

Abstract
Decellularized extracellular matrix (dECM) is a promising biomaterial for repairing cardiovascular tissue, as dECM most effectively captures the complex array of proteins, glycosaminoglycans, proteoglycans, and many other matrix components that are found in native tissue, providing ideal cues for regeneration and repair of damaged myocardium. dECM can be used in a variety of forms, such as solid scaffolds that maintain native matrix structure, or as soluble materials that can form injectable hydrogels for tissue repair. dECM has found recent success in many regeneration and repair therapies, such as for musculoskeletal, neural, and liver tissues. This review focuses on dECM in the context of cardiovascular applications, with variations in tissue and species sourcing, and specifically discusses advances in solid and soluble dECM development, in vitro studies, in vivo implementation, and clinical translation.
Keywords: cardiac patches, decellularized, extracellular matrices, injectable hydrogels
1. Introduction
Biomaterials, used either as a standalone therapy or with the incorporation of cellular or biological components, have the potential to improve function of the damaged myocardium by induction of endogenous repair or by replacement through tissue grafts.[1–4] The effectiveness of biomaterial therapy comes from the ability of the material to recapitulate healthy tissue mechanics and chemical cues that drive repair and regeneration. While synthetic materials such as polymers have been explored for cardiovascular regeneration, natural materials may more effectively capture tissue properties and induce repair, as seen with materials such as alginate and collagen moving toward clinical testing.[1–5] Biomaterials have also been evaluated with a variety of cell sources, such as cardiac progenitor cells (CPCs), embryonic stem cells (ESCs), and induced pluripotent stem cells (iPSCs), and have successfully improved cellular function, engraftment, and survival, which is otherwise limited in cell therapy.[2–5]
Decellularized extracellular matrix (dECM) is a promising biomaterial for repairing cardiovascular tissue, as dECM most effectively captures the complex array of proteins, glycosaminoglycans (GAGs), proteoglycans, and many other matrix components that are found in native tissue, providing ideal cues for regeneration, repair, and remodeling of damaged myocardium.[2–4,6] dECM has found success in many tissue regeneration and repair contexts, such as for musculoskeletal, neural, and liver tissues.[2–4] dECM has been used in a variety of forms for cardiac therapy, described broadly as either solid scaffolds that maintain native vasculature structure or soluble materials that can form injectable hydrogels for tissue repair, summarized in Figure 1.[2,3,7]
Figure 1.
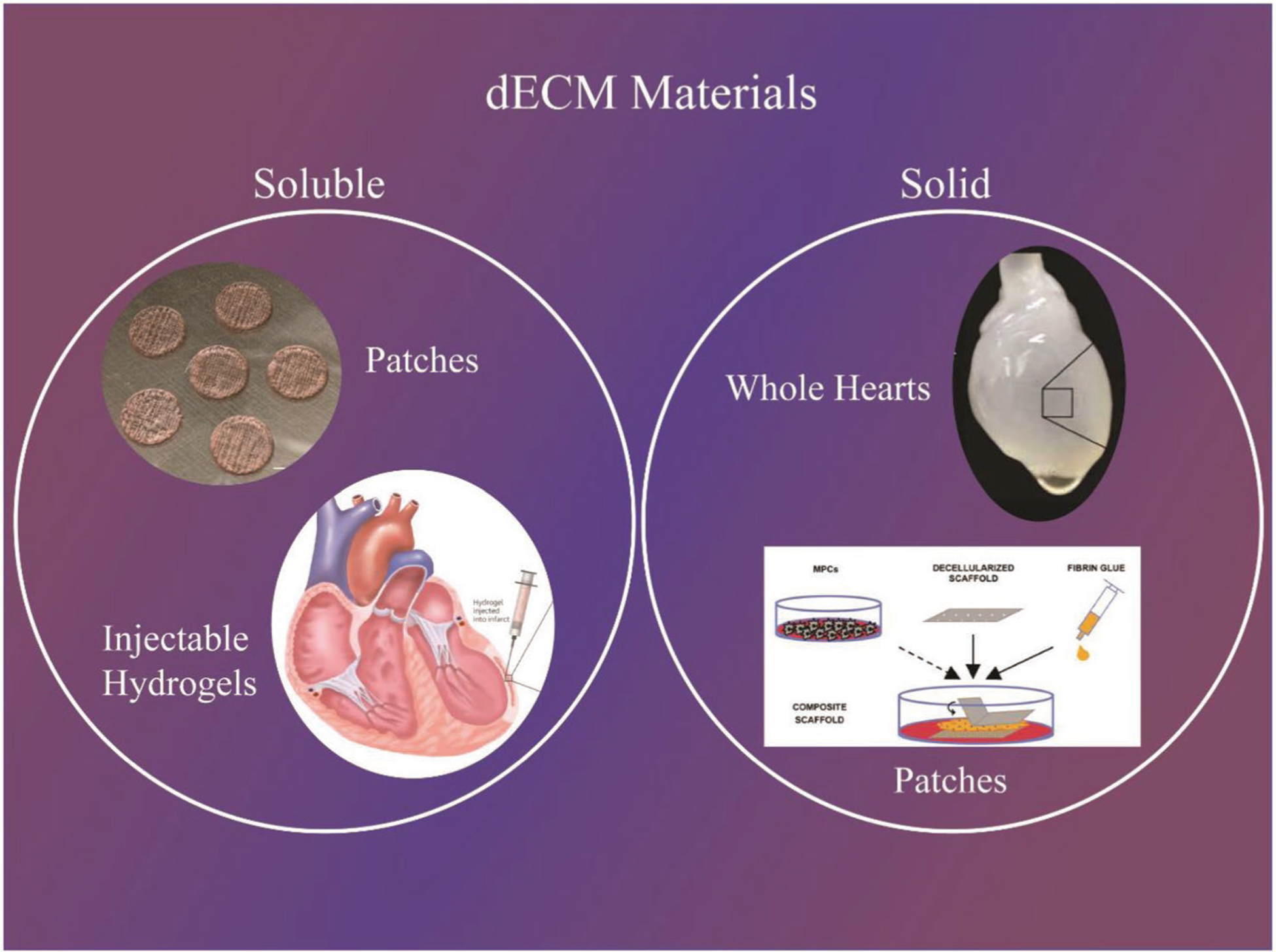
Main terminology used to differentiation dECM technologies. Examples: (Top left - patches) Reproduced with permission.[103] Copyright 2008, Springer Nature. (Bottom left - injectable hydrogels) Reproduced with permission.[131] Copyright 2011, National Academy of Sciences. (Top right - whole hearts) Reproduced with permission.[37] Copyright 2018, Wiley-VCH. (Bottom right - patches) Reproduced with permission.[73] Copyright 2016, Elsevier.
Solid dECM are scaffolds that have been decellularized and used as a biomaterial directly without further breakdown in the dECM microstructure. Solid scaffolds can be categorized based on application, which includes tissue engineered dECM patches/sheets and whole hearts. Soluble dECM are materials that have been decellularized followed by additional steps to break down the ECM structure and solubilize the ECM into a liquid form. Soluble materials can be categorized based on method of reconstitution or application, which includes injectable hydrogels, 2D and 3D hydrogels, and combinatorial patches composed of dECM and additional biomaterials.
This review will focus on dECM in the context of cardiovascular applications, with variations in tissue and species sourcing, and will specifically discuss advances in solid and soluble dECM development, in vitro studies, in vivo implementation, and clinical translation.
2. Structure and Function of Native Cardiac Extracellular Matrix
2.1. cECM Chemical, Physical, and Mechanical Properties
Organs and tissues are the combinations of multiple cell types and biochemical components such as soluble factors and ECM proteins.[6–14] The ECM is critical in establishing tissue structure, transducing signals from cells to cells or from the ECM to cells, and maintaining homeostasis.[7] In most tissues and in the heart, the ECM is composed of a complex combination of proteins, sugars, and soluble factors.[9–14] Quantification of decellularized adult human cECM by Johnson et al., among others, has determined that ≈70% of human cECM is composed of fibrillar collagens, mainly collagen types I and V.[10] The basement membrane comprises 20% of human cECM, composed mainly of collagen IV, but with additional proteins such as laminin, agrin, perlecan, and nidogen. Structural ECM comprises 4% of cECM, which include mainly proteoglycans such as biglycan and decorin, and fibrous glycoproteins such as fibrillin 1, all of which play a key role in secondary structural support and induction of intracellular signaling. Matricellular components compose roughly 3% of cECM, including collagen VI, fibronectin, dermatopontin, emilin 1, fibulin 5, lumican, periostin, prolargin, and thrombospondin 2. cECM is a complex system of matrix components that play a role in cardiac function, with the relative concentrations of matrix components within cECM being highly conserved and regulated in homeostasis.
The cECM components found in cardiac tissue are organized in a specific manor to support cells and maintain tissue function.[12–14] In healthy adult cardiac tissue, the ECM is organized as a heterogeneous structure, with areas of formed collagen fibers, basement membrane, and large spaces where clusters of cardiomyocytes (CMs) reside, seen in Figure 2.[14] While uninterrupted interactions and cell junctions between CMs are critical for contraction, the ECM also dictates CM contractility and endothelium-CM coupling.[15] These requirements result in ECM components surrounding mature CMs, seen by intertwined collagen nanofibrils in an organized manor with CM alignment. Cells also rest on homogeneous and matured basement membranes with honeycomb-shaped areas of laminin and site-specific collagen IV. The basement membranes of cECM show complete assembly of laminin and collagen IV in thick microfibers, possibly due to the basement membrane forming around vasculature. While the mechanical properties of both solid and soluble dECM are much different than native myocardium, the mechanical modulus of healthy myocardium has been measured as anywhere from 3 to 100 kPa, based on method of analysis and location of measurement within the myocardium itself.[16–20] This heterogeneity in tissue mechanics is seen within dECM as well and studies are needed to better understand how changes in various ECM and cellular components, such as collagen and titin, modulate myocardial stiffness.[21,22]
Figure 2.
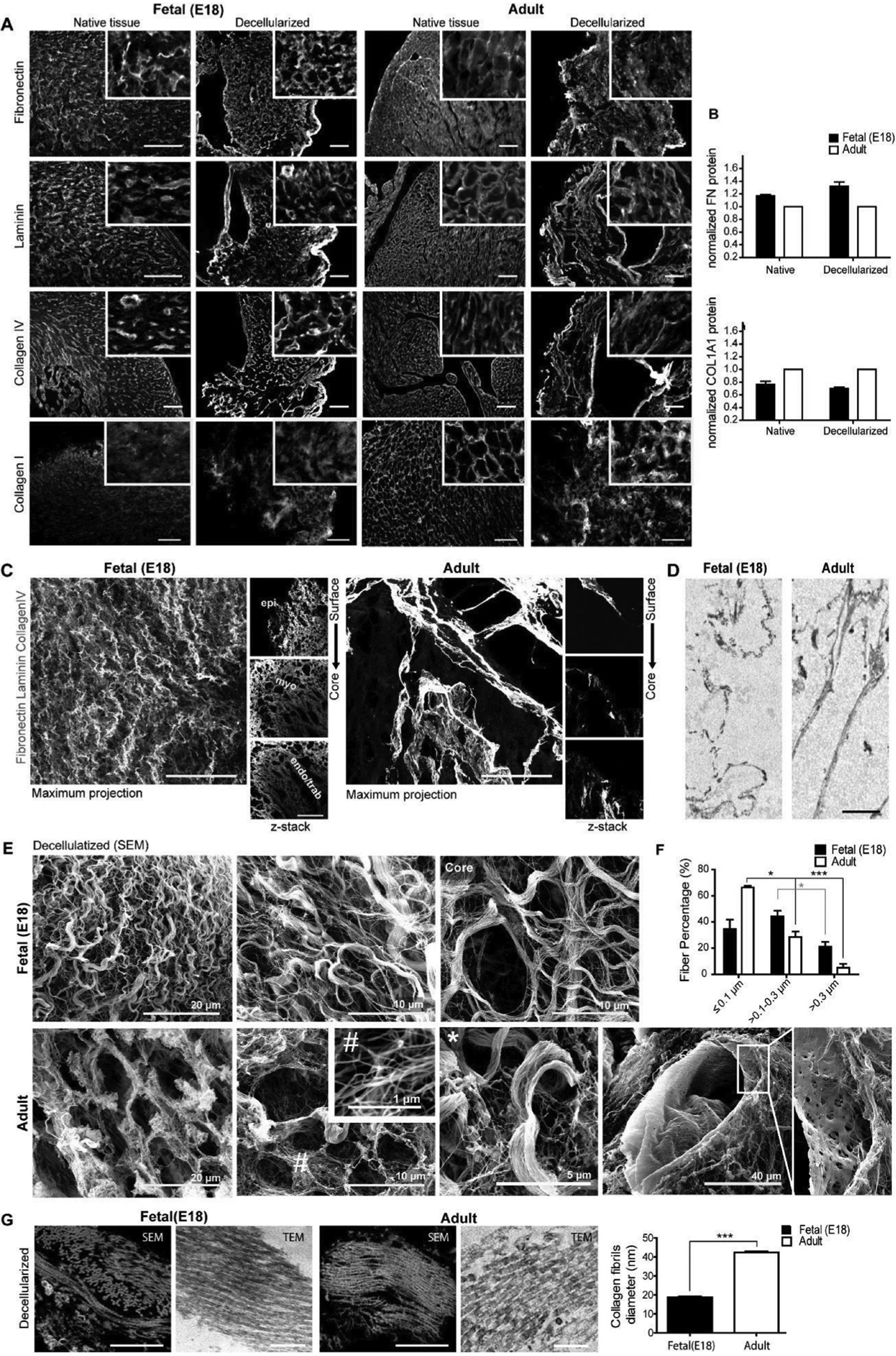
Biochemical and structural characterization of fetal (E18) and adult bioscaffolds. A) Side-by-side comparison of immunofluorescence for main ECM components before and after decellularization. Scale bar, 100 μm. B) Quantification of fibronectin and mature collagen type I by Western blot. C,D) Detail of the basement membrane and pericellular matrix obtained following C) whole mount immunostaining and optical clearing and by D) TEM. Scale bar: C) 50 μm and D) 2 μm. E) Representative images of fetal (E18) and adult decellularized scaffolds surface by SEM. Scale bars are identified in the image. # nanofibrils (<0.1 μm), *thick microfibers (>0.3 μm). F) Bar graph shows the percentage of fibrils with different diameters following quantification on SEM images. G) Representative images of collagen fibers obtained by SEM (left) and TEM (right) on tissue ultrathin sections. Bar graph shows the mean diameter of fibril units. Scale bar: 1 μm. Representative images and quantitative data were obtained from two or three (Western blot) independent experiments. Data are expressed as mean ± SEM. Student’s t-test, two-tailed, *p < 0.05, **p < 0.01, ***p < 0.001. Reproduced with permission.[14] Copyright 2016, Elsevier.
2.2. Changes in cECM due to Age, Disease, and Damage
cECM undergoes both physiological and pathological changes driven by cells and matrix remodeling enzymes such as matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). As cardiac tissue forms during development, cECM composition changes drastically as cells differentiate from proliferative, immature CPCs to mature, contractile CMs.[12] There are substantial differences in macrostructure and biochemical composition between fetal, neonatal, and adult cECM, which has been investigated by Silva et al. and Williams et al. through tissue decellularization, summarized in Table 1.[13,14,23] Fibronectin and periostin substantially decreased in aged compared to young cECM, as both proteins drive expansion and differentiation of neonatal CMs.[13] Components such as collagen IV, collagen VI, fibrillin 1, and perlecan also decreased in abundance with age, but not as substantially as fibronectin and periostin. Emulin-1 and fibrillin 2 were only found in fetal and neonatal cECM, while collagen III and V were only found in adult cECM. Collagen I and laminin were substantially increased in older compared to younger cECM, indicating the formation of a supporting system for mature CMs. The differences in ECM with aging is further demonstrated when comparing the structural changes between collagen IV, laminin, fibronectin, and collagen I in fetal and adult decellularized hearts.[14] Fetal cECM had extensive fibronectin in fibrillar arrangements, while adult cECM had less fibronectin with discrete distributions. Fetal cECM was composed a loose meshwork with a thin and irregular basement membrane, while adult cECM had thick basement membranes with complete assembly of laminin and collagen IV. While the overall fiber percent in both tissues was similar, fetal scaffolds mainly consisted of nanofibers and slender microfibers, while adult cECM had intertwined nanofibrils where mature CMs resided. These changes in biochemical profiles and structural expression represent changes to cardiac function and growth related to modulation of cellular phenotype and expression. Aging seems to represent the production of mature tissue and formation of stiff collagenous matrix with mature basement membrane. The change in matrix may be a factor in the inability for aging myocardium to repair, with stiffer components in aged myocardium and lack of factors such as fibronectin or periostin, which would otherwise push CMs toward a proliferative phenotype.
Table 1.
Differences in cECM due to age and disease.
| Condition | ECM composition | Structural properties | References |
|---|---|---|---|
| Adult (healthy) | High collagen I and laminin Uniquely contains collagen III and collagen V | Intertwined nanofibrils and thick microfibers Thick/mature basement membrane Complete laminin/collagen IV assembly in honeycomb-shape Discrete distribution of fibronectin | [12–14] |
| Fetal/neonatal | High fibronectin, periostin, collagen IV, collagen VI, fibrillin 1, perlecan Uniquely contains emulin-1, fibrillin 2 | Loose meshwork of nanofibers and slender microfibers Thin/irregular basement membrane Fibrillar fibronectin | [13,14,23] |
| Post-MI | Increase in collagen overall Initial increase in collagen IV and periostin Decrease in collagen IX | Change in stiffness (possible decrease) Increased fiber alignment Decreased crosslinking | [22,31] |
There are extensive changes to cECM after ischemia or damage.[11,15,24,25] The balance of normal cECM turnover in the heart is disrupted with disease and causes accumulation of collagen (fibrosis), which impairs cardiac function, increases myocardial stiffness, and drives heart failure.[26] ECM regulatory factors, growth factors (GFs) such as tumor necrosis factor-α (TNF-α) and transforming growth factor-β (TGF-β), and cells such as myofibroblasts drive ECM changes and hinder cardiac function in pathological situations. Fibroblasts and myofibroblasts become hyperactive and excessively deposit ECM with reduced production of MMPs and increased production of TIMPs, resulting in less ECM turnover.[27–29] Cardiovascular disease also affects other heart cells, including CMs, endothelial cells (ECs), CPCs, smooth muscle cells (SMCs), and transient and tissue-resident immune cells.[30] Fibroblasts dynamically interact with these cells, by modulating the ECM itself, as well as through cell-cell coupling and paracrine effects.
Studies by Sullivan et al. and Quinn et al. on solid decellularized cECM have attempted to elucidate the post-MI changes on the biochemical and macrostructure level, comparing healthy cECM to cECM after 1–8 weeks following infarction, summarized in Table 1.[22,31] Although stiffness changes to decellularized myocardium after infarction seems to have conflicting values based on measurement technique used, optical measurements show a decrease in stiffness postinfarct, which may be more supported since the methodology overcomes errors in mechanical measurements by nondestructively evaluating tissue stiffness.[22,31] Collagen content was significantly increased at 4 and 8 weeks compared to the healthy cECM. The infarcted cECM showed an increase in fiber alignment and decrease in crosslinking, which supports a decrease in myocardial stiffness and indicates that crosslinking plays a key role in stiffness along with total collagen content following infarction. Collagen III, collagen V, collagen VI, fibronectin, laminin, and elastin content did not change from healthy to 8 weeks after infarction. Collagen IV seemed to increase between 1 week to 2 weeks after infarct, then decreased again at 4 and 8 weeks, possibly due to initial upregulation of basement membrane formation that was reduced as the tissue remodeled. Collagen XV decreased significantly in the infarcted cECM at all time points compared to the healthy cECM. Finally, periostin content increased 1 week after infarct, but then decreased to preinfarct levels after 4 and 8 weeks, possibly indicating that the tissue was in a reparative state after infarction, although further studies are necessary. Studies on decellularized cECM show a substantial change in cECM mechanics and composition following infarction, pointing toward pathological changes that result in hindrance to cardiac function, which may be remedied with replacement or addition of healthy dECM.
