

Abstract
Prized for their ability to reliably forge stereocenters with precise regiocontrol from simple and abundant starting materials, substrate-directable enantioselective reactions are widely used in modern organic synthesis. As such, enantioselective C(sp3)−H functionalization reactions directed by innate functional groups could provide new routes to introduce molecular complexity within the inert hydrocarbon moiety, but so far this approach has been met with little success. While free primary aliphatic amines are common, versatile intermediates in synthesis, they are traditionally unreactive in C(sp3)−H activation reactions. Herein we report the Pd-catalyzed enantioselective C(sp3)−H functionalization of free aliphatic amines (cyclopropylmethylamines) enabled by a chiral bidentate thioether ligand. This ligand’s privileged bidentate coordination mode and thioether motif favor the generation of the requisite mono(amine)-Pd(II) intermediate, thus enabling the enantioselective C−H activation of free amines. The resulting C−Pd(II) species could engage in either Pd(II)/Pd(IV) or Pd(II)/Pd(0) catalytic cycles, enabling access to a diverse range of products through (hetero)arylation, carbonylation and olefination reactions. Consequently, this versatile reactivity offers medicinal chemists a general strategy to rapidly prepare and functionalize biologically relevant amines.
Graphical Abstract
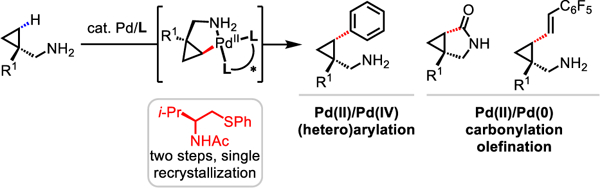
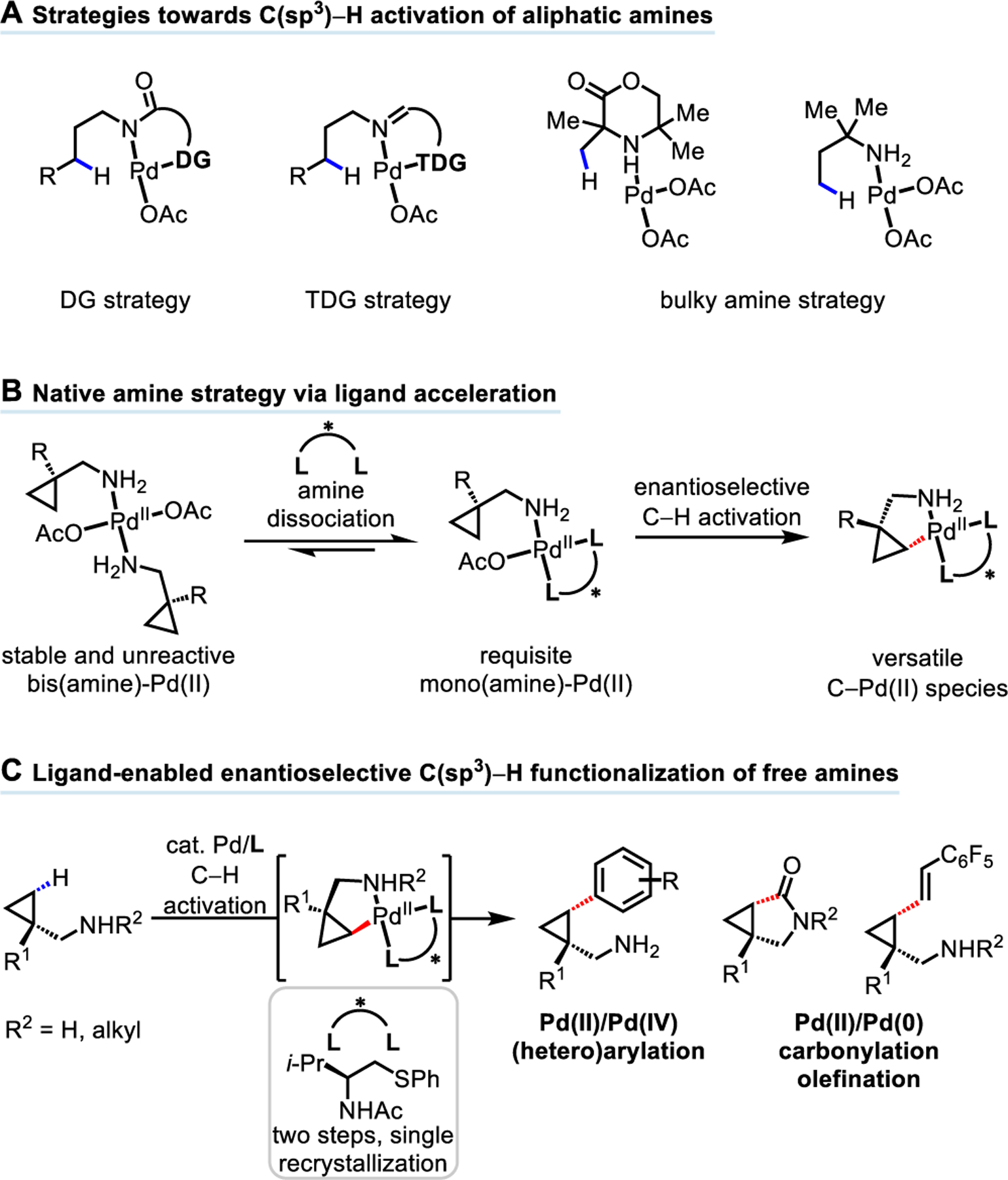
Ligand-controlled enantioselective reactions directed by innate functional groups (e.g. carboxylic acids, amines, and alcohols) comprise a classic toolkit to control the regio- and stereochemical outcomes of organic reactions1, as exemplified by the Sharpless’s oxidation2, Noyori and Knowles’s hydrogenation3,4 reactions of alkenes. While such strategies highlight significant developments in the functionalization of unsaturated moieties, only recently has serious attention been directed toward complementary strategies for the functionalization of saturated hydrocarbons5. Despite advances in C−H functionalization directed by weakly coordinating free carboxylic acids6, aliphatic amines bearing free NH2 groups − ubiquitous structural motifs among compounds of pharmaceutical, agrochemical, and societal importance − are less amenable to Pd-catalyzed C−H functionalization reactions7. This is due to the formation of stable but unreactive bis(amine)-Pd(II) species, stemming from the strong metal binding properties of the amine8. Additionally, free amines are susceptible to oxidative degradation through β-hydride elimination and N-substitution reactions towards electrophiles under the conditions of C−H activation. In this context, extensive studies have been focused on the design of preinstalled9 or transient10 bidentate directing groups (DGs) on account of their ability to accelerate cyclometalation and suppress β-hydride elimination (Scheme 1A). Despite the elegance of DG or TDG strategies, these reactions are difficult to render enantioselective due to the persistent racemic background reactions driven by strong coordination, as well as the lack of available coordination sites for a chiral ligand. Bidentate coordination from the substrate also confines the reactivity to a Pd(II)/Pd(IV) catalytic cycle, owing to the lack of a vacant coordination site to engage the coupling partner in a Pd(II)/Pd(0) cycle. Most importantly, the requirement for substrate DGs precludes the use of native amino groups to direct C−H activation, which affords superior atom and step economies in the context of protecting-group-free synthesis. In fact, reported free aliphatic amine-directed C−H functionalization reactions are largely limited to bulky amines; a bulky group at the α position of the free amine is required to favor dissociation of bis(amine)-Pd(II) complexes through steric repulsion, hamper oxidative degradation by blocking the β position, as well as weaken the nucleophilicity of amino group (Scheme 1A)11.
Scheme 1.
C(sp3)−H Functionalization of Aliphatic Amines
