

Abstract
Analyses of individual atherosclerotic plaques are mostly descriptive, relying, for example, on histological classification by spectral analysis of ultrasound waves or staining and observing particular cellular components. Such passive methods have proved useful for characterizing the structure and vulnerability of plaques but have little quantitative predictive power. Our aim is to introduce and discuss a computational framework to provide insight to clinicians and help them visualize internal plaque dynamics. We use partial differential equations (PDEs) with macrophages, necrotic cells, oxidized lipids, oxygen concentration, and platelet-derived growth factor (PDGF) as primary variables coupled to a biomechanical model to describe vessel growth. The model is deterministic, providing mechanical, morphological, and histological characteristics of an atherosclerotic vessel at any desired future time point. We use our model to create computer-generated animations of a plaque evolution that are in qualitative agreement with published serial ultrasound images and hypothesize possible atherogenic mechanisms. A systems biology model consisting of five differential equations is able to capture the morphology of necrotic cores residing within vulnerable atherosclerotic plaque. In the context of the model, the distribution of oxidized low-density lipoprotein (Ox-LDL) particles, endothelial inflammation, plaque oxygenation (via the presence of vasa vasora), and intimal oxygenation are four important factors that drive changes in core morphology.
NEW & NOTEWORTHY In this article, we propose a quantitative framework to describe the evolution of atherosclerotic plaque. We use partial differential equations (PDEs) with macrophages, necrotic cells, oxidized lipids, oxygen concentration, and PDGF as primary variables coupled to a biomechanical model to describe vessel growth. A feature of our method is that it outputs color-coded vessel sections corresponding to regions of the plaque that are necrotic and fibrous, qualitatively similar to images generated by enhanced intravascular ultrasound.
Keywords: mathematical model, morphoelasticity, necrotic core, simulation, vulnerable plaque
INTRODUCTION
Cardiovascular disease affected more than 121 million people in 2016 in the United States and ~48% of adults (4). It is often due to atherosclerosis, which manifests itself as a buildup of fatty deposits mainly in the intima of medium-sized and large arteries. Plaques exhibit considerable variability in their internal structure and histology. When they develop a thin cap and a large necrotic core, they are prone to mechanical rupture (49), which often results in thrombosis and acute events such as myocardial infarction (MI). The prediction of plaque progression and rupture remains one of the most important open problems in cardiovascular disease today.
Figure 1 shows the main actors involved in atherogenesis. Healthy endothelial cells produce a certain amount of nitric oxide (NO), which is a vasodilator. A decrease in laminar shear stress reduces the production of this chemical, which could lead to endothelial dysfunction, increased uptake of low-density lipoproteins (LDLs), and upregulation of vascular cell adhesion molecule-1 (VCAM-1). This adhesion molecule starts an inflammatory process by binding with intercellular adhesion molecule-1 (ICAM-1) on the surface of leukocytes present in the bloodstream. Attached to the endothelium, monocytes penetrate the vessel wall in response to chemoattractants such as monocyte chemoattractant protein 1 (MCP1) and transform into macrophages in the presence of macrophage colony-stimulating factor (M-CSF). LDLs in the intima go through oxidization, turning into oxidized LDLs (Ox-LDLs). Macrophages may release more chemoattractant, thereby starting a cascade of inflammation. Although they preferentially consume Ox-LDLs and turn into foam cells, smooth muscle cells (SMCs) can also migrate into the plaque from the underlying media and consume Ox-LDLs, albeit at a slower rate. The death of SMCs, foam cells, and macrophages all contribute to a necrotic core, one of the defining characteristics of a vulnerable (rupture prone) plaque (32, 50).
Fig. 1.

A summary of atherosclerosis microbiology. Low-density lipoproteins (LDLs) that enter the intima become oxidized LDLs (Ox-LDLs) and are consumed by macrophages, leading to accumulation of foam cells. Foam cells later undergo necrosis and form a necrotic core. ICAM-1, intercellular adhesion molecule-1; MCP1, monocyte chemoattractant protein 1; M-CSF, macrophage colony-stimulating factor; SMC, smooth muscle cell; VCAM-1, vascular cell adhesion molecule-1.
Atherosclerosis has been studied from cellular (3), genetic (15), and clinical (6) perspectives. Animal models have also proved valuable in elucidating the main aspects of early-stage atherosclerosis (44). However, one set of tools that remains underutilized is the application of deterministic mathematical models. Historically, the analysis of plaque has been descriptive or statistical. Scientists may observe an individual plaque at a single time point or gather statistics from large cross-sectional studies. In either case, it is difficult to make predictions from observations at a single time point. In cross-sectional studies individual risk factors are assessed with statistical methods, but this approach may not be able to establish causal relationships. In this article, we advocate a paradigm shift to study the natural history of individual plaques with mathematical models. Combined with advances in imaging technology, we believe that such quantitative approaches will be pivotal in enhancing our understanding of the mechanisms of atherogenesis and plaque rupture. We believe that the best way to characterize the data being generated by imaging modalities such as ultrasound, optical coherence tomography (OCT), and palpography is by connecting them to, and concurrently developing, mathematical models.
In this article we first review some paradigmatic models of atherosclerotic plaque that are popular in the engineering and mathematical communities. We then show how elements of these models can be combined into a biologically motivated, computer-generated animation that illustrates plaque progression in terms of thickening of arterial layers, deformation of the vessel wall, and changes in plaque histology.
METHODS
Plaque histology.
Plaque internal structure has been gradually elucidated since around the 1960s (22). Researchers now believe that the internal structure of a plaque primarily determines its stability. Early plaques start life as “intimal cushions,” “intimal thickenings,” or “intimal xanthomas” (48). These are relatively innocuous lesions. Over time, however, they can progress into “fatty streaks,” which have a higher lipid content. Atheromas have regions of interior necrosis and are considered more dangerous since they are associated with cardiovascular events, e.g., stroke or myocardial infarction.
Enhanced ultrasound (virtual histology) protocols use machine learning to analyze the frequency content in ultrasound waves and classify atherosclerotic tissue into four different types: fibrous, fatty, necrotic, and calcific (39). The resulting patterns are fascinating and thought provoking. For example, Kubo et al. (2010) (30) tried to gain insight into the dynamic evolution of plaque histology and morphology. Figure 2 shows serial intravascular ultrasound (IVUS) images of plaque at baseline and after a 12-mo follow-up. Data specific to individual plaques at these two time points were also collected. For example, morphological characteristics such as the area occupied by the lumen and the fraction of the plaque occupied by the necrotic core were observed. This data set raises several important quantitative questions: How long does it take for a plaque to become vulnerable to rupture? How quickly do plaques grow in size? How quickly do regions of necrosis grow? How does necrosis affect the likelihood of rupture? All these questions can, in principle, be answered by a mathematical model of plaque development, properly calibrated against suitable data sets.
Fig. 2.

Serial images reprinted from Ref. 30 with permission from Elsevier. Baseline and follow-up were 12 mo apart. A: thin-cap fibroatheroma (TCFA) turns into a thick-cap fibroatheroma (ThCFA). B: TCFA turns into a fibrous plaque. C: plaque cap rotates from 7 to 5 o'clock. D: transition from pathological intimal thickening to TCFA. E: ThCFA becomes a TCFA.