2.3. Properties and Function of cECM Compared to dECM from Secondary and Nonhuman Sources
In addition to cECM, other types of dECM from different sources have been used for heart-based applications, summarized in Table 2.[3] The sourcing of tissue can be separated based on species and tissue (primary cardiac or secondary tissue) variance. Decellularized human skin, porcine SIS, and porcine urinary bladder matrix (UBM) were investigated in the late 2000s, with focus on restoring cardiac function through use of solid dECM, modulated with GFs, stem cells, or additional polymers.[32–35] Since 2008, methods to directly decellularize heart tissue have shifted dECM research and applications toward using heart-specific dECM.[36,37] Although there have been recent advances using various dECM sources for cardiovascular engineering, the question remains as to the ideal sourcing for dECM-based therapies for cardiovascular tissue. Secondary dECM sources such as SIS and UBM may be easier to obtain and process than cECM, with improved batch-to-batch variability for clinical application, as seen in commercial products such as MatriStem and AlloDerm.[38] Primary cardiac-derived dECM, both as cECM and pECM, may have a tissue-specific biochemical profile and structure composition that induces more effective cardiac repair compared to secondary dECM, where batch to batch variability is improving as these therapies move toward the clinic.[39]
Table 2.
Comparison of various dECM versus cECM.
| Form | Tissue | Species | Differences compared to cECM | References |
|---|---|---|---|---|
| Solid sheet | Skeletal | Murine | Similar fiber structure | [40] |
| Liver | Murine | Lower fibrilin-1, microfibrillar-associated protein 2/5 Higher collagen II, arginase-1 Decreased ESC cardiac commitment | [41] | |
| Pericardium | cECM—porcine pECM—human |
Similar microstructure Larger pore size Better cell infiltration pECM expressed 25 unique components cECM expressed 14 unique components |
[42] | |
| Hydrogel | Adipose | Porcine | No change in cardiac and fibroblast commitment of CPCs compared to collagen | [43] |
| Skeletal | Porcine | sECM uniquely expressed heparin sulfate, decorin cECM uniquely expressed collagen IV, elastin, fibrinogen, fibrillin-1 | [44] | |
| Lung | Porcine | lECM uniquely expressed collagen II and collagen IX cECM uniquely expressed collagen VII, fibrinogen, heparan sulfate | [47] | |
| Milled powder within fibrin | Liver | Murine | Promoted in vivo fibroblast migration, neovascularization, cell infiltration | [46] |
The review of the numerous sources and implementations of dECM solid scaffolds until 2009 by Badylak et al. shows that many companies have attempted to commercialize dECM as solid dry or hydrated sheets, generated through natural processing or crosslinking methods.[38] An example of dECM that moved toward clinical use is bovine pericardium-derived heart valves, which are often crosslinked to improve mechanical functionality as valves. The implementation of solid dECM for myocardial repair has been limited, and although clinical functionality is being assessed, most companies have moved forward with valve, vasculature, or pericardial patches rather than myocardial replacement or repair. Direct comparisons of differences in composition and function between tissue source, species, and decellularization method of dECM for cardiovascular applications have not been explored and recent solid dECM studies have mainly focused on whole heart decellularization and recellularization. Since SIS, UBM, and other tissues are commercially available, their applications may be best compared in clinical systems.[38] Hong et al. decellularized murine cECM and skeletal ECM (sECM) solid scaffolds and found that both cECM and sECM had similar fiber structure after decellularization.[40] Higuchi et al. compared solid scaffolds of cECM and liver dECM in terms of their relative protein concentrations and cellular responses to ESCs.[41] cECM had lower collagen II and arginase-1 content and higher fibrillin-1, microfibrillar-associated protein 2, and microfibrillar-associated protein 5 than liver dECM. ESCs showed higher cardiac differentiation when cultured on cECM compared to liver dECM. Perea-Gil et al. compared two decellularized scaffolds based on either porcine cECM or human pECM repopulated with adipose tissue-derived mesenchymal stem cells (MSCs).[42] The general structure and mechanical properties of the two grafts were preserved after decellularization and recellularization, although the decellularized cECM was stiffer than the native myocardium. The decellularized pECM showed much higher expression of major ECM components, better cell infiltration and retention, and larger pore size. The study evaluated the protein expression of both scaffolds, with 14 distinct components found in the human cECM (such as fibrillin-2 and nidogen-2) and 25 distinct components found in porcine pECM (such as galectin-1, biglycan, and GFs). It is important to mention that since sourcing of both tissue and species source were variables in the study, it was difficult to determine which factor influenced the cellular effects.
Soluble porcine cECM has moved toward translation, with clinical trials currently ongoing in post-MI patients (NCT02305602). While human, rat, and goat myocardium, human and ovine pericardium, and porcine omentum have undergone in vitro analysis and/or histological assessment for soluble hydrogels, they are the least therapeutically evaluated using in vivo models.[3] Human placenta, porcine SIS, and porcine pECM have undergone functional studies, but may be lacking in vitro analysis and/or biocompatibility and histological assessment.[3] An analysis of the general soluble dECM hydrogels toward in vivo implementation is seen in the review by Spang and Christman.[3] There are, however, limited studies as to the differences in effectiveness of these soluble dECM toward heart repair. French et al. showed that CPCs grown on cECM significantly increased proliferation and expression of cardiac markers and decreased expression of fibroblast markers, compared to CPCs grown on collagen or adipose dECM.[43] These results suggest that tissue-specific cECM may drive CM differentiation of CPCs more effectively. DeQuach et al. characterized differences in the biochemical composition of sECM and cECM through mass spectrometry.[44] While there was overlap in component expression, such as collagen I and V, fibrinogen, and fibrillin-1, there were marked differences, with cECM expressing collagen IV, elastin, fibronectin, and laminin exclusively, while sECM expressed several unique collagens, heparin sulfate, and decorin. Ungerleider et al. showed that similar methods can be used to develop soluble porcine sECM and cECM, although the difference in protein expression was not qualified.[45] Tabuchi et al. compared rat liver dECM and cECM milled powders within injectable fibrin material for the treatment of acute myocardial infarction (MI) in rats.[46] They found that both dECM powders, especially liver dECM, promoted fibroblast migration into the materials in vitro. While both dECM powders induced neovascularization in the infarct area and cell infiltration into the materials in vivo, the liver powder was more effective, although there was limited direct comparisons on dECM composition and function. Merna et al. grew heart and lung fibroblasts on cECM and lung ECM (lECM) and found that fibroblast source and integrin expression were more important than dECM source in myofibroblast differentiation potential.[47] They also characterized the differences in dECM composition, where lECM expressed collagen II and IX exclusively and cECM expressed collagen VII, fibrinogen, and heparan sulfate proteoglycan. Overall, tissue quantification of dECM scaffolds compared to cECM seem to be contradictory, and more sophisticated techniques such as proteomics are required to better elucidate differences.
Both tissue and species variances may be important characteristics of dECM that drive effective regeneration and repair. Johnson et al. investigated the differences between human and porcine sourcing of cECM. Both soluble cECM materials could gel and spread in vivo after injection.[48] In terms of biochemical composition, both matrices were similar, where porcine cECM had a higher sulfated GAG (sGAG) content, while human cECM had components such as periostin, fibulin-2, and differential collagens. Most importantly, generating human cECM was problematic due to difficulty in obtaining healthy human myocardium, patient-to-patient variability in composition due to age, and increased difficulty in processing human tissue. This patient variably was assessed in further studies by the same laboratory, which showed marked differences in pECM composition and mechanical properties between patients.[39] It seems that overall it is much more difficult to scale human cECM toward clinical application, while porcine cECM is easier to obtain and process, while being composed of similar components as human cECM.
While these studies have attempted to compare differences in tissue and species sourcing between cECM and other dECM through quantification of chemical composition or assessments in cellular responses, comparison between implementing differently sourced dECM in the treatment of cardiovascular diseases is critical in determining which sourcing allows for the most effective therapy.[38–48] The studies discussed in this section point toward diverse differences between dECM sources based on tissue or species source. It may turn out that the sourcing is not as important as the method of decellularization, functional modification, cell sourcing, or any other factor involved in dECM therapy. Alternatively, tissue-specific dECM may be the most critical factor in cardiac regeneration and repair. Regardless, progress toward the clinic is promising.
3. Solid dECM
This section focuses on solid dECM scaffolds, which are not solubilized into dECM powder or liquid, and are used directly after decellularization. This methodology preserves the native tissue structure and vasculature while expressing dECM components in their tissue-specific location. Therapies that use this method of solid decellularization focus on direct use of the material as a solid patch or attempt to recellularize the dECM toward functional tissue development.
3.1. Material Development
3.1.1. Decellularization Methods
The first whole-heart decellularization through a perfusion approach was implemented by Ott et al. in 2008, and since then, there have been many studies on decellularizing heart tissue to maintain vasculature, structural mechanics, and functional ECM components.[37] Perfusion decellularization is the most common approach as it minimizes ECM damage and leaves ECM structure and vasculature intact throughout the myocardium or whole heart.[2,4] Methods of removing cells through perfusion rely on chemical treatment, enzymatic reaction, or mechanical degradation, although no method has been found to be ideal due to difficulty in comparing tissues and sourcing.[2,49] The review by Taylor et al. explores advances in solid dECM manufacturing for all tissues, including a table of various decellularization methods for myocardial tissue.[2] Chemical decellularization involves using acidic or basic solutions, alcohols, or detergents to lyse and remove cells.[2,37,50–59] An example of this method is seen in Figure 3, as was used by Ott et al.[37] The methodology can be applied to sections of cECM, which can be sectioned into 3D patches before or after decellularization.[53,60,61] Enzymatic decellularization uses biological enzymes such as MMPs, proteases, or nucleases to interrupt cell-cell and cell-matrix attachments and lyse cells through membrane cleavage.[2] Enzymatic methods are often combined with chemical methods to allow for better cell removal before detergent implementation.[62,63] Physical methods rely on using freeze-thaw cycles, agitation, or application of pressure to damage cells and allow for cell removal, and are commonly used in conjunction with physical or enzymatic methods to improve decellularization.[2,38,64–68] Regardless of method, complete decellularization is readily available for applications of dECM materials in cardiac tissue engineering.[69–75]
Figure 3.
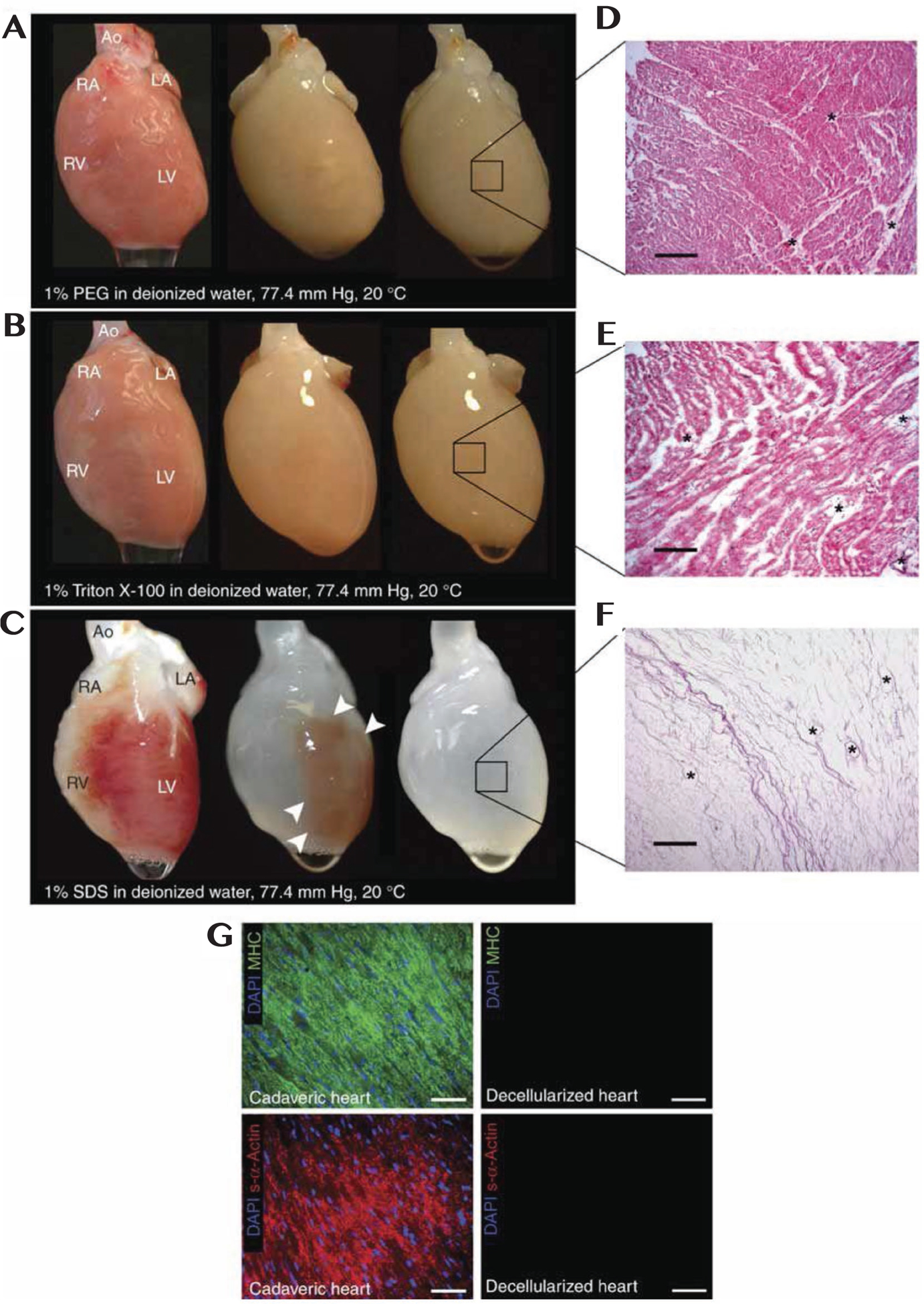
a–c) Photographs of cadaveric rat hearts mounted on a Langendorff apparatus. Ao, aorta; LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle. Retrograde perfusion of cadaveric rat heart using A) PEG, B) Triton-X-100 or C) SDS over 12 h. The heart becomes more translucent as cellular material is washed out from the right ventricle, then the atria and finally the left ventricle. d,e) Corresponding H&E staining of thin sections from LV of rat hearts perfused with D) PEG or E) Triton-X-100, showing incomplete decellularization. Hearts treated with PEG or Triton-X-100 retained nuclei and myofibers. Scale bars: 200 μm. F) H&E staining of thin section of SDS-treated heart showing no intact cells or nuclei. Scale bar: 200 μm. All three protocols maintain large vasculature conduits (black asterisks). G) Immunofluorescent staining of cadaveric and SDS-decellularized rat heart thin sections showing the presence or absence of DAPI-positive nuclei (purple), cardiac α-myosin heavy chain (green) or sarcomeric α-actin (red). Nuclei and contractile proteins were not detected in decellularized constructs. Scale bars: 50 μm. Reproduced with permission.[37] Copyright 2008, Springer Nature.
Recently, research toward creating controllable or analytic decellularization systems have improved overall decellularization of cardiac tissue. Momtahan et al. used automation of pressure during the decellularization process to effectively remove 98% of total DNA from a whole porcine heart using only 6 h of SDS treatment.[66] The system measured and controlled tissue pressure during decellularization to maintain pressure at physiological values, which reduced DNA content, but maintained collagen, elastin, and GAG composition, as well as compressive modulus compared to native tissue. Lee et al. used an inverted orientation of decellularization to help maintain integrity of thinner areas of the heart while effectively removing cells from the thickest components.[68] Compared to venting the heart apex during decellularization, the inversion method showed higher coronary perfusion efficiency, better cellular and DNA removal, higher collagen and elastin content in the aortic valve, and better shape retention. Seo et al. used a decellularization method based on supercritical CO2 and ethanol to preserve collagen, laminin, GAGs, fibronectin, and angiogenic GFs more effectively than detergent methods.[71] Merna et al. used multiphoton microscopy and image correlation spectroscopy to noninvasively characterize the mechanical and structural properties of whole hearts during decellularization.[72] They were able to use this system to compare combination and individual methods of decellularization, and found that only Triton-X decellularization preserved tissue modulus, collagen fiber density, and elastin fiber density.
3.1.2. Characterization Methods
Common characterization techniques used to quantify decellularization and effectiveness of different methods in generating solid dECM involve measuring cellular content, ECM organization/composition, and general macrostructures such as vasculature and pore size.[37,53,70] As the goal of decellularization is to remove cellular components to open scaffolds for healthy cell seeding and reduction of immune response, quantifying left-over cell debris is important. Cellular remains can be observed by using fluorescent immunostaining for CM markers or histology to evaluate overall cellular locations. It is also important to analyze the ECM composition after decellularization, since decellularization methods may damage ECM components and macrostructure. Fluorescent immunostaining and histology can be used to determine locations and densities of key ECM proteins such as collagen, while overall GAG concentration can be measured by assays that determine negative charges. Since GFs within the matrix are critical for modulating cardiac repair and detergents can release GFs during perfusions, the analysis of GF release through assays such as ELISA or Luminex is important.[65] Scanning electron microscopy (SEM) is effective in analyzing the microstructure of dECM to evaluate fiber size, density, and distribution, pore size, and variations in macrostructure throughout the heart. Finally, mechanical properties of dECM after decellularization can be determined through compressive or tensile strain measurements and transmural pressure analysis. These methods must be employed to ensure complete decellularization and preservation of ECM composition, structure, and mechanics.
3.2. Tissue Engineered Constructs
3.2.1. Tissue Engineered Patches Derived from Solid cECM
A goal of generating whole decellularized hearts is the ultimate recellularization of the hearts to produce functional off-the-shelf organs for transplantation into patients. Although many recent studies have attempted to recellularize whole hearts with a variety of cell sources and bioreactor setups, these studies are not the focus of this review and interested readers are recommended to read the recent review by Taylor et al.[2,49,76–80] Cardiac patches derived from sections of decellularized heart tissue may be just as effective as whole hearts in repairing damaged myocardium, without the need for full transplantation. Studies that focus on cardiac patches follow classic tissue engineering methods, where 3D scaffolds are generated from decellularized cECM followed by seeding with stem cells or differentiated cells generated from stem cells. A summary of these studies is seen in Table 3. Sánchez et al. seeded a combination of human MSCs, CPCs, HUVECS, and CMs on dECM scaffolds, with varying effects after 21 d based on cell type.[53] MSCs infiltrated the matrix without complete alignment, while CPCs stayed on the scaffold without infiltration. HUVECs grew around large-diameter vessels without angiogenesis, while CMs attached to the matrix and aligned within the fibers. This differential cellular propensity is seen in the work by Oberwallner et al., which used slices of human cECM, Matrigel, and Geltrex, and grew murine ESCs, iPSCs, and MSCs on the scaffolds.[81] Compared to the nonspecific ECM materials, the cECM supported proliferation and reduction of apoptosis in ESCs and iPSCs. They also saw cardiac commitment on cECM only from ESCs and iPSCs, while MSCs showed no CM differentiation. Garreta et al. compared CMs derived from iPSCs and human ESCs grown on cECM after 10 d in culture.[82] Compared to Matrigel, CMs showed expression of cardiac transcription factor Nkx2.5, Cx43, and TnT, improved conduction velocity, decreases in Ca2+ upstroke, and increases in ion channel formation.
Table 3.
