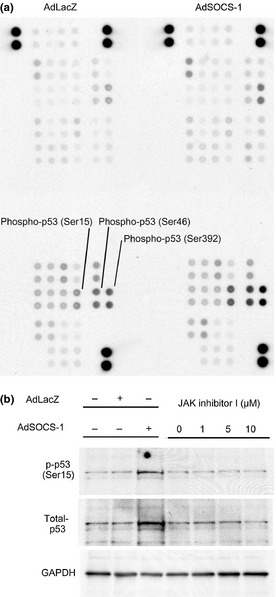
Protein p53 is upregulated by adenovirus vector containing suppressor of cytokine signaling‐1 (AdSOCS‐1) in A549 lung cancer cells. (a) A549 cells were cultured in RPMI‐1640 medium containing 10% FBS with AdSOCS‐1 at an MOI of 40. After a 24‐h culture, protein extracts were examined with a phospho‐kinase array with each phosphorylated protein identified in duplicate. The double‐labeled spots in the upper right corner represent the positive controls. (b) Cell lysates were prepared 48 h after infection with AdLacZ or AdSOCS‐1 at an MOI of 40. Cell lysates were immunoblotted with anti‐phospho‐53 (p‐p53 [Ser15]) and anti‐p53 antibodies. (c) Cell lysates were prepared 48 h after exposure to 5 μM JAK inhibitor I and immunoblotted with anti‐p‐p53 (Ser15) and anti‐p53 antibodies.
Antitumor activity of SOCS‐1 in a lung cancer xenograft model
We also evaluated the therapeutic effect of AdSOCS‐1 injection on the growth of NSCLC cells in vivo. We established a xenograft model of ICR nu/nu mice in which A549 cells were s.c. implanted. Injection of AdSOCS‐1 vector (4 × 108 pfu) intratumorally twice per week significantly suppressed tumor growth compared to control AdLacZ injection (Fig. 6a). AdSOCS‐1 in vivo could modulate intracellular signaling in NSCLC cells as in vitro, as Western blot analysis showed that phosphorylation levels of STAT3 were decreased in the A549 tissues from AdSOCS‐1 injected animals (Fig. 6b). Furthermore, few Ki‐67‐positive nuclei were detected by immunohistochemical analysis in AdSOCS‐1 infected tissues compared to AdLacZ, indicating that proliferating cells are decreased by overexpression of SOCS‐1 (Fig. 6c). Additionally, induction of apoptosis was detected in AdSOCS‐1 infected A549 tissue compared to AdLacZ by TUNEL analysis (Fig. 6c).
Figure 6.
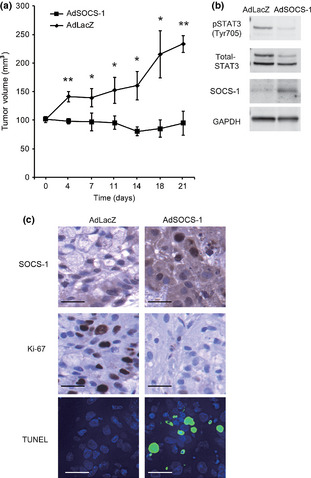
Suppressor of cytokine signaling‐1 (SOCS‐1) shows antitumor activity in a lung cancer xenograft model. (a) Female ICR nu/nu mice were s.c. implanted with 2 × 106 A549 cells in their flank. When the calculated tumor volumes reached to approximately 100 mm3, 4 × 108 pfu AdSOCS1 or AdLacZ adenoviral vectors were intratumorally treated twice per week. Tumor volumes were determined twice per week. The mean volume ± SEM of five tumors in each group, and were analyzed by Student's t‐test (*P < 0.05, **P < 0.01). (b) Western blot analysis of phosphorylated signal transducer and activator of transcription 3 (pSTAT3), STAT3, SOCS‐1, and GAPDH in A549 tissues from AdSOCS‐1 or AdLacZ injected animals. Lysates from tumors were analyzed by Western blotting. (c) Immunohistochemical analysis of SOCS‐1, Ki‐67, and TUNEL (blue fluorescence, DAPI staining for nuclei; cyan fluorescence, TUNEL positivity) in A549 tissues from animals injected with AdSOCS‐1 or AdLacZ. Scale bar = 25 μm.
