

Abstract
Cycloaddition reactions provide an expeditious route to construct ring systems in a highly convergent and stereoselective manner. For a typical cycloaddition reaction to occur, however, the installation of multiple reactive functional groups (π-bonds, leaving group, etc.) are required within the substrates, compromising the overall efficiency or scope of the cycloaddition reaction. Here, we report a palladium-catalyzed [3+2] reaction that utilizes two-fold C(sp3)–H activation to generate the three-carbon unit for formal cycloaddition. The initial β-C(sp3)–H activation of aliphatic amide, followed by maleimide insertion, triggers a relayed, second C(sp3)–H activation to complete a formal [3+2] cycloaddition. The key to success was the use of weakly coordinating amide as the directing group, as previous studies have shown that Heck or alkylation pathways are preferred when stronger-coordinating directing groups are used with maleimide coupling partners. To promote the amide-directed C(sp3)–H activation step, the use of pyridine-3-sulfonic acid ligands is crucial. This method is compatible with a wide range of amide substrates, including lactams, which lead to spiro-bicyclic products. The [3+2] product is also shown to undergo a reductive desymmetrization process to access chiral cyclopentane bearing multiple stereocenters with excellent enantioselectivity.
Graphical Abstract
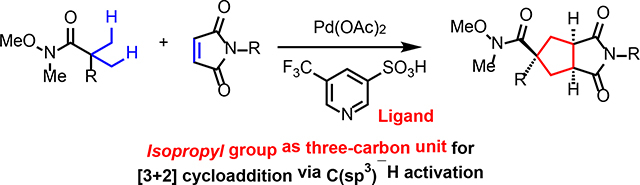
Synthetic methods that allow the facile construction of cyclic structures are of great importance in organic synthesis. In particular, cycloaddition reactions can serve as powerful tools to expedite the synthesis of ring-containing molecules,1 as witnessed by the numerous applications of the [4+2] Diels-Alder reaction in the total synthesis of natural products.2 While the direct analogue of the Diels-Alder reaction for the synthesis of cyclopentanes would be a [3+2] reaction, the generation of an appropriate three-carbon unit and the subsequent control of its reactivity for cycloaddition is significantly challenging (Scheme 1A).3 Indeed, methods that directly generate free trimethylenemethane (TMM) intermediates as three-carbon units for [3+2] reactions require specifically designed substrates with limited applicability.4 Strategies to generate TMM-equivalents with transition metal catalysts have also been developed.5 For instance, Trost and coworkers pioneered the use of Pd-TMM complexes as intermediates for various [3+2] reactions,6 which has been further expanded to asymmetric variants for the preparation of chiral cyclopentanes.7 However, the generation of these TMM-equivalents still requires the precursors to be highly activated, either by ring-strain or multiple pre-functionalization. While other elegant cycloaddition approaches for cyclopentane synthesis have also been developed,8 multiple π-bonds or leaving groups are required to be installed within the substrates. Given the abundance of five-membered rings in complex natural products and bioactive molecules,9 a complementary [3+2] method that can directly use simple, abundant substrates, such as carboxylic acids or amides, would be highly attractive.
Scheme 1.

[3+2] Cycloaddition reaction
Pd-catalyzed diverse C–H functionalization can provide transformative disconnections via directly converting inert C–H bonds into versatile C–Pd bonds.10 In this regard, we questioned whether C(sp3)–H activation can be applied to allow a simple, unfunctionalized three-carbon propane skeleton to be used for [3+2] cycloaddition, where the C(sp3)–H bonds of a gem-dimethyl moiety are formally stitched into a double bond (Scheme 1B). Inspired by the norbornene-relayed 1,2-C(sp2)–H activation,11 we envisioned that an analogous olefin-relayed two-fold 1,3-C(sp3)–H activation process could be harnessed to achieve the desired [3+2] cycloaddition. However, it is noteworthy that while the norbornene-relay involves a favorable five-membered cyclopalladation with C(sp2)–H bonds, the proposed 1,3-C(sp3)–H activation process would require a six-membered cyclopalladation with inert C(sp3)–H bonds, which is highly challenging.