A popular method in the mathematical modeling community is the application of differential equations. This approach describes how concentrations of certain cell types and metabolites change in time. One well-known example is Hao and Friedman’s model (24). They described the movement of macrophages, T cells, and smooth muscle cells into the intima, which promotes intimal thickening. Their model, however, did not consider the mechanical properties of the intima and neglected the other two layers of the vessel wall. Chalmers et al. (10) used differential equations to explore the dynamics of early atherosclerosis. Their model considered the concentration of LDLs, chemoattractants, endothelium-stimulating (ES) cytokines, macrophages, and foam cells. All of their simulations were done in one dimension, and their results provided qualitative and quantitative insight into the effect of LDL in the inflammatory response. In another paper, Chalmers et al. (9) further investigated the effect of high-density lipoproteins (HDLs) in plaque regression. They observed that increasing HDL influx regresses plaques with a low density of foam cells and slows the growth of plaques with a high density of foam cells. El Khatib et al. (18) suggested that inflammation propagates in the intima as a reaction diffusion wave. They concluded that in the case of intermediate LDL concentrations there are two stable equilibria: one corresponding to the disease-free state and another for the inflammatory state, while a traveling wave connects these two states. Fok (19) investigated the effect of the spatial distribution of Ox-LDL on the location and size of the necrotic core . He predicted the location and size of the necrotic core due to the chemotaxis of macrophages toward sources of Ox-LDLs and their death due to lack of oxygen. Finally, Cobbold et al. (14) investigated the multiple stages of LDL oxidation using a system of ordinary differential equations. In their model, they considered the concentration of antioxidants (specifically vitamin E and vitamin C), HDL, LDL, and free radicals. They modeled how HDL and vitamin C were crucial for slowing down the oxidation of LDL particles.
Cardiovascular research is not limited to processes occurring in the arterial wall. Blood flow also plays an important role in atherogenesis. Changes in the laminar shear stress or the concentration of different chemicals within the bloodstream are some of the crucial factors that can affect the composition of arteries. Therefore, investigating the early stages of atherosclerosis as a fluid-structure interaction (FSI) problem has attracted many researchers (8, 12, 16). As well as coronary arteries, there are studies that utilize FSI in other types of arteries. For example, Pozzi and Vergara (40) investigate the carotids, whereas Thon et al. (46) study aortic arteries. Their approaches can be adapted for coronary arteries with minor modifications.
In this report we consider a simple system of partial differential equations (PDEs) that could describe the formation of hypoxia-induced plaque cores:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
These equations are supplemented with boundary conditions (BCs):
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
The boundaries ∂ω1, ∂ω2, and Γ and the vectors n1 and n2 are indicated in Fig. 3. Variables N, M, Q, C, and P represent concentrations of necrotic cells (NCs), macrophage cells (MCs), a chemokine (CK) such as monocyte chemoattractant protein 1 (MCP1), molecular oxygen, and platelet-derived growth factor (PDGF); D1–D5 are their diffusivities; μ is a chemotactic coefficient for macrophages; β1–β4 are decay coefficients; λ1 is a production rate of MCP1; λ2 is the consumption rate of oxygen by macrophages; and κ is the outflux rate of macrophages from the intima to media (see Table 1 for a summary). Furthermore, we take the concentration of Ox-LDL (L) to be a known function in space and time. In our problem, most of the histological dynamics occur much more quickly than the growth of the plaque. Therefore, Eqs. 1–5 are all solved at steady state. On the other hand, we assume that the timescales associated with endothelial inflammation, arterial oxygenation, and changes in Ox-LDL density are on par with that of the plaque growth, and the time dependence of M0(t), C0(t), and f(x,y,t) is explicitly accounted for.
Fig. 3.

PDGF produced by platelets promotes cell proliferation at the site of an injury.
Table 1.
| Symbol | Meaning | Case A | Case B | Case C | Case D | Case E | Units |
|---|---|---|---|---|---|---|---|
| μ | Chemotactic coefficient of MCs | 2.26 | 7.77 | 4.41 | 9.17 | 6.02 | (mm2/day)·(cm3/mg) |
| D1 | Diffusivity of NCs | 3.2 × 10−3 | 3.3 × 10−3 | 3.6 × 10−3 | 3.9 × 10−3 | 4.3 × 10−3 | cm2/day |
| D2 | Diffusivity of MCs | 1.72 | 1.74 | 1.92 | 2.06 | 2.30 | cm2/day |
| D3 | Diffusivity of CKs | 80.8 | 81.6 | 89.7 | 96.3 | 107.6 | cm2/day |
| D4 | Diffusivity of O2 | 172.5 | 174.3 | 191.6 | 205.8 | 229.9 | cm2/day |
| D5 | Diffusivity of PDGF | 19.4 | 0 | 32.3 | 15.4 | 21.5 | cm2/day |
| β1 | Clearance rate of NCs | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | day−1 |
| β2 | Clearance rate of CKs | 10 | 10 | 10 | 10 | 10 | day−1 |
| β3 | Background O2 consumption rate | 1.2 × 104 | 1.2 × 104 | 1.2 × 104 | 1.2 × 104 | 1.2 × 104 | day−1 |
| β4 | Decay rate of PDGF | 2 | 0 | 2 | 2 | 2 | day−1 |
| λ1 | Production rate of CKs | 2.5 × 10−6 | 2.5 × 10−6 | 2.5 × 10−6 | 2.5 × 10−6 | 2.5 × 10−6 | cm3/day |
| λ2 | Consumption rate of O2 by MCs | 2.5 × 10−7 | 2.5 × 10−7 | 2.5 × 10−7 | 2.5 × 10−3 | 2.5 × 10−7 | cm3/day |
| γmin | Normoxic MC death rate | 3 × 10−3 | 3 × 10−3 | 3 × 10−3 | 3 × 10−3 | 3 × 10−3 | day−1 |
| γmax | Hypoxic MC death rate | 1.2 | 1.2 | 1.2 | 1.2 | 1.2 | day−1 |
| P0 | PDGF BC | 1.4 × 10−3 | 0 | 10−3 | 0.28 × 10−3 | 0.45 × 10−3 | mg/cm3 |
| n | No. of injury points | 1 | 0 | 1 | 2 | 1 | None |
| p | Lumen pressure | 120 | 120 | 120 | 120 | 120 | mmHg |
BC, boundary conditions; CK, chemokine; MC, macrophage cell; NC, necrotic cell; O2, oxygen; PDGF, platelet-derived growth factor.
In Eq. 1, the term on the left-hand side represents the time rate of change of necrotic cells at a given point x = (x,y,z) in the plaque. This equation specifies that the rate of change of NCs at x has three contributions. First, NCs from nearby points x+δx can be moved to x. This diffusion could arise from immune cells in the plaque exerting random forces on dead cells through their own random motion. Second, macrophage cells at x can die and become necrotic, essentially converting from vital cells to necrotic ones. The death rate γ depends on the oxygen concentration at that point, C(x,y,z), via the equation (19)
| (16) |
The normoxic death rate γmin is the death rate for macrophages in an oxygen-sufficient environment, and γmax is the hypoxic death rate in an oxygen-limited environment. The last term in parentheses is called a Hill function, and the term m is called the Hill coefficient (we take m = 4). Equation 16 states that macrophages die quickly when oxygen levels are low (C < Ccrit) but slowly when levels are high (C > Ccrit). We see that Ccrit acts as a hypoxic threshold for macrophage cells and the larger the value of m, the more abrupt the switch. Finally, NCs at x can be cleared by leukocytes, and the clearance rate is proportional to the number of NCs. The minus sign signifies that the clearance term reduces the rate of generation of NCs.
In Eq. 2, the first term on the left-hand side represents the time rate of change of MCs. The second term represents chemotaxis, the directed movement of cells toward chemoattractants such as Ox-LDL (L) and MCP1 (Q). Macrophages chemotax along the vector ∇(L+Q) with associated “flux” μM∇(L+Q) and the coefficient μ capturing the speed of taxis. Note that ∇(L+Q) is a vector that points from small values of L+Q to large values, so MCs are modeled to move from low to high concentrations of total chemoattractant. There are two contributions on the right-hand side. Similar to Eq. 1, the first term represents the diffusion of the cells. The final term represents the death of macrophages with a death rate dependent on the local oxygen concentration C as given by Eq. 16. The +γ(C)M in Eq. 1 and the −γ(C)M in Eq. 2 couple the equations together: when a macrophage cell dies, it converts to a necrotic cell.