In light of our approach using the combination of monodentate practical DGs and bidentate ligands to enable enantioselective C−H activation reactions12, we were particularly interested in applying this concept to the otherwise challenging chemical feedstocks such as aliphatic amines (Scheme 1B). We reasoned that due to the bidentate chelation mode of the ligand, unreactive bis(amine)-Pd(II) species would readily dissociate as a result of strong coordination from the ligand, generating the desired reactive mono(amine)-Pd(II) intermediate for cyclopalladation. The bidentate ligand also creates a rigid framework that is ideal for the transfer of chiral information to the substrate during C−H activation. Moreover, the privileged amidate group (N-acyl) on the ligand can not only participate as an internal base to facilitate C(sp3)−H activation over β-hydride elimination but is also labile enough to generate a vacant coordination site for coupling partners used in Pd(II)/Pd(0) chemistry. Herein we report the design of a thioether-based chiral bidentate ligand that enables Pd(II)-catalyzed enantioselective C−H functionalization reactions of free cyclopropylmethylamines (Scheme 1C). Arylation via a Pd(II)/Pd(IV) catalytic cycle afforded chiral free aliphatic amines bearing aryl groups, including aza-heterocycles, at the γ-position. Preliminary results also showed that this catalyst enables the olefination and carbonylation reactions of primary and secondary free amines through a Pd(II)/Pd(0) catalytic cycle, providing valuable γ-olefinated free amines and γ-lactams respectively.
Our previous research has disclosed two enantioselective C(sp3)−H functionalization reactions using a monodentate triflamide DG: the γ-C(sp3)−H arylation of cyclopropylmethylamine enabled by a mono-N-protected amino acid (MPAA) ligand13 and the γ-C(sp3)−H cross-coupling of 3-aminopentane enabled by acetyl-protected aminomethyl oxazoline ligands (APAO) ligand14. Bidentate picolinamide-directed benzylic C(sp3)−H arylation has also been reported using a BINOL phosphoric acid ligand with limited scope15. Due to the widespread presence of cyclopropyl moieties among drug and agrochemical molecules16, we selected free cyclopropylmethylamine 1a as a model substrate for our initial investigations. Upon extensive studies of the reaction conditions in the absence of ligand, a combination of Pd(TFA)2, 4-iodotoluene 2a, Ag2O, and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) solvent produced a 6% 1H NMR yield of the desired cis-γ-arylation product 3a with a considerable amount of the degradation product cyclopropane aldehyde. To test our hypothesis, chiral bidentate ligands that previously promoted enantioselective C(sp3)−H activation, including MPAA L15c,13, O-methylhydroxamic acid L212c, dimethylamine L36b, oxazoline L412b,14, and quinoline L512a, were investigated under the standard reaction conditions (Table 1). To our delight, these ligands generally promoted the arylation reaction, delivering the arylation product 3a in 30% to 40% yield with MPAA ligand L1 being the superior ligand (40% yield, 85:15 er). However, further modifications of the ligand structure and optimization of the reaction conditions failed to improve the yield and er. Therefore, we turned our attention to using thioethers as soft σ-donors, believing that thioethers might facilitate dissociation of bis(amine)-Pd(II) complexes, as well as stabilize the Pd catalysts through their strong coordination. The racemic ligand L7 developed for the olefination of free carboxylic acids17 significantly improved the reaction to 57% yield (see Supporting Information Table S5). Through systematic modifications of the backbone of the thioether ligand we found that introduction of an isopropyl group gave optimal reactivity with 97.5:2.5 er in 68% yield (see Supporting Information Table S5). Notably, this optimal thioether ligand L6 could be easily synthesized in two steps with a single recrystallization in 92% yield from the commercially available Evans’ oxazolidinone chiral auxiliary (see Supporting Information preparation of ligand section).