To test this hypothesis, we turned our attention to using maleimide as the relaying olefin instead of norbornene for two reasons. First, the resulting product from maleimide would have significant synthetic utility compared to the resulting product from norbornene. Second, maleimides are reactive cyclic olefins that have been used as coupling partners for Pd-catalyzed C(sp3)–H functionalization. However, the key maleimide-inserted intermediate (I) can preferably undergo undesired formal β-hydride elimination (Heck product II) or protonation (alkylation product III) pathways instead of the relayed C(sp3)–H activation pathway (Scheme 1C). For instance, the reaction between pivalic acid and maleimide has been demonstrated to exclusively form the Heck product via formal β-hydride elimination.12 Also, it has been shown that the formation of net alkylation product via a [C(sp3)–Pd] protonation pathway is favored with strongly coordinating bidentate directing groups.13 These divergent reactivities imply that the fate of the maleimide-inserted species (I) can be altered by employing either a new directing group or a ligand. We speculated that using a weakly coordinating directing group might promote the relayed C(sp3)–H activation due to the more facile release of the Pd center from the directing group, which would be essential for the Pd center to re-orient for the second C(sp3)–H activation. In this sense, our recent development of the pyridinesulfonic acid ligand allowed us to exploit the weakly coordinating carbonyl group of amides as the directing group.14 To our delight, using amide substrates led to the exclusive formation of [3+2] products (V), presumably via the intermediacy of the dialkylpalladacycle intermediate (IV). Here, we report the development of a catalytic [3+2] cycloaddition between amide substrates and maleimides that operates via Pd-catalyzed two-fold C(sp3)–H activation.
We first evaluated the ligand effect for the reaction between pivalic amide 1 and N-(4-nitrophenyl)maleimide (Table 1). Without using any ligand, 10% yield of the cyclopentane product was observed along with trace amounts of the Heck product. While using N-acetylglycine (L1) or pyridone (L2) ligands led to improved results compared to the ligand-free conditions, the use of pyridine-3-sulfonic acid (L3) as the ligand gave the desired product in significantly increased yield. In accordance with the previous result in the Pd-catalyzed C(sp3)–H olefination reaction,14b more electron-deficient L4 proved to be the optimal ligand for the [3+2] reaction. As shown in Table 1, the Heck product was only observed as the minor product in all cases, indicating that the [3+2] pathway is predominant regardless of the nature of the ligand. It is also noteworthy that when pivalic acid was subjected to the identical [3+2] reaction conditions with L4, only the Heck product was observed, implying that the use of the weakly coordinating amide directing group is crucial to achieve the [3+2] pathway (See Supplementary information). The high level of diastereoselectivity observed in the [3+2] product prompted us to further analyze its stereochemistry. First, regarding the fused bicyclic core, the [3+2] product displayed a cis-ring junction, strongly supporting our mechanistic hypothesis that the second C(sp3)–H activation occurs in a relayed manner. Second, the configuration of the major diastereomer exhibited a cis-relationship between the imide group and the directing group, whereas the minor diastereomer exhibited a trans-relationship, indicating that the second C(sp3)–H activation step is selective between the two remaining methyl groups. Overall, the diastereoselectivity profile of the [3+2] reaction highly resembles that of a typical pericyclic cycloaddition reaction, in a sense that the relationships between multiple stereocenters are controlled.
Table 1.
