

Abstract
Insulin-like growth factor-1 (IGF-1) decreases atherosclerosis in apolipoprotein E (Apoe)-deficient mice when administered systemically. However, mechanisms for its atheroprotective effect are not fully understood. We generated endothelium-specific IGF-1 receptor (IGF1R)-deficient mice on an Apoe-deficient background to assess effects of IGF-1 on the endothelium in the context of hyperlipidemia-induced atherosclerosis. Endothelial deficiency of IGF1R promoted atherosclerotic burden, when animals were fed on a high-fat diet for 12 wk or normal chow for 12 mo. Under the normal chow feeding condition, the vascular relaxation response to acetylcholine was increased in the endothelial IGF1R-deficient aorta; however, feeding of a high-fat diet substantially attenuated the relaxation response, and there was no difference between endothelial IGF1R-deficient and control mice. The endothelium and its intercellular junctions provide a barrier function to the vasculature. In human aortic endothelial cells, IGF-1 upregulated occludin, claudin 5, VE-cadherin, JAM-A, and CD31 expression levels, and vice versa, specific IGF1R inhibitor, picropodophyllin, an IGF1R-neutralizing antibody (αIR3), or siRNA to IGF1R abolished the IGF-1 effects on junction and adherens proteins, suggesting that IGF-1 promoted endothelial barrier function. Moreover, endothelial transwell permeability assays indicated that inhibition of IGF-1 signaling elevated solute permeability through the monolayer of human aortic endothelial cells. In summary, endothelial IGF1R deficiency increases atherosclerosis, and IGF-1 positively regulates tight junction protein and adherens junction protein levels and endothelial barrier function. Our findings suggest that the elevation of the endothelial junction protein level is, at least in part, the mechanism for antiatherogenic effects of IGF-1.
NEW & NOTEWORTHY Endothelial insulin-like growth factor-1 (IGF-1) receptor deficiency significantly elevated atherosclerotic burden in apolipoprotein E-deficient mice, mediated at least in part by downregulation of intercellular junction proteins and, thus, elevated endothelial permeability. This study revealed a novel role for IGF-1 in supporting endothelial barrier function. These findings suggest that IGF-1’s ability to promote endothelial barrier function may offer a novel therapeutic strategy for vascular diseases such as atherosclerosis.
Keywords: atherosclerosis, endothelium, IGF, insulin like, vascular permeability
INTRODUCTION
The endothelium, a single layer of endothelial cells (ECs) comprising the inner surface of the vascular wall, is a major regulator of vascular functions. It regulates vascular tone, proinflammatory cell recruitment by expressing adhesion molecules, and platelet activation and thrombogenesis (51). Dysregulation of such endothelial functions (termed “endothelial dysfunction”), causing reduced vasodilation, a proinflammatory state, and prothrombic properties, has been linked to vascular complications including atherosclerosis [reviewed in (7, 8, 12, 19)].
Insulin-like growth factor-1 (IGF-1) is a growth factor produced by a variety of cells and tissues throughout the body, exerting local (autocrine/paracrine) or systemic (endocrine) effects via the circulation. These include stimulation of tissue growth and differentiation, cell proliferation, survival, and anabolism. In the artery wall, both ECs and vascular smooth muscle cells express IGF-1 and its cognate receptor (IGF1R) (21). Moreover, macrophages, which play a key role in inflammation and are integral to the pathogenesis of atherosclerosis, also express high levels of IGF-1 and IGF1R. Thus, IGF-1 has multiple sources and can potentially target multiple types of cells in the setting of inflammatory arterial diseases such as atherosclerosis.
In our previous reports, we demonstrated that systemic infusion of IGF-1 has antioxidant and anti-inflammatory effects and reduces atherosclerosis in apolipoprotein E-deficient (Apoe-deficient) mice. However, smooth muscle cell-specific overexpression of IGF-1 in Apoe-deficient mice did not alter total atherosclerotic burden but induced features consistent with a more stable plaque (40), suggesting that smooth muscle cells do not mediate the reduction of atherosclerosis induced by an increase in systemic IGF-1. Moreover, IGF1R deficiency in smooth muscle cells (as well as in fibroblasts) led to enhanced atherosclerosis in Apoe-deficient mice (46). Thus, IGF-1, via its ability to promote cell survival, proliferation, and extracellular protein deposition, is essential to support the integrity of the smooth muscle cell layer and fibrous cap in atherosclerotic plaques, and loss of smooth muscle cell integrity may lead to atheroprogression (46). On the other hand, IGF-1’s potential effects on the endothelium in the context of atherosclerosis largely remain obscure. Here we generated a mouse model of atherosclerosis with partial depletion of IGF1R expression in the endothelium to assess potential effects of the loss of function of IGF-1 signaling on endothelial function and ultimately on the development of atherosclerosis.