Solid dECM scaffolds with cells.
| dECM source | Cell source | Findings | References |
|---|---|---|---|
| Human cECM | Human MSCs, CPCs, HUVECs, CMs | MSCs—infiltrated, no alignment CPCs—no infiltration HUVECs—grew around vessels CMs—attached in matrix, aligned with fibers |
[53] |
| Human iPSC-derived CMs, ESCs | Improved cardiac commitment, conduction velocity, decreased calcium upstroke, ion channel formation of CMs in cECM compared to Matrigel | [82] | |
| Human iPSC-derived CMs | Sarcomere formation, cell/matrix deformation, contractile force, electrical conduction | [52] | |
| Murine ESCs, iPSCs, MSCs | Improved proliferation, cardiac commitment of ESCs and iPSCs only compared to Matrigel/Geltrex | [81] | |
| Human cECM and fibrin hydrogel | Human MSCs | Improved angiogenic GF release, enhanced vascular formation in a murine infarction model | [73] |
| Porcine cECM treated with HA | Human MSCs, HUVECs | Thick scaffolds showed high cell density, survival, angiogenesis, vascular formation | [83] |
| Porcine SIS | Porcine MSCs | MSC addition reduced immune response following pericardial/epicardial implantation compared to SIS alone | [84] |
| Murine cECM | Human iPSC-derived CMs, iPSC-derived CD90+ cells | Improved cardiac commitment, higher myofilament width of cells in patches compared to CM aggregates | [61] |
| Murine cECM (Fetal) | Murine neonatal CMs and CPCs | Improved cell migration, cardiac commitment, paracrine release, adhesion in fetal compared to adult cECM | |
| Murine sECM | Murine ESCs and ESC-derived CMs | Adherence, survival, proliferation, electrical response, cardiac commitment of both cell types CMs formed gap junctions and synchronized contraction |
[40] |
| Mouse FDM | Rat CMs | Improved cardiac commitment compared to gelatin or fibronectin, with further improvement in cross-linked scaffolds | [75] |
| Human cardiac FDM | Human CDCs | Improved cardiac commitment, release of reparative factors on healthy versus infarct derived FDM | [86] |
The trend of thin solid dECM scaffolds modulating cell phenotype is further seen in models of thick patches and composite cell-biomaterial patches. Sarig et al. generated 1.7 mm thick cECM constructs, treated with hyaluronic acid (HA) to improve cell retention, and seeded with HUVECs and MSCs.[83] After 21 d of growth in a perfusion bioreactor, the patches showed high cell density, cell survival, angiogenesis, and vasculature formation. Godier-Furnémont et al. generated a composite cardiac patch composed of a cECM sheet and fibrin hydrogel loaded with preconditioned MSCs.[73] The cell delivery platform, along with changes in cECM structure based on tissue directionality, is seen in Figure 4.[73] The system allowed for codelivery of cECM and paracrine factor releasing MSCs, and showed increased release of angiogenic GFs and enhanced vascular network formation in an infarction model. The MSCs migrated into the ischemic, but not healthy, myocardium. This combination system represents a step forward in functional cardiac patches derived from solid cECM. Wang et al. seeded CMs differentiated form iPSCs and CD90+ cells onto rat cECM patches.[61] Compared to CM aggregates, cells grown on the patches had higher cardiac differentiation and myofilament width, although the effect of CD90+ cell addition was not separated quantitatively.
Figure 4.
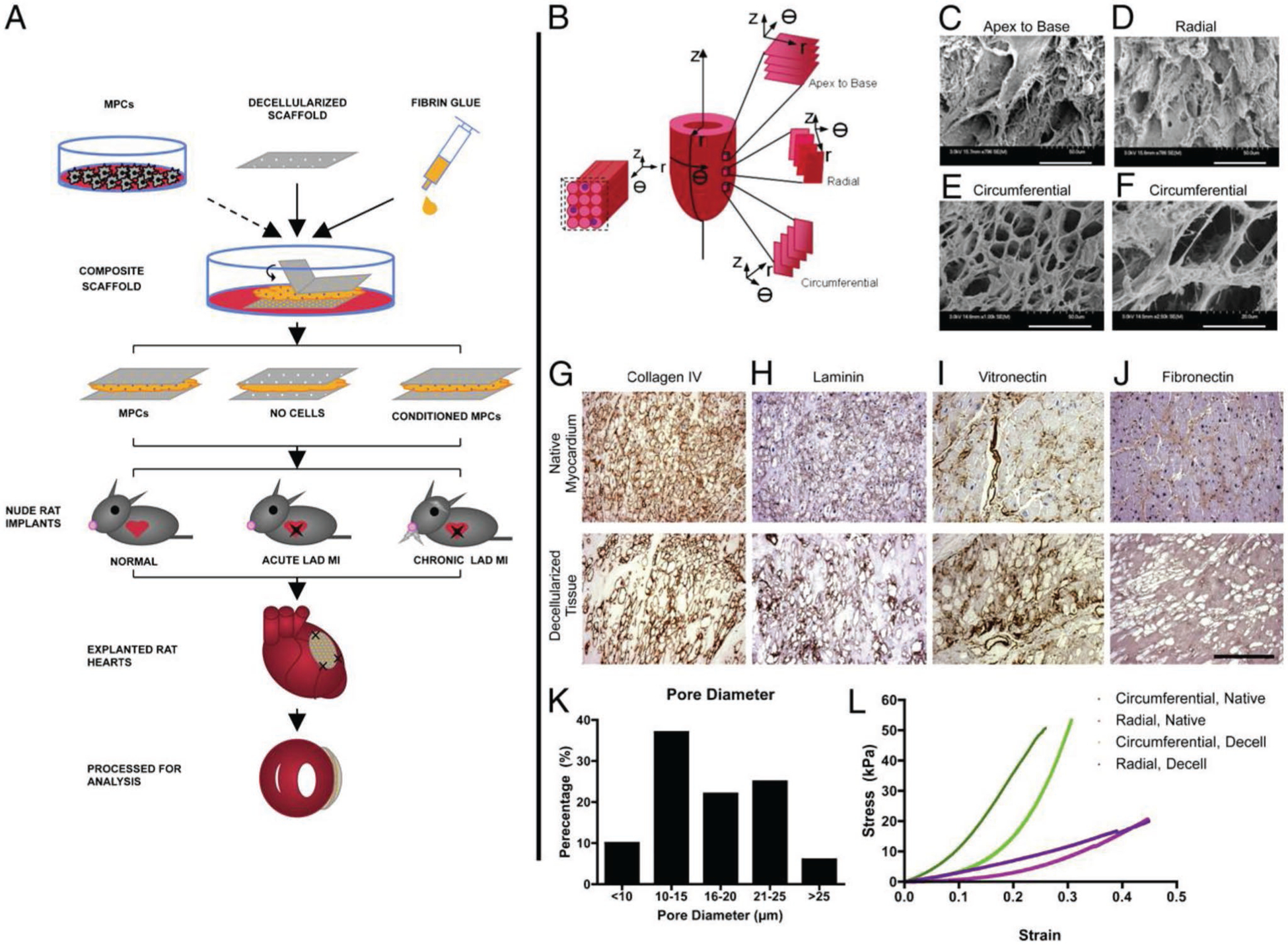
Cell delivery platform. A) Composite scaffolds assembled from thin sheets of decellularized human myocardium and fibrin hydrogel were seeded with MPCs, cultured in vitro with or without TGF-β conditioning, and implanted into nude rat models of acute and chronic ischemia. After 4 week, heart function was evaluated by echocardiography ex vivo analyses. B) The three axes of the heart (r, q, and z), the directions for sectioning (apex to base, circumferential, and radial), and the short axis of myocytes seen in circumferential sections. C–F) Scanning electron micrographs of decellularized scaffolds: apex to base sections (C; 796×), radial sections (D; 986×), and circumferential sections (E; 1000×; F, 2500 ×). Staining of extracellular matrix components circumferential sections of native and decellularized tissue: G) collagen IV, H) laminin, I) vitronectin, and J) fibronectin. (Scale bar: 250 μm.) K) Average pore diameters in decellularized scaffolds. L) Similar properties of native (circumferential, blue; radial, pink) and decellularized (circumferential, green; radial, purple) tissue measured in uniaxial tensile tests (tensile moduli calculated at 20% strain). Reproduced with permission.[73] Copyright 2011, National Academy of Sciences.
In developing in vitro analysis tools, Schwan et al. generated thin strips of cECM, which were mounted on chips that could be mechanically controlled.[60] They harvested different areas and fiber orientations of the myocardium through laser cutting to form the cECM strips. Seeded neonatal rat CMs on the chips beat together and developed length-dependent activation by day 21, where fiber orientation affected peak stress and production of brain natriuretic peptide (BNP), indicating that ECM orientation is critical in guiding cells and growing cardiac tissue in vitro. In a model of implementing human iPSC-derived CMs seeded on human cECM, Guyette et al. grew CMs in a bioreactor for 3 months and showed that cells formed sarcomere structure, cell and matrix deformation, contractile force, and electrical conduction.[52] Overall, the use of solid dECM scaffolds with cells may be a powerful tool in therapy for cardiac repair.
3.2.2. Differential Solid Scaffolds for Cardiovascular Tissue Engineering
Solid dECM derived from secondary tissue sources, such as SIS, sECM, fetal cECM, and FDM, have recently been investigated for cardiovascular applications. Chang et al. used SIS seeded with porcine MSCs in a porcine model to determine ideal seeding density and potential immunological effects.[84] After seeding cells, SIS structure, mechanics, and ECM composition were maintained, and MSCs remained viable. Following pericardial or epicardial implantation, MSC addition reduced T-cell response regardless of cell dose and implantation site location compared to SIS alone. Hong et al. investigated sECM recellularized with ESCs and ESC-derived CMs.[40] Both ESCs and CMs could adhere, survive, proliferate, and differentiate into CMs with electrically stimulated response. CMs also formed Cx43 gap junctions and generated synchronized contraction within 6 d of recellularization.
The age of cECM may also play a role in scaffold effectiveness for cardiac repair, as fetal and adult cECM have different structural and biochemical properties. Silva et al. evaluated growth of CPCs and neonatal CMs on fetal and adult cECM.[14] While both cell types migrated toward the center of scaffolds, more cells were adhered inside fetal scaffolds, and more CPCs relied on β1 integrin independent binding on fetal compared to adult cECM. CPCs were better able to express cardiogenic and reparative factors on fetal cECM, such as fibronectin, troponin C (TnC), vascular endothelial growth factor (VEGF), TGF-β, and periostin, while only collagen 1 expression was improved on adult cECM. These results point the way to potentially modulating the ECM composition of cECM derived from adult porcine or human sources, to induce better differentiation with fetal components.
In addition to tissue derived dECM, there have been studies on cardiac-specific dECM derived from cardiac fibroblasts grown in sheets, called fibroblast-derived matrix (FDM).[74,75,85,86] These scaffolds can be used as patches, either directly or with additional cell seeing. Schmuck et al. generated FDM be seeding rat fibroblasts at high density for 14 d, at which point the cells were removed.[74] The scaffolds contained over 80% fibronectin, 15% collagen, and a variety of other ECM proteins such as elastin, biglycan, and nidogen. Following MSC seeding onto the patches and implantation in a mouse infarct model for 2 d, the MSCs migrated out of the FDM and throughout the infarcted tissue. Although FDM chemical composition contains much more fibronectin compared to native cECM, FDM may play a comparable role to other dECM in treating cardiovascular disease. Suhaeri et al. evaluated the cardiogenic potential of FDM compared to gelatin or fibronectin.[75] At 7 d, cardiomyoblasts seeded on FDM had higher levels of myosin light chain 2 (Myl2), troponin T (TnT), and connexin 43 (Cx43) compared to cells grown on gelatin or fibronectin. In addition, by altering crosslinking of the FDM to increase stiffness, there were further increases in cardiogenic differentiation, with α5 integrin expression increased on crosslinked compared to non-crosslinked FDM. In a cardiac-specific method, Pagano et al. grew CPCs isolated as cardiosphere-derived cells (CDCs) on FDM derived from fibroblasts harvested from healthy and infarcted hearts.[86] After 7 d in culture, both systems showed expression of cardiac differentiation, although CDCs on healthy FDM secreted higher levels of reparative factors such as osteopontin, FGF, IGF, and TIMP-2, modeling the decrease in reparative and antiremodeling potential of CDCs when injected in an infarct environment. While this system was used as an in vitro model, application of a cardiac fibroblast derived FDM patch seeded with cardiac stem cells could be beneficial therapy in cardiac tissue engineering.
4. Soluble dECM
This section focuses on soluble dECM scaffolds derived from myocardium, pericardium or secondary sources such as SIS or omentum. These dECM materials have been decellularized, followed by additional steps to break down the ECM structure and solubilize the complex ECM material into liquid form. The solubilized dECM can form 2D and 3D hydrogels either in vitro or in vivo after injection into the myocardium. Studies using dECM for cardiac tissue engineering applications either focus on generating and modifying pure dECM for direct injection into damaged myocardium, or by combining dECM with cells and/or additional biomaterials to produce bioactive and cell-laden injectable gels or cardiac patches. Soluble dECM is much more versatile than solid dECM scaffolds, maintaining ECM composition but lacking structural and mechanical similarity to solid dECM scaffolds.
4.1. Material Development
4.1.1. Synthesis Methods
Methods of solubilizing dECM based on SIS was first developed by Freytes et al. in 2008, with the development of solubilizing cECM following shortly after by Singelyn et al. in 2009.[36,87] Since then, a variety of methods have been developed to generate soluble dECM for cardiac applications. Solid dECM scaffolds have benefits such as structural and mechanical similarity to cardiac tissue but suffer from limitations such as the requirement of surgical implantation of patches. Additionally, combining dECM solid scaffolds with cells requires seeding and bioreactor growth, while combining dECM scaffolds with additional biomaterials have been exceptionally limited. Soluble dECM contains the ECM profile of native tissue but can be injected into the myocardium without invasive surgery. In addition, various fabrication techniques can be employed for generating cardiac patches with cellular or additional biomaterial integration. Soluble dECM has been generated from cECM, pECM, sECM, SIS, placenta, and omentum from a variety of animal sources for cardiovascular applications.[3,71,88,89] Decellularization of organ tissue is followed using similar protocols as implemented for solid dECM, where detergent methods are most often used. Specific areas of hearts are isolated, sectioned into small pieces, and allowed to decellularize in solution rather than through perfusion. The dECM is then lyophilized and milled into a fine powder, followed by pepsin digestion to break down the dECM further and improve solubility. The dECM is digested with a low concentration of pepsin relative to cECM in HCl for several days, followed by modification of the solution to basic pH to inactivate the pepsin. The solution is then adjusted to physiological pH and salt concentration, diluted in buffer, frozen, and finally lyophilized. The lyophilized dECM solution can be resuspended at varying concentrations, most commonly 1–10 mg mL−1, and most often in water. The soluble dECM derived in this manor self-assembles into a nanofibrous hydrogel after 30 min to 1 h at physiological temperature. Since dECM materials are homogenized tissues that are derived from specific tissue areas and sources, the materials have similar properties if batch-to-batch variability is reduced through combination of multiple animals. This is commonly performed with porcine or murine dECM, such as for cECM or pECM, but becomes problematic when isolating human dECM. Interestingly, human cECM requires additional steps of lipid removal after formation and requires harsher decellularization protocols, possibly denaturing components and increasing patient to patient variability.[48]
4.1.2. Characterization Methods and Properties
Decellularization extent, mechanical properties, and ECM composition are all important characterization properties used to analyze soluble dECM materials.[1,3,88] Decellularization can be determined through histology to determine cell remains and fluorescence immunochemistry to evaluate presence of cellular components. ECM composition can be analyzed using assays such as dimethylmethylene blue (DMMB) to measure sGAG content, polyacrylamide gel electrophoresis (PAGE) to measure overall protein size distribution, and fluorescent immunochemistry to identify ECM components. Mass spectrometry and proteomics are more powerful systems in determining dECM material composition.[10] SEM analysis after gel formation is useful in analyzing structural properties such as porosity and fiber diameter, where cECM generally has a fiber range between 40 and 100 nm. Soluble dECM requires gelation to form solid structures, based on temperature, pH, ionic concentration, and time.[90] Gelation mechanics and time can be measured through assays such as turbidity and rheological measurements. Since the dECM hydrogels are viscoelastic, characterization of both the storage and loss modulus though parallel plate rheology is key in evaluating hydrogel mechanics. Soluble cECM is much softer than solid cECM, having a storage modulus of approximately 5 Pa based on concentration.[88] Since thicker prepolymer solutions are more difficult to inject through a syringe or catheter within tissues, prepolymer viscosity is an important mechanical property for cardiovascular applications. cECM is a shear thinning material, which decreases in viscosity as shear rate increases, indicating ideal properties for catheter injection.
4.1.3. Modification Methods
Soluble dECM synthesis allows for formation of hydrogels with complex combinations of natural components, which would be exceptionally difficult to generate in the lab due to the high cost and difficulty of purchasing specific ECM components, derived from human or porcine sources exclusively. Although soluble dECM materials have been used directly after formation for in vivo repair, soluble dECM suffers from exceptionally low mechanical modulus upon gelation, which may cause tissue-hydrogel mechanical mismatch when injected into the myocardium or used as a hydrogel patch. This mismatch can lead to unwanted effects such as immune responses, reduction in cellular recruitment, or fast material degradation. Recent techniques have attempted to remedy these problems and modulate dECM properties through crosslinking or material addition.
Johnson et al. quantified the gelation properties of cECM hydrogels for the first time by modulating pH, temperature, ionic strength, and concentration to improve mechanics and gelation kinetics.[90] cECM solutions at 4 °C and 22 °C did not gel and exhibited globular morphologies after 24 h, while solutions developed at 37 °C formed nanofibrous gels at both 6 and 8 mg mL−1, although across all groups 8 mg mL−1 gels were twice as stiff. Increasing salt concentration decreased fiber formation and scaffold stiffness, while modulating pH did not affect gelation compared to standard conditions. It was expected that the cECM material would behave as collagen does to modulations in gelation parameters, since cECM is mainly composed of collagen I, but collagen does gel at 22 °C after a long enough time and collagen gelation is significantly affected by pH modulation. These differences may be due to the other matrix components in the cECM. Regardless, modulating simple factors for gelation proves to be an excellent method of tailoring material properties of cECM, although most other studies focus on changing stiffness and degradation through crosslinking.
Singelyn et al. used glutaraldehyde, a common crosslinking agent for collagen and pericardium-derived heart valves, to increase the storage modulus of cECM gels from 5 to 136 Pa.[91] The crosslinked scaffolds showed effective catheter injection, decreased in vitro degradation, and slowed cellular migration through gels. Using in vitro and in vivo analysis of degradation, Wassenaar et al. used glutaraldehyde, genipin, and transglutaminase to crosslink cECM hydrogels, or loaded the gels with the doxycycline to inhibit MMPs.[92] When injected into rat myocardium, only the doxycycline loaded cECM prolonged hydrogel degradation compared to nonmodified cECM, indicating its effectiveness in modulating in vivo degradation without modifying cECM properties. Although crosslinking agents may improve mechanical properties, none of the commonly used methods allowed both biocompatibility and effectiveness in vivo. Rather than crosslinking, Grover et al. used polyethylene glycol (PEG) to improve cECM properties.[93] NHS acrylate was incubated with cECM solutions, which binds to amine groups and allows for a quick acrylation method for protein-based materials. The cECM solutions were mixed with varying concentrations of PEG-NHS, PEG-acrylate, and PEG-diacrylate (PEGDA), allowed to crosslink using UV light, and finally incubated at physiological temperature to induce cECM polymerization. Only PEG-acrylate combinations increased the storage modulus of gels to 125 or 700 Pa, using low and high concentrations of PEG-acrylate, respectively. All formulations prolonged degradation and increased fiber diameter (except low concentration PEGDA) compared to nonmodified cECM. These studies show that cECM mechanics, gelation, and degradation can be modulated through crosslinkers, polymers, and MMP-inhibiting drugs.
4.1.4. Advanced Fabrication Methods
Recent studies have shown improved dECM functionality and device fabrication with advanced methods such as GF-tethering, dECM microparticle development, and 3D bioprinting. An important function of native ECM is GF sequestration and release, induced by natural GF binding domains found on sGAGs. Native ECM may be an ideal material for modulating release of growth factors into damaged tissue. Seif-Naraghi et al. used pECM loaded with FGF to investigate release from soluble dECM.[94] FGF release was slowed in pECM loaded scaffolds compared to collagen scaffolds, both after 6 d and after collagenase treatment. In a rodent infarct model, injection of FGF loaded pECM induced neovascularization significantly compared to FGF loaded collagen, with new vasculature forming from existing vessels. A follow-up study by Sonnenberg et al. used pECM loaded with an engineered hepatocyte growth factor (HGF) in a rat model of infarction and showed the effectiveness of dECM-GF tethering to treat LV remodeling postinfarction.[95] Park et al. loaded milled SIS matrix (CorMatrix) with FGF, which showed extended release from the material and reduction in myofibroblast contraction on collagen gels.[96] Overall, these studies point toward an effective use of ECM GF-tethering properties as tools for cardiac repair.
Milled and partially solubilized dECM has been used directly for cardiovascular applications. These methods rely on direct injection of semisolubilized dECM powders or manufacture of dECM microparticles. Williams et al. partially digested adult cECM to liberate crosslinked components and promote CM proliferation, based on the idea that the effectiveness of fetal over adult cECM comes from reduced crosslinking or differential composition in fetal cECM.[97] By growing neonatal rat cardiac cells on substrates coated with adult cECM that was solubilized in pepsin for a variety of time lengths, they found that CM proliferation was highest on cECM digested for 1 and 3 h, while CMs matured on cECM digested for 24 and 48 h. Protein analysis showed that less solubilized cECM had exposure of fibrillin-1, fibrinogen, and laminin, compared to fully solubilized cECM, which mainly contained collagen I. Partially solubilized cECM may contain more reparative components than fully solubilized cECM. Another novel direction for using milled dECM is through the generation of dECM microparticles, which may retain ECM structure and protein composition better than soluble ECM, while allowing for injection into the myocardium.[98] Kappler et al. developed a method of generating uniform cECM particles derived from human left ventricle.[99] The particles had native-like fibrous structures, contained a large variety of ECM proteins, and were easily resuspended in buffer and gelatin solutions. Murine CMs cultured in gelatin hydrogels containing the particles expressed increased metabolic activity compared to gelatin alone.