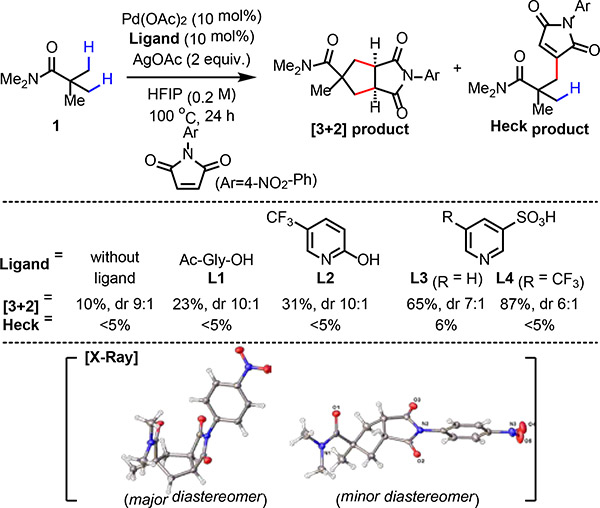 |
Conditions: 1 (0.1 mmol), Pd(OAc)2 (10 mol%), Ligand (10 mol%), N-(4-nitrophenyl)maleimide (0.15 mmol), AgOAc (0.2 mmol), HFIP (0.5 mL), 100 °C, 24 h.
The yield and diastereomeric ratio were determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
With the optimal ligand in hand, the substrate scope of the [3+2] reaction was examined (Table 2). First, various substituents on the amide carboxyl group were tested (Table 2A). Simple alkyl (2–3), aryl (4), and heteroatom-containing functional groups (5–8) were all tolerated to give the desired cyclopentane products. Substrates containing saturated heterocycles (9–12) were also suitable substrates for the [3+2] reaction. To our disappointment, substrates bearing α-hydrogens were incompatible. Given that C(sp3)–H olefination process works with these substrates under nearly identical reaction conditions,14b it is supposed that the second C–H activation step (six-membered palladacycle formation without any quaternary carbon center in between) becomes problematic. Next, the scope of the amino group of the substrate was investigated (Table 2B). Substrates bearing dialkylamino groups with varying sterics (13–15) and cyclic amino groups (16–21) successfully provided the corresponding products. Amino acid-derived substrates (22–23) also gave the products in good yields, as well as Weinreb amide (24) and N-Aryl (25) substrates. While secondary amides (26–28) also underwent the desired reaction, electron-deficient maleimide was required to achieve high yields. In general, simple N-phenylmaleimide is less reactive compared to electron-deficient maleimides which are more activated Michael acceptors, especially with secondary amides which afford lower yields compared to tertiary amides due to the Thorpe-Ingold effect. The effect of the maleimide N-substitution was also systematically studied (Table 2C). Among the N-arylmaleimides, both electron-rich (29–30) and electron-deficient (31–35) maleimides gave high yields and diastereoselectivities. Ortho- and meta-substituted N-arylmaleimides (36–37) were suitable coupling partners as well. Electron-rich N-heteroarylmaleimides (38–39) also provided the products in high yields, although slightly lower diastereoselectivities were observed. N-alkylmaleimides proved to be highly reactive coupling partners, regardless of the nature of the N-substituents (40–44). When other related electron-deficient olefins, such as maleic anhydride, were tested, [3+2] products were not observed (See Supplementary Information). Finally, lactams were tested as substrates for the [3+2] reaction (Table 2D). Such a process would provide a unique disconnection towards the elusive spiro-bicyclic products bearing cyclopentane motifs. To our delight, the six-membered lactam (45) provided the desired bicyclic product in 47% yield. Seven-membered or larger lactams (46–48) served as highly reactive substrates to give the bicyclic products in good yields. Functionalized seven-membered lactams (49–51), as well as 1,4-diazepanone derived substrate (52) were also suitable for the [3+2] reaction. Overall, the major diastereomers in all cases showed cis-relationship between the imide group and the amide directing group. Since the diastereomeric ratios cannot be rationalized by sterics alone, we hypothesize that attractive interactions through dispersion between the two polar groups could be one important factor.
Table 2.