Discussion
In this study, we investigated the possibility that SOCS‐1 could be used in LC therapies. Previous reports showed that PC9 harbors a deletion mutation in EGFR and that A549 and LU65 cells possess wild‐type EGFR.34, 39 Although EGFR mutation in NSCLC was previously reported to activate AKT, MAPK, and STAT3 signaling,40 our research showed that STAT3 was more strongly expressed in A549 and LU65 than in PC9 cells, and that sensitivity to overexpression of SOCS‐1 was also higher in A549 and LU65 cells than in PC9 (Fig. 1). In addition, JAK inhibitor I significantly suppressed proliferation of A549 and LU65 cells, but not of PC9. Therefore, we consider that the marked antiproliferative effect by overexpression of SOCS‐1 on A549 and LU65 cells, but not on PC9, was attributable to the inhibition of JAK/STAT3 pathway in vitro. As SOCS‐1 also shows an antiproliferative effect in vivo (Fig. 6), AdSOCS1 gene therapy might be effective for patients with NSCLC, in which the JAK/STAT3 signaling pathway is constitutively activated. It has been reported that approximately 50% of NSCLC tumors showed elevated phosphorylation levels of STAT3 (Tyr705) by immunohistochemical analysis.4 There is a further possibility that LC patients harboring STAT3 dependence, detected by immunostaining analysis of phosphorylation levels of STAT3 (Tyr705) in specimens obtained surgically or bronchoscopically, could be selected for treatment with SOCS‐1 overexpression.
Comparative analyses of the antiproliferative effects of SOCS‐1 gene introduction and JAK inhibitor I treatment suggest that overexpression of SOCS‐1 may have a stronger effect than that of the JAK inhibitor I on A549 and LU65 cells (Fig. 2e). In fact, SOCS‐1, but not JAK inhibitor I, inhibited FAK and EGFR, which are important for the survival of A549 cells. In addition, the combined effect of FAK siRNA and JAK inhibitor I, or that of PD153035 and JAK inhibitor I, was superior to the antiproliferative effect of JAK inhibitor alone (Figs 3, 4). These findings suggest that the potent antiproliferative effect of SOCS‐1 depends not only on JAK inhibition but also on the suppression of other distinct signal transduction pathways, such as FAK and EGFR. In addition, phosphorylation of p53 at Ser15 was enhanced by the overexpression of the SOCS‐1 gene in A549 cells (Fig. 5). Because phosphorylation of p53 at Ser15 contributes to antitumor effects under certain experimental conditions,35, 36, 37, 38 and A549 cells express wild‐type p53,41 activation of p53 by SOCS‐1 overexpression seems to be involved in the antitumor effects.42
In conclusion, the findings of our study suggest that SOCS‐1 gene therapy is potentially effective for at least a subset of NSCLC both in vitro and in vivo. It was shown that SOCS‐1 had a potent antiproliferative effect on JAK‐dependent NSCLC cells by targeting the JAK/STAT3 pathway. In addition, SOCS‐1 successfully targeted many factors such as FAK, EGFR, and p53 in NSCLC cells. It is thus possible that SOCS‐1 gene therapy could have a unique advantage over JAK inhibitor for the treatment of NSCLC.
Further studies will be needed to elucidate the mechanism of JAK/STAT3 pathway‐dependence in NSCLC, and to validate the benefits that SOCS‐1 gene therapy could provide for NSCLC treatment in clinical practice.
Abbreviations
- AdSOCS‐1
adenovirus vector containing SOCS‐1
- EGFR
epidermal growth factor receptor
- FAK
focal adhesion kinase
- LC
lung cancer
- NSCLC
non‐small‐cell lung cancer
- SOCS
suppressor of cytokine signaling
- STAT
signal transducer and activator of transcription
- TYK
tyrosine kinase
Supporting information
Data S1. Methods.
Acknowledgments
This work was supported by a Grant‐in‐Aid from the Ministry of Health, Labour and Welfare, Japan (T. Naka) and a grant from the Kansai Biomedical Cluster Project in Saito, which is promoted by the Knowledge Cluster Initiative of the Ministry of Education, Culture, Sports, Science and Technology, Japan (T. Naka). The authors are grateful to Ms. M. Urase for experimental assistance and Ms. Y. Kanazawa and Ms. J. Yamagishi for secretarial assistance.
Disclosure Statement
The authors have no conflict of interest.