In Eq. 3, the term on the left-hand side represents the time rate of change of CKs. Similarly, we have three terms on the right-hand side. The first term corresponds to the diffusion of the CKs. The next term represents the natural decay of CKs at a rate β2. The last term represents the production of CKs by MCs after consumption of Ox-LDL at a rate λ1. Motivated by the principle of mass action, the production rate is proportional to the product of macrophage and Ox-LDL concentrations, LM. The net production is greater if there are more MCs or Ox-LDL particles.
In Eq. 4, the term on the left-hand side represents the time rate of change of the oxygen concentration. The first term on the right-hand side corresponds to the diffusion of oxygen in tissue. The second term represents the background consumption of oxygen by all cells (excluding MCs) with rate β3. The third term represents the consumption of the oxygen concentration by macrophages at a rate λ2. Again, motivated by mass action principles, the net consumption rate is proportional to the product of oxygen concentration and macrophage density, CM. The final term represents contributions from oxygen sources such as microvessels that could be present in advanced plaques.
In Eq. 5, the term on the left-hand side represents the time rate of change of the PDGF concentration. The first term on the right-hand side represents the diffusive spread of PDGF. The second term corresponds to the natural (thermal) degradation of PDGF at a rate β4.
BC Eqs. 8, 12, and 14 prescribe the concentration of macrophages, oxygen, and PDGF at the endothelium, whereas BC Eqs. 6 and 10 account for the flux of necrotic cells and MCP1 across the endothelium ∂ω1. Equations 7, 9, 11, 13, and 15 prescribe the flux of necrotic cells, macrophages, MCP1, oxygen, and PDGF from the intima into the media through the boundary ∂ω2.
So far, we have explained the physical and biological origins of each of the terms in Eqs. 1–5. The validity of these equations relies on the continuum assumption: rather than describing the behavior of individual cells, we are calculating their aggregate or average behavior, using smooth functions. Although this can be a limitation, there are more advanced methods that can be used to capture individual cell behavior (17). Furthermore, the transport properties of the vessel wall have to be homogeneous for the equations to hold. At a molecular level, the random motion of cells and chemicals must follow a Brownian motion, which results in diffusive PDEs. Finally, we have only focused on a small set of cell-cell, cell-cytokine, and cell-substrate interactions, ignoring for example M-CSF-induced monocyte-macrophage transformations and durotactic effects.
In principle, the solution of Eqs. 1–5 produces N, M, Q, C, and P as smooth functions of space and time, representing densities of NCs, MCs, CKs, O2, and PDGF. When the governing equations are solved in practice, these quantities all find their equilibrium levels very quickly compared with the observed rate of plaque progression, so the steady-state, time-independent versions of Eqs. 1–5 are actually computed by setting all time derivatives to zero. An example of the function N is shown in Fig. 4, along with a thresholding method to formally distinguish between necrotic and fibrous tissue. To solve the equations, one needs the values of all the constants D1–D5, β1–β4, μ, λ1, λ2, γmin, and γmax and the Hill coefficient m. These constants can be found from experiments or estimated independently.
Fig. 4.
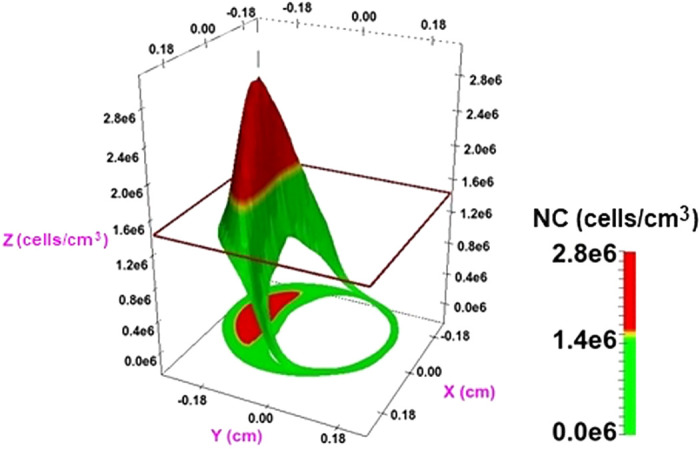
Solving the partial differential equations Eqs. 1–5 produces smooth functions for biological quantities such as the necrotic cell (NC) density, N(x,y,t). By introducing a threshold value (here N = 1.4 × 106), the function can be used to explain regions of necrosis in advanced fibroatheromas: N < 1.4 × 106 indicates the presence of fibrotic tissue (green), and N > 1.4 × 106 indicates the presence of necrotic tissue (red). The 3-dimensional surface N(x,y,t) can be projected onto the xy plane to recreate the 2-dimensional intravascular ultrasound (IVUS) cross sections in Fig. 2.
Plaque morphology and mechanics.
In addition to describing the plaque’s histology through PDEs, it is also important to describe the stresses within a plaque. Virmani et al. (49) have shown that plaques are more likely to rupture if their caps are thin (<65 μm). This observation becomes intuitive after we understand that caps in fibroatheromas rupture when they are stressed beyond a threshold yield stress (33). As cap morphology evolves over time, so do the associated stress fields and the propensity for rupture. Therefore an integrated model of vulnerable plaque should also explain how stress and strain fields evolve and couple these fields to tissue growth and atrophy.
Mechanics and deformation.
Leonhard Euler (1707–1783) was possibly the first person to use mathematics to formulate models for arterial mechanics. His calculations described how the pulsatility of blood flow affects the expansion of arterial cross sections. By the 1970s arterial hemodynamics was a mature field, and in the 1990s the mechanical properties of tissues became a core focus. Aided by developments in numerical methods for fluid mechanics and solid-fluid interactions, simulation of the cardiovascular system became a highly evolved enterprise (21, 41, 45). The ubiquity and power of computers allowed researchers to predict the deformations of the arterial wall and the hemodynamics contained within to an unprecedented level of detail. For example, Simon et al. (43) developed a computational method based on the poroelasticity of arterial tissues. Their model coupled the wall deformation, fluid mechanics, and associated transport phenomena in the arterial wall. Auricchio et al. (2) investigated the biomechanical reaction of a stenotic artery wall to an expandable stent. Their aim was to understand the mechanisms underlying stent-related restenosis. Finally, Akyildiz et al. (1) studied the effect of intima stiffness and plaque morphology on maximum plaque stress. They found that reducing cap thickness and increasing the size of the necrotic core increased the peak stress.
The finite element method is a very versatile method that can be used for solving both dynamic and static problems. Most of the approaches outlined above utilize this method for computing the equilibrium configuration of an elastic artery under a given load. This framework can be adapted to accommodate a model of tissue growth called morphoelasticity. The underlying assumption is that growth occurs so slowly that mechanical equilibrium is maintained at all times. Below, we describe the elastostatics problem and then explain how morphoelasticity can be used to evolve the plaque dynamically.
Elastostatics.
The deformation vector tracks the position of every material point in the artery under a given load and maps a reference configuration to a deformed configuration. The starting point for our method is an energy integral that accounts for all the ways that deformations affect the total energy. This usually consists of three parts. First, there is the stored potential energy associated with deformations from an unstressed, reference state. Second, there is the energy associated with the deformations doing work against body forces (such as gravity). Finally, the deformations also do work against surface forces (such as a lumen pressure). Although deformations of the elastic body increase the energy, any work done against body and surface forces requires energy and is subtracted from the total budget. In the absence of growth we have
| (17) |
where Π is the total energy, Φ is the deformation vector, f and g are body and surface force densities, Ω is the “reference domain” (points in space occupied by the unloaded artery), and ∂Ω is the mathematical boundary between the lumen and the undeformed vessel wall. The strain energy density of the elastic artery is W and depends on the deformation gradient ∇Φ. A quick way to understand this is that the energy should depend on the change in the dimensions of an infinitesimal cuboid relative to its dimensions in the reference configuration. In other words, the energy does not depend on the deformation vector Φ but rather on how Φ changes when applied to neighboring points in the reference configuration. To find the arterial deformation, one first defines the mechanical properties of the vessel wall by specifying W. For example, it may be mechanically anisotropic (collagen fibers in the vessel make it harder to stretch radially than axially), or it may be layer dependent (mechanical properties of the intima are different from the media or adventitia). We employ a layer-specific strain energy following Ref. 27 that comes from ex vivo stress tests of arterial tissue. For more details on the strain energy see Ref. 37.