Table 1.
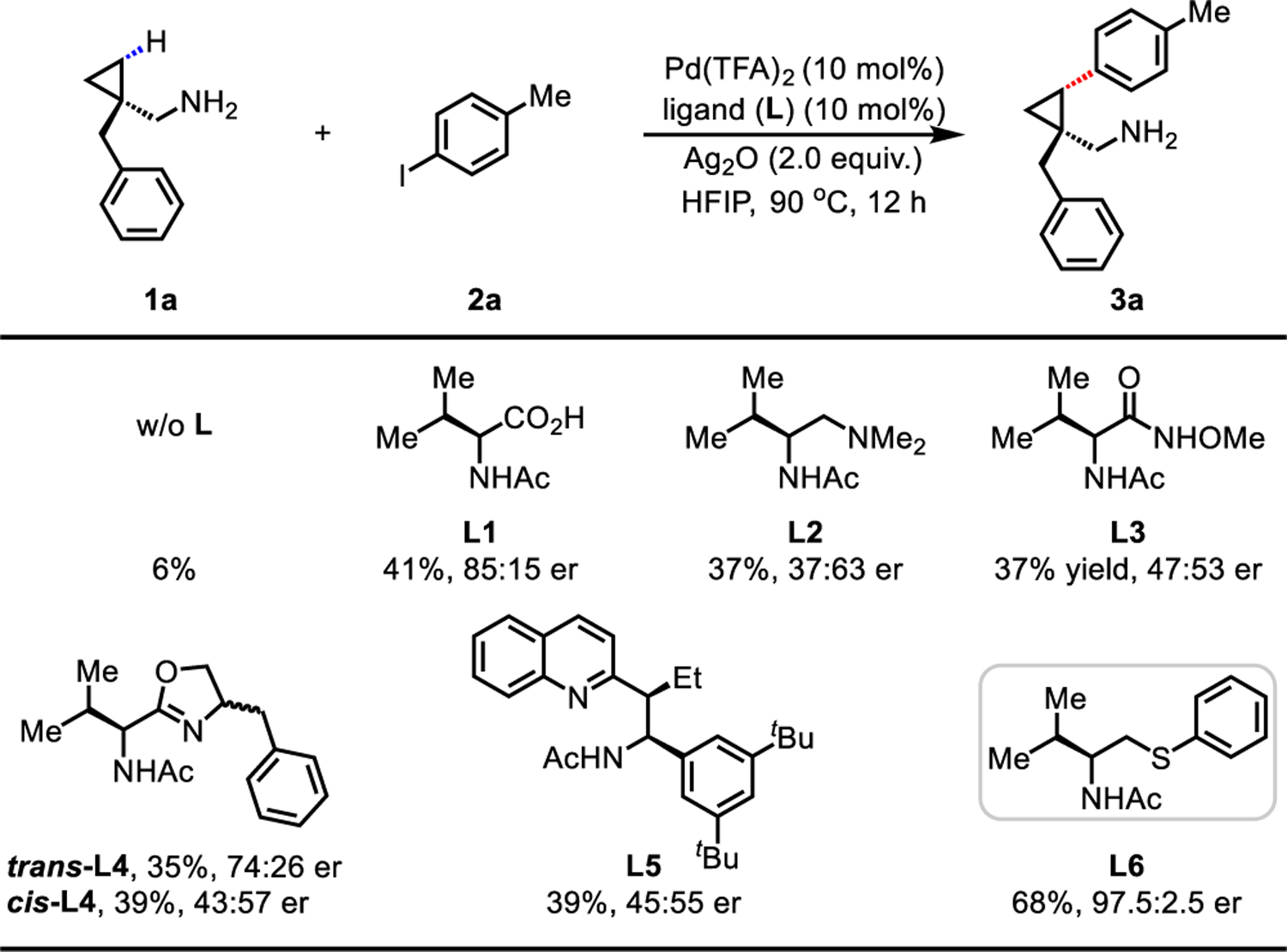 |
Conditions: 1a (0.1 mmol), Pd(TFA)2 (10 mol%), ligand(L) (10 mol%), 4-iodotoluene 2a (2.0 equiv.), Ag2O (2.0 equiv.), HFIP (0.2 mL), 90 °C, 12 h.
The yields were determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard. The er values of the corresponding Boc-protected amine 3a’ were determined on the SFC system using commercially available chiral columns.