(Cancer Sci 2013; 104: 1483–1491)
References
- 1. Hattori M, Fujita M, Ito Y, Ioka A, Katanoda K, Nakamura Y. Use of a population‐based cancer registry to calculate twenty‐year trends in cancer incidence and mortality in Fukui Prefecture. J Epidemiol 2010; 20: 244–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goffin J, Lacchetti C, Ellis PM, Ung YC, Evans WK. First‐line systemic chemotherapy in the treatment of advanced non‐small cell lung cancer: a systematic review. J Thorac Oncol 2010; 5: 260–74. [DOI] [PubMed] [Google Scholar]
- 3. Rossi A, Di Maio M, Chiodini P et al Carboplatin‐ or cisplatin‐based chemotherapy in first‐line treatment of small‐cell lung cancer: the COCIS meta‐analysis of individual patient data. J Clin Oncol 2012; 30: 1692–8. [DOI] [PubMed] [Google Scholar]
- 4. Gao SP, Mark KG, Leslie K et al Mutations in the EGFR kinase domain mediate STAT3 activation via IL‐6 production in human lung adenocarcinomas. J Clin Invest 2007; 117: 3846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lai SY, Johnson FM. Defining the role of the JAK‐STAT pathway in head and neck and thoracic malignancies: implications for future therapeutic approaches. Drug Resist Updat 2010; 13: 67–78. [DOI] [PubMed] [Google Scholar]
- 6. Song L, Rawal B, Nemeth JA, Haura EB. JAK1 activates STAT3 activity in non‐small‐cell lung cancer cells and IL‐6 neutralizing antibodies can suppress JAK1‐STAT3 signaling. Mol Cancer Ther 2011; 10: 481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin‐6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev 2012; 38: 904–10. [DOI] [PubMed] [Google Scholar]
- 8. Jiang R, Jin Z, Liu Z, Sun L, Wang L, Li K. Correlation of activated STAT3 expression with clinicopathologic features in lung adenocarcinoma and squamous cell carcinoma. Mol Diagn Ther 2011; 15: 347–52. [DOI] [PubMed] [Google Scholar]
- 9. Verstovsek S, Kantarjian H, Mesa RA et al Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 2010; 363: 1117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Naka T, Narazaki M, Hirata M et al Structure and function of a new STAT‐induced STAT inhibitor. Nature 1997; 387: 924–9. [DOI] [PubMed] [Google Scholar]
- 11. Starr R, Willson TA, Viney EM et al A family of cytokine‐inducible inhibitors of signalling. Nature 1997; 387: 917–21. [DOI] [PubMed] [Google Scholar]
- 12. Endo TA, Masuhara M, Yokouchi M et al A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997; 387: 921–4. [DOI] [PubMed] [Google Scholar]
- 13. Piessevaux J, Lavens D, Peelman F, Tavernier J. The many faces of the SOCS box. Cytokine Growth Factor Rev 2008; 19: 371–81. [DOI] [PubMed] [Google Scholar]
- 14. Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. SOCS, Inflammation, and Autoimmunity. Front Immunol 2012; 3: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Narazaki M, Fujimoto M, Matsumoto T et al Three distinct domains of SSI‐1/SOCS‐1/JAB protein are required for its suppression of interleukin 6 signaling. Proc Natl Acad Sci USA 1998; 95: 13130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yasukawa H, Misawa H, Sakamoto H et al The JAK‐binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J 1999; 18: 1309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valentino L, Pierre J. JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol 2006; 71: 713–21. [DOI] [PubMed] [Google Scholar]
- 18. Naka T, Fujimoto M, Tsutsui H, Yoshimura A. Negative regulation of cytokine and TLR signalings by SOCS and others. Adv Immunol 2005; 87: 61–122. [DOI] [PubMed] [Google Scholar]
- 19. Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007; 7: 454–65. [DOI] [PubMed] [Google Scholar]
- 20. Zhang J, Li H, Yu JP, Wang SE, Ren XB. Role of SOCS1 in tumor progression and therapeutic application. Int J Cancer 2012; 130: 1971–80. [DOI] [PubMed] [Google Scholar]
- 21. Iwahori K, Serada S, Fujimoto M et al Overexpression of SOCS3 exhibits preclinical antitumor activity against malignant pleural mesothelioma. Int J Cancer 2011; 129: 1005–17. [DOI] [PubMed] [Google Scholar]
- 22. Souma Y, Nishida T, Serada S et al Antiproliferative effect of SOCS‐1 through the suppression of STAT3 and p38 MAPK activation in gastric cancer cells. Int J Cancer 2012; 131: 1287–96. [DOI] [PubMed] [Google Scholar]
- 23. Iwahori K, Serada S, Fujimoto M et al SOCS‐1 gene delivery cooperates with cisplatin plus pemetrexed to exhibit preclinical antitumor activity against malignant pleural mesothelioma. Int J Cancer 2013; 132: 459–71. [DOI] [PubMed] [Google Scholar]