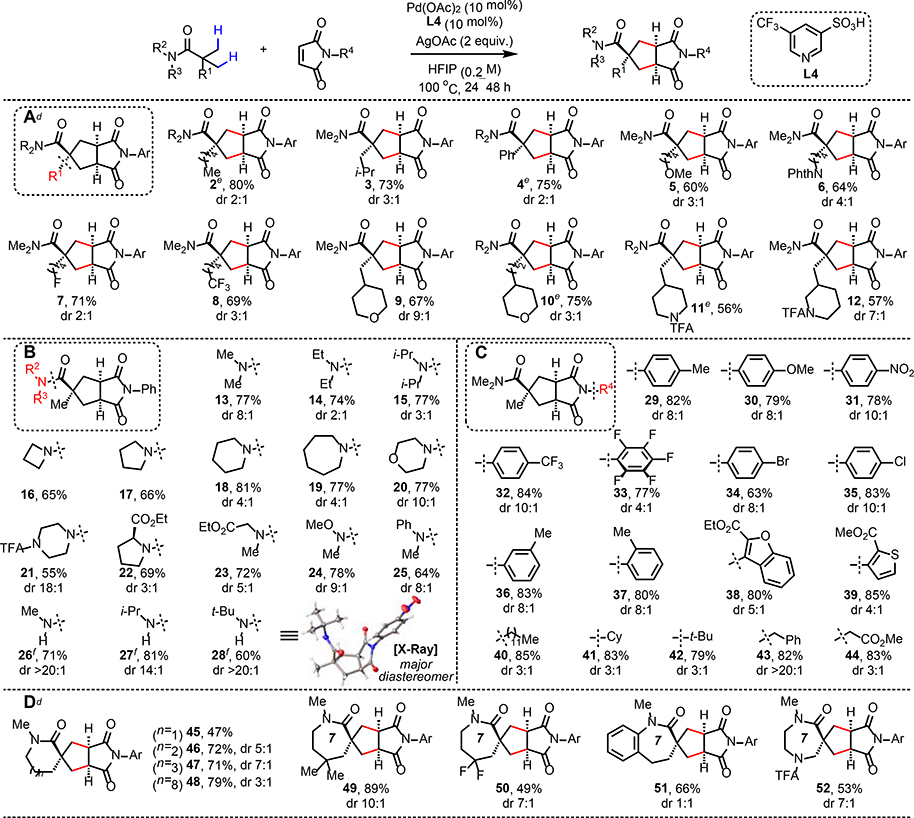 |
Conditions: amide substrate (0.1–0.2 mmol), Pd(OAc)2 (10 mol%), L4 (10 mol%), maleimide (1.5 equiv.), AgOAc (2 equiv.), HFIP (0.2 M), 100 °C, 24–48 h. See Supplementary Information for details.
Isolated yield/diastereomeric ratio.
The isolated diastereomeric ratio does not necessarily reflect the diastereoselectivity of the reaction.
N-(4-(trifluoromethyl)phenyl)maleimide was used.
R2NH = piperidine.
N-(4-nitrophenyl)maleimide was used.
To further demonstrate the synthetic utility, the [3+2] reaction was carried out on gram-scale without any loss of efficiency (Scheme 3A). As a cheaper alternative for the silver oxidant, copper salt could also serve as the terminal oxidant for the gram-scale [3+2] reaction, albeit in diminished yields (See Supplementary Information for oxidant screening). We also attempted to access chiral cyclopentanes via reductive desymmetrization of the [3+2] products using chiral oxazaborolidine catalysis (Scheme 3B).15 Following the procedures developed by Jones and coworkers,16 the [3+2] product 20 underwent desymmetrization with (1S,2R)-cis-1-aminoindan-2-ol derived catalyst to yield the hydroxylactam 53. Then, 53 was directly converted into lactam 54 for analytical purpose. To our delight, 98% ee was observed with 54,17 although only 45% yield was obtained based on recovered starting material. The low yield is due to the over-reduction that leads to the formation of pyrrolidine byproduct. Nevertheless, the combined sequence of diastereoselective [3+2] cycloaddition and enantioselective reduction allows one to rapidly synthesize novel chiral cyclopentane compounds with multiple stereocenters.
Scheme 3.