MATERIALS AND METHODS
Materials.
IGF-1 ELISA kits were obtained from Diagnostic Systems Laboratories (Webster, TX). Cholesterol assay kit and OxiSelect Hydroxyl Radical Antioxidant Capacity Activity Assay Kit were obtained from Cell Biolabs (San Diego, CA). Picropodophyllin (PPP) was purchased from Selleck Chemicals (Houston, TX). In Vitro Vascular Permeability Assay kit was purchased from Millipore Sigma (Cat. No. ECM644). Biotin-labeled concanavalin A (ConA) was purchased from Vector Laboratories. Alexa Fluor 488 phalloidin was purchased from Invitrogen. Human aortic endothelial cells (AoECs), growth medium (EGM-2), and basal endothelial cell medium (EBM-2) were purchased from Lonza (Basel, Switzerland). Information about antibodies used for immunofluorescence or Western blot analysis is indicated in Table 1.
Table 1.
Antibodies used for immunofluorescence or Western blot analysis
| Target Antigen | Vendor | Catalog Number | Dilution | Application |
|---|---|---|---|---|
| VCAM1 | Abcam | ab134047 | 1/2,000 | WB |
| ICAM1 | Santa Cruz Biotechnology | sc-8439 | 1/200 | WB |
| CD31 | Abcam | ab28364 | 1/500 | WB |
| VE-cadherin | Santa Cruz Biotechnology | sc-9989 | 1/200 | WB |
| VE-cadherin | Abcam | ab33168 | 1/1,250 | IF |
| β-Catenin | Cell Signaling Technology | 2677 | 1/300 | IF |
| Occludin | Thermo Fisher Scientific | 33–1500 | 1/1,000 | WB/human cells |
| Claudin 5 | Thermo Fisher Scientific | 35–2500 | 1/250 | WB |
| Claudin 5 | Abcam | ab15106 | 1/500 | IF |
| ZO-1 | Thermo Fisher Scientific | 33–9100 | 1/300 | IF |
| JamA | Abcam | ab89203 | 1/500 | WB |
| JamC | Sigma-Aldrich | HPA003417 | 1/200 | WB |
| IGF1R | Cell Signaling Technology | 3027 | 1:1,000 | |
| α-Mouse IgG | GE Healthcare | NA931 | 1/1,000 | WB |
| α-Rabbit IgG | GE Healthcare | NA934 | 1/1,000 | WB |
| α-Mouse IgG-Alexa Fluor 594 | Thermo Fisher Scientific | A21201 | 1/400 | IF |
| α-Rabbit IgG-DyLight 488 | Vector Laboratories | DI-1488 | 1/300 | IF |
Animals.
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee. Igf1rflox/flox/Apoe−/− mice were generated by cross-breeding Apoe−/− mice (C57Bl/6 background, Jackson Laboratory) with Igf1Rflox/flox mice (C57Bl/6 background, a generous gift from Dr. Brüning, University of Leipzig, Germany). Cre recombinase expression was introduced selectively into the vascular endothelium by crossing B6;129-Tg(Cdh5-cre)1Spe/J mice (Jackson Laboratories), which was also done on an Apoe−/− background. Successful IGF-1R gene truncation and knockdown were confirmed in isolated lung ECs (Fig. 1). Atherosclerotic lesion development was assessed in mice fed on a normal chow diet until 32 wk or 52 wk of age. In a separate experiment, 8-wk-old mice were fed a high-fat diet (42% of total calories from fat, 0.15% cholesterol; ENVIGO, Cat. No. TD.88137) for 12 wk and assessed for atherosclerosis. Animals’ systolic blood pressure was measured once a week using the BP-2000 Blood Pressure Analysis System (Visitech Systems). We took 10 measurements on each mouse per time point and used the average value as the representative value.
Fig. 1.