- 24. Baltayiannis G, Baltayiannis N, Tsianos EV. Suppressors of cytokine signaling as tumor repressors. Silencing of SOCS3 facilitates tumor formation and growth in lung and liver. J BUON 2008; 13: 263–5. [PubMed] [Google Scholar]
- 25. Zhang S, Guo D, Jiang L, Zhang Q, Qiu X, Wang E. SOCS3 inhibiting migration of A549 cells correlates with PYK2 signaling in vitro. BMC Cancer 2008; 8: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin YC, Lin CK, Tsai YH et al Adenovirus‐mediated SOCS3 gene transfer inhibits the growth and enhances the radiosensitivity of human non‐small cell lung cancer cells. Oncol Rep 2010; 24: 1605–12. [DOI] [PubMed] [Google Scholar]
- 27. Mizuguchi H, Kay MA. A simple method for constructing E1‐ and E1/E4‐deleted recombinant adenoviral vectors. Hum Gene Ther 1999; 10: 2013–7. [DOI] [PubMed] [Google Scholar]
- 28. Sakurai H, Tashiro K, Kawabata K et al Adenoviral expression of suppressor of cytokine signaling‐1 reduces adenovirus vector‐induced innate immune responses. J Immunol 2008; 180: 4931–8. [DOI] [PubMed] [Google Scholar]
- 29. Verma A, Kambhampati S, Parmar S, Platanias LC. Jak family of kinases in cancer. Cancer Metastasis Rev 2003; 22: 423–34. [DOI] [PubMed] [Google Scholar]
- 30. Schmidmaier R, Baumann P. ANTI‐ADHESION evolves to a promising therapeutic concept in oncology. Curr Med Chem 2008; 15: 978–90. [DOI] [PubMed] [Google Scholar]
- 31. Liu E, Cote JF, Vuori K. Negative regulation of FAK signaling by SOCS proteins. EMBO J 2003; 22: 5036–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kornberg LJ, Grant MB. Adenoviruses increase endothelial cell proliferation, migration, and tube formation: partial reversal by the focal adhesion kinase inhibitor, FRNK. Microvasc Res 2007; 73: 157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quesnelle KM, Boehm AL, Grandis JR. STAT‐mediated EGFR signaling in cancer. J Cell Biochem 2007; 102: 311–9. [DOI] [PubMed] [Google Scholar]
- 34. Zhang D, Takigawa N, Ochi N et al Detection of the EGFR mutation in exhaled breath condensate from a heavy smoker with squamous cell carcinoma of the lung. Lung Cancer 2011; 73: 379–80. [DOI] [PubMed] [Google Scholar]
- 35. Lai JM, Chang JT, Wen CL, Hsu SL. Emodin induces a reactive oxygen species‐dependent and ATM‐p53‐Bax mediated cytotoxicity in lung cancer cells. Eur J Pharmacol 2009; 623: 1–9. [DOI] [PubMed] [Google Scholar]
- 36. Amin AR, Wang D, Zhang H et al Enhanced anti‐tumor activity by the combination of the natural compounds (‐)‐epigallocatechin‐3‐gallate and luteolin: potential role of p53. J Biol Chem 2010; 285: 34557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oh HL, Lee DK, Lim H, Lee CH. HY253, a novel decahydrofluorene analog, from Aralia continentalis, induces cell cycle arrest at the G1 phase and cytochrome c‐mediated apoptosis in human lung cancer A549 cells. J Ethnopharmacol 2010; 129: 135–9. [DOI] [PubMed] [Google Scholar]
- 38. Yamada C, Ozaki T, Ando K et al RUNX3 modulates DNA damage‐mediated phosphorylation of tumor suppressor p53 at Ser‐15 and acts as a co‐activator for p53. J Biol Chem 2010; 285: 16693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagai Y, Miyazawa H, Huqun et al Genetic heterogeneity of the epidermal growth factor receptor in non‐small cell lung cancer cell lines revealed by a rapid and sensitive detection system, the peptide nucleic acid‐locked nucleic acid PCR clamp. Cancer Res 2005; 65: 7276–82. [DOI] [PubMed] [Google Scholar]
- 40. Zimmer S, Kahl P, Buhl TM et al Epidermal growth factor receptor mutations in non‐small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling. J Cancer Res Clin Oncol 2009; 135: 723–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kashii T, Mizushima Y, Monno S, Nakagawa K, Kobayashi M. Gene analysis of K‐, H‐ras, p53, and retinoblastoma susceptibility genes in human lung cancer cell lines by the polymerase chain reaction/single‐strand conformation polymorphism method. J Cancer Res Clin Oncol 1994; 120: 143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mallette FA, Calabrese V, Ilangumaran S, Ferbeyre G. SOCS1, a novel interaction partner of p53 controlling oncogene‐induced senescence. Aging (Albany NY) 2010; 2: 445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Methods.