Compared to developing soluble cECM materials for injection, generating solid scaffolds of pure soluble cECM can be challenging due to soft mechanical properties and difficulty in handling. Advanced techniques, such as extrusion bioprinting, have attempted to remedy these problems through higher cECM concentrations, crosslinking modifications, and combination with additional biomaterials. Extrusion bioprinting relies on stacking layers of structurally supportive bioink on top of one another, requiring high viscosity solutions for proper printing and scaffold fabrication.[100–103] At concentrations commonly used when cECM is injected, from 6 to 8 mg mL−1, cECM pre-polymers cannot form layers or print due to very low viscosity. Pati et al. developed a system to print soluble cartilage dECM, adipose dECM, and cECM scaffolds using polymeric support materials.[101] However, cECM scaffolds were printed without support, possibly due to high concentrations used, although it is not mentioned in the study and the print structure of the cECM scaffolds was not as defined as the other dECM materials printed with support. In a second study, Jang et al. developed a system to 3D print cECM scaffolds without support materials using vitamin B2 UV-light induced crosslinking.[102] It is clear in the study that 20 and 30 mg mL−1 solutions of cECM were used to print structures, which had storage moduli of 0.33 and 0.95 kPa, significantly higher than the moduli seen in 6 mg mL−1 solutions of 0.05 kPa. The 20 mg mL−1 cECM solutions were printed in successive layers by extrusion deposition, where each layer was cured with UV light for 3 min before additional deposition. The 20 mg mL−1 cECM 0.02% vitamin B2 combination showed viable CPCs that differentiated more effectively than when grown in pure cECM scaffolds, although VEGF release was lower in crosslinked scaffolds. Although this methodology allowed for printing of cECM, it suffers from limitations in using an exceptionally high concentration of cECM and reduced paracrine release. Bejleri et al. developed a system of printing human neonatal CPCs with porcine cECM at 8 mg mL−1 by incorporating gelatin methacrylate (GelMA), which allows for a viscous bioink upon cooling.[103] The inclusion of cECM improved the printability of the bioinks and modulus of the patches over pure GelMA, showing a further use for dECM in developing reparative devices. Through this method, described in Figure 5, cardiac patches were generated and investigated as a therapy for paracrine release toward the damaged myocardium, developing a key step toward printing cECM materials.
Figure 5.
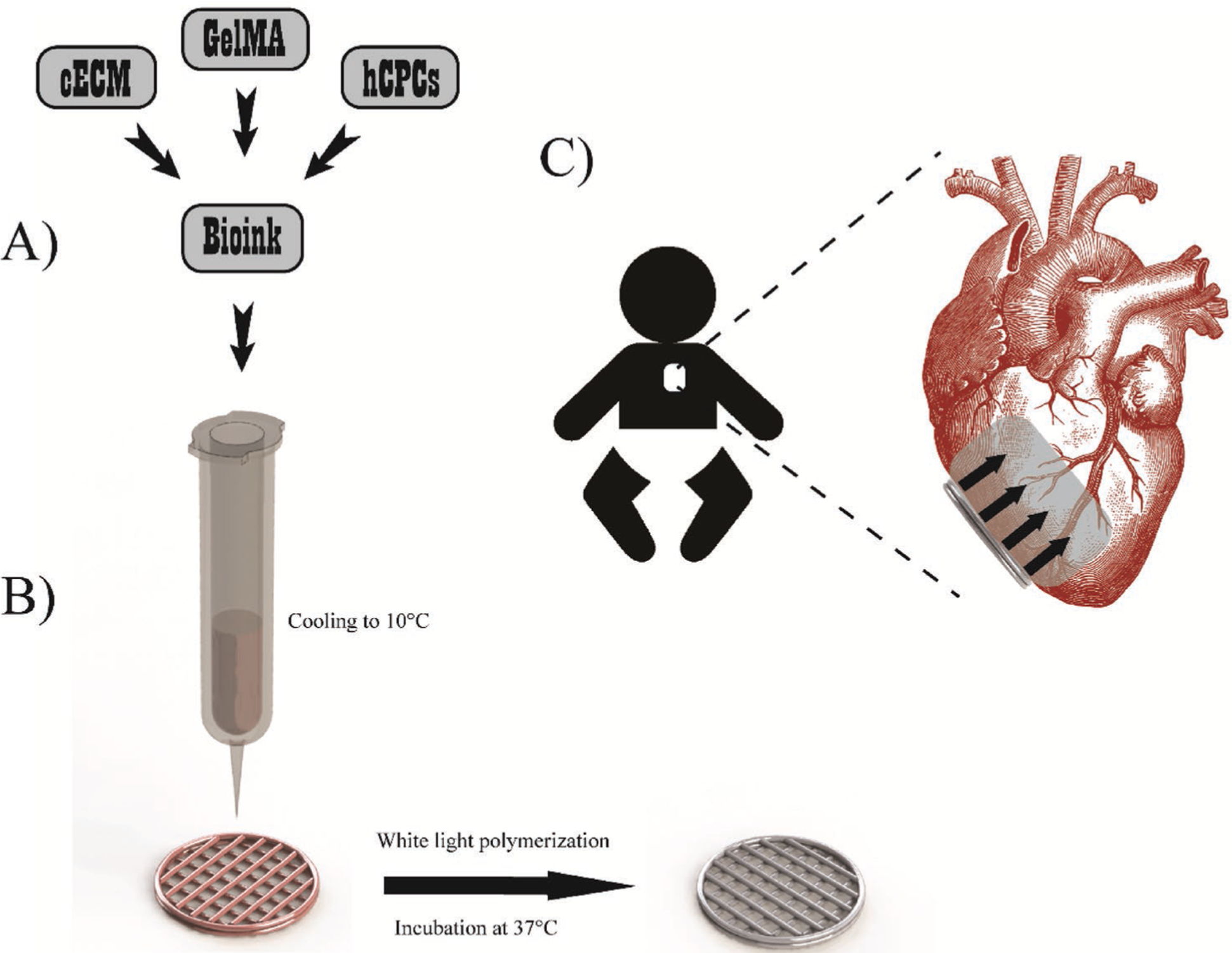
Printing overview. A) Bioink preparation involved combining cECM, hCPCs, and GelMA to form naturally derived and cell-laden materials for printing. B) Printing methodology involved cooling the bioink to 10 °C in the 3D bioprinter barrels to allow GelMA polymerization for improved printability. Patches were printed with infill patterns of 90° intersecting filaments and contour. Patches were polymerized via white light to induce radical polymerization of GelMA, followed by incubation at 37 °C for at least 1 h to induce cECM polymerization. C) Patch implementation will involve pericardially inserting the patch to the RV of pediatric patients, where the patch will release key regenerative paracrine factors. Reproduced with permission.[103] Copyright 2018, Wiley-VCH.
4.2. Cellular Responses to Soluble dECM
4.2.1. 2D Coatings and Hydrogels
Platforms using dECM scaffolds through 2D coating or hydrogel systems provide an excellent method to evaluate cellular responses to different dECM, allowing for high-throughput analysis of multiple parameters and evaluation of rare or difficult to obtain pathological dECM. Tissue-specific dECM can be used to coat cell culture dishes in similar methods employed for coating dishes with natural materials such as collagen, or can be polymerized as a thin hydrogel for cell seeding on top of the scaffold.[44] These soluble dECM platforms have been used in modeling biological niches, infarct environments, matrix stiffness, and cell-matrix binding, while also evaluating the ways in which dECM modulates stem cell phenotype and matrix binding.
Sarig et al. grew HUVECs and human MSCs on porcine cECM and evaluated tissue properties and phenotypic changes during cell growth through mechanical testing, molecular biology assays, and principal component analysis.[104] They found that MSCs remodeled the cECM, HUVECs improved the tissue-specific recognition of differentiation, and coculture improved tissue integration, angiogenesis, and differentiation. Stem cells grown on tissues at different ages can help understand factors involved in cardiac development, such as mechanotransduction. Gershlak et al. grew MSCs on cECM scaffolds derived from fetal, neonatal, or adult myocardium, which were further modified with polyacrylamide gels of varying stiffness as a system to evaluate contractile forces of MSCs across cECM development.[23] MSC responses to varying stiffness were different based on cECM age, and MSC differentiation induced toward early cardiomyogenesis was only seen in stiff neonatal substrates.
Modulations to cardiac ECM properties after infarction can change therapeutic outcomes for cell therapy compared to testing with healthy tissue. Sullivan et al. decellularized and solubilized healthy and infarcted cECM at 1 and 4 weeks after infarct, which were then combined with hard and soft polyacrylamide gel to grow MSCs in 2D culture.[22] MSC differentiation toward the cardiac lineage, based on Nkx2.5 expression, was only evident on soft healthy and soft 1 week infarct cECM. Interestingly, MSC-induced release of GFs such as HGF and stromal cell-derived factor 1 were highest in 4-week infarct cECM. In follow-up work, Sullivan and Black modified the in vitro model to more accurately replicate an in vivo infarct environment by increasing dECM stiffness, decreasing oxygen tension, removing serum from culture media, and incorporating common GFs found in infarct environments such as FGF and TGF-β.[105] The model determined that expression of Isl-1 by CPCs was the main factor required for differentiation in an infarct environment.
In vitro systems using cardiac specific cells or stem cells on cardiac specific dECM have also been used to determine the biology of cell-ECM binding in the heart. Merna et al. grew human cardiac and lung fibroblasts on intact cECM, collagen gels, and coatings of cECM, lECM, and ECM components.[47] Cardiac fibroblasts had higher expression of β3 and β4 integrins compared to lung fibroblasts, and inhibition of only β3 integrin resulted in substantial increases to myofibroblast differentiation of cardiac fibroblasts cultured on either lECM, cECM, or fibronectin. The integrin profile and source of fibroblasts were more important than dECM type in generating myofibroblast phenotypes, although dECM and fibronectin binding through β3 integrin seems to reduce the instance. A similar study by Gershlak and Black investigated how MSCs interact with cECM scaffolds to modulate traction forces.[106] They found that once again, MSCs grown on cECM did not respond to changes in substrate stiffness, although MSCs grown on single ECM components such as collagen did modulate traction forces based on stiffness. When β1 integrin was inhibited, the MSCs grown on cECM responded to substrate stiffness by modulated traction forces similarly to cells grown on single ECM components.
Stem cell-ECM interactions in modulating stem cell differentiation and reparative potential are of most interest in quantifying cellular modifications through dECM materials on 2D substrates. CPC differentiation toward a reparative phenotype, whether by improved differentiation, paracrine release, or both, has been studied with many materials. French et al. grew rat neonatal CPCs on collagen, porcine cECM, and adipose dECM substrates to evaluate the CPCs differentiation potential.[43] Compared to collagen and adipose dECM, CPCs grown on cECM expressed an increase in early CM markers and protein expression, improved proliferation, resistance to apoptosis, and improved adhesion. The CPCs expressed an increased profile of MMP and TIMP production compared to cells grown on collagen. A follow-up study by French et al. evaluated the effects on CPCs on cECM or single ECM components with cyclic strain.[107] The study found that strain improved VEGF production and decreased Cx43 expression of CPCs grown on cECM, indicating that combined strategies may improve CPC release profiles. Baghalishahi et al. grew human adipose derived stem cells (ADSCs) on rat cECM with and without a cardiac inductive cocktail to analyze cardiac differentiation potential after 3 weeks in culture.[108] Expression of cardiac genes was significantly increased when ADSCs were grown on cECM compared to standard tissue culture or addition of the inductive cocktail, with a combination of both cECM and cocktail further driving cardiac differentiation.
4.2.2. 3D Hydrogels
In therapy, cells are exposed to 3D scaffolds and environments, which may significantly alter cell properties and reparative potential compared to 2D growth. CPCs and iPSCs have been investigated within pure cECM and pECM scaffolds, summarized in Table 4. Gaetani et al. encapsulated human fetal and adult CPCs in porcine cECM for up to 1 week in culture.[109] Adult and Fetal CPCs had an increased gene expression for cardiac and endothelial lineages up to 1 week in culture, remained viable, proliferated more, and survived more effectively after H2O2 treatment compared to cells grown in collagen. Fetal CPCs seemed to proliferate more than adult CPCs and adult CPCs seemed to survive in higher numbers than fetal CPCs when grown in cECM. Rajabi-Zeleti et al. grew human CPCs in human pECM gels and found that CPCs were able to migrate, survive, proliferate, and differentiate toward a cardiac phenotype more effectively than cells grown on 2D pECM membranes or 3D collagen scaffolds, mirroring the results seen in cECM materials.[110] Fong et al. grew human iPSC-derived CMs on 2D and 3D cECM scaffolds derived from fetal or adult bovine cECM.[111] While both 3D cECM scaffolds induced CM expression of calcium-handling genes, CMs grown in 3D adult cECM had higher expression of these genes in comparison to 3D fetal cECM, although there was no significant difference when comparing fetal and adult 2D cECM. The CMs in 3D adult cECM showed increase calcium signaling and kinetics compared to CMs grown in 2D and were more responsive to calcium inducing drugs. Jeffords et al. used genipin crosslinking to modulate mechanical properties of porcine cECM hydrogels for induction of human MSC endothelial differentiation.[112] Genipin crosslinking increased cECM hydrogel storage modulus, decreased swelling ratio, and prolonged degradation. MSCs showed maintained viability, downregulation of early EC markers, and upregulation of mature EC markers. Taken together, these studies show that 3D cECM and pECM scaffolds are effective in driving stem cell differentiation toward cardiac lineages and may be more effective than 2D cardiac dECM models.
Table 4.
Pure Decm-stem cell interactions.
| dECM source | Formulation | Cell source | Findings | References |
|---|---|---|---|---|
| Porcine cECM | 2D coating | Rat neonatal CPCs | Improved cardiac commitment, proliferation, adhesion, reduction in apoptosis compared to collagen or adipose dECM Improved production of MMPs/TIMPs compared to collagen |
[43] |
| 2D coating | Rat neonatal CPCs | Improved VEGF production, decreased Cx43 expression with strain | [107] | |
| 3D hydrogel | Human fetal and adult CPCs | Improved cardiac commitment, endothelial commitment, proliferation, survival compared to collagen | [109] | |
| 3D hydrogel with genipin | Human MSCs | Improved mature EC commitment, reduced early EC commitment with genipin crosslinking | [112] | |
| Murine cECM | 2D coating | Human ADSCs | Improved cardiac commitment compared to standard tissue culture, further improved by combination of inductive cocktail and cECM | [108] |
| Human pECM | 3D hydrogel | Human CPCs | Improved migration, survival, proliferation, cardiac commitment compared to 2D pECM or 3D collagen | [110] |
| Bovine cECM (fetal and adult) | 2D coating and 3D hydrogel | Human iPSC-derived CMs | Improved expression of calcium-handling genes in 3D adult cECM compared to fetal Improved calcium signaling and kinetics in 3D adult cECM compared to 2D adult cECM |
[111] |
4.3. Combination Patches
Many advanced methodologies for using dECM in cardiovascular applications follow similar methods explored for other tissue engineering technologies, such as developing multimaterial scaffolds, 3D printed constructs, electrospinning, and many more. These methodologies are challenging to implement using solid dECM scaffolds, which have limited modification opportunities. Soluble dECM allows for a more flexible material system for use in developing combination biomaterial scaffolds generated through several methods, with the option of stem cell loading. Pure cECM hydrogels are soft and difficult to handle as cellular patches for myocardial implantation. Due to this limitation, studies using combination systems and dECM (most often cECM) rely on addition of natural ECM components, non-mammalian derived biomaterials, or synthetic materials, summarized in Table 5.
Table 5.
Combinatorial soluble dECM-biomaterial scaffolds.
| dECM source | Additional material | Cell source | Findings | References |
|---|---|---|---|---|
| Porcine cECM | GelMA | Human neonatal CPCs | Improved cardiac commitment, endothelial commitment, angiogenic potential compared to pure GelMA patches Neovascularization following 14 d implantation | [103] |
| Collagen I | Human ESCs | Higher cECM content improved cardiac commitment | [112,113] | |
| Chitosan | Murine CMs | Improved cardiac commitment, retention, conduction velocities, contractile stress compared to gelatin-chitosan | [116] | |
| Chitosan, PCL core | None | Induced M2 macrophages in vivo | [117] | |
| Silk | Human ESCs and ESC-derived CMs | Anisotropic, aligned fibers formed via oriented freezing Improved cardiac commitment compared to aligned or isotropic silk cECM inclusion improved cell infiltration and vascularization in vivo |
[120] | |
| Silk | Murine cardiac fibroblasts | Silk/cECM concentration tailors mechanical properties and fibroblast proliferation, viability, integrin expression | [121] | |
| PEG-acrylate | Murine fibroblasts | Increased cECM scaffold modulus Fibroblasts remained viable with inclusion of PEG | [93] | |
| PLGA | Human MSCs | Tissue papers induced MSC proliferation | [122] | |
| PCL and VEGF | Human CPCs and MSCs | Patterned patches improved angiogenesis and ejection fraction in rat MI model | [123] | |
| Human cECM | Amniotic membrane | Human cardiac fibroblasts, epicardial cells, CMs | CMs showed improved adhesion and survival compared to pure amniotic membrane Reduced monocyte secretion of inflammatory cytokines and induction of M1 macrophages |
[114] |
| Murine cECM (fetal and adult) | Fibrin and transglutaminase | Human CPCs | CPCs remained viable and showed cardiac commitment | [115] |
| Bovine cECM | Chitosan | Human CPCs | Higher cECM ratio improves CPC viability | [118] |
| Porcine pECM | Chitosan | Human MSCs | Cardiac preservation and increase in cardiac function 8 weeks postinjection in MI model | [119] |
| Ovine pECM | CNTs | Murine CMs | CNTs suppressed CM cytotoxicity Improved proliferation, gap junction expression, and contraction compared to pECM hydrogels or gelatin-fibronectin-coated plates | [124] |
4.3.1. Natural ECM Component—dECM Cardiac Patches
Several groups have attempted to augment the bioactivity or device fabrication properties of dECM by adding single ECM components such as collagen to maintain the developed patches as fully bioactive devices composed of only natural materials. Bejleri et al. developed bioprinted human neonatal CPC laden patches composed of cECM and GelMA.[103] CPCs in cECM-GelMA patches showed improved cardiac and endothelial differentiation, decreased proliferation, and improved angiogenic potential compared to GelMA patches. Patches were retained in vivo in a rat model for up to 14 d, and showed neovascularization formation within the patches themselves, indicating that the CPCs may survive and release reparative factors toward the myocardium during therapy. Duan et al. developed hydrogels composed of cECM and collagen I at varying ratios to evaluate the material effectiveness in inducing ESC cardiac differentiation, with and without FGF and VEGF.[113] ESC embryoid bodies grown in 75% cECM with no GFs expressed higher levels of cTnT, compared to hydrogels with 25% cECM or 0% cECM with GFs. The high cECM content hydrogels induced formation of striated cTnI and expression of Cx43, although the effect may be due to the varying storage modulus of the low (8 Pa) and high (60 Pa) cECM scaffolds rather than the cECM composition. Instead of combining cECM with single ECM components, Becker et al. dry coated cell-free amniotic membrane with cECM, combining two complex natural materials.[114] While the cECM coating did not change scaffold mechanical properties, CMs showed improved adhesion and survival on cECM coated substrates compared to pure amniotic membrane. On cECM coated scaffolds, monocytes secreted less inflammatory cytokines and macrophages polarized toward the proinflammatory M1 type. Williams et al. used fibrin, adult or fetal cECM, and transglutaminase to generate CPC loaded injectable hydrogels.[115] Transglutaminase effectively modulated the scaffold stiffness from 2 to 32 kPa and CPCs grown in scaffolds remained viable and expressed markers for cardiac tissue differentiation such as titin, vWF, and calponin 1.
4.3.2. Nonmammalian Biomaterial—dECM Cardiac Patches
Nonmammalian matrix components, such as chitosan, alginate, or silk, have found relative success in tissue engineering applications, where alginate has moved toward clinical translation for cardiac treatment. Of these, chitosan and silk have been explored in combination with dECM for cardiovascular applications.