For most arterial problems, body forces such as gravity are negligible, so f = 0. For a lumen pressure p, one can show that the surface force density is in fact
| (18) |
where J = det ∇Φ and N is the unit outward normal vector to ∂Ω. The ultimate objective is to find the deformation Φ that minimizes Π in Eq. 17. This high-dimensional optimization problem is solved computationally. Once Φ is known, the displacement of every point in the reference configuration determines the deformed configuration: see Fig. 5 for how the finite element mesh deforms under the effect of Φ. More details can be found in textbooks such as Ref. 27. In this report we take p to be the average of systolic and diastolic blood pressure. Since atherosclerosis is always associated with high blood pressure we take p = 120 mmHg, which according to the American Heart Association corresponds to stage II hypertension.
Fig. 5.

Minimizing the energy (Eq. 17) produces a displacement field (indicated by arrows) that deforms the arterial cross section.
Tissue growth and morphoelasticity.
The way we account for tissue growth in our model is by employing the theory of morphoelasticity (23, 51). Equation 17 above depends on the deformation gradient F = ∇Φ, which captures how an infinitesimal cuboid of tissue changes dimensions under a deformation Φ. Since biological tissue mostly consists of water, the volume of the cuboid is approximately conserved as it is strained. The consequence is that the total volumes of the reference and deformed arteries are almost identical. However, in morphoelasticity we assume that tissue first undergoes a stress-free volumetric growth (characterized by a growth tensor) before being strained. Specifically, an element first responds to a pure growth, meaning that it increases its volume by a specified amount. This will cause an “overlap” with neighboring elements resulting in an incompatible configuration. Therefore, by itself the growth tensor does not give a physical configuration and needs to be followed by a corrective strain, characterized by an elastic tensor. The resulting compound process guarantees that the outcome is a continuous grown domain with no overlaps. Mathematically the process assumes that the deformation gradient can be written as a product of the growth tensor (Fg) and the elastic tensor (Fe):
| (19) |
(see Fig. 6). The energy in Eq. 17 also needs to be modified under this assumption of growth; for details, see Ref. 20.
Fig. 6.

Decomposition of the deformation gradient F into a pure growth (Fg) and an elastic (Fe) response. This is the fundamental assumption in morphoelasticity theory; see Eq. 19.
How does morphoelasticity help with plaque modeling? The growth tensor Fg can be informed by the biology of atherosclerosis. From murine models the initial stages of plaque growth are characterized by an increase in intima mass, resulting from a migration of smooth muscle cells from the media (13). One possible trigger for this migration is PDGF (29). In our model, we simplify this process by assuming that PDGF is directly responsible for growth and take : increments of arterial tissue increase their radial and circumferential dimensions by g1 and g2, respectively and we let g1 and g2 depend on PDGF concentration (as predicted by Eq. 5) through a Hill function so that
| (20) |
where α1 = 1, α2 = 0.25, m = 4, and P0 = 1.12 × 10−5 mol/L (these constants were chosen for convenience). The growth tensor is a crucial element of the model because it allows the histological and morphoelastic models to communicate with each other. However, one has to be careful about constructing the growth tensor from the PDGF distribution. The growth tensor is always applied to the reference domain: recall Eq. 19. However, the PDGF Eq. 5, like the other PDEs, is solved in the deformed domain. Because there is a one-to-one correspondence between mesh elements in the reference and deformed configurations (see Fig. 5), the PDGF concentration in a reference element is simply taken as the value that was computed for the corresponding deformed element. Using this method, we define a spatially varying PDGF distribution in the reference domain that can be used to update Eq. 20 at each time step.
The resulting integrated model of atherosclerosis allows us to understand how morphological features of plaque (e.g., lumen size, stenosis, necrotic fraction) could be affected by microbiology (e.g., Ox-LDL density, glucose concentrations, PDGF density) and can be summarized by the following processes:
-
•
Oxidized LDLs infiltrate the intima and diffuse.
-
•
Macrophages chemotax toward oxidized LDLs, consume them, and release MCP1, attracting more macrophages.
-
•
Macrophages die if oxygen levels are low, producing necrotic cells.
-
•
PDGF released from platelets induces growth of neointima.
Together, these four processes are able to produce a vast range of morphological and histological changes in the vessel. Our simulations are done in two dimensions since plaque structure is often presented in two-dimensional arterial cross sections. Our computations are carried out in the FEniCS finite element computational environment using the University of Delaware’s Caviness cluster. The coupling of PDEs 1–4 is done by introducing a “mixed” finite element space (31), consisting of the Cartesian product of individual (first order) finite element spaces. Specifically, Newton’s iteration finds a vector (N,M,Q,C) that makes the weak form of Eqs. 1–4 and Eqs. 6–13 stationary for all test functions drawn from the mixed space; see Fig. 7 for a flowchart of the algorithm. The time step used for our simulations is Δt = 0.004 mo. With this value of Δt, the growth functions gk(t) (Eq. 20) are approximated with Simpson’s integration rule at t = tj = jΔt, j = 0, 1, 2, . . . and the time-dependent functions C0(t), M0(t), f(x,y,t), and L(x,y,t) in Eqs. 12, 8, 4, 2, and 3 are directly evaluated at t = tj. One possible realization of the model at a single time point is shown in Fig. 8. We see that oxygen and macrophages are localized near the arterial lumen; a single localized injury to the endothelium has released PDGF, leading to elevated levels of the growth factor near the lower part of the lumen; and a large necrotic core has resulted from the death of many MCs deep in the intima.
Fig. 7.
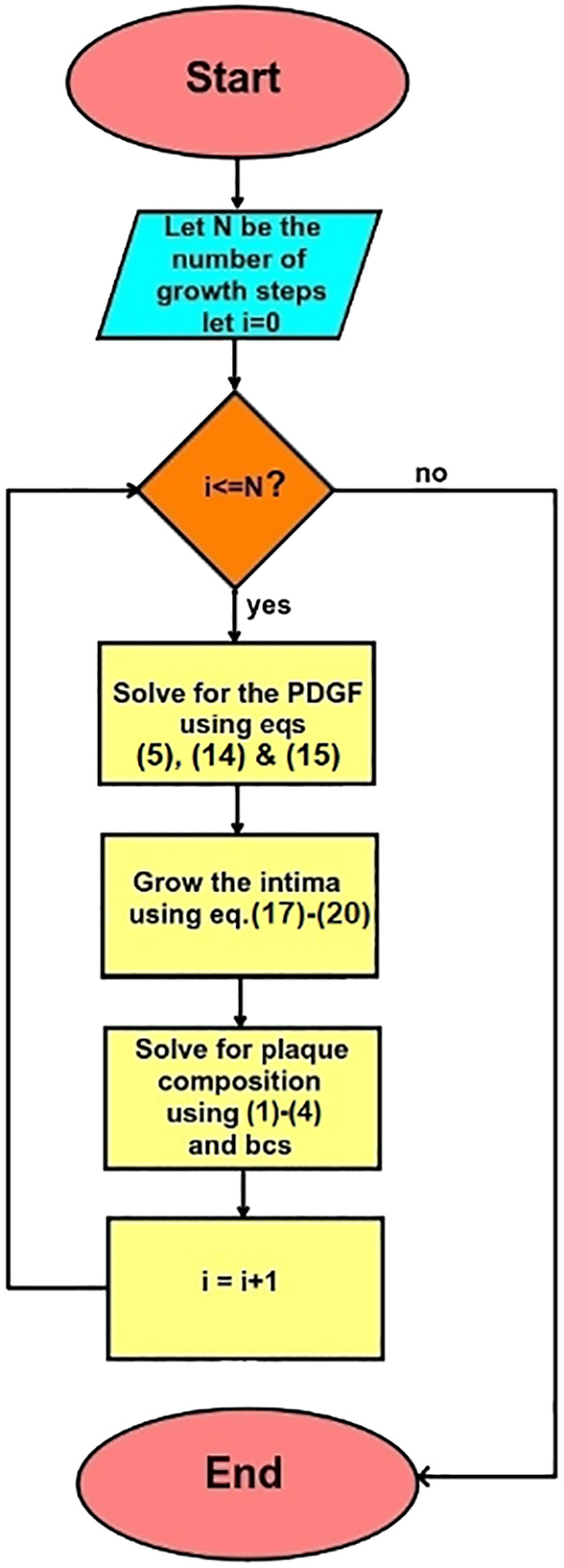
Flowchart of atherosclerosis simulation algorithm. bc, Boundary condition.