Endothelial cell-specific knockdown of IGF1R in Apoe-deficient mice. Mice were created by cross-breeding Cdh5-Cre transgenic mouse to Igf1rflox/flox mouse, both on an Apoe-deficient background. A: lung endothelial cells (ECs) were isolated by immunomagnetic separation (CD31) of enzymatically digested lung cell suspension. Purity was confirmed using staining for von Willebrand factor (98%) and VE-cadherin (97%). IGF1R β-chain was quantified by Western blot analysis. There was a 60% decrease of IGF1R β-chain expression levels, compared with control (lung ECs isolated from IGF1R-flox mouse, *P < 0.01, n = 4–5). B: antioxidant capacity in aorta tissue was determined and expressed as gallic acid equivalents (GAE). C and D: body weight (C) and systolic blood pressure (D) over 12 wk on a high-fat diet. E and F: relaxation response to acetylcholine (ACh) assessed by aortic ring assay (see materials and methods). Four segments (~5 mm in length) of thoracic aorta were used to assess relaxation responses to increasing doses of ACh after contraction to phenylephrine (10−6 M). Animals were fed on a normal chow diet (E) or a high-fat diet (F) for 4 wk before the assay. *P < 0.05 (n = 6) and **P < 0.01 (n = 6) vs. IGF1R-flox with the same dose of ACh.
Cell culture and tissue preparation for protein and mRNA analysis.
Mouse ECs were isolated from the lung of En-IGF1R-KD or IGF1R-flox mice according to the method of Kobayashi et al. (26) and subcultured in Dulbecco’s modified Eagle’s medium supplemented with 20% fetal bovine serum (Invitrogen), 80 μg/mL streptomycin, 80 U/mL penicillin, 100 μg/mL heparin, and 100 μg/mL EC growth supplement (Sigma-Aldrich, Cat. No. E2759). Greater than 90% purity of the murine EC isolations was confirmed by positive staining for von Willebrand factor (Invitrogen, Cat. No. PA5-16634) and VE-cadherin (Abcam, Cat. No. ab33168).
Human AoECs were cultured in the endothelial cell growth medium-2 (EGM-2; supplemented with 2% fetal bovine serum, antibiotics, and supplements including IGF-1 (Lonza, Cat. No. CC-3162) at 37°C in a humidified 5% CO2 atmosphere with the medium replaced approximately every 48 h. Actively dividing cells (passage 4 to 8) were used for experiments. Fully confluent culture was exposed to 100 ng/mL human recombinant IGF-1, 1 µM PPP, or 5 µg/mL anti-IGF1R neutralizing antibody (clone αIR3, Millipore Sigma, Cat. No. MABS192) to assess effects of elevated IGF-1 signaling (exposure to IGF-1) or inhibition of IGF-1 signaling (PPP or αIR3) on junction protein expression levels (serum and IGF-1-free EGM-2 or complete EGM-2, i.e., supplemented with serum and IGF-1, were used as control, respectively). Alternatively, AoECs were transfected with a silencing RNA against Igf1r mRNA by using Xfect RNA transfection reagent (Takara Bio USA, Mountain View, CA) to lower IGF1R expression levels and assess the effects of loss of IGF1 signaling. Predesigned silencing RNA (Cat. No.; H00003480-R04) and its control (R0017) were purchased from Abnova and transfected at 50 nM by following the manufacturer’s instruction.
To assess tissue specificity of IGF1R deficiency, we determined IGF1R levels in isolated aortic smooth muscle cells and Igf1r mRNA levels in circulating leukocytes and in bone marrow tissue. Aortic smooth muscle cells were isolated by enzymatic digestion. Mouse aortas cleaned of adventitial fat and connective tissue were chopped into small pieces (3–5 mm diagonal). The tissue pieces were incubated at 37°C in 1 mg/mL 1,4-dithioerythritol and 27 U/mL papain in Hank’s balanced salt solution (HBSS) for 30 min and then in 1 U/mL collagenase II, 1 mg/mL soybean trypsin inhibitor, and 40 U/mL elastase in HBSS for an additional 20 min. Liberated cells were washed using HBSS and then cultured in DMEM/F-12 media (Thermo Fisher), supplemented with 10% fetal bovine serum, 4 mmol/L l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cell identity was confirmed by staining for calponin and α-smooth muscle actin (α-SMA), which are smooth muscle cell markers. More than 90% of the cells were double positive for calponin and α-SMA. Circulating leukocytes were isolated from whole blood collected by cardiac puncture. Red blood cells were lysed by using BD Pharm Lyse lysing buffer (BD Biosciences, Cat. No. 555899), and then leukocytes were sedimented by centrifugation at 200 g for 5 min. Sedimented leukocytes were immediately lysed in TriPure Isolation reagent (Roche). Bone marrow tissue was collected from femurs and tibias by flushing out using phosphate-buffered saline and immediately lysed in TriPure isolation reagent. Total RNA was isolated using RNeasy Mini Kit (Qiagen) and tested for Igf1r mRNA expression levels.