With the optimal ligand and conditions in hand, we next explored the scope of aliphatic amines (Table 2). The γ-arylated products were isolated in the form of tert-butyloxycarbonyl (Boc) or benzoyl (Bz)-protected amines for their ease of purification and analysis. Cyclopropylmethylamines (1b−1g) bearing various aliphatic chains at the β position, including cyclobutanes (1e), cyclopentanes (1f), and cyclohexanes (1g), were all compatible with the optimal protocol, affording the arylation products (3b−3g) in yields ranging from modest to good (54% to 70%) with excellent ers (greater than 97.5:2.5). A range of functionalities such as fluoro (3i), trifluoromethyl (3j), tetrahydropyran (3h), benzyl (Bn)-protected hydroxyl (3k), methoxy (3l), and phenolic ether (3m) were all well tolerated, and products were obtained in moderate yields (45% to 66%) and high ers (greater than 96:4). Phenyl groups (3a and 3n−3s) were compatible under the current conditions, and remained intact despite the potentially reactive aryl C(sp2)−H or benzylic C(sp3)−H bonds. A range of substituents on the aryl ring from electron-withdrawing (fluoro and chloro) (3n, 3o, and 3p) to electron-donating (methoxy) (3q) groups were also well tolerated, providing arylation products in good yields (up to 73%) and excellent ers (up to 99:1 er). The less reactive unsubstituted cyclopropylmethylamine 1t also reacted in a synthetically useful yield (42%) and correspondingly high er (96.5:3.5). Extension of this desymmetrization protocol to isobutylamine and 3-pentanamine also afforded desired arylation products in good yields and moderate ers (see Supporting Information Table S9). However, cyclobutylmethylamine substrates are not reactive under these conditions.
Table 2.
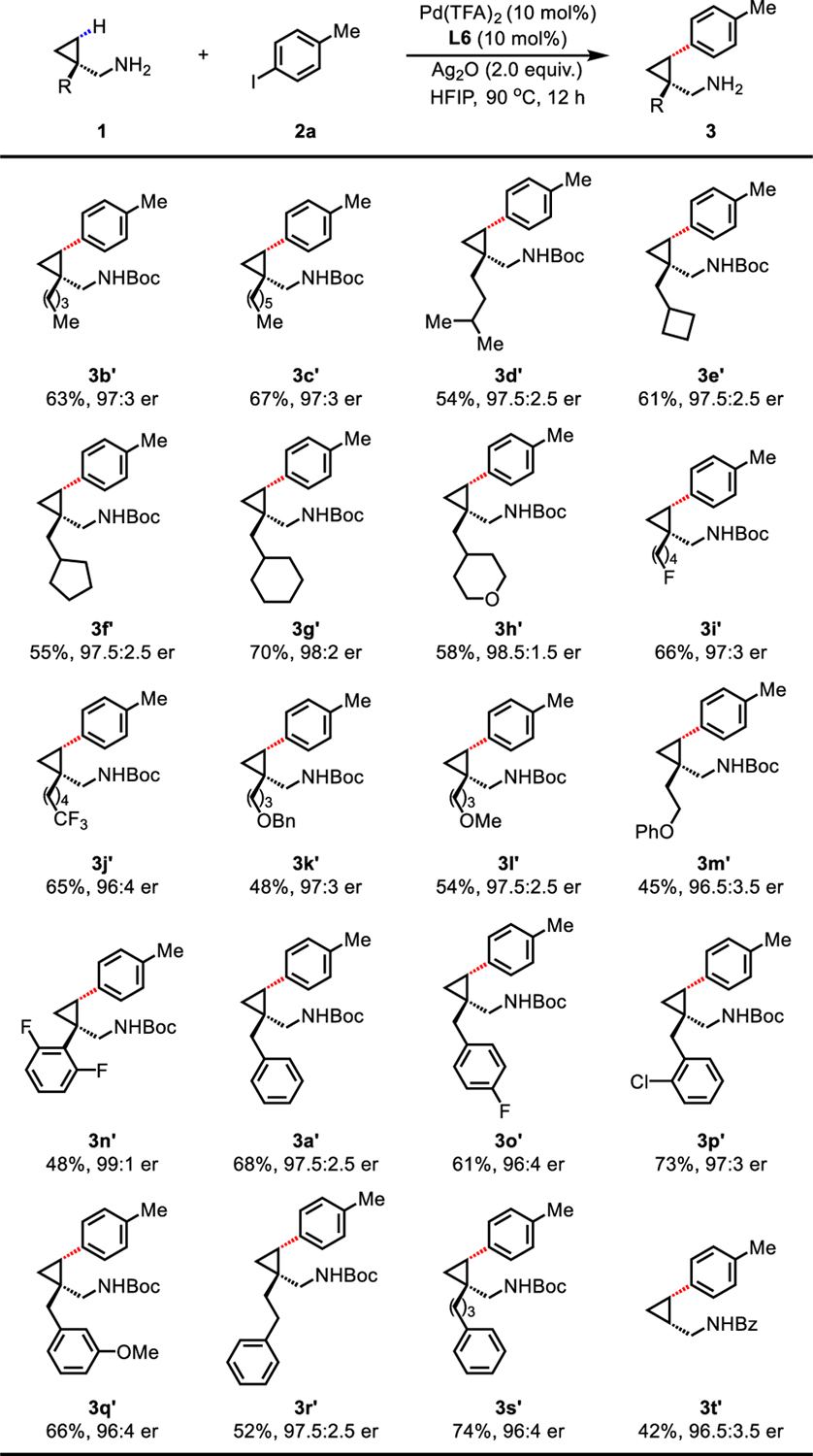 |
Conditions: 1 (0.1 mmol), Pd(TFA)2 (10 mol%), L6 (10 mol%), 4-iodotoluene 2a (3.0 equiv.), Ag2O (2.0 equiv.), HFIP (0.2 mL), 90 °C, 12 h.
Isolated yields of the corresponding Bz or Boc-protected amines. The er values were determined on the SFC system using commercially available chiral columns.