Fig. 8.

Typical plaque model results. Concentrations of necrotic cells (NC), chemoattractant, oxidized low-density lipoprotein (Ox-LDL), macrophage cells (MC), and oxygen are outputted as spatially dependent fields distributed within the intima. For PDGF concentration, the media and adventitia are also indicated along with the finite element mesh.
RESULTS
Now we illustrate the results of our model. Although there are many ways to produce different plaque outcomes, we focus on changing four parameters over time, reflecting four main mechanisms that are thought to be associated with inflamed plaques:
Mechanism 1: changes in Ox-LDL distribution, L(x,y,t).
It has long been hypothesized that modified LDLs are atherogenic since they are mainly responsible for the appearance of the foam cell phenotype (7). The Ox-LDL density inside a plaque is governed by the function L whose time evolution we control directly in the model.
Mechanism 2: changes in inflammation, M0(t).
We define inflammation in the endothelium as the density of macrophages adsorbed on the layer. Mathematically we control this density through the boundary condition Eq. 8. Biologically, an increasing M0(t) corresponds to an endothelium that becomes more inflamed over time.
Mechanism 3: development of vasa vasora, f(x,y,t).
Vasa vasora are a network of small vessels that supply the arterial walls with resources such as oxygen and glucose. Sources of oxygen are controlled in our model through the function f(x,y,t) in Eq. 4.
Mechanism 4: increase in oxygenation, C0(t).
Arterial oxygenation can depend on intercirculatory mixing, hemoglobin concentration, and other systemic factors (34). For example, exercise can increase hemoglobin levels and blood flow to tissues (5) and has also been shown to increase arterial oxygenation in patients with chronic heart failure (25, 42). We control C0(t) using the Dirichlet boundary condition (Eq. 12).
A summary of these functions is provided in Table 2. In addition to these four mechanisms, intimal growth depends on PDGF (P) through Eq. 20. Mathematically, we impose P = P0 for x ∈ ∂ω1, with P0 > 0 on n = 0, 1, or 2 small segments of ∂ω1, representing discrete injury points on the endothelium, and P0 = 0 otherwise. The number of injury points n is fixed in each case, and how quickly the plaque grows can be controlled through n.
Table 2.
Time-dependent parameters for mechanisms 1–4
| Meaning | Case A | Case B | Case C | Case D | Case E | Units | |
|---|---|---|---|---|---|---|---|
| L(x,y,t) | Ox-LDL concentration | Static | Decreasing in time | Location changes in time | Increasing in time | Static | mg/cm3 |
| M0(t) | Macrophage density in endothelium | Increases from 4.6 × 105 to 4.6 × 105 | Decreases from 5.2 × 105 to 1.2 × 105 | 4 × 105 | Increases from 0 to 1.7 × 105 | 4.2 × 105 | cells/cm3 |
| C0(t) | O2 density in endothelium | Increases from 5.6 × 10−5 to 11.2 × 10−5 | 5.6 × 10−5 | 5.6 × 10−5 | N/A | N/A | mol/L |
| f(x,y,t) | O2 sources within intima | 0 | 0 | 0 | 6.7 | Sum of Gaussian functions* | mol·L−1·day−1 |
N/A, not applicable (a zero Neumann condition was used instead of Eq. 12); Ox-LDL, oxidized low-density lipoprotein.
Specifically, for some constants ak, bk, N, xk, and yk and time-dependent functions fk(t).
Now we make connections to the enhanced IVUS images from the paper of Kubo et al. (30). In this study, the authors imaged the same plaques 12 mo apart to gain insight into their natural history (see Fig. 2). There are five cases: A–E. To recreate these evolutions, the following protocols were used. Our parameters are separated into two types: those that can potentially change in time and reflect the four mechanisms described above and those that remain static in time (but can differ from case to case). The quantities L, M0, C0 and f are in the first category (see Table 2), and all other parameters are in the second (see Table 1). The virtual plaques are evolved over a period of 12 mo (up to t = 3 in simulation time), and tissue was defined as necrotic when N exceeded a threshold of 1.4 × 106 cells/cm3; otherwise the tissue was classified as fibrotic (see Fig. 4). Other components (calcific, lipidic) were not directly accounted for by the model. Our simulation results are shown in Fig. 9, which should be compared to Fig. 2. Parameter values for each of cases A–E are given in Tables 1 and 2. Computer-generated animations of all five plaque evolutions are available as Supplemental Material for this article (available at https://doi.org/10.6084/m9.figshare.11968722.v3).
Fig. 9.
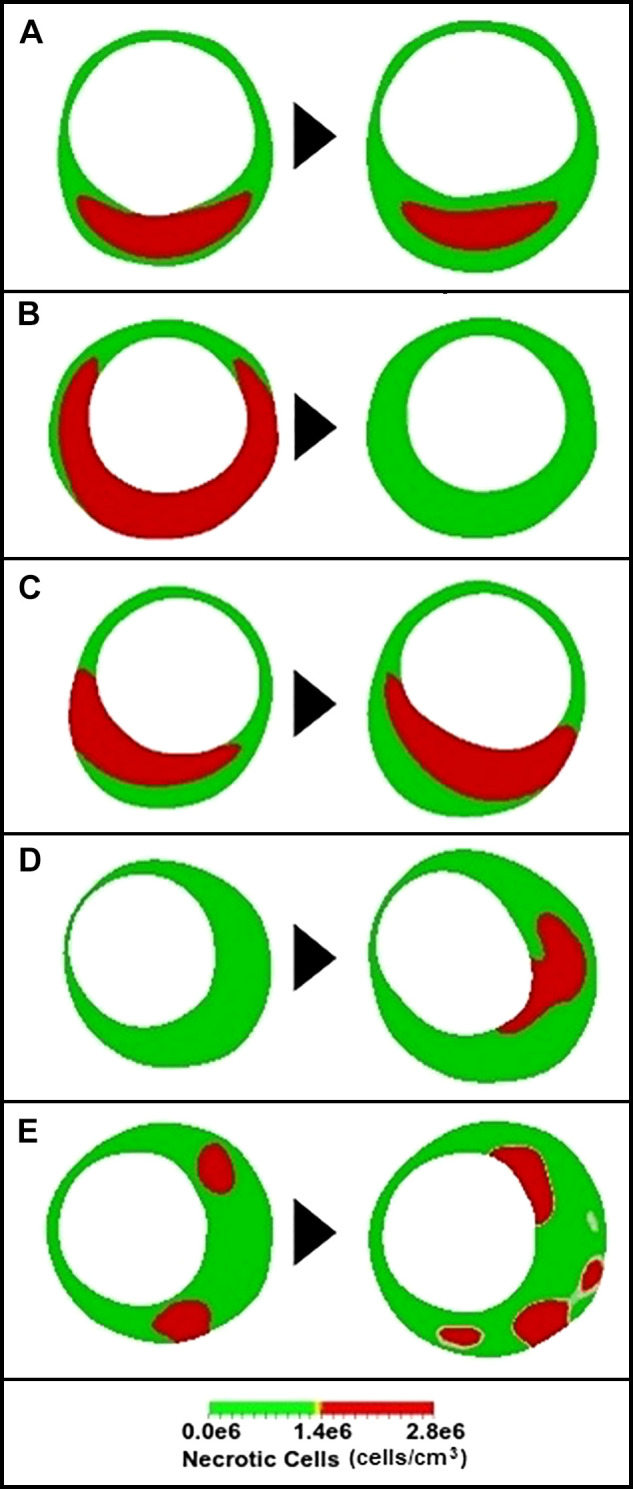
Simulations of necrotic core development in a coronary artery (compare with Fig. 2). Simulations follow the same qualitative behavior as in Fig. 2. A: thin-cap fibroatheroma (TCFA) turns into a thick-cap fibroatheroma (ThCFA). B: TCFA turns into a fibrous plaque. C: plaque cap rotates from 7 to 5 o’clock. D: transition from pathological intimal thickening to TCFA. E: ThCFA becomes a TCFA.