Quantitative real-time reverse transcription PCR.
Quantitative real-time PCR was performed as previously described (45). Briefly, complementary DNA was synthesized using RT2 Easy First Strand Kit (Qiagen) and used for 40-cycle, 2-step PCR with sequence-specific primer pairs in CFX Connect Real-Time Detection System (Bio-Rad). Primers used for detecting Igf1r mRNA and Actb mRNA were obtained from Qiagen (RT2 qPCR Primer Assay).
Atherosclerosis quantification.
Atherosclerosis burden was quantified by measuring the surface area of Oil Red O-positive lesions on en face preparations of whole aortae as previously described (47). In addition, serial sections (6 μm) were taken throughout the entire aortic valve area and stained with hematoxylin-eosin for quantitation of plaque cross-sectional area as previously described (47). The α-SMA-positive area and Mac3-positive area were measured using anti-α-SMA-stained and anti-Mac3-stained aortic valve sections, respectively. For α-SMA staining, sections were incubated with anti-α-smooth muscle actin antibody (clone ASM-1, Millipore, Cat. No. CBL171), followed by incubation with secondary antibody labeled with Alexa Fluor 594. Sections were then counterstained with DAPI. For Mac-3 staining, sections were incubated with anti-Mac-3 antibody (BD Biosciences, Cat. No. 553142), followed by incubation with biotinylated secondary antibody and avidin-peroxidase complex (Vectastain Elite ABC kit, Vector Laboratories, Inc.). Sections were developed with DAB substrate kit and counterstained with hematoxylin. Antibody specificity was verified by staining of a serial section with nonimmune IgG of the same species as that of the primary antibody. Images were captured using the Olympus IX71 microscope and quantified using cellSens Dimension software (version 1.18).
Biochemical assays.
Serum IGF-1 levels were determined using Mouse/Rat IGF-I/IGF-1 Quantikine ELISA Kit (R&D systems, Minneapolis, MN). Serum cholesterol levels were determined using Total Cholesterol and Cholesteryl Ester Colorimetric/Fluorometric Assay Kit (BioVision, Milpitas, CA). Serum nitrate/nitrite levels were assessed using Nitrate/Nitrite Fluorometric Assay Kit (Cayman Chemical, Ann Arbor, MI). The capacity of tissue antioxidant systems to quench hydroxyl radicals was measured using OxiSelect HORAC Activity assay (Cell Biolabs, San Diego, CA). The antioxidant capacity of the cells was calculated compared with an antioxidant standard curve obtained with gallic acid and normalized to protein concentration of sample.
Western blot analysis.
Western blot analysis was performed as described previously (22). In brief, cells were washed with PBS and lysed in RIPA buffer, containing 150 mmol/L NaCl, 20 mmol/L Tris·Cl (pH 7.2), 1 mmol/L EDTA, 1% Nonidet P-40, 5 mmol/L dithiothreitol, 0.1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L sodium orthovanadate, 0.1 mol/L okadaic acid, 0.1 mol/L aprotinin, 10 g/mL leupeptin, and 10 mmol/L NaF. Lysates were quantified via bicinchoninic acid protein assay (Thermo Fisher Scientific) and subjected to 10% SDS-PAGE, after addition of loading buffer containing β-mercaptoethanol and after Western blot analysis. Proteins of interest were detected using an antibody from the list in Table 1, and immunopositive bands were visualized by enhanced chemiluminescence (Pierce ECL Western Blotting Substrate, Thermo Fisher Scientific). Images were captured and quantified using a Bio-Rad Gel Doc XR+ Gel Documentation System and software (Image Laboratory, version 5.2.1). Blots were stripped and reprobed with monoclonal anti-β-actin antibody or anti-α-tubulin antibody as a control for equal loading.
Immunofluorescence.