The first chitosan-cECM patch was developed by Pok et al., who combined cECM powder with chitosan, followed by lyophilization to form 3D scaffolds.[116] cECM-chitosan scaffolds had similar porosity and elastic modulus (4–13 kPa) to gelatin-chitosan scaffolds. However, cECM-chitosan scaffolds had higher retention of seeded rat CMs compared to gelatin-chitosan scaffolds, in addition to increased α-MHC and Cx43 expression, conduction velocities, and contractile stresses. The patch had ideal potential for direct use as a thick patch for myocardial replacement. Pok et al. modified the patch by adding a polycaprolactone (PCL) core, and tested the therapeutic potential of the patch in a rat infarction model.[117] Pourfarhangi et al. evaluated the optimized ratio of cECM and chitosan for improving CPC function, and determined that higher cECM composition in composite patches improved CPC viability.[118] Efraim et al. evaluated pECM with genipin crosslinking and chitosan addition, with the intention of therapeutic injection rather than patch implantation.[119] The combined material system showed improved MSC viability, organization, and remodeling on 2D coated systems compared to noncoated plates. The material was injected into acute and long term chronic infarct rat models, which showed preservation and increase in cardiac function eight weeks posttreatment compared to nontreated animals.
Stoppel et al. developed anisotropic silk-cECM scaffolds using an oriented freezing method to form aligned fibers within the scaffolds.[120] The scaffolds had tailorable structures, degradation rates, and mechanical properties based on alignment and composition. In vitro, both primary and ESC-derived CM showed improved expression of cardiac markers cTnI and Cx43 in aligned cECM-silk scaffolds compared to aligned or isotropic silk. The patches were implanted subcutaneously in rats, where addition of cECM in silk scaffolds significantly improved cell infiltration and vascularization. A follow-up study by Stoppel et al. showed that changing silk and cECM concentration could modulate scaffold mechanical properties and cardiac fibroblast proliferation, viability, and integrin expression.[121]
4.3.3. Synthetic Biomaterial—dECM Cardiac Patches
Synthetic materials such as PEG, PCL, and carbon nanotubes have been used in cardiovascular tissue engineering as bioinert scaffolds or as supports for bioactive materials. These materials have been combined with cECM and pECM to improve material properties or provide support through similar methods. Grover et al. used PEG-acrylate materials to modulate mechanical properties of cECM scaffolds, and showed that fibroblast viability was not hindered by polymer inclusion.[93] Jakus et al. used a variety of dECM materials, including cECM, to create thin, large “tissue papers” for use in patching applications via the incorporation of poly-lactic-co-glycolic acid (PLGA).[122] The cECM-derived paper showed effective MSC proliferation, although the technology was not evaluated further in the study. Using the vitamin B2 methodology for bioprinting cECM, Jang et al. generated cardiac patches composed of PCL, CPCs, MSCs, VEGF, and cECM. CPC-cECM strands and MSC-VEGF-cECM strands were printed separately for endothelial induction of MSCs and improved functionality of CPCs.[123] The patch development method is seen in Figure 6.[123] Alternating PCL layers were required to form printed patches, which in turn improved patch mechanical properties and handling for surgical implantation and may significantly increase degradation time of the patch. The methodology of combining cells, GFs, cECM, and PCL into a complex tissue scaffold is a significant step toward multicomponent patches for cardiac repair. Roshanbinfar et al. developed a unique application of using dECM via the incorporation of carbon nanotubes (CNT) within injectable pECM materials to improve pECM electrical conductivity and mechanical properties.[124] Dispersion of CNTs within the pECM materials was achieved through carbodihydrazide modification of CNTs, which also suppressed cytotoxicity of cultured CMs. In addition, CMs in the pECM-CNT scaffolds had higher proliferation and expression of Cx43 compared to pECM hydrogels or gelatin-fibronectin coated plates.
Figure 6.
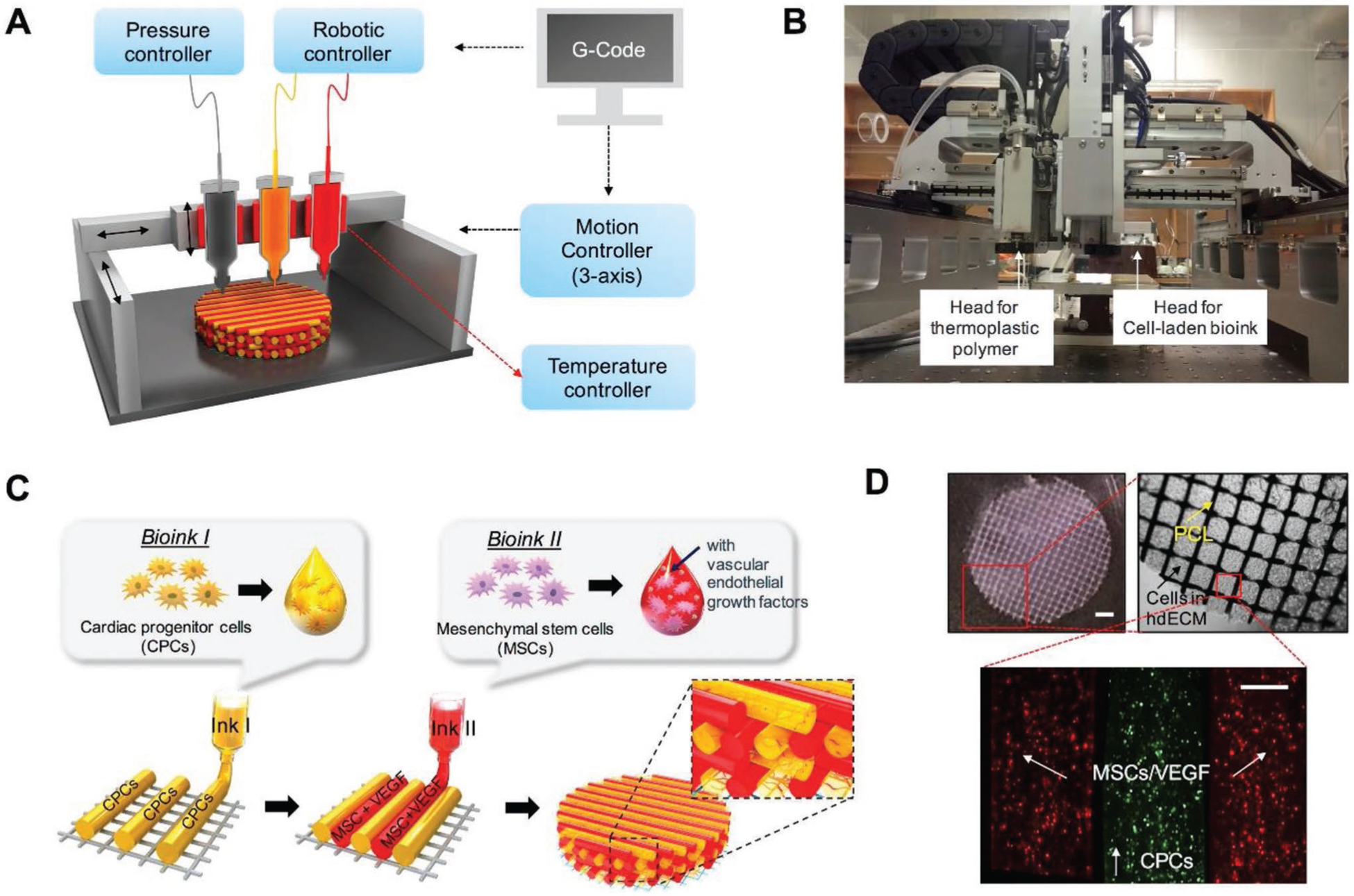
Schematic of prevascularized stem cell patch. A) Illustration of 3D cell printing system, and B) macroscopic view of the printer. C) Illustration of prevascularized stem cell patch including multiple cell-laden bioinks and supporting PCL polymer. D) Fabricated patch including the two types of cell-laden bioink and PCL supporting layer [Scale bar (top left), 1 mm; Scale bar (bottom), 200 μm]. All bioinks were composed of cECM. Reproduced with permission.[123] Copyright 2017, Elsevier.
Combinational devices use a wide variety of materials and methods to modulate or supplement cECM and pECM mechanical, bioactive, or formation properties. While investigations on novel materials may pave the way for enhanced dECM therapy in cardiac repair, in vivo analysis in animal models is critical for truly evaluating the effects of differential therapies.
5. In Vivo Evaluation of dECM Therapies for Cardiovascular Tissue Engineering
Ultimately, the effectiveness of dECM technology in treating cardiovascular disease rests on in vivo implementation of dECM therapies and assessing functional outcomes. This section discusses recent in vivo studies using dECM in treating cardiovascular disease, most often in cases of MI, in murine, large animal, and clinical cases.
5.1. Murine Models
5.1.1. Cardiac Patches
Cardiac patches hold the promise of myocardial regeneration and repair through paracrine release from biomaterials and cells loaded within the devices or use as myocardial replacement when developed as contractile tissue. Cardiac patches have the benefit of improved cell retention, device customization before implantation, and improved mechanical support for the myocardium compared to injectable systems. However, patches require invasive surgical implantation and nondegradable materials used for patches may result in increased stress on the myocardium due to permanent retention. dECM-based patches may remedy this later point through degradation, while additionally improving repair through the bioactive properties of dECM itself. Early studies using dECM have shown that solid scaffolds of SIS and pECM can be used to improve function of myocardium and RV outflow tract in rat models, and many SIS and pECM scaffolds were commercially available by 2008.[35,38,125] Since then, a variety of dECM scaffolds have been tested extensively in vivo for cardiovascular repair.
Sarig et al. evaluated the use of a solid cECM scaffold in the treatment of both acute and chronic MI, compared to sham and nontreated MI controls.[126] In both acute and chronic infarction, cECM decreased infarct size, improved left ventricle diameter (LVD), and increased LV wall thickness 30 d after treatment compared to non-treated groups. In the acute model, cECM treatment improved fractional area change (FAC), fractional shortening (FS), and EF immediately after treatment, with gradual improvements approaching sham values 60 d after treatment. For both cases, cECM treatment improved progenitor and myocyte recruitment, with progenitors expressing early and late CM markers along with Cx43. Vasculature was evident in cECM patches, and patch treatment increased M2/M1 macrophage ratios in the acute model. This evaluation of both acute and chronic MI shows the effectiveness of the pure cECM scaffolds in therapy for post-MI at multiple timepoints of tissue damage.
dECM-based patches may further improve functional outcomes when combined with cells, secondary biomaterials, or GFs. Jang et al. developed a cardiac patch composed of alternating layers of PCL and CPC/MSC/VEGF-loaded cECM.[123] The group tested three groups with variations in the cECM layer—CPC only, mixed CPC/MSC/VEGF, and patterned CPC and MSC/VEGF strands. While the CPC-laden patch showed little development of vasculature in the infarct, the mixed group showed improved vasculature formation and the patterned patch showed even higher degrees of vessel formation. This trend was also seen in cell migration from the patches to the myocardium. Epicardial activated gene expression, indicated by WT1 and Raldh2, was seen in the myocardium with implanted patches compared to non-treated groups. At 8 weeks, all patches induced increased wall thickness, improvement of EF, and decreased fibrosis in the infarct, where the patterned patch induced the highest repair. Godier-Furnémont et al. developed a combination scaffold generated from layers of cECM and TGF-β conditioned MSCs in fibrin and tested the scaffolds efficacy in both acute and chronic models of MI.[73] Four weeks after implantation, composite patches showed improvements in FS, left ventricle systolic diameter (LVSD), left ventricle systolic area (LVSA), and FAC compared to MSC injection and non-treated controls, which showed no improvements. Wang et al. seeded iPSC-derived CMs and CD90+ cells in a solid cECM scaffold for treatment of MI. Patches were attached to the LV wall through a layer of fibrin glue and cardiac functionality was measured at 1–4 weeks for nontreated, pure cECM, and cell-laden cECM groups.[61] EF, FS, and LVSD were improved at all time points (except for EF at week 1) in hearts treated with cell-laden patches compared to pure cECM and nontreated groups. In addition, infarct size decreased, and wall thickness and vascular density increased in the LV of groups treated with cell laden patches compared to all other groups. Using a solid scaffold derived from decellularized human heart valve tissue (hHVS), Wan et al. evaluated the application of the patch in a rat infarct model.[127] The hHVS patches, both bare and loaded with bone marrow-derived c-kit+ stem cells, showed improved cardiac function and reduced infarct size compared to control groups, where c-kit+ cell loaded scaffolds improved cardiac functionality over bare hHVS scaffolds. D’Amore et al. evaluated the effectiveness of incorporation of cECM within a polyurethane cardiac patch in a rat chronic infarction model.[128] Eight weeks after implantation, the patches with cECM decreased LV mechanical compliance, inhibited myocardial deterioration, reduced scar formation, and improved angiogenesis compared to pure polyurethane patches.
Mewhort et al. used commercially available SIS matrix (CorMatrix) for treatment of MI, where the SIS matrix contained no FGF compared to FGF preloaded SIS.[129] Sixteen weeks after treatment, both SIS and FGF-SIS groups showed improved LV volume and EF compared to sham controls, where the FGF-SIS group also improved LV end diastolic volume (EDV) compared to both the SIS and sham groups. In a follow-up study, Mewhort et al. used a rehydrated SIS scaffold (CorMatrix) to treat MI, where scaffolds were implanted 3 weeks after infarction, without FGF pretreatment.[130] In vitro, cardiac fibroblasts grown on intact scaffolds showed increase in FGF-2, VEGF, and HGF release compared to scaffolds inactivated with glutaraldehyde. Fourteen weeks post-MI, hearts implanted with intact scaffolds showed increases in EF, end systolic pressure volume relationship (ESPVR), and pre-load stroke work (PRSW) compared to both inactive scaffolds and sham controls. The myocardium of intact scaffold treated groups had significantly increased expression of FGF-2 and vascular density compared to both inactive and sham groups. It seems that the commercially available SIS had FGF natively bound to the scaffold without pretreatment, although some of the results of the earlier study by Sonnenberg et al.[95] seem to be contradictory in terms of untreated scaffold FGF loading extent and functional outcomes. Regardless, the study related the reparative properties of the intact scaffold to FGF-2 release from the SIS, which upregulated growth factor release from endogenous cardiac fibroblasts to improve angiogenesis and neovascularization within infarct regions.
While most in vivo studies for cardiac repair focus on treating MI, congenital heart diseases represent another class of cardiovascular problems with an incidence/mortality of 8–9/1000 live births. In surgical treatments for Tetralogy of Fallot, patches most often composed of nondegradable, bioinert materials such as nylon or noncardiac specific materials such as SIS are placed across the right ventricular outflow tract (RVOT). Pok et al. generated a bioactive, degradable full-thickness cardiac patch composed cECM/chitosan and PCL layers for use as a RVOT patch and compared the system to patches composed of gelatin/chitosan/PCL and pure bovine pericardium.[117] By week 8, both gelatin and cECM patches showed similar decreases in PCL thickness. LV EF was decreased at 2 weeks after surgery in all groups compared to sham controls, although there was no difference between groups at weeks 4 and 8. However, cECM patches showed improved M2/M1 macrophage ratios compared to gelatin patches, while both patches showed improved vascular cell density and cell invasion into patches along with decreased scar tissue formation at week 8 compared to week 4 within groups. Although limited, these results show that cECM patches can be used for treatment of congenital heart diseases.
5.1.2. Injectable Materials
Injectable materials can be therapeutically administered through catheters into the myocardium, which do not require invasive surgery and limit the potential of infection.[3] The importance and interest in implementing catheter-based cardiovascular intervention can be seen in the current high demand for transcatheter valve replacement compared to surgical procedures. In addition, injectable materials can be directly incorporated within the myocardium or infarct, interacting at all sides with the damaged tissue, compared to single-sided patch interactions. However, large islands of pure materials without CMs coupled to healthy myocardium may induce arrhythmias. Nondegradable bioinert materials injected into the heart would remain for an extended time, increasing the likelihood of tissue mechanical failure and immune responses. Rane et al. showed that injecting bioinert PEG into rat myocardium postischemia did not produce any significant improvements in myocardial function over saline groups, even though the PEG injection allowed for structural support.[131] Bioactive materials are needed to truly repair damaged myocardium, of which dECM has shown promise, particularly soluble cECM.
Singelyn et al. tested injectable cECM (6 mg mL−1) in a rat model of ischemia.[132] The hydrogel was injected via catheter 2 weeks after ischemia reperfusion. One-week postinjection, cECM did not induce increased arrhythmia potential compared to saline, showing safety in use. There was an increased number of CM islands and proliferative cells in the infarct of cECM injected hearts compared to saline controls, and there was no change in the number of infiltrating M2 macrophages between both groups. Four weeks postinjection, saline injected animals showed a decrease in EF and increases in end systolic volume (ESV) and EDV, while cECM injected animals showed no changes in EF, ESV, or EDV compared to baseline measurements 1 week after infarct. Wassenaar et al. attempted to analyze the tissue mechanisms driving therapeutic benefits of injectable cECM within the myocardium of a rat ischemia model.[133] cECM was injected 1 week after ischemia, and myocardial functionality was evaluated 5 weeks after injection, showing similar results to the study by Singelyn et al. Using whole transcriptome microarrays performed on RNA taken from infarct tissue at 3 d and 1 week after injection, principal component analysis showed that the cECM injected infarcts were distinct from saline controls. Analysis of pathways revealed that the cECM injected myocardium exhibited reduction in cardiac fibrosis/hypertrophy, recruitment of progenitor cells, induction of cardiac transcription factors, alterations to tissue metabolism, increased angiogenesis and vascular development, reduction in CM apoptosis, and altered immune responses, especially at 1 week after injection, as summarized in Figure 7. Sonnenberg et al. modulated soluble pECM by addition of a bioengineered HGF fragment, for combined material and GF therapy after injection.[95] In vitro, the HGF-pECM system protected CMs from serum-starvation and reduced fibrotic marker expression. When injected 1 week after MI into a rat model, the HGF-pECM showed significant improvements in FAC, EF, and arteriole number compared to pECM, HGF, or saline control injections. Interestingly, the HGF and saline groups increased CM area compared to healthy, pECM, and HGF-pECM treated myocardium, indicating that the pECM is a critical component in the combined therapy.
Figure 7.
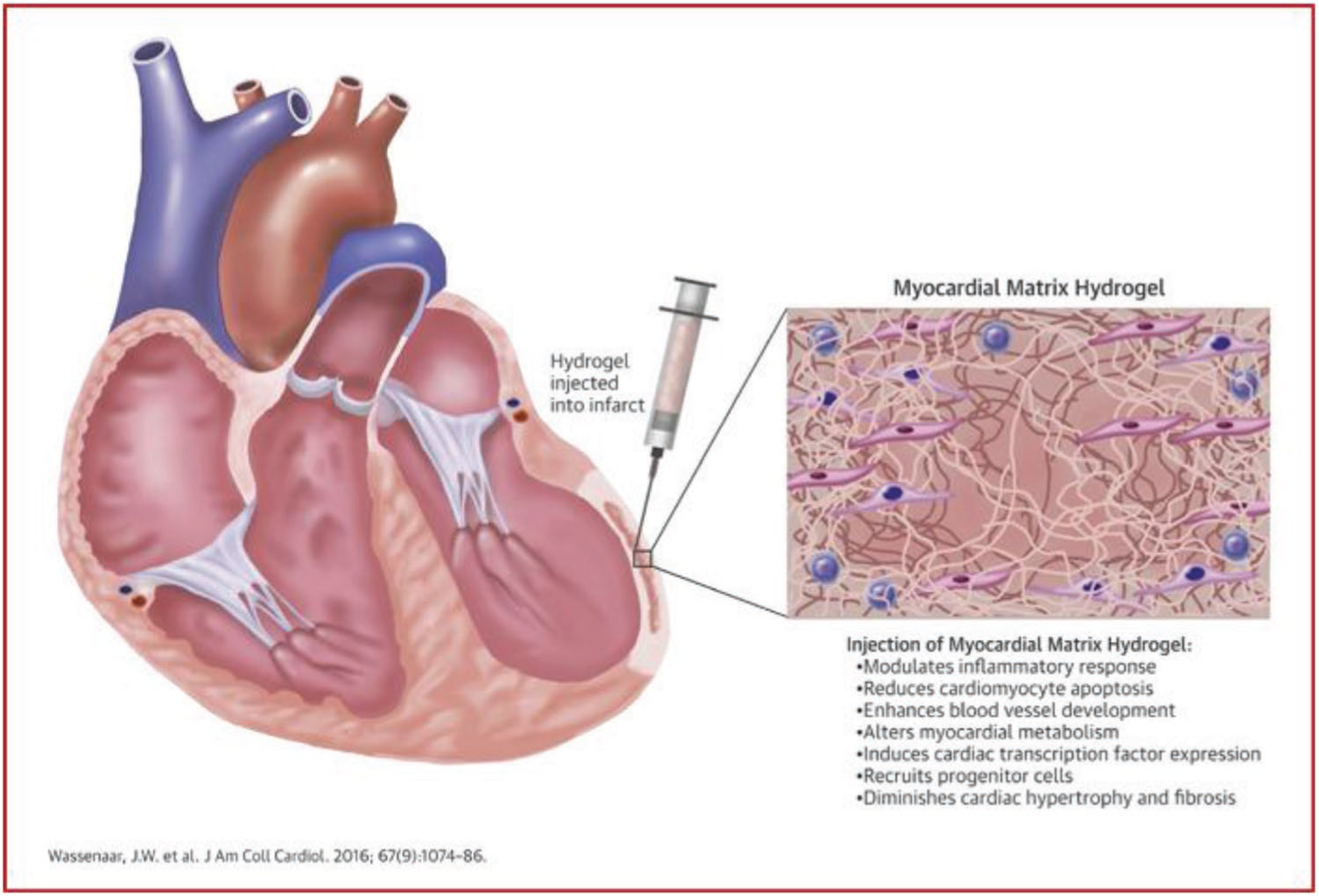
Effects of myocardial matrix hydrogel after myocardial infarction: mechanisms underlying the functional benefits. Injection of myocardial matrix 1 week after myocardial infarction (MI) into the infarcted area induced various tissue level changes that reduced negative left ventricular remodeling and improved hemodynamics. Altering these key pathways created a pro-regenerative environment, potentially preventing or slowing development of heart failure. Reproduced with permission.[132] Copyright 2016, Elsevier.