Case A results from a combination of mechanisms 2 and 4. Endothelial inflammation increases slightly, and macrophages chemotax toward sources of Ox-LDL and chemokine. Regions of the intima close to the lumen are more oxygenated, and more oxygen in the intima reduces macrophage death from hypoxia. The result is plaque growth along with a necrotic core that “drifts” deeper into the plaque, in rough qualitative agreement with the IVUS results in Fig. 2A.
Case B is achieved through mechanisms 1 and 2. For our simulations we assumed that endothelial injury is quickly healed (so there are no sources of PDGF) and that both the Ox-LDL concentration in the intima and macrophage density at the endothelium reduce over time. These effects lead to a vanishing necrotic core.
Case C results from mechanism 1. By changing the location of Ox-LDLs, we were able to alter the direction of macrophage chemotaxis. Because NCs are continuously produced by MC death, the necrotic core follows the macrophage density and moves to the right when the Ox-LDLs move to the right.
Case D results from a combination of mechanisms 1 and 2. The Ox-LDL concentration follows a combination of Gaussian functions with peak densities that increase in time (37). The location of Gaussians was chosen to produce the crescent-shaped necrotic core in Fig. 2D. Endothelial inflammation also increases in time. There are low levels of oxygen throughout the intima, and PDGF concentrations are maximal at 1 and 7 o’clock. The NC appears in a crescent shape adjacent to the lumen from 3 to 5 o’clock.
Finally, case E results from mechanism 4. Changes in the spatial distribution of microvessels are key to producing the pair of images in Fig. 2E (37). There are several regions of necrosis in the baseline state, but they are dynamic and increase in number and size in the follow-up case. The seemingly random spatial distribution of necrosis in the follow-up could result from changes in the number and density of vasa vasora in the intima, reflected by taking f to be a function that changes both temporally and spatially.
For more details on dynamics of other concentrations such as macrophages, Ox-LDLs, MCP1, oxygen, and PDGF for cases A–E, see Ref. 37.
DISCUSSION
Plaques present considerable interpatient variability, depending on location and age of the plaque, local and global cell biology, personal genetics, and environmental and lifestyle factors. It is not surprising that the associated evolutions seem hopelessly complicated. We advocate that integrated mathematical models may help with understanding how plaque complexity results from microbiology.
In this paper we assumed that Eqs. 1–17 are universal, applying to all plaques (this assumption is further discussed below), and different plaque states and evolutions are characterized by different parameters. Studying plaques through these equations is still daunting because there are 21 different parameters and functions in Tables 1 and 2. Although we estimated them for the purposes of matching the IVUS images in Fig. 2, in principle each one should be measured in vivo for a particular patient. Although this may not be practical or even possible, there are two reasons why the model could still be valuable. First, even though the parameters and driving functions are unknown, they could be estimated from in vitro experiments or from animal models. Knowledge of more parameters gives the model more predictive power and puts the study of plaque on a more rigorous scientific footing. Second, and more importantly, the integrated model can be systematically tested and recalibrated to accommodate new data. Rather than passively describing plaques and accumulating observations, the integrated model puts clinicians in the driving seat, enabling them to formulate and test new hypotheses.
There is the remaining issue that Eqs. 1–17 are probably not universal. However, this does not pose any added conceptual difficulty providing experimentalists, clinicians, and theorists closely collaborate. Instead of updating parameter values, individual terms in the equations can be modified to reflect the latest data. New equations can even be included (or removed) if required. The power of a mathematical model is that it can be continuously refined and updated in light of new scientific developments.
Caveats and limitations.
The model presented in this paper focused on six main quantities: necrotic cells, macrophage cells, monocyte chemotactic protein, platelet-derived growth factor, oxygen, and oxidized LDLs. This is a minimal model: for example, we have not distinguished between M1 and M2 macrophages (M1s are thought to be proinflammatory whereas M2s are atheroprotective); neither have we accounted for hemodynamics, the effect of glucose, or the intricate biology of cell death. However, the extension to these more complex cases is not conceptually more difficult in terms of PDE modeling. In principle one can write down as many equations as the biology demands. Physical laws realized through a mathematical framework provide systematic ways to do this that are consistent with principles of mass conservation and continuum mechanics. The main technical hurdle is not in the mathematical modeling but in finding parameter values and making comparisons with data. To do this requires careful calibration of the model output with serial images such as Fig. 2. It also requires close collaboration between mathematicians/engineers and clinicians/imaging specialists.
Our model was mostly focused on a structural model of plaque that can be affected by local intimal growth. Therefore, we focused on a two-dimensional (2-D) domain, following IVUS images. We are aware that an arterial model is not complete without blood flow dynamics. In reality, endothelial permeability is impacted by laminar wall shear stress that changes Ox-LDL deposition rates and triggers the intimal inflammatory cascade; all other atherosclerotic biomechanical and chemical responses follow, including platelet production of PDGF. However, in this paper the locations of endothelial dysfunction, (Γ in Fig. 3) as well as the Ox-LDL distribution were imposed without considering any hemodynamics. If we considered a three-dimensional (3-D) arterial geometry and included hemodynamics in our model, the tissue mechanics simulation would be more computationally intensive and Γ would be a 2-D surface rather than a curve segment and determined as a result of blood flow rather than decided a priori. Solution of the blood flow equations in a 3-D lumen and coupling them to the tissue mechanics would certainly increase the complexity of the model.
When lumen pressure is removed from arterial segments ex vivo, a stress called a residual stress still remains: this can be seen when a radial cut is made, usually causing the segment to spring open. Another limitation of the present model is that we have not accounted for residual stresses (11) in the arterial segments at baseline. The residual stress can alter the total Cauchy stress after pressurization and growth, so predictions of stress must be made very carefully with this model. Incorporating the effects of residual stress can be done by modifying the reference configuration (47).
Other chemical messengers can be incorporated into the model depending on what is suggested by imaging data. Currently, enhanced IVUS provides spatially resolved maps of necrosis, cholesterol, and calcium phosphate in vivo. Although other cytokines such as interleukins (28, 35) also play an important role in the development of plaques, including them in our model does not provide additional insight if there are no data to indicate their spatial distribution and temporal evolution; in fact, including their effect would only increase the number of unknown parameters in our model and therefore the uncertainty of the predictions. Histological stains such as Movat stain and hematoxylin and eosin (H&E) constitute the “gold standard” in terms of determining spatial data within the plaque. If these stains can be used in an animal study in which animals are periodically euthanized and particular arteries analyzed in terms of cell positions and cell types, this experimental protocol could provide more information about the dynamics within a plaque and further inform our integrated model.