Next, we surveyed the scope of aryl and heteroaryl iodides using 1a as the model substrate (Table 3). γ-Arylation of 1a with simple iodobenzene 2b resulted in good yield (71%) and excellent er (97.5:2.5). A wide range of para substituents on the aryl iodides from electron-donating (Me and isopropyl) to electron-withdrawing (CF3, ester, and halide) groups were well tolerated, delivering γ-arylated products (3a and 4c−4g) with high ers (greater than 96:4 er) in moderate to good yields (46% to 72%). Meta-substituted aryl iodides bearing methyl or coordinating OCF3 and NO2 groups consistently provided useful yields (51% to 68%) and ers greater than 94.5:5.5 (4h−4j). Indeed, the above products featured additional reactive groups such as esters (in 4e) and halogens (chloro in 4g) that could serve as useful synthetic handles for subsequent derivatizations. Given the ubiquity of heteroaromatics, especially pyridine-containing heterocycles, in small molecule drug discovery18, we examined the reactivity of aza-heteroaryl iodides under the aforementioned conditions. Although heteroaryl iodides were incompatible with the previously reported MPAA-enabled arylation of triflamide13, we reasoned that when switching to a strongly coordinating thioether ligand, catalyst poisoning by coordination of these aza-heterocycles might be dramatically suppressed. While unsubstituted 4-iodopyridine 2t failed to give any desired arylation product under the standard conditions, we were pleased to find that a broad range of 2-substituted iodopyridines (2k−2r) proceeded smoothly with excellent ers (up to 97.5:2.5). A variety of functional groups including fluoro (4k), chloro (4l and 4m), bromo (4n), methoxy (4o), and CF3 (4p−4r) were compatible with the optimal conditions, yielding valuable cis-γ-heteroaryl cyclopropylmethylamines. Electron-rich 5-iodoindole 2s was also a suitable coupling partner, affording product 4s in moderate yield (51%) with excellent er (96.5:3.5).
Table 3.
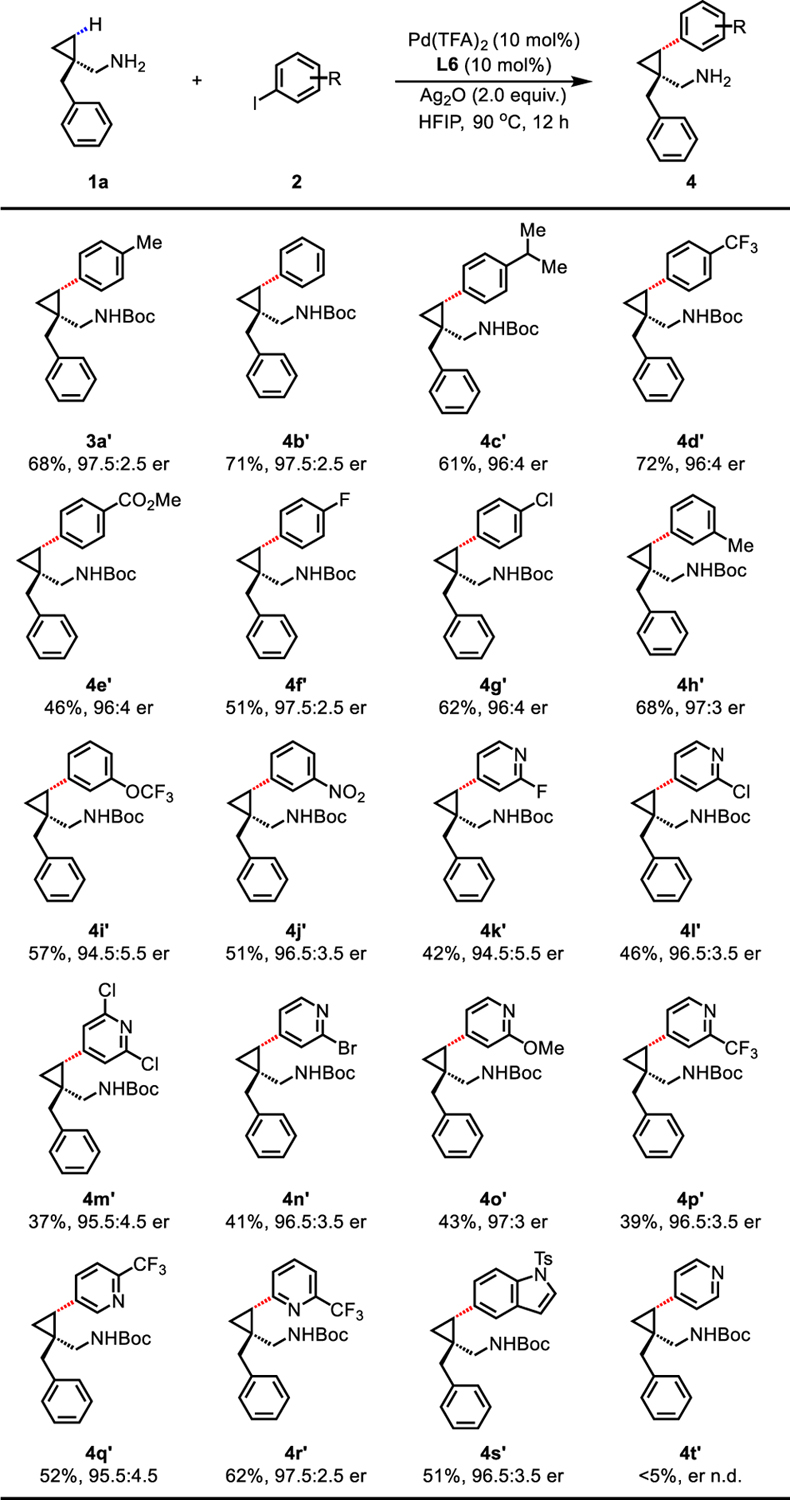 |
Conditions: 1a (0.1 mmol), Pd(TFA)2 (10 mol% or 15 mol% for heteroaryl iodides), L6 (10 mol% % or 15 mol% for heteroaryl iodides), aryl or heteroaryl iodide 2 (3.0 equiv.), Ag2O (2.0 equiv.), HFIP (0.2 mL), 90 °C, 12 h.