Efraim et al. developed injectable pECM combined with genipin alone or with genipin and chitosan to improve the mechanical properties of injectable dECM gels.[119] The gels were injected into an acute MI model and evaluated 4 weeks after treatment. Both genipin and genipin/chitosan gels were nonimmunogenic and improved cardiac EF, LV thickness, and FS compared to saline groups. In a chronic model of MI both gels improved ESPVR and PRSW compared to nontreated saline. This evaluation of both acute and chronic MI shows the effectiveness of the modulated scaffolds in repairing cardiovascular tissue for both immediate and extended treatment. Francis et al. created soluble dECM based on human placenta (hpECM) for use as an injectable scaffold for MI treatment.[134] The hpECM was shown to contain a complex composition of reparative ECM components such as agrin, sGAGs, fibronectin, vitronectin, and various collagen and laminin forms. At 4 and 8 weeks after hpECM treatment, there was no change in ESV, EF, or infarct surface area in hpECM treated groups compared to saline, although infarct volume and change in conduction velocity area were reduced in hpECM treated groups compared to saline. SIS matrix is another noncardiac injectable dECM that has been studied in vivo. Toeg et al. injected SIS matrix laden with circulating angiogenic cells (CACs) into a mouse model of MI.[135] Three weeks after treatment, EF increased and infarct size decreased in groups treated with SIS-CAC and pure SIS compared to CAC and saline treated groups. In addition, arteriolar density and progenitor cell infiltration in the infarct area was increased in both SIS and SIS-CAC groups. While most studies see an improved effect with cellular addition, it is clear from the results of this work that the type of cell or cell-ECM interaction may be important in determining combinations that improve cardiac function.
Ravi et al. used ground bone marrow-derived ECM (BM-ECM) as an alternative injectable scaffold for cardiac repair, with and without addition of methylcellulose.[136] The BM-ECM contained reparative factors such as FGF-2, IGF-1, TGF-β, VEGF, and sGAGs. When injected into a rat infarct model, the BM-ECM-methylcellulose and pure BM-ECM groups improved FS compared to nontreated groups at 7 d, where the combined group also showed infarct area reduction compared to nontreated groups at day 21. Decreases in apoptotic cell number and immune cell count as well as increases in progenitor cell infiltration and blood vessel count were seen in only the BM-ECM-methylcellulose group compared to untreated, methylcellulose, and BM-ECM groups, indicating the importance of a carrier material for injection of ground, non-soluble dECM.
Employing a nonmammalian source of dECM, Chen et al. used milled healthy and healing zebrafish myocardial ECM (zECM) as an injectable treatment for mouse acute MI.[137] Zebrafish hearts have a high reparative capacity, potentially driven by cardiac ECM composition/dynamics. While both normal and healing zECM improved cardiac functional recovery and repair of mouse hearts after MI, the healing zECM induced better improvements on heart function. In addition, zECM treated groups had reduced LV dilatation and more elastic myocardium compared to saline or mouse cECM treatment, and showed CM proliferation, increased CPC cell populations, and expression of ErbB2 in CMs. Inhibition of ErbB2 reduced the improved effects of zECM treatment compared to controls, indicating that ErbB2 may play a role in regeneration and repair induced by zECM. Overall, injectable dECM materials for the treatment of MI have been explored in vivo using murine models, with varying dECM sourcing such as cECM, pECM, hpECM, SIS, BM-ECM, and zECM.
5.2. Large Animal Models
Large animal models using dECM for cardiovascular therapy have been limited but effective. Early studies using dECM materials attempted to fix myocardial defects using UBM in pig and dog models, with and without addition of MSCs.[138–140] Since then, large animal models have been carried out for evaluation of dECM patch or injectable gel efficacy in treating MI, summarized in Table 6.
Table 6.
Large animal models in dECM cardiac therapy.
| dECM source | Formulation | Animal model | Findings | References |
|---|---|---|---|---|
| Porcine cECM | Injectable hydrogel | Pig healthy | cECM remained in myocardium with no pericardial effusion 2 h after injection | [132] |
| Pig MI | Treatment with cECM 2 weeks after MI, followed through 3 months post-treatment, saline/no treatment controls Increased EF, wall motion index, endocardial layer Decreased ESV, EDV, infarct size, collagen content in infarct No induction of arrhythmia, spreading to peripheral tissues, change in blood chemistry | [141] | ||
| Porcine SIS | Solid dECM patch—CorMatrix | Pig coronary ischemia | Treatment with SIS patch epicardially 75 min postischemia, followed through 6 weeks, compared outcomes to 1 week baselines and sham Improved wall and epicardial thickness, angiogenesis in infarct No change in fibrosis | [142] |
| Sheep RVOT defect | Followed through 8 months postimplantation Patch showed contractility, organized electrical signal, aligned cells with mature cardiac proteins Integration into RVOT by 5 months | [143] | ||
| Pig subcutaneous implantation | Gradual reduction in initial inflammation and fibrosis by 12 months | [144] | ||
| Solid dECM patch with human MSCs | Pig healthy | Implantation epicardially, followed for 2 weeks, compared to SIS alone Reduced T-cell response at high and low MSC doses, no change in B-cell response | [85] | |
| Porcine cECM and human pECM | Solid dECM patch with adipose-derived MSCs | Pig MI | Followed for 40 d, compared both patches with and without cells Increased LV EF, decreased infarct size in both cell loaded patches Integration with myocardium, angiogenesis, induced nerve sprouting with both patches | [42] |
| Rabbit cECM | Injectable microparticles with FGF and BMSCs | Rabbit MI | Followed for 6 weeks, compared injection of BMSCs, cECM-FGF, and all combined Decreased infarct area, LVEDV, increased LV thickness, EF in all groups |
[145] |
Singelyn et al. evaluated cECM hydrogel delivery in a porcine model, which was the first study that delivered an in situ gelling material by endocardial injection.[132] Hearts were excised 2 h after injection and the cECM material was observed within the healthy myocardium with no pericardial effusion. A follow-up study by Seif-Naraghi et al. in a porcine model of MI showed the effectiveness of injectable cECM as a therapy for myocardial repair.[141] Pigs were treated with cECM 2 weeks after infarct induction where control groups consisted of pigs treated with saline or no treatment. Three months after injection, cECM treated groups saw significantly increased EF and cardiac global wall motion index, as well as decreased ESV and EDV compared to control groups. Infarct size and collagen content of infarcts decreased, with a larger endocardial layer evident in cECM treated groups compared to controls at 3 months after injection.[141] In addition, cECM injection did not affect peripheral tissue, blood chemistry, or cardiac rhythm in pigs. To assess biocompatibility, cECM treated rats did not show increased instance of myocardial inflammation, embolism, or ischemia compared to controls. In addition, cECM did not clot human plasma compared to saline, and degraded within 28 d when injected into rat hearts. These findings paved the way for clinical trials using porcine cECM in treating MI.
In a continuation of the studies on solid SIS (CorMatrix) patches, Mewhort et al. tested patches on a model of ischemia, where SIS patches were applied to porcine epicardium 75 min after ischemia, and cardiac functionality was assessed at 6 weeks relative to 1 week baseline.[142] Wall thickness was improved and epicardial thickness increased in SIS treated groups compared to sham groups, while patch application did not cause myocardial constriction or fibrotic changes. Most importantly, the number of blood vessels, arterioles, and capillaries within the infarcted region significantly increased compared to sham groups. The study proposes a similar mechanism for repair presented in the earlier studies by Mewhort et al., where GF factor release from the SIS patch induces activation of the epicardium, although quantification of this effect was not fully assessed. Chang et al. developed an SIS patch seeded with MSCs for cardiac repair.[85] While tested only on healthy pigs, patch implantation reduced T-cell response at both high and low MSC doses when compared to implantation of SIS alone, although B cell response was similar across all groups. Scully et al. tested SIS (CorMatrix) in ovine model of RVOT defect, where the patch was implanted and evaluated at 5 and 8 months.[143] The patch showed contractility, organized electrical signal, and aligned cells positive for mature cardiac proteins. The SIS patches integrated into the RVOT by 5 months, showing reduced stiffness that matched the native myocardium compared to patch stiffness before implantation. While these studies evaluated porcine and ovine models using SIS patches, a careful analysis on potential host response to the scaffolds must be performed, such as was evaluated by Seif-Naraghi et al. for soluble cECM. Nezhad et al. subcutaneously implanted CorMatrix, porcine pericardium (Vascutek), bovine pericardium (SJM), and Gore-Tex in growing pig models and evaluated host reactions and structural changes over 12 months.[144] CorMatrix showed gradual and full reduction in inflammation and fibrosis, which was mild by 12 months. While only bovine pericardium showed signs of early degradation at 6 months, Gore-Tex and glutaraldehyde fixed porcine pericardium did not degrade. The xenopericardial patches induced encapsulation and limited remodeling, indicating the need for further studies evaluating these materials epicardially.
Combinatorial therapy of cell-seeded or GF-modified dECM materials has been evaluated in porcine and rabbit models, using either cardiac patches or injectable dECM material. Perea-Gil et al. tested the effects of solid porcine cECM or human pECM repopulated with adipose tissue-derived MSCs in a porcine infarction model.[42] Both patches showed LV functional recovery after 40 d, where LV EF increased and infarct size decreased in pECM and cECM groups when MSCs were added compared to bare scaffolds. Both patches integrated with the myocardium, improved angiogenesis, and induced nerve sprouting, seen in Figure 8. Zhang et al. evaluated treatment in a full rabbit model of MI by injecting 100 μm sized particles of solid rabbit cECM bound with FGF, suspended in saline, and loaded with rabbit bone marrow mesenchymal stem cells (BMSCs).[145] While injection of BMSCs, cECM-FGF, and cECM-FGF-BMSC groups all decreased infarct area and LVEDV and increased LV thickness and EF, the cECM-FGF-BMSC group had the strongest effect compared to control groups. The combinatorial therapy of matrix, GF, and stem cells, all with allogenic sourcing, may be an effective therapy for myocardial repair.
Figure 8.
Histological and immunohistochemical analysis after in vivo scaffold implantation. A) Representative images (top to bottom) of H/E, Movat’s pentachrome, Movat’s pentachrome for simultaneous collagen and mucopolysaccharide acid staining and Gallego’s modified trichrome. Scale bar = 100 μm. B) Movat’s pentachrome of the scaffolds from the Per-ATMSCs, Myo-ATMSCs, Per-MI, and Myo-MI groups displaying the presence of arterial blood vessels (red arrows), veins (blue arrows), and nerve fibers (yellow arrows). Scale bar = 50 μm. C) Representative SEM images of scaffolds from Per-MI and Myo-MI experimental groups after sacrifice. D) Immunohistochemical images of the scaffolds against IsoB4 (green), SMA (red), and elastin (white) antibodies confirming the presence of arteriolar blood vessels positive for SMA. Nuclei appear counterstained in blue. Scale bars = 50 μm. Abbreviations: Per-MI (cell-free pericardial scaffold), Myo-MI (cell-free myocardial scaffold), Per-ATMSCs (adipose tissue mesenchymal stem cell-enriched pericardial scaffold), Myo-ATMSCs (adipose tissue mesenchymal stem cell-enriched myocardial scaffold). Reproduced with permission.[42] Copyright 2018, Springer Nature.
5.3. Clinical Advances
Clinical applications of dECM materials for cardiovascular treatment have generally been limited to repair or replacement of heart valves, large vasculature, and congenital heart defects.[146,147] These applications rely on patches and valves such as PhotoFix, CryoValve, SJM pericardial patch, and Tutopatch, with many additional products implemented extensively in clinical treatment. In contrast, tissue engineered dECM scaffolds and injectable materials have been studied extensively in vitro since 2008, with a modest number of in vivo rat studies of MI, and even less large animal studies. Two main dECM technologies are being evaluated in clinical testing—injectable cECM (VentriGel) and SIS cardiac patches (CorMatrix).
Injectable cECM therapy is the only soluble dECM therapy that has moved from in vitro analysis, to in vivo testing, and finally to clinical studies. This work has been developed since the original paper by Singelyn et al., which was followed immediately by several studies on cECM hydrogel analysis in vitro.[88] In vivo success through rat models (Singelyn et al., Wassenaar et al.) and porcine preclinical models (Singelyn et al., Seif-Naraghi et al.) have paved the way for a phase 1 clinical trial (NCT02305602) that will evaluate the effectiveness of VentriGel in treating MI.[132,133,141] Alternatively, the SIS-based solid dECM patch, CorMatrix, has found commercial use in carotid repair, pericardial reconstruction, and cardiac tissue repair, although cardiac closure is the most common use of the product. Studies by Mewhort et al. on both rat and porcine preclinical models have showed success in treating MI in animal models, with phase 1 clinical trials having completed in 2017 (NCT02887768), although results have not been published.[129,130,142]
Two studies on individual human patients have recently been published that use dECM materials for treatment of cardiovascular dysfunction. Avery et al. used human amniotic fluid-derived MSCs loaded within a micronized human liquid matrix derived from amniotic membrane for treatment of a 59 year old male patient with refractory angina, heart failure, and ischemic cardiomyopathy from chronic diabetes.[148] Laser guided transmyocardial revascularization (TMR) was performed on the LV walls and the MSC-liquid matrix was added to several of the TMR channels in the infarct. On day 6 postoperation, there was a large, stable, transmural infarct, and the patient showed reduction in LV EF. On day 27 postoperation, the patient showed improvement in LV wall thickness and EF of 34%. By day 91, the patient showed a decrease in angina and MRI showed confluence areas of healthy myocardium with improved wall thickening and EF of 40%. Thallium imaging of the infarct showed new areas of thallium labeling in the LV, suggesting formation of new, viable myocardium and ventricular remodeling. Ferng et al. implanted CorMatrix in a 62 year old male with history of diabetes, atrial fibrillation, class IV heart failure, and hypertension.[149] The patient underwent removal of a left ventricular explant device (LVAD) due to persistent infection, at which point a single CorMatrix scaffold was used to reconstruct the aortic graft anastomosis site and LV apex defect. After 3 months, the patient had worsening heart failure and underwent implantation of a new LVAD, at which time the CorMatrix graft was removed and evaluated histologically. The apex graft showed striations like native myocardium, presence of mature CMs, and fibrotic stroma possibly due to resolution of initial inflammation. Sections of apex graft, but not aortic graft, stained positive for HOP, a gene important in the developing heart. These results may support the hypothesis presented in the studies by Mewhort et al., where CorMatrix was thought to promote cardiac repair through remodeling. Regardless, these clinical studies have shed interesting findings on dECM based materials for heart repair, whether by improvement of cardiac function using MSC-loaded amniotic membrane or by evaluation of remodeling potential, but possibly limited therapeutic potential, of solid SIS grafts. With time, the results from current and future clinical trials may be most helpful in understanding how dECM can be used in cardiovascular tissue engineering for treatment of cardiovascular disease.
6. Challenges and Future Directions
6.1. Challenges and Considerations
dECM-based tissue engineering therapies for cardiovascular applications have been developed significantly since 2008, with many types of dECM, therapeutic methodologies, and combination systems moved toward preclinical testing. In addition, solid SIS patches (CorMatrix) and injectable cECM hydrogels (Ventrigel have moved toward clinical treatment of cardiovascular dysfunction, although challenges and considerations remain.[3,150]
Improvements in generating clinically effective dECM therapies include development of technologies to reduce problems such as sterilization, potential arrhythmia from dECM injection, requirement of surgical implantation of cardiac patches, and immune reactions from xenogeneic scaffolds. Harsh treatment methods such as detergents used to decellularize cECM scaffolds result in removal of not only cells, but of contaminant pathogens as well.[3,76] This sterilization is further improved in soluble scaffolds, where pepsin can remove most remnant pathogens and reduce the risk of infection. Although these methods result in nearly sterile materials, the only method of further sterilization in most studies is to maintain sterile conditions throughout the entire fabrication and implantation/injection process. Additional methods used for dECM applications, such as supercritical carbon dioxide application or antibiotic cocktails, may be beneficial when used with dECM for cardiovascular applications.[71,76]
Injectable dECM materials have benefits due to catheter implantation and direct injection within the infarct site, although these same benefits could be potential problems. Islands of noncontractile materials within the myocardium may induce arrhythmia and cause worsening cardiac function. While in vivo models show no arrhythmia with cECM systems, arrhythmia induction by injectable dECM materials must be evaluated with each new system.[141] Suarez et al. assessed the effects of PEG injection with varying degrees of interstitial spread on the cardiac electrophysiology of healthy and infarcted rat hearts.[151] While there was a delayed propagation of LV action potentials in hydrogel groups compared to saline or nontreated groups, hydrogels with high spread did not induce conduction problems, while low spread materials caused LV activation delay and reduced connexin density at the injection site. These results are mirrored in animal models of injectable cECM gels.
Conversely, cardiac patch treatment suffers from requirements of surgical attachment, which may not be feasible in all patients based on cardiac condition and age. Montgomery et al. developed shape memory polymer-based scaffolds for delivery through injection without thoracotomy.[152] While this advancement was made with synthetic materials, modifying dECM properties or combining dECM with shape memory polymers may mimic this effect. Another important factor is degradation of cardiac patches, as therapeutic timeframes for cardiovascular repair may require varying times of patch retention. Several studies have attempted to improve patch retention through crosslinking, biomaterial addition, or inhibition of MMP degradation. While injectable and patch-based dECM therapies rely on simple intervention for therapy, recellularization requires bioreactor designs for growth of functional myocardial tissue. Bioreactor issues such as oxygen/nutrient distribution through hearts, temperature control, system input response, and sterility all must be evaluated.[153]
Many sources of dECM for cardiovascular applications use xenogeneic materials, mainly from porcine sources, in generating dECM. Human tissue, especially cardiac-specific tissue, is much rarer, more expensive, and suffers from more patient-to-patient variability than xenograft dECM. However, nonhuman materials may cause increased inflammation and rejection after dECM treatment, due to the complex composition of ECM components that can activate immune cascades. Early studies evaluated dECM scaffolds in mice models and saw that exogenically implanted dECM induced acute inflammatory responses followed by chronic inflammation and ultimate graft necrosis and rejection.[154] However, most recent studies using in vivo models of dECM therapy in rats and pigs saw little immune response with implanted xenogeneic scaffolds and showed that degradation products from variety of dECM materials activate constructive M2 macrophage phenotype.[155,156] Mendoza-Novelo et al. showed that leaching of basement membrane components may be responsible for promoting faster macrophage stimulation and decreased inflammation. While dECM materials may not be inflammatory, the response with each new therapeutic material and device must be thoroughly evaluated. To allow for evaluation of human immune response without clinical treatment of patients, Wang et al. developed a humanized mouse model for improved testing of naturally derived biomaterial immune responses and compared injectable porcine or human cECM therapy using the mouse model.[157] Three weeks after injection, the allogenic matrix reduced total T helper cell infiltration compared to xenogeneic ECM, which was not observed in wild-type mice. In both models, the injected dECM materials showed constructive M2 macrophage infiltration and Th2 T helper cell polarization, indicating that both xenogeneic and allogenic dECM materials may be used in vivo without negative immune responses.