Gram-scale reaction/Reductive desymmetrization of cyclopentane product
An alternative mechanistic pathway where the Heck product is formed first, followed by a second C(sp3)–H activation/intramolecular migratory insertion is also worth consideration. Although such a process would lead to a trans-fused bicyclic core due to the syn-addition of [C–Pd], rapid epimerization to the thermodynamic cis-fused product may allow us to obtain the observed [3+2] product. To check the viability of this mechanistic pathway, Heck product 55 was prepared and subjected to the reaction conditions (Scheme 4). Regardless of the presence of maleimide, the formation of [3+2] product 20 was not observed, implying that the Heck product is not an intermediate for the [3+2] pathway.
Scheme 4.

Attempted reaction with Heck product 55
In summary, a catalytic [3+2] reaction between gem-dimethyl-containing amide substrates and maleimides via two-fold C(sp3)–H activation is developed. The employment of the weak-coordinating amide directing groups was essential to unlock the [3+2] pathway, as other directing groups have been previously reported to give either Heck or alkylation products. To enable such an amide-directed C(sp3)–H activation, the use of an electron-deficient pyridine-3-sulfonic acid was crucial. A diverse array of cyclopentane products were obtained in a diastereoselective manner, including spiro-bicyclic compounds derived from lactam substrates. Using chiral oxazaborolidine catalysis, we demonstrated that the [3+2] product can be desymmetrized to give a chiral cyclopentane in excellent enantioselectivity. Given that both C(sp3)–C(sp3) bonds are directly forged from C(sp3)–H bonds without the need for pre-functionalization, we anticipate that this method would complement the conventional strategies for cyclopentane synthesis.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge The Scripps Research Institute and the NIH (NIGMS, R01GM084019) for their financial support. We thank the Korea Foundation for Advanced Studies for a predoctoral fellowship (H.P.). We thank Maximilian Bernbeck, Dr. Milan Gembicky, and Dr. Arnold L. Rheingold (UCSD) for X-ray crystallographic analysis. We thank Dr. Jason Chen, Brittany Sanchez, and Emily Sturgell (Scripps Automated Synthesis Facility) for assistance with HRMS.
Footnotes
The authors declare no competing financial interests.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and spectral data for all new compounds (PDF)
REFERENCES
- (1).Cycloaddition Reactions in Organic Synthesis; Kobayashi S, Jørgensen KA, Eds., WileyVCH: Weinheim, Germany, 2002. [Google Scholar]
- (2).Nicolaou KC; Snyder SA; Montagnon T; Vassilikogiannakis G The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed 2002, 41, 1668–1698. [DOI] [PubMed] [Google Scholar]
- (3).Yamago S; Nakamura E [3+2] Cycloaddition of Trimethylenemethane and its Synthetic Equivalents. Org. React 2003, 61, 1–217. [Google Scholar]
- (4).(a) Berson JA The chemistry of trimethylenemethanes, a new class of biradical reactive intermediates. Acc. Chem. Res 1978, 11, 446–453. [Google Scholar]; (b) Yamago S; Nakamura E Use of methylenecyclopropanone ketals for cyclopentane synthesis. A new efficient thermal [3 + 2] cycloaddition. J. Am. Chem. Soc 1989, 111, 7285–7286. [Google Scholar]
- (5).(a) Trost BM; Chan DMT New conjunctive reagents. 2-Acetoxymethyl-3-allyltrimethylsilane for methylenecyclopentane annulations catalyzed by palladium(0). J. Am. Chem. Soc 1979, 101, 6429–6432. [Google Scholar]; (b) Binger P; Wedemann P Regioselektive [3+2] cycloadditionen von methylencyclopropan mit elektronenarmen olefinen an R3P-nickel(O)-katalysatoren. Tetrahedron Lett. 1985, 26, 1045–1048. [Google Scholar]