Isolated yields of the corresponding Boc-protected amines. The er values were determined on the SFC system using commercially available chiral columns.
To test the feasibility of extending this Pd(II)/thioether catalyst system to a Pd(II)/Pd(0) catalytic cycle, we embarked on the enantioselective carbonylation and olefination of free primary and secondary aliphatic amines (Table 4). Reported works on C(sp3)−H carbonylation19 and olefination20 are largely limited to DG strategies, and no asymmetric version has been achieved to date. Using Mo(CO)6 as a nonhazardous and air-stable solid source of CO, carbonylation of primary amine 1a delivered bicyclic γ-lactam 5a in a synthetically useful yield (45%) and moderate er (89.5:10.5). A range of secondary amines (1u−1x) were also reactive using the optimal protocol, affording the valuable cyclopropane fused pyrrolidones (5b−5e) in moderate yields (up to 63%) and good ers (up to 97:3 er). Olefination of primary amines (1a and 1i) and secondary amines (1u−1w) with pentafluorostyrene proceeded smoothly, delivering the γ-olefinated aliphatic amines (6a−6e) with good to excellent ers (94:6 to 98.5:1.5). Under current conditions, other olefins such as acrylate and styrene failed to deliver desired olefination product.
Table 4.
Free Primary and Secondary Cyclopropylmethylamine Scope for γ-C(sp3)−H Carbonylation and Olefinationa,b
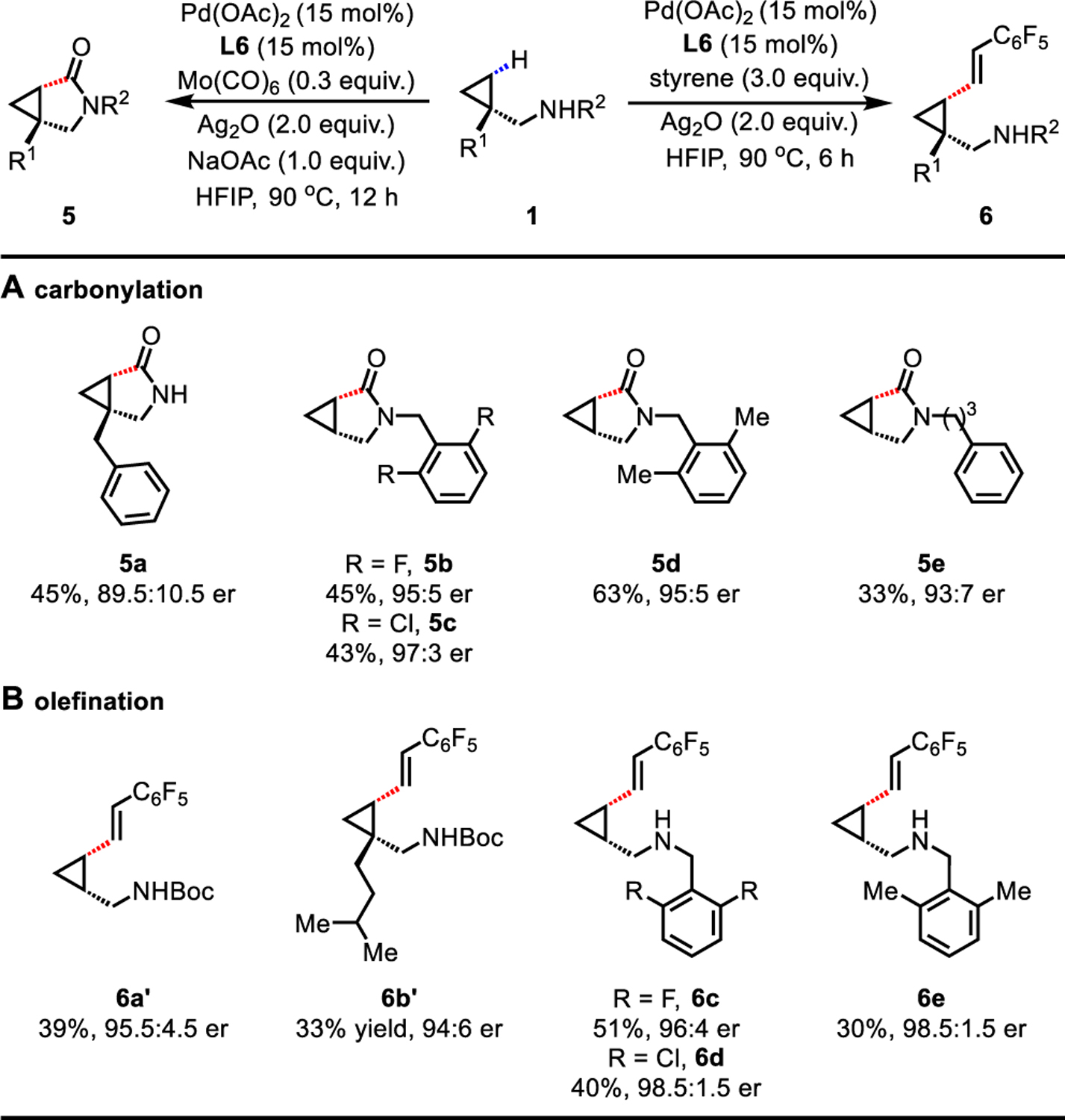 |
Conditions for carbonylation: 1 (0.1 mmol), Pd(OAc)2 (15 mol%), L6 (15 mol%), Mo(CO)6 (0.3 equiv.), Ag2O (2.0 equiv.), NaOAc (1.0 equiv.), HFIP (0.1 mL), 90 °C, 12 h. Conditions for olefination: 1 (0.1 mmol), Pd(OAc)2 (15 mol%), L6 (15 mol%), pentafluorostyrene (3.0 equiv.), Ag2O (2.0 equiv.), HFIP (0.1 mL), 90 °C, 6 h.