As therapy using dECM materials continues to improve in overcoming the challenges presented, answers to basic question about dECM materials in terms of their efficacy in tissue repair may continue to be uncovered. An important question is one asked for every tissue engineered therapy—what aspects of therapy, such as dECM sourcing, processing, fabrication methodology, or combination with cells/GFs, is clinically relevant? dECM materials have varying compositions and functionalities based on tissue source, age, species, and processing, all of which may or may not be important aspects that need to be controlled before treatment. Studies comparing overall dECM materials are limited, with very few studies investigating differences between cardiac specific dECM such as cECM and pECM.[42,144,158] Additional studies that compare the structural composition, cellular effects, and cardiac repair for several dECM materials are critical to uncover what aspects in dECM design are important. Even when comparing studies that evaluate composition of one dECM type, such as porcine cECM, protein analysis shows differences in dECM composition which confounds results. In combination therapies, the testing groups can be endless, with fabrication methodology, cell source, GF type, and secondary biomaterial adding to the list of testable variables. Regardless, with increasing value, number, and evaluation of studies involving dECM for cardiovascular tissue engineering, these questions may be answered more effectively.
6.2. Future Directions
dECM materials have shown effectiveness toward phase 1 clinical trials as both an injectable gel and cardiac patch. These pure dECM materials have improved cardiac functionality in large animal models post-MI or ischemia, with improved angiogenesis, reduction in inflammatory response, increased progenitor cell recruitment, and stimulation of positive remodeling. The numerous in vivo studies that combined pure dECM materials with cells, additional biomaterials, or paracrine factors and showed improvements in cardiac function compared to pure dECM materials point toward future directions being focused on combinational therapies, through novel formulations, generation methodologies, and therapeutic applications.
Countless studies have been undertaken involving dECM materials for treatment of diseases in tissues such as liver, brain, and skeletal muscle using novel sources of dECM, summarized for dECM hydrogels in Spang and Christman.[3] A limited amount of dECM materials have been investigated in cardiovascular applications, where testing of novel sources of dECM may be more beneficial than systems currently studied. For example, hpECM has only been evaluated in one study for cardiovascular repair with limited but observable functional improvements in murine treatment. However, one of the few clinical investigations on using cell-loaded dECM involved the use of amniotic fluid matrix, which may be functionally similar to hpECM, and showed recovery of cardiac function in a human patient for the first time using dECM materials. Additional sources of dECM may also be found in the heart, besides cECM and pECM. The epicardium has been shown to be critical in cardiac regeneration, with epicardium-resident cells and growth factors inducing both endogenous and therapeutic cardiac repair. Although technically difficult to isolate, epicardial-derived ECM may be an effective source of tissue specific dECM.
Similar to Bejleri et al.,[103] 3D printing methodologies have been employed to generate solid structures using dECM for other tissue applications, such as for skin and placenta.[159,160] These methods have high degrees of device design, effectively printing vasculature, location-specific GFs, and alternating layers and areas of cell and material composition. This control may be helpful in producing aligned dECM materials and patches, which can modulate CM induction and stem cell differentiation, especially for cardiac-specific stem cells. Aligned ECM scaffolds have been developed for vasculature applications and can be implemented for cardiac treatment.[161]
Several specific ECM components have been shown to induce differentiation of stem cells toward cardiogenic lineages, as well as directly used in treatment of cardiac dysfunction.[1,4,6,51] These regenerative components, such as agrin, periostin, fibronectin, sGAGs, and laminin, have been observed in dECM scaffolds used in cardiovascular repair.[10,14] While studies have not identified the specific ECM components in dECM materials are that drive cardiac improvements and stem cell differentiation, methods to analyze fractionalized dECM or combinational ECM components may prove helpful. Li et al. fractionalized SIS matrix by protein size to determine that a specific fraction of SIS matrices possess chemoattractant properties for ECs derived from liver, heart, and kidney tissue.[162] This method can be applied to cardiac-specific ECM to identify regenerative components. Combinatorial methods use multiple ECM-derived components in 2D or 3D to analyze which combination of materials induces functional changes in stem cells.[163–165] These systems can also be used to analyze components that may be beneficial to supplement dECM scaffolds and improve reparative outcomes. An example system is seen in a study by Turner et al. that incorporated HA with UBM to improve ESC-derived CM and EC function and alignment, where matrix component (HA) addition improved function of the dECM material.[165] The functional outcomes for cardiovascular treatment using additional regenerative matrix components along with dECM materials may be improved by fabrication of multilayer material systems or in materials with modulated mechanical properties.[166,167]
GF addition to biomaterials can significantly improve regenerative outcomes in vivo.[94,96] Functional improvement from GF loading is seen with dECM therapies for other tissues, such as for diabetic wound healing.[168] GFs induce tissue remodeling, reduce inflammation, enhance angiogenesis in vivo, and modulate stem cell response based on different GF levels.[169] dECM scaffold components contain high degrees of functional sites that bind GFs to sequester factors and modulate release rates, which would otherwise need to be tailored through functionalization in synthetic or non-ECM-based materials. As more and more studies incorporate GFs within scaffolds, functional improvements may increase in treating damaged cardiac tissue using dECM and dECM native GF-binding properties.[170] Exosomes, another type of regenerative paracrine factor released by cells, have recently gained critical interest in the cardiovascular field due to their powerful effects in driving cardiac repair with direct injection. Exosomes may be more beneficial compared to GF treatment, as exosomes show cardiac specific induction of repair through direct cellular phenotypic modification. Potentially, exosome loaded dECM materials may show further improvements in cardiac repair over other methods.
While dECM materials may be improved by developing new sources of dECM or by addition of purified regenerative ECM components, GFs, and/or exosomes, stem cell loading may show similar or improved therapeutic functionality compared to all other combination methods. Most studies using stem cells and dECM evaluate the differentiation of stem cells into functional CMs but fail to measure paracrine release profiles from stem cells. It is becoming clear that cardiac regeneration via stem cell therapy is driven by release of reparative cytokines, chemokines, growth factors, and exosomes, rather than direct differentiation and integration of implanted stem cells within the myocardium.[171] Compared to GF therapy, healthy cells produce a wide variety of GFs, exosomes, MMPs, and additional paracrine signals that would be difficult and expensive to recapitulate with purified paracrine components alone. Cell-loaded dECM therapy for cardiovascular disease has tremendous potential in effectively and specifically repairing damaged tissue and may be improved with additional biomaterial or GF incorporation.
7. Conclusions
dECM materials have found an important place in the treatment of cardiovascular disease through tissue engineering applications. In vitro analysis of dECM materials, developed as solid scaffolds and soluble hydrogels derived from a variety of tissue sources, has shown that dECM is a complex material composed of a multitude of key regenerative components, which in turn regulates cellular responses toward tissue repair. dECM materials can be generated and modified through many methods and sophisticated devices derived from dECM have been recently developed. In vivo models of treating MI with both solid and injectable dECM therapies have shown functional cardiac improvements in both small and large animal models, and dECM materials are moving toward treating human cardiovascular diseases where two products (Ventrigel and CorMatrix) are in clinical trials. While the results of these trials are still unknown, it is hopeful that these therapies move forward in the clinic, with a variety of additional therapies using dECM materials combined with secondary biomaterials, stem cells, and/or GFs following closely behind. dECM-based cardiovascular therapy has improved significantly in the past 10 years, with a growing number of studies pointed toward overcoming challenges, determining dECM biological effects, and generating sophisticated therapies for next generation cardiovascular tissue engineering.
Acknowledgements
D.B. was supported by the Cell and Tissue Engineering NIH Biotechnology Training Grant (T32 GM-008433).
Biographies

Donald Bejleri received his M.S. in biomedical engineering from the University of Florida in 2016. He is currently a Ph.D. candidate in the joint Coulter Department of Biomedical Engineering at Georgia Institute of Technology and Emory University, where he plans to graduate in 2020. His doctoral research, under the supervision of Dr. Michael Davis, focuses on developing bioprinted cardiac stem cell-extracellular matrix patches for cardiac repair and regeneration.

Michael E. Davis holds positions as an associate professor in both Cardiology and Biomedical Engineering at the Wallace H. Coulter Department of Biomedical Engineering at Georgia Tech and Emory University. Additionally, he serves as director of the Children’s Heart Research and Outcomes (HeRO) Center. He received his Ph.D. in molecular and systems pharmacology at Emory University in 2003 working on molecular regulation of eNOS expression by shear stress. From 2003 to 2006, he completed his postdoctoral fellowship at Brigham and Women’s Hospital working on cardiac tissue engineering with collaborators at the Massachusetts Institute of Technology. He moved back to Emory in 2006 to join the faculty of the Biomedical Engineering Department and Division of Cardiology.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Radisic M, Christman KL, Mayo Clin. Proc 2013, 88, 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Taylor DA, Sampaio LC, Ferdous Z, Gobin AS, Taite LJ, Acta Biomater. 2018, 74, 74. [DOI] [PubMed] [Google Scholar]
- [3].Spang MT, Christman KL, Acta Biomater. 2018, 68, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Garreta E, Oria R, Tarantino C, Pla-Roca M, Prado P, Fernandez-Aviles F, Campistol JM, Samitier J, Montserrat N, Mater. Today 2017, 20, 166. [Google Scholar]
- [5].Landa N, Miller L, Feinberg MS, Holbova R, Shachar M, Freeman I, Cohen S, Leor J, Circulation 2008, 117, 1388. [DOI] [PubMed] [Google Scholar]
- [6].Gaffney L, Wrona EA, Freytes DO, ACS Biomater. Sci. Eng 2018, 4, 1208. [Google Scholar]
- [7].Mecham R, The Extracellular Matrix: an Overview, Springer Science & Business Media, Berlin, Germany: 2011. [Google Scholar]
- [8].Valiente-Alandi I, Schafer AE, Blaxall BC, J. Mol. Cell. Cardiol 2016, 91, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kim BS, Kim H, Gao G, Jang J, Cho D-W, Biofabrication 2017, 9, 034104. [DOI] [PubMed] [Google Scholar]
- [10].Johnson TD, Hill RC, Dzieciatkowska M, Nigam V, Behfar A, Christman KL, Hansen KC, Proteomics: Clin. Appl 2016, 10, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rienks M, Papageorgiou A-P, Frangogiannis NG, Heymans S, Circ. Res 2014, 114, 872. [DOI] [PubMed] [Google Scholar]
- [12].Williams C, Black LD, in Biomaterials for Cardiac Regeneration (Eds: Suuronen EJ, Ruel M), Springer, New York City, USA: 2015, pp. 1–35. [Google Scholar]
- [13].Williams C, Quinn KP, Georgakoudi I, Black III LD, Acta Biomater. 2014, 10, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Silva A, Rodrigues S, Caldeira J, Nunes A, Sampaio-Pinto V, Resende T, Oliveira M, Barbosa M, Thorsteinsdóttir S, Nascimento D, Biomaterials 2016, 104, 52. [DOI] [PubMed] [Google Scholar]
- [15].Mishra PK, Givvimani S, Chavali V, Tyagi SC, Biochim. Biophys. Acta, Mol. Basis Dis 2013, 1832, 2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bronshtein T, Au-Yeung GCT, Sarig U, Nguyen EB-V, Mhaisalkar PS, Boey FYC, Venkatraman SS, Machluf M, Tissue Eng., Part C 2013, 19, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wen H, Bennett E, Epstein N, Plehn J, Magn. Reson. Med 2005, 54, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pislaru C, Urban MW, Pislaru SV, Kinnick RR, Greenleaf JF, Ultrasound Med. Biol 2014, 40, 1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hiesinger W, Brukman MJ, McCormick RC, Fitzpatrick III JR, Frederick JR, Yang EC, Muenzer JR, Marotta NA, Berry MF, Atluri P, J. Thorac. Cardiovasc. Surg 2012, 143, 962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ghista DN, Vayo WH, Sandler H, Med. Biol. Eng 1975, 13, 151. [DOI] [PubMed] [Google Scholar]
- [21].Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Circulation 2015, 131, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sullivan KE, Quinn KP, Tang KM, Georgakoudi I, Black LD, Stem Cell Res. Ther 2014, 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gershlak JR, Resnikoff JI, Sullivan KE, Williams C, Wang RM, Black LD, Biochem. Biophys. Res. Commun 2013, 439, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cleutjens JP, Creemers EE, Card J. Failure 2002, 8, S344. [DOI] [PubMed] [Google Scholar]
- [25].Bonnans C, Chou J, Werb Z, Nat. Rev. Mol. Cell Biol 2014, 15, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kwak H-B, J. Exercise Rehabil 2013, 9, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fan D, Takawale A, Lee J, Kassiri Z, Fibrog. Tissue Repair 2012, 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Spinale FG, Physiol. Rev 2007, 87, 1285. [DOI] [PubMed] [Google Scholar]
- [29].Kassiri Z, Khokha R, Thromb. Haemostasis 2005, 93, 212. [DOI] [PubMed] [Google Scholar]
- [30].Howard CM, Baudino TA, J. Mol. Cell. Cardiol 2014, 70, 19. [DOI] [PubMed] [Google Scholar]
- [31].Quinn KP, Sullivan KE, Liu Z, Ballard Z, Siokatas C, Georgakoudi I, Black LD, Sci. Rep 2016, 6, 35823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ye L, Zimmermann W-H, Garry DJ, Zhang J, Circ. Res 2013, 113, 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ota T, Gilbert TW, Schwartzman D, McTiernan CF, Kitajima T, Ito Y, Sawa Y, Badylak SF, Zenati MA, J. Thorac. Cardiovasc. Surg 2008, 136, 1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang B, Borazjani A, Tahai M, de Jongh Curry AL, Simionescu DT, Guan J, To F, Elder SH, Liao J, J. Biomed. Mater. Res., Part A 2010, 94, 1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhao Z-Q, Puskas JD, Xu D, Wang N-P, Mosunjac M, Guyton RA, Vinten-Johansen J, Matheny R, J. Am. Coll. Cardiol 2010, 55, 1250. [DOI] [PubMed] [Google Scholar]
- [36].Freytes DO, Martin J, Velankar SS, Lee AS, Badylak SF, Biomaterials 2008, 29, 1630. [DOI] [PubMed] [Google Scholar]
- [37].Ott HC, Matthiesen TS, Goh S-K, Black LD, Kren SM, Netoff TI, Taylor DA, Nat. Med 2008, 14, 213. [DOI] [PubMed] [Google Scholar]
- [38].Badylak SF, Freytes DO, Gilbert TW, Acta Biomater. 2009, 5, 1. [DOI] [PubMed] [Google Scholar]
- [39].Seif-Naraghi SB, Horn D, Schup-Magoffin PA, Madani MM, Christman KL, J. Cardiovasc. Transl. Res 2011, 4, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hong X, Yuan Y, Sun X, Zhou M, Guo G, Zhang Q, Hescheler J, Xi J, Cell. Physiol. Biochem 2018, 45, 319. [DOI] [PubMed] [Google Scholar]
- [41].Higuchi S, Lin Q, Wang J, Lim TK, Joshi SB, Anand GS, Chung MC, Sheetz MP, Fujita H, J. Biosci. Bioeng 2013, 115, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Perea-Gil I, Gálvez-Montón C, Prat-Vidal C, Jorba I, Segú-Vergés C, Roura S, Soler-Botija C, Iborra-Egea O, Revuelta-López E, Fernández MA, Sci. Rep 2018, 8, 6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].French KM, Boopathy AV, DeQuach JA, Chingozha L, Lu H, Christman KL, Davis ME, Acta Biomater. 2012, 8, 4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].DeQuach JA, Mezzano V, Miglani A, Lange S, Keller GM, Sheikh F, Christman KL, PLoS One 2010, 5, e13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ungerleider J, Johnson T, Rao N, Christman K, Methods 2015, 84, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tabuchi M, Negishi J, Yamashita A, Higami T, Kishida A, Funamoto S, Mater. Sci. Eng., C 2015, 56, 494. [DOI] [PubMed] [Google Scholar]
- [47].Merna N, Fung KM, Wang JJ, King CR, Hansen KC, Christman KL, George SC, Tissue Eng., Part A 2015, 21, 2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Johnson TD, DeQuach JA, Gaetani R, Ungerleider J, Elhag D, Nigam V, Behfar A, Christman KL, Biomater. Sci 2014, 2, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Momtahan N, Sukavaneshvar S, Roeder BL, Cook AD, Tissue Eng., Part B 2015, 21, 115. [DOI] [PubMed] [Google Scholar]
- [50].Agmon G, Christman KL, Curr. Opin. Solid State Mater. Sci 2016, 20, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Robertson MJ, Dries-Devlin JL, Kren SM, Burchfield JS, Taylor DA, PLoS One 2014, 9, e90406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Guyette JP, Charest JM, Mills RW, Jank BJ, Moser PT, Gilpin SE, Gershlak JR, Okamoto T, Gonzalez G, Milan DJ, Circ. Res 2016, 118, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sánchez PL, Fernández-Santos ME, Costanza S, Climent AM, Moscoso I, Gonzalez-Nicolas MA, Sanz-Ruiz R, Rodríguez H, Kren SM, Garrido G, Biomaterials 2015, 61, 279. [DOI] [PubMed] [Google Scholar]