- (6).Trost BM [3+2] Cycloaddition Approaches to Five-Membered Rings via Trimethylenemethane and Its Equivalents [New Synthetic Methods (55)]. Angew. Chem. Int. Ed. Engl 1986, 25, 1–20. [Google Scholar]
- (7).Trost BM; Mata G Forging Odd-Membered Rings: Palladium-Catalyzed Asymmetric Cycloadditions of Trimethylenemethane. Acc. Chem. Res 2020, 53, 1293–1305. [DOI] [PubMed] [Google Scholar]
- (8).(a) Gibson SE; Mainolfi N The Intermolecular Pauson–Khand Reaction. Angew. Chem. Int. Ed 2005, 44, 3022–3037. [DOI] [PubMed] [Google Scholar]; (b) Zhou Y-Y; Uyeda C Catalytic reductive [4 + 1]-cycloadditions of vinylidenes and dienes. Science 2019, 363, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Trost BM Centenary Lecture. Cyclopentanoids: a challenge for new methodology. Chem. Soc. Rev 1982, 11, 141–170. [Google Scholar]
- (10).(a) Lyons TW; Sanford MS Palladium-Catalyzed Ligand-Directed C−H Functionalization Reactions. Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daugulis O; Roane J; Tran LD Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon–Hydrogen Bonds. Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-Catalyzed Transformations of Alkyl C–H Bonds. Chem. Rev 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Wang X-C; Gong W; Fang L-Z; Zhu R-Y; Li S; Engle KM; Yu J-Q Ligand-enabled meta-C–H activation using a transient mediator. Nature 2015, 519, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Della Ca’ N; Fontana M; Motti E; Catellani M Pd/Norbornene: A Winning Combination for Selective Aromatic Functionalization via C–H Bond Activation. Acc. Chem. Res 2016, 49, 1389–1400. [DOI] [PubMed] [Google Scholar]; (c) Ye J; Lautens M Palladium-catalysed norbornene-mediated C–H functionalization of arenes. Nature Chem. 2015, 7, 863–870. [DOI] [PubMed] [Google Scholar]
- (12).(a) Wasa M; Engle KM; Yu J-Q Pd(II)-Catalyzed Olefination of sp3 C−H Bonds. J. Am. Chem. Soc 2010, 132, 3680–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhuang Z; Yu C-B; Chen G; Wu Q-F; Hsiao Y; Joe CL; Qiao JX; Poss MA; Yu J-Q Ligand-Enabled β-C(sp3)–H Olefination of Free Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Zhan B-B; Li Y; Xu J-W; Nie X-L; Fan J; Jin L; Shi B-F Site-Selective δ-C(sp3)−H Alkylation of Amino Acids and Peptides with Maleimides via a Six-Membered Palladacycle. Angew. Chem. Int. Ed 2018, 57, 5858–5862. [DOI] [PubMed] [Google Scholar]; (b) He Q; Ano Y; Chatani N The Pd-catalyzed C–H alkylation of ortho-methyl-substituted aromatic amides with maleimide occurs preferentially at the ortho-methyl C–H bond over the ortho-C–H bond. Chem. Commun 2019, 55, 9983–9986. [DOI] [PubMed] [Google Scholar]
- (14).(a) Park H; Chekshin N; Shen P-X; Yu J-Q Ligand-Enabled, Palladium-Catalyzed β-C(sp3)–H Arylation of Weinreb Amides. ACS Catal. 2018, 8, 9292–9297. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park H; Li Y; Yu J-Q Utilizing Carbonyl Coordination of Native Amides for Palladium-Catalyzed C(sp3)−H Olefination. Angew. Chem. Int. Ed 2019, 58, 11424–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Corey EJ; Helal CJ Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew. Chem. Int. Ed 1998, 37, 1986–2012. [DOI] [PubMed] [Google Scholar]
- (16).Barker MD; Dixon RA; Jones S; Marsh BJ The crucial role of the nitrogen substituent in the desymmetrisation of cyclic meso-imides using B–Me and B–OMe oxazaborolidine catalysts. Tetrahedron 2006, 62, 11663–11669. [Google Scholar]
- (17).Stereochemistry of 54 is assigned based on ref. 16.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.