Future challenges.
We believe that the future for quantitative models of atherosclerosis is promising. Imaging technologies such as OCT and infrared spectroscopy are giving us the ability to visualize the evolution of plaque morphology and composition. The great challenge at the moment is the way the data are being collected. Scientific prediction is concerned with quantifying future states from past states. The more informed we are of past states, the better our prediction of future states. The few existing “natural history” studies of human atherosclerosis have an extremely low time resolution. For example, the plaques in Ref. 30 were scanned at just two time instances (a baseline and a follow-up) within a year from each other. Serial studies in single patients need to be performed more frequently and at greater time resolution. The hurdle here is clinical indication for the procedure. Nevertheless, we believe that high-time-resolution serial studies hold the key to a better understanding of atherosclerosis. Providing modelers and quantitative scientists with more snapshots, or even a movie, of disease progression will greatly enhance the modeling and possibly prediction of atherosclerotic development.
Another advance that will greatly help in aiding prediction is an improvement in virtual histology technology and integration with other imaging methods. Currently the master algorithm classifies tissues into four categories: fibrous, fatty, necrotic, and calcific. In reality, a given region can share characteristics from each category. For example, necrotic cells could be interspersed with flecks of calcium. What is really needed is a continuous version of the four categories. What could such a scale look like? One possibility is to use a vector that represents weights of each category: p = (p1, p2, p3, p4), where . For example, (0,0,0,1) corresponds to a region that is completely calcified and (0,0,1,0) corresponds to a region that is completely necrotic. However, an area where calcium and necrosis are present in equal amounts can be represented as (0,0,0.5,0.5). Constructing the algorithms to provide this level of detail will prove challenging, since even the current four-color method is not universally accepted and does not always match the histological gold standard.
Overall, the mathematical community is well positioned to make inroads in understanding the patterns seen in enhanced IVUS images or other imaging modalities of atherosclerotic plaque. Although the underlying biology in plaque development is intricate and complex, we believe that mathematics provides the conceptual bridge that connects the plaque environment to the spatial patterns that ultimately result.
GRANTS
P.-W.F was supported by Simons Foundation Collaboration Grant 282579. N.M.M. and P.-W.F were supported by DE-CTR ShoRe Pilot Grant NIGMS IdeA U54 GM-104941.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.M.M., W.S.W., and P.-W.F. conceived and designed research; N.M.M. performed experiments; N.M.M. analyzed data; N.M.M. and P.-W.F. interpreted results of experiments; N.M.M. and P.-W.F. prepared figures; P.-W.F. drafted manuscript; W.S.W. and P.-W.F. edited and revised manuscript; N.M.M., W.S.W., and P.-W.F. approved final version of manuscript.
ACKNOWLEDGMENTS
Preprint is available (36).
REFERENCES
- 1.Akyildiz AC, Speelman L, van Brummelen H, Gutiérrez MA, Virmani R, van der Lugt A, van der Steen AF, Wentzel JJ, Gijsen FJ. Effects of intima stiffness and plaque morphology on peak cap stress. Biomed Eng Online 10: 25, 2011. doi: 10.1186/1475-925X-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auricchio F, Di Loreto M, Sacco E. Finite-element analysis of a stenotic artery revascularization through a stent insertion. Comput Methods Biomech Biomed Engin 4: 249–263, 2001. doi: 10.1080/10255840108908007. [DOI] [Google Scholar]
- 3.Bendeck MP, Zempo N, Clowes AW, Galardy RE, Reidy MA. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res 75: 539–545, 1994. doi: 10.1161/01.RES.75.3.539. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MS, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UK, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart Disease and Stroke Statistics–2019 Update: a report from the American Heart Association. Circulation 139: e56–e528, 2019. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 5.Boone T. Introduction to Exercise Physiology. Burlington, MA: Jones & Bartlett Publishers, 2014. [Google Scholar]
- 6.Bourantas CV, Garcia-Garcia HM, Farooq V, Maehara A, Xu K, Généreux P, Diletti R, Muramatsu T, Fahy M, Weisz G, Stone GW, Serruys PW. Clinical and angiographic characteristics of patients likely to have vulnerable plaques: analysis from the PROSPECT study. JACC Cardiovasc Imaging 6: 1263–1272, 2013. doi: 10.1016/j.jcmg.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem 52: 223–261, 1983. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 8.Calvez V, Houot JG, Meunier N, Raoult A, Rusnakova G. Mathematical and numerical modeling of early atherosclerotic lesions. ESAIM Proc 30: 1–14, 2010. doi: 10.1051/proc/2010002. [DOI] [Google Scholar]
- 9.Chalmers AD, Cohen A, Bursill CA, Myerscough MR. Bifurcation and dynamics in a mathematical model of early atherosclerosis: how acute inflammation drives lesion development. J Math Biol 71: 1451–1480, 2015. doi: 10.1007/s00285-015-0864-5. [DOI] [PubMed] [Google Scholar]
- 10.Chalmers AD, Bursill CA, Myerscough MR. Nonlinear dynamics of early atherosclerotic plaque formation may determine the efficacy of high density lipoproteins (HDL) in plaque regression. PLoS One 12: e0187674, 2017. doi: 10.1371/journal.pone.0187674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuong CJ, Fung YC. Residual stress in arteries. In: Frontiers in Biomechanics, edited by Schmid-Schönbein GW, Woo SL, Zweifach BW. New York: Springer, 1986, p. 117–129. [Google Scholar]
- 12.Cilla M, Pena E, Martinez MA. Mathematical modelling of atheroma plaque formation and development in coronary arteries. J R Soc Interface 11: 20130866, 2014. doi: 10.1098/rsif.2013.0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clowes AW, Reidy MA, Clowes MM. Kinetics of cellular proliferation after arterial injury. I. Smooth muscle growth in the absence of endothelium. Lab Invest 49: 327–333, 1983. [PubMed] [Google Scholar]
- 14.Cobbold CA, Sherratt JA, Maxwell SR. Lipoprotein oxidation and its significance for atherosclerosis: a mathematical approach. Bull Math Biol 64: 65–95, 2002. doi: 10.1006/bulm.2001.0267. [DOI] [PubMed] [Google Scholar]
- 15.Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J Clin Invest 103: 1597–1604, 1999. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Tomaso G, Díaz-Zuccarini V, Pichardo-Almarza C. A multiscale model of atherosclerotic plaque formation at its early stage. IEEE Trans Biomed Eng 58: 3460–3463, 2011. doi: 10.1109/TBME.2011.2165066. [DOI] [PubMed] [Google Scholar]
- 17.DiMilla PA, Barbee K, Lauffenburger DA. Mathematical model for the effects of adhesion and mechanics on cell migration speed. Biophys J 60: 15–37, 1991. doi: 10.1016/S0006-3495(91)82027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Khatib N, Génieys S, Volpert V. Atherosclerosis initiation modeled as an inflammatory process. Math Model Nat Phenom 2: 126–141, 2007. doi: 10.1051/mmnp:2008022. [DOI] [Google Scholar]