- [54].Ng SL, Narayanan K, Gao S, Wan AC, Biomaterials 2011, 32, 7571. [DOI] [PubMed] [Google Scholar]
- [55].Weymann A, Patil NP, Sabashnikov A, Jungebluth P, Korkmaz S, Li S, Veres G, Soos P, Ishtok R, Chaimow N, PLoS One 2014, 9, e111591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hodgson MJ, Knutson CC, Momtahan N, Cook AD, Methods Mol. Biol 2017, 1, 95. [DOI] [PubMed] [Google Scholar]
- [57].Ferng AS, Connell AM, Marsh KM, Qu N, Medina AO, Bajaj N, Palomares D, Iwanski J, Tran PL, Lotun K, Johnson K, Khalpey Z, J. Clin. Transl. Res 2017, 3, 260. [PMC free article] [PubMed] [Google Scholar]
- [58].Weymann A, Loganathan S, Takahashi H, Schies C, Claus B, Hirschberg K, Soós P, Korkmaz S, Schmack B, Karck M, Circ. J 2011, 75, 852. [DOI] [PubMed] [Google Scholar]
- [59].Schulte JB, Simionescu A, Simionescu DT, Tissue Eng., Part C 2013, 19, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schwan J, Kwaczala AT, Ryan TJ, Bartulos O, Ren Y, Sewanan LR, Morris AH, Jacoby DL, Qyang Y, Campbell SG, Sci. Rep 2016, 6, 32068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang Q, Yang H, Bai A, Jiang W, Li X, Wang X, Mao Y, Lu C, Qian R, Guo F, Biomaterials 2016, 105, 52. [DOI] [PubMed] [Google Scholar]
- [62].Lu T-Y, Lin B, Kim J, Sullivan M, Tobita K, Salama G, Yang L, Nat. Commun 2013, 4, 2307. [DOI] [PubMed] [Google Scholar]
- [63].de Castro Brás LE, Ramirez TA, DeLeon-Pennell KY, Chiao YA, Ma Y, Dai Q, Halade GV, Hakala K, Weintraub ST, Lindsey ML, J. Proteomics 2013, 86, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hülsmann J, Aubin H, Kranz A, Godehardt E, Munakata H, Kamiya H, Barth M, Lichtenberg A, Akhyari P, Artif J. Organs 2013, 16, 294. [DOI] [PubMed] [Google Scholar]
- [65].Methe K, Bäckdahl H, Johansson BR, Nayakawde N, Dellgren G, Sumitran-Holgersson S, BioRes. Open Access 2014, 3, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Momtahan N, Poornejad N, Struk JA, Castleton AA, Herrod BJ, Vance BR, Eatough JP, Roeder BL, Reynolds PR, Cook AD, Tissue Eng., Part C 2015, 21, 1148. [DOI] [PubMed] [Google Scholar]
- [67].Kitahara H, Yagi H, Tajima K, Okamoto K, Yoshitake A, Aeba R, Kudo M, Kashima I, Kawaguchi S, Hirano A, Interact. Cardiovasc. Thorac. Surg 2016, 22, 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lee P-F, Chau E, Cabello R, Yeh AT, Sampaio LC, Gobin AS, Taylor DA, Acta Biomater 2017, 49, 181. [DOI] [PubMed] [Google Scholar]
- [69].Remlinger NT, Wearden PD, Gilbert TW, J. Visualized Exp 2012, 70, e50059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang B, Tedder ME, Perez CE, Wang G, de Jongh Curry AL, To F, Elder SH, Williams LN, Simionescu DT, Liao J, J. Mater. Sci.: Mater. Med 2012, 23, 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Seo Y, Jung Y, Kim SH, Acta Biomater 2018, 67, 270. [DOI] [PubMed] [Google Scholar]
- [72].Merna N, Robertson C, La A, George SC, Tissue Eng., Part C 2013, 19, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Godier-Furnémont AF, Martens TP, Koeckert MS, Wan L, Parks J, Arai K, Zhang G, Hudson B, Homma S, Vunjak-Novakovic G, Proc. Natl. Acad. Sci. USA 2011, 108, 7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Schmuck EG, Mulligan JD, Ertel RL, Kouris NA, Ogle BM, Raval AN, Saupe KW, Cardiovasc. Eng. Technol 2014, 5, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Suhaeri M, Subbiah R, Van SY, Du P, Kim IG, Lee K, Park K, Tissue Eng., Part A 2015, 21, 1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fidalgo C, Iop L, Sciro M, Harder M, Mavrilas D, Korossis S, Bagno A, Palù G, Aguiari P, Gerosa G, Acta Biomater. 2018, 67, 282. [DOI] [PubMed] [Google Scholar]
- [77].Crawford B, Koshy ST, Jhamb G, Woodford C, Thompson CM, Levy AS, Rush JW, Guillemette JG, Lillicrap D, Jervis E, Can. J. Chem. Eng 2012, 90, 1457. [Google Scholar]
- [78].Eitan Y, Sarig U, Dahan N, Machluf M, Tissue Eng., Part C 2010, 16, 671. [DOI] [PubMed] [Google Scholar]
- [79].Rajabi S, Pahlavan S, Ashtiani MK, Ansari H, Abbasalizadeh S, Sayahpour FA, Varzideh F, Kostin S, Aghdami N, Braun T, Biomaterials 2018, 154, 99. [DOI] [PubMed] [Google Scholar]
- [80].Yasui H, Lee J-K, Yoshida A, Yokoyama T, Nakanishi H, Miwa K, Naito AT, Oka T, Akazawa H, Nakai J, Biomaterials 2014, 35, 7839. [DOI] [PubMed] [Google Scholar]
- [81].Oberwallner B, Brodarac A, Anić P, Šarić T, Wassilew K, Neef K, Choi Y-H, Stamm C, Eur. J. Cardiothorac. Surg 2015, 47, 416. [DOI] [PubMed] [Google Scholar]
- [82].Garreta E, De Oñate L, Fernández-Santos ME, Oria R, Tarantino C, Climent AM, Marco A, Samitier M, Martínez E, Valls-Margarit M, Biomaterials 2016, 98, 64. [DOI] [PubMed] [Google Scholar]
- [83].Sarig U, Nguyen EB-V, Wang Y, Ting S, Bronshtein T, Sarig H, Dahan N, Gvirtz M, Reuveny S, Oh SK, Tissue Eng., Part A 2015, 21, 1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chang CW, Petrie T, Clark A, Lin X, Sondergaard CS, Griffiths LG, PLoS One 2016, 11, e0153412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhang L, Qian Z, Tahtinen M, Qi S, Zhao F, Tissue Eng J. Regener. Med 2018, 12, e1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pagano F, Angelini F, Castaldo C, Picchio V, Messina E, Sciarretta S, Maiello C, Biondi-Zoccai G, Frati G, Meglio F. d., Stem Cells Int. 2017, 2017, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Singelyn JM, DeQuach JA, Seif-Naraghi SB, Littlefield RB, Schup-Magoffin PJ, Christman KL, Biomaterials 2009, 30, 5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wang RM, Christman KL, Adv. Drug Delivery Rev 2016, 96, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Shevach M, Zax R, Abrahamov A, Fleischer S, Shapira A, Dvir T, Biomed. Mater 2015, 10, 034106. [DOI] [PubMed] [Google Scholar]
- [90].Johnson TD, Lin SY, Christman KL, Nanotechnology 2011, 22, 494015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Singelyn JM, Christman KL, Macromol. Biosci 2011, 11, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wassenaar JW, Braden RL, Osborn KG, Christman KL, J. Mater. Chem. B 2016, 4, 2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Grover GN, Rao N, Christman KL, Nanotechnology 2014, 25, 014011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Seif-Naraghi SB, Horn D, Schup-Magoffin PJ, Christman KL, Acta Biomater. 2012, 8, 3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sonnenberg SB, Rane AA, Liu CJ, Rao N, Agmon G, Suarez S, Wang R, Munoz A, Bajaj V, Zhang S, Biomaterials 2015, 45, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Park DS, Mewhort HE, Teng G, Belke D, Turnbull J, Svystonyuk D, Guzzardi D, Kang S, Fedak PW, Tissue Eng., Part A 2018, 24, 128. [DOI] [PubMed] [Google Scholar]
- [97].Williams C, Sullivan K, Black III LD, Adv. Healthcare Mater 2015, 4, 1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hoganson DM, Owens GE, Meppelink AM, Bassett EK, Bowley CM, Hinkel CJ, Finkelstein EB, Goldman SM, Vacanti JP, J. Biomed. Mater. Res., Part A 2016, 104, 1728. [DOI] [PubMed] [Google Scholar]
- [99].Kappler B, Anic P, Becker M, Bader A, Klose K, Klein O, Oberwallner B, Choi Y-H, Falk V, Stamm C, J. Mater. Sci.: Mater. Med 2016, 27, 120. [DOI] [PubMed] [Google Scholar]
- [100].Gaetani R, Feyen DA, Verhage V, Slaats R, Messina E, Christman KL, Giacomello A, Doevendans PA, Sluijter JP, Biomaterials 2015, 61, 339. [DOI] [PubMed] [Google Scholar]
- [101].Pati F, Jang J, Ha D-H, Kim SW, Rhie J-W, Shim J-H, Kim D-H, Cho D-W, Nat. Commun 2014, 5, 3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Jang J, Kim TG, Kim BS, Kim S-W, Kwon S-M, Cho D-W, Acta Biomater. 2016, 33, 88. [DOI] [PubMed] [Google Scholar]
- [103].Bejleri D, Gaetani R, Streeter BW, Nachlas ALY, Brown ME, Christman KL, Davis ME, Adv. Healthcare Mater 2018, 7, 1800672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Sarig U, Sarig H, Gora A, Krishnamoorthi MK, Au-Yeung GCT, de-Berardinis E, Chaw SY, Mhaisalkar P, Bogireddi H, Ramakrishna S, Sci. Rep 2018, 8, 3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Sullivan K, Black L, Tissue Eng., Part A 2015, 21, S69. [Google Scholar]
- [106].Gershlak JR, Black LD, Exp. Cell Res 2015, 330, 311. [DOI] [PubMed] [Google Scholar]
- [107].French KM, Maxwell JT, Bhutani S, Ghosh-Choudhary S, Fierro MJ, Johnson TD, Christman KL, Taylor WR, Davis ME, Stem Cells Int. 2016, 2016, 8364382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Baghalishahi M, Efthekhar-Vaghefi SH, Piryaei A, Nematolahi-Mahani SN, Mollaei HR, Sadeghi Y, Biochem. Biophys. Res. Commun 2018, 502, 215. [DOI] [PubMed] [Google Scholar]
- [109].Gaetani R, Yin C, Srikumar N, Braden R, Doevendans PA, Sluijter JP, Christman KL, Cell Transplant. 2016, 25, 1653. [DOI] [PubMed] [Google Scholar]
- [110].Rajabi-Zeleti S, Jalili-Firoozinezhad S, Azarnia M, Khayyatan F, Vahdat S, Nikeghbalian S, Khademhosseini A, Baharvand H, Aghdami N, Biomaterials 2014, 35, 970. [DOI] [PubMed] [Google Scholar]
- [111].Fong AH, Romero-López M, Heylman CM, Keating M, Tran D, Sobrino A, Tran AQ, Pham HH, Fimbres C, Gershon PD, Tissue Eng., Part A 2016, 22, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Jeffords M, Wu J, Shah M, Hong Y, Zhang G, ACS. Appl. Mater. Inter 2015, 7, 11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Duan Y, Liu Z, O’Neill J, Wan LQ, Freytes DO, Vunjak-Novakovic G, J. Cardiovasc. Transl. Res 2011, 4, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Becker M, Maring JA, Schneider M, Herrera Martin AX, Seifert M, Klein O, Braun T, Falk V, Stamm C, Int. J. Mol. Sci 2018, 19, 1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Williams C, Budina E, Stoppel WL, Sullivan KE, Emani S, Emani SM, BlackIII LD, Acta Biomater. 2015, 14, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Pok S, Benavides OM, Hallal P, Jacot JG, Tissue Eng., Part A 2014, 20, 1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Pok S, Stupin IV, Tsao C, Pautler RG, Gao Y, Nieto RM, Tao ZW, Fraser CD Jr., Annapragada AV, Jacot JG, Adv. Healthcare Mater 2017, 6, 1600549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Pourfarhangi KE, Mashayekhan S, Asl SG, Hajebrahimi Z, Biologicals 2018, 53, 10. [DOI] [PubMed] [Google Scholar]
- [119].Efraim Y, Sarig H, Anavy NC, Sarig U, de Berardinis E, Chaw S-Y, Krishnamoorthi M, Kalifa J, Bogireddi H, Duc TV, Acta Biomater. 2017, 50, 220. [DOI] [PubMed] [Google Scholar]
- [120].Stoppel WL, Hu D, Domian IJ, Kaplan DL, Black III LD, Biomed. Mater 2015, 10, 034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Stoppel WL, Gao AE, Greaney AM, Partlow BP, Bretherton RC, Kaplan DL, Black III LD, J. Biomed. Mater. Res., Part A 2016, 104, 3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Jakus AE, Laronda MM, Rashedi AS, Robinson CM, Lee C, Jordan SW, Orwig KE, Woodruff TK, Shah RN, Adv. Funct. Mater 2017, 27, 1700992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Jang J, Park H-J, Kim S-W, Kim H, Park JY, Na SJ, Kim HJ, Park MN, Choi SH, Park SH, Biomaterials 2017, 112, 264. [DOI] [PubMed] [Google Scholar]
- [124].Roshanbinfar K, Hilborn J, Varghese OP, Oommen OP, RSC Adv. 2017, 7, 319808. [Google Scholar]
- [125].Wainwright JM, Hashizume R, Fujimoto KL, Remlinger NT, Pesyna C, Wagner WR, Tobita K, Gilbert TW, Badylak SF, Cells Tissues Organs 2012, 195, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Sarig U, Sarig H, de-Berardinis E, Chaw S-Y, Nguyen EB, Ramanujam VS, Thang VD, Al-Haddawi M, Liao S, Seliktar D, Acta Biomater. 2016, 44, 209. [DOI] [PubMed] [Google Scholar]
- [127].Wan L, Chen Y, Wang Z, Wang W, Schmull S, Dong J, Xue S, Imboden H, Li J, Sci. Rep 2017, 7, 39988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].D’Amore A, Yoshizumi T, Luketich SK, Wolf MT, Gu X, Cammarata M, Hoff R, Badylak SF, Wagner WR, Biomaterials 2016, 107, 1. [DOI] [PubMed] [Google Scholar]
- [129].Mewhort HE, Turnbull JD, Meijndert HC, Ngu JM, Fedak PW, J. Thorac. Cardiovasc. Surg 2014, 147, 1650. [DOI] [PubMed] [Google Scholar]
- [130].Mewhort HE, Svystonyuk DA, Turnbull JD, Teng G, Belke DD, Guzzardi DG, Park DS, Kang S, Hollenberg MD, Fedak PW, JACC: Basic Transl. Sci 2017, 2, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Rane AA, Chuang JS, Shah A, Hu DP, Dalton ND, Gu Y, Peterson KL, Omens JH, Christman KL, PLoS One 2011, 6, e21571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Singelyn JM, Sundaramurthy P, Johnson TD, Schup-Magoffin PJ, Hu DP, Faulk DM, Wang J, Mayle KM, Bartels K, Salvatore M, J. Am. Coll. Cardiol 2012, 59, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Wassenaar JW, Gaetani R, Garcia JJ, Braden RL, Luo CG, Huang D, DeMaria AN, Omens JH, Christman KL, J. Am. Coll. Cardiol 2016, 67, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Francis MP, Breathwaite E, Bulysheva AA, Varghese F, Rodriguez RU, Dutta S, Semenov I, Ogle R, Huber A, Tichy A-M, Acta Biomater. 2017, 52, 92. [DOI] [PubMed] [Google Scholar]
- [135].Toeg HD, Tiwari-Pandey R, Seymour R, Ahmadi A, Crowe S, Vulesevic B, Suuronen EJ, Ruel M, Ann. Thorac. Surg 2013, 96, 1686. [DOI] [PubMed] [Google Scholar]
- [136].Ravi S, Caves JM, Martinez AW, Xiao J, Wen J, Haller CA, Davis ME, Chaikof EL, Biomaterials 2012, 33, 7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Chen WC, Wang Z, Missinato MA, Park DW, Long DW, Liu H-J, Zeng X, Yates NA, Kim K, Wang Y, Sci. Adv 2016, 2, e1600844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Ota T, Gilbert TW, Badylak SF, Schwartzman D, Zenati MA, J. Thorac. Cardiovasc. Surg 2007, 133, 979. [DOI] [PubMed] [Google Scholar]
- [139].Potapova IA, Doronin SV, Kelly DJ, Rosen AB, Schuldt AJ, Lu Z, Kochupura PV, Robinson RB, Rosen MR, Brink PR, Am. J. Physiol.: Heart Circ. Physiol 2008, 295, H2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Kochupura PV, Azeloglu EU, Kelly DJ, Doronin SV, Badylak SF, Krukenkamp IB, Cohen IS, Gaudette GR, Circulation 2005, 112, I144. [DOI] [PubMed] [Google Scholar]
- [141].Seif-Naraghi SB, Singelyn JM, Salvatore MA, Osborn KG, Wang JJ, Sampat U, Kwan OL, Strachan GM, Wong J, Schup-Magoffin PJ, Sci. Transl. Med 2013, 5, 173ra125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Mewhort HE, Turnbull JD, Satriano A, Chow K, Flewitt JA, Andrei A-C, Guzzardi DG, Svystonyuk DA, White JA, Fedak PW, J. Heart Lung Transplant 2016, 35, 661. [DOI] [PubMed] [Google Scholar]
- [143].Scully BB, Fan C, Grigoryan B, Jacot JG, Vick III G, Kim JJ, Fraser CD Jr., Grande-Allen KJ, Morales DL, J. Biomed. Mater. Res., Part B 2016, 104, 1713. [DOI] [PubMed] [Google Scholar]
- [144].Nezhad ZM, Poncelet A, Fervaille C, Gianello P, J. Thorac. Cardiovasc. Surg 2017, 67, 44. [DOI] [PubMed] [Google Scholar]
- [145].Zhang G-W, Gu T-X, Guan X-Y, Sun X-J, Qi X, Li X-Y, Wang X-B, Yu L, Jiang D-Q, Tang R, Biomed. Mater 2015, 10, 065018. [DOI] [PubMed] [Google Scholar]
- [146].Parmaksiz M, Dogan A, Odabas S, Elçin AE, Elçin YM, Biomed. Mater 2016, 11, 022003. [DOI] [PubMed] [Google Scholar]
- [147].Neethling WM, Strange G, Firth L, Smit FE, Interact. Cardiovasc. Thorac. Surg 2013, 17, 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Avery RJ, Yu S, Cherukuri G, Runyan RB, Konhilas J, Khalpey ZI, ASAIO J 2018, 64, e130. [DOI] [PubMed] [Google Scholar]
- [149].Ferng A, Connell A, Nunez M, Johnson K, Braunhut B, Lick S, Desai A, Kazui T, Runyan R, Khalpey Z, Ann. Thorac. Surg 2017, 104, e239. [DOI] [PubMed] [Google Scholar]
- [150].Ogle BM, Bursac N, Domian I, Huang NF, Menasché P, Murry CE, Pruitt B, Radisic M, Wu JC, Wu SM, Sci. Transl. Med 2016, 8, 342ps313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Suarez SL, Rane AA, Muñoz A, Wright AT, Zhang SX, Braden RL, Almutairi A, McCulloch AD, Christman KL, Acta Biomater 2015, 26, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Montgomery M, Ahadian S, Huyer LD, Rito ML, Civitarese RA, Vanderlaan RD, Wu J, Reis LA, Momen A, Akbari S, Nat. Mater 2017, 16, 1038. [DOI] [PubMed] [Google Scholar]
- [153].Singelyn JM, Christman KL, J. Cardiovasc. Transl. Res 2010, 3, 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Allman AJ, McPherson TB, Badylak SF, Merrill LC, Kallakury B, Sheehan C, Raeder RH, Metzger DW, Transplantation 2001, 71, 1631. [DOI] [PubMed] [Google Scholar]
- [155].Sicari BM, Dziki JL, Siu BF, Medberry CJ, Dearth CL, Badylak SF, Biomaterials 2014, 35, 8605. [DOI] [PubMed] [Google Scholar]
- [156].Mendoza-Novelo B, Castellano LE, Padilla-Miranda RG, Lona-Ramos MC, Cuéllar-Mata P, Vega-González A, Murguía-Pérez M, Mata-Mata JL, Ávila EE, J. Biomed. Mater. Res., Part A 2016, 104, 2810. [DOI] [PubMed] [Google Scholar]
- [157].Wang RM, Johnson TD, He J, Rong Z, Wong M, Nigam V, Behfar A, Xu Y, Christman KL, Biomaterials 2017, 129, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Kuo S, Kim HM, Wang Z, Bingham EL, Miyazawa A, Marcelo CL, Feinberg SE, Tissue Eng J. Regener. Med 2018, 12, 983. [DOI] [PubMed] [Google Scholar]
- [159].Kim BS, Kwon YW, Kong J-S, Park GT, Gao G, Han W, Kim M-B, Lee H, Kim JH, Cho D-W, Biomaterials 2018, 168, 38. [DOI] [PubMed] [Google Scholar]
- [160].Kuo C-Y, Eranki A, Placone JK, Rhodes KR, Aranda-Espinoza H, Fernandes R, Fisher JP, Kim PC, ACS Biomater. Sci. Eng 2016, 2, 1817. [DOI] [PubMed] [Google Scholar]
- [161].Xing Q, Qian Z, Tahtinen M, Yap AH, Yates K, Zhao F, Adv. Healthcare Mater 2017, 6, 1601333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Li F, Li W, Johnson S, Ingram D, Yoder M, Badylak SF, Endothelium 2004, 11, 199. [DOI] [PubMed] [Google Scholar]
- [163].Flaim CJ, Chien S, Bhatia SN, Nat. Methods 2005, 2, 119. [DOI] [PubMed] [Google Scholar]
- [164].Hou L, Coller J, Natu V, Hastie TJ, Huang NF, Acta Biomater. 2016, 44, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Turner WS, Wang X, Johnson S, Medberry C, Mendez J, Badylak SF, McCord MG, McCloskey KE, J. Biomed. Mater. Res. B 2012, 100B, 2060. [DOI] [PubMed] [Google Scholar]
- [166].Dai B, Pan Q, Li Z, Zhao M, Liao X, Wu K, Ma X, Stem Cells Int. 2016, 2016, 4796578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Yahalom-Ronen Y, Rajchman D, Sarig R, Geiger B, Tzahor E, eLife 2015, 4, e07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Yan W, Liu H, Deng X, Jin Y, Wang N, Chu J, J. Tissue Eng. Regener. Med 2018, 12, e1461. [DOI] [PubMed] [Google Scholar]
- [169].Almeida HV, Mulhall KJ, O’Brien FJ, Kelly DJ, Tissue Eng J. Regener. Med 2017, 11, 2979. [DOI] [PubMed] [Google Scholar]
- [170].Shi C, Zhao Y, Yang Y, Chen C, Hou X, Shao J, Yao H, Li Q, Xia Y, Dai J, Biomater. Sci 2018, 6, 356. [DOI] [PubMed] [Google Scholar]
- [171].Gnecchi M, Zhang Z, Ni A, Dzau VJ, Circ. Res 2008, 103, 1204. [DOI] [PMC free article] [PubMed] [Google Scholar]