Isolated yields for secondary amines or isolated yields of the corresponding Boc-protected amines for primary amines. The er values were determined on the SFC system using commercially available chiral columns.
In summary, we have realized effective and general Pd-catalyzed enantioselective C(sp3)−H functionalizations of free aliphatic amines enabled by a single Pd(II) catalyst bearing a bidentate chiral thioether ligand. Both Pd(II)/Pd(IV) and Pd(II)/Pd(0) catalytic cycles are compatible with this chiral ligand, as demonstrated by (hetero)arylation, carbonylation and olefination reactions respectively. A broad range of chiral γ-(hetero)aryl, γ-olefinated free amines and γ-lactams could be easily accessed without installing exogenous directing groups.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge The Scripps Research Institute and NIH (NIGMS, R01GM084019) for financial support. Z. Z. thanks Alastair N. Herron for proofreading.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Full experimental details and characterization of new compounds (PDF)
Notes
The authors declare no competing financial interests.
REFERENCES
- (1).Hoveyda AH; Evans DA; Fu GC Substrate-directable chemical reactions. Chem. Rev 1993, 93, 1307–1370. [Google Scholar]
- (2).Katsuki T; Sharpless KB The first practical method for asymmetric epoxidation. J. Am. Chem. Soc 1980, 102, 5974–5976. [Google Scholar]
- (3).Noyori R Asymmetric catalysis: science and opportunities (Nobel lecture). Angew. Chem., Int. Ed 2002, 41, 2008–2022. [PubMed] [Google Scholar]
- (4).Knowles WS Asymmetric hydrogenations (Nobel lecture). Angew. Chem., Int. Ed 2002, 41, 1998–2007. [PubMed] [Google Scholar]
- (5). For reviews, see:; (a) Giri R; Shi B-F; Engle KM; Maugel N; Yu J-Q Transition metal-catalyzed C−H activation reactions: diastereoselectivity and enantioselectivity. Chem. Soc. Rev 2009, 38, 3242–3272. [DOI] [PubMed] [Google Scholar]; (b) Newton CG; Wang S-G; Oliveira CC; Cramer N Catalytic enantioselective transformations involving C−H bond cleavage by transition-metal complexes. Chem. Rev 2017, 117, 8908–8976. [DOI] [PubMed] [Google Scholar]; (c) Shao Q; Wu K; Zhuang Z; Qian S; Yu J-Q From Pd(OAc)2 to chiral catalysts: the discovery and development of bifunctional mono-N-protected amino acid ligands for diverse C−H activation reactions. Acc. Chem. Res 2020, 53, 833–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Shi B-F; Zhang Y-H; Lam JK; Wang D-H; Yu J-Q Pd(II)-catalyzed enantioselective C−H olefination of diphenylacetic acids. J. Am. Chem. Soc 2010, 132, 460–461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shen P-X; Hu L; Shao Q; Hong K; Yu J-Q Pd(II)-catalyzed enantioselective C(sp3)−H arylation of free carboxylic acids. J. Am. Chem. Soc 2018, 140, 6545–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Giri R; Maugel N; Li J-J; Wang D-H; Breazzano SP; Saunder LB; Yu J-Q Palladium-catalyzed methylation and arylation of sp2 and sp3 C−H bonds in simple carboxylic acids. J. Am. Chem. Soc 2007, 129, 3510–3511. [DOI] [PubMed] [Google Scholar]; (d) Zhuang Z; Yu J-Q Lactonization as a general route to β-C(sp3)−H functionalization. Nature 2020, 577, 656–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7). For reviews, see:; (a) Daugulis O; Roane J; Tran LD Bidentate, monoanionic auxiliary-directed functionalization of carbon−hydrogen bonds. Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He G; Wang B; Nack WA; Chen G Syntheses and transformations of α-amino acids via palladium-catalyzed auxiliary directed sp3 C−H Functionalization. Acc. Chem. Res 2016, 49, 635–645. [DOI] [PubMed] [Google Scholar]
- (8).Ryabov AD Mechanisms of intramolecular activation of C−H bonds in transition-metal complexes. Chem. Rev 1990, 90, 403–424. [Google Scholar]
- (9).(a) Zaitsev VG; Shabashov D; Daugulis O Highly regioselective arylation of sp3 C−H bonds catalyzed by palladium acetate. J. Am. Chem. Soc 2005, 127, 13154–13155. [DOI] [PubMed] [Google Scholar]; (b) He G; Chen G A practical strategy for the structural diversification of aliphatic scaffolds through the palladium-catalyzed picolinamide-directed remote functionalization of unactivated C(sp3)−H Bonds. Angew. Chem. Int. Ed 2011, 50, 5192–5196. [DOI] [PubMed] [Google Scholar]; (c) Chan KSL; Wasa M; Chu L; Laforteza BN; Miura M; Yu J-Q Ligand-enabled cross-coupling of C(sp3)−H bonds with arylboron reagents via Pd(II)/Pd(0) catalysis. Nat. Chem 2014, 6, 146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Liu