- 19.Fok PW. Growth of necrotic cores in atherosclerotic plaque. Math Med Biol 29: 301–327, 2012. doi: 10.1093/imammb/dqr012. [DOI] [PubMed] [Google Scholar]
- 20.Fok PW, Gou K. Finite element simulation of intimal thickening in multi-layered arterial cross sections by morphoelasticity. Comput Methods Appl Mech Eng 363: 112860, 2020. doi: 10.1016/j.cma.2020.112860. [DOI] [Google Scholar]
- 21.Franzone PC, Pavarino LF, Scacchi S. Mathematical Cardiac Electrophysiology. Cham, Switzerland: Springer, 2014. [Google Scholar]
- 22.Geer JC, McGill HC Jr, Strong JP. The fine structure of human atherosclerotic lesions. Am J Pathol 38: 263–287, 1961. [PMC free article] [PubMed] [Google Scholar]
- 23.Goriely A. The Mathematics and Mechanics of Biological Growth. New York: Springer, 2017. [Google Scholar]
- 24.Hao W, Friedman A. The LDL-HDL profile determines the risk of atherosclerosis: a mathematical model. PLoS One 9: e90497, 2014. doi: 10.1371/journal.pone.0090497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herrlin B, Sylvén C. Increased arterial oxygen content—an important compensatory mechanism in chronic moderate heart failure. Cardiovasc Res 25: 384–390, 1991. doi: 10.1093/cvr/25.5.384. [DOI] [PubMed] [Google Scholar]
- 26.Holzapfel GA, Sommer G, Gasser CT, Regitnig P. Determination of layer-specific mechanical properties of human coronary arteries with nonatherosclerotic intimal thickening and related constitutive modeling. Am J Physiol Heart Circ Physiol 289: H2048–H2058, 2005. doi: 10.1152/ajpheart.00934.2004. [DOI] [PubMed] [Google Scholar]
- 27.Holzapfel GA. Nonlinear Solid Mechanics: A Continuum Approach for Engineering. Chichester, UK: John Wiley and Sons, 2000. [Google Scholar]
- 28.Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol 19: 2364–2367, 1999. doi: 10.1161/01.ATV.19.10.2364. [DOI] [PubMed] [Google Scholar]
- 29.Jackson CL, Raines EW, Ross R, Reidy MA. Role of endogenous platelet-derived growth factor in arterial smooth muscle cell migration after balloon catheter injury. Arterioscler Thromb 13: 1218–1226, 1993. doi: 10.1161/01.ATV.13.8.1218. [DOI] [PubMed] [Google Scholar]
- 30.Kubo T, Maehara A, Mintz GS, Doi H, Tsujita K, Choi SY, Katoh O, Nasu K, Koenig A, Pieper M, Rogers JH, Wijns W, Böse D, Margolis MP, Moses JW, Stone GW, Leon MB. The dynamic nature of coronary artery lesion morphology assessed by serial virtual histology intravascular ultrasound tissue characterization. J Am Coll Cardiol 55: 1590–1597, 2010. doi: 10.1016/j.jacc.2009.07.078. [DOI] [PubMed] [Google Scholar]
- 31.Langtangen HP, Logg A. Solving PDEs in Python: The FEniCS Tutorial I. Cham, Switzerland: Springer International Publishing, 2016. [Google Scholar]
- 32.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 105: 1135–1143, 2002. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 33.Loree HM, Kamm RD, Stringfellow RG, Lee RT. Effects of fibrous cap thickness on peak circumferential stress in model atherosclerotic vessels. Circ Res 71: 850–858, 1992. doi: 10.1161/01.RES.71.4.850. [DOI] [PubMed] [Google Scholar]
- 34.Mair DD, Ritter DG. Factors influencing systemic arterial oxygen saturation in complete transposition of the great arteries. Am J Cardiol 31: 742–748, 1973. doi: 10.1016/0002-9149(73)90009-X. [DOI] [PubMed] [Google Scholar]
- 35.Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, Soubrier F, Esposito B, Duez H, Fievet C, Staels B, Duverger N, Scherman D, Tedgui A. Protective role of interleukin-10 in atherosclerosis. Circ Res 85: e17–e24, 1999. doi: 10.1161/01.RES.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 36.Mohammad Mirzaei N, Fok PW, Weintraub WS. An integrated approach to simulating the vulnerable atherosclerotic plaque. arXiv 1910.00734, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohammad Mirzaei N. Finite Element Simulation of Atherosclerotic Plaque through Morphoelasticity (PhD dissertation). Newark, DE: University of Delaware, 2020. https://cpb-us-w2.wpmucdn.com/sites.udel.edu/dist/e/8224/files/2020/07/thesis.pdf. [Google Scholar]
- 39.Nair A, Kuban BD, Obuchowski N, Vince DG. Assessing spectral algorithms to predict atherosclerotic plaque composition with normalized and raw intravascular ultrasound data. Ultrasound Med Biol 27: 1319–1331, 2001. doi: 10.1016/S0301-5629(01)00436-7. [DOI] [PubMed] [Google Scholar]
- 40.Pozzi S, Vergar C. Mathematical and numerical models of atherosclerotic plaque progression in carotid arteries. European Numerical Mathematics and Advanced Applications Conference 2019 Egmond aan Zee, The Netherlands September 30–October 4, 2019. [Google Scholar]
- 41.Quarteroni A, Manzoni A, Vergara C. The cardiovascular system: mathematical modelling, numerical algorithms and clinical applications. Acta Numerica 26: 365–590, 2017. doi: 10.1017/S0962492917000046. [DOI] [Google Scholar]
- 42.Rubin SA, Brown HV, Swan HJ. Arterial oxygenation and arterial oxygen transport in chronic myocardial failure at rest, during exercise and after hydralazine treatment. Circulation 66: 143–148, 1982. doi: 10.1161/01.CIR.66.1.143. [DOI] [PubMed] [Google Scholar]
- 43.Simon BR, Kaufmann MV, McAfee MA, Baldwin AL. Finite element models for arterial wall mechanics. J Biomech Eng 115: 489–496, 1993. doi: 10.1115/1.2895529. [DOI] [PubMed] [Google Scholar]
- 44.Stadius ML, Rowan R, Fleischhauer JF, Kernoff R, Billingham M, Gown AM. Time course and cellular characteristics of the iliac artery response to acute balloon injury. An angiographic, morphometric, and immunocytochemical analysis in the cholesterol-fed New Zealand white rabbit. Arterioscler Thromb 12: 1267–1273, 1992. doi: 10.1161/01.ATV.12.11.1267. [DOI] [PubMed] [Google Scholar]
- 45.Taylor CA, Figueroa CA. Patient-specific modeling of cardiovascular mechanics. Annu Rev Biomed Eng 11: 109–134, 2009. doi: 10.1146/annurev.bioeng.10.061807.160521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thon MP, Hemmler A, Glinzer A, Mayr M, Wildgruber M, Zernecke-Madsen A, Gee MW. A multiphysics approach for modeling early atherosclerosis. Biomech Model Mechanobiol 17: 617–644, 2018. doi: 10.1007/s10237-017-0982-7. [DOI] [PubMed] [Google Scholar]
- 47.Vandiver R. Effect of residual stress on peak cap stress in arteries. Math Biosci Eng 11: 1199–1214, 2014. doi: 10.3934/mbe.2014.11.1199. [DOI] [PubMed] [Google Scholar]
- 48.Virmani R, Burke AP, Kolodgie FD, Farb A. Pathology of the thin-cap fibroatheroma: a type of vulnerable plaque. J Interv Cardiol 16: 267–272, 2003. doi: 10.1034/j.1600-0854.2003.8042.x. [DOI] [PubMed] [Google Scholar]
- 49.Virmani R, Burke AP, Kolodgie FD, Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol 15: 439–446, 2002. doi: 10.1111/j.1540-8183.2002.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 50.Virmani R, Narula J, Leon MB, Willerson JT (Editors). The Vulnerable Atherosclerotic Plaque: Strategies for Diagnosis and Management. Malden, MA: Wiley-Blackwell, 2008. [Google Scholar]
- 51.Yang Y, Jäger W, Neuss-Radu M, Richter T. Mathematical modeling and simulation of the evolution of plaques in blood vessels. J Math Biol 72: 973–996, 2016. doi: 10.1007/s00285-015-0934-8. [DOI] [PubMed] [Google Scholar]