Y; Ge H Site-selective C−H arylation of primary aliphatic amines enabled by a catalytic transient directing group. Nat. Chem 2017, 9, 26–32. [Google Scholar]; (b) Wu Y; Chen Y-Q; Liu T; Martin DE; Yu J-Q Pd-catalyzed γ-C(sp3)−H arylation of free amines using a transient directing group. J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li B; Seth K; Niu B; Pan L; Yang H; Ge H Transient-ligand-enabled ortho-arylation of five-membered heterocycles: facile access to mechanochromic materials. Angew. Chem., Int. Ed 2018, 57, 3401–3405. [DOI] [PubMed] [Google Scholar]; (d) Li B; Lawrence B; Li G; Ge H Ligand-controlled direct γ-C−H arylation of aldehydes. Angew. Chem., Int. Ed 2020, 59, 3078–3082. [DOI] [PubMed] [Google Scholar]
- (11).(a) Lazareva A; Daugulis O Direct palladium-catalyzed ortho-arylation of benzylamines. Org. Lett 2006, 8, 5211–5213. [DOI] [PubMed] [Google Scholar]; (b) McNally A; Haffemayer B; Collins BSL; Gaunt MJ Palladium-catalysed C−H activation of aliphatic amines to give strained nitrogen heterocycles. Nature 2014, 510, 129–133. [DOI] [PubMed] [Google Scholar]; (c) Calleja J; Pla D; Gorman TW; Domingo V; Haffemayer B; Gaunt MJ A steric tethering approach enables palladium-catalysed C−H activation of primary amino alcohols. Nat. Chem 2015, 7, 1009–1016. [DOI] [PubMed] [Google Scholar]; (d) Chen K; Wang D; Li Z-W; Liu Z; Pan F; Zhang Y-F; Shi Z-J Palladium catalyzed C(sp3)−H acetoxylation of aliphatic primary amines to γ-amino alcohol derivatives. Org. Chem. Front 2017, 4, 2097–2101. [Google Scholar]
- (12).(a) Chen G; Gong W; Zhuang Z; Andrä MS; Chen Y-Q; Hong X; Yang Y-F; Liu T; Houk KN; Yu J-Q Ligand-accelerated enantioselective methylene C(sp3)−H bond activation. Science 2016, 353, 1023–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu Q-F; Shen P-X; He J; Wang X-B; Zhang F; Shao Q; Zhu R-Y; Mapelli C; Qiao JX; Poss MA; Yu J-Q Formation of α-chiral centers by asymmetric β-C(sp3)−H arylation, alkenylation, and alkynylation. Science 2017, 355, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xiao K-J; Lin DW; Miura M; Zhu R-Y; Gong W; Wasa M; Yu J-Q Palladium(II)-catalyzed enantioselective C(sp3)−H activation using a chiral hydroxamic acid ligand. J. Am. Chem. Soc 2014, 136, 8138–8142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chan KSL; Fu H-Y; Yu J-Q Palladium(II)-catalyzed highly enantioselective C−H arylation of cyclopropylmethylamines. J. Am. Chem. Soc 2015, 137, 2042–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shao Q; Wu Q-F; He J; Yu J-Q Enantioselective γ-C(sp3)−H activation of alkyl amines via Pd(II)/Pd(0) catalysis. J. Am. Chem. Soc 2018, 140, 5322–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang H; Tong HR; He G; Chen G An enantioselective bidentate auxiliary directed palladium-catalyzed benzylic C−H arylation of amines using a BINOL phosphate ligand. Angew. Chem., Int. Ed 2016, 55, 15387–15391. [DOI] [PubMed] [Google Scholar]
- (16).Rubin M; Rubina M; Gevorgyan V Transition metal chemistry of cyclopropenes and cyclopropanes. Chem. Rev 2007, 107, 3117–3179. [DOI] [PubMed] [Google Scholar]
- (17).Zhuang Z; Yu C-B; Chen G; Wu Q-F; Hsiao Y; Joe CL; Qiao JX; Poss MA; Yu J-Q Ligand-enabled β-C(sp3)−H olefination of free carboxylic acids. J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen Y-Q; Wang Z; Wu Y; Wisniewski SR; Qiao JX; Ewing WR; Eastgate MD; Yu J-Q Overcoming the limitations of γ- and δ-C−H arylation of amines through ligand development. J. Am. Chem. Soc 2018, 140, 17884–17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Wang C; Zhang L; Chen C; Han J; Yao Y; Zhao Y Oxalyl amide assisted palladium-catalyzed synthesis of pyrrolidones via carbonylation of γ-C(sp3)−H bonds of aliphatic amine substrates. Chem. Sci 2015, 6, 4610–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang P-L; Li Y; Wu Y; Li C; Lan Q; Wang X-S Pd-catalyzed C(sp3)−H carbonylation of alkylamines: a powerful route to γ-lactams and γ-amino acids. Org. Lett 2015, 17, 3698–3701. [DOI] [PubMed] [Google Scholar]; (c) Hernando E; Villalva J; Martínez ÁM; Alonso I; Rodríguez N; Arrayás RG; Carretero JC Palladium-catalyzed carbonylative cyclization of amines via γ-C(sp3)−H activation: late-stage diversification of amino acids and peptides. ACS Catal 2016, 6, 6868–6882. [Google Scholar]
- (20).Jiang H; He J; Liu T; Yu J-Q Ligand-enabled γ-C(sp3)−H olefination of amines: en route to pyrrolidines. J. Am. Chem. Soc 2016, 138, 2055–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.