Human AoECs were grown in monolayer on collagen I-coated, 8-well culture chamber slides (Corning, Cat. No. 354630). The cells were treated with PPP solution (1 µM) in DMSO or with 0.1% DMSO (control) or with IGF-1 (100 ng/mL) (control, serum-free and IGF-1-depleted medium) for 24 h. After treatment, cells were washed with HBSS, fixed with cold 95% ethanol for 30 min at 4°C, permeabilized with 0.2% Triton X-100 for 10 min at room temperature, and blocked in Protein Block solution (Abcam, Cat. No. 64226) for 1 h at room temperature. Cells were incubated overnight at 4°C with a mixture of primary antibodies (rabbit anti-VE-cadherin and mouse anti-β-catenin or rabbit anti-claudin 5 and mouse anti-ZO-1). Images were developed with goat anti-rabbit DyLight 488 and chicken anti-mouse Alexa Fluor 594 secondary antibodies for 1 h at room temperature. Slides were counterstained with DAPI and mounted with ProLong Diamond Antifade mountant (Thermo Fisher Scientific, Cat. No. P36961). Antibody specificity was verified by cell staining with nonimmune IgG of the same species as that of the primary antibody. Images were visualized and captured with an Olympus fluorescent microscope (IX71) equipped with a camera system (DP80), and immunopositivity was quantified using cellSens Dimension software (version 1.18).
To quantify coimmunofluorescent signals, “green” (DyLight 488-positive) and “red” (Alexa Fluor 594 positive) images were merged. The master file containing merged images of both experimental and control cells was generated, and the sum area of merged color (i.e., yellow) was quantified as pixels2 using cellSens Dimension software.
Endothelial permeability.
In vitro permeability assay was performed using In Vitro Vascular Permeability assay kit from Millipore Sigma (Cat. No. ECM644) by following the kit’s instructions. Briefly, human AoECs were grown to 100% confluence on culture inserts and treated with 1 µM PPP or 0.1% DMSO (solvent control) for 24 h. FITC-labeled dextran was added in the upper chamber, and its permeation into the lower chamber was assessed in 20 min and expressed as relative fluorescence units.
For in vivo permeability assessments, mice were systemically perfused in situ with biotin-labeled ConA with physiologic pressure (100 mmHg) to assess solute permeability. In brief, mice were anesthetized to a surgical plane, and the heart tissue was exposed. Perfusate was introduced into the left ventricle, flowing out from an open incision in the right atrium. Perfusion was performed in four steps, initiated with saline for 5 min, followed by 1% BSA-20 µg/mL biotin-labeled ConA in saline for 5 min, saline for 5 min, and finally 4% PFA-8% sucrose in saline for 10 min. The aorta was dissected and immerse fixed for additional 2–4 h and quenched by l-glycine. The tissue was then immunostained and observed using a multiphoton microscope.
THP-1 cell adhesion on monolayer of human AoECs.
Human AoECs were grown to 100% confluence on culture plates and then treated with IGF-1 or PPP for 24 h. THP-1 monocytic cells were labeled with 2 µM CellTracker Green CMFDA (Invitrogen) and added into human AoEC culture at a density of 5 × 105 cells/mL. After 40 min of incubation, the culture was extensively washed three times using PBS to remove THP-1 cells that were floating or settled on the monolayer. Adherent green THP-1 cells were photographed (viewing area = 696 × 524 nm) and counted. The average cell count of three random views represents one sample in each treatment group.
Multiphoton microscopy.
Mouse thoracic aortae were immunostained and cleared by the method reported in Hama et al.’s study (16). Briefly, a piece of thoracic aorta (3–4 mm in length) was incubated with anti-CD31 antibody (R&D Systems, Cat. No. AF3628, diluted 1:200) at 37°C for 24 h in 1% bovine serum albumin, 0.88% sodium chloride, 0.44% sodium citrate, and 0.05% Triton X-100 (pH 7.0). After being washed, the tissue was further incubated with anti-rabbit IgG-Alexa Fluor 555 (Invitrogen, Cat. No. A-21431) and streptavidin-Atto 647 (Rockland, Cat. No. S000-56) at 37°C for 24 h, followed by staining with Alexa Fluor 488 phalloidin (Invitrogen, Cat. No. A12379). Stained tissue was cleared by adopting the ScaleS method (16) for aorta tissue: adaptation with ScaleS0 for 30–60 min at 37°C; permeabilization by sequential incubation in ScaleS1, S2, and S3 for 30–60 min per each step at 37°C; descaling with PBS for 30–60 min at 4°C; and clearing with ScaleS4 for 60 min at 37°C. Confocal images were taken using Leica TCS SP8 STED and MP system and rendered to three-dimensional images and quantified using Leica LAS X Core software. We were able to acquire images only from two samples per day, owing to lengthy time requirement for confocal imaging and capturing. Therefore, we assessed one tissue from each of two groups, i.e., En-IGF1R-KD and IGF1R-flox mice, in a day, and a comparison of ConA-positive volume was made between them. To make a comparison, ConA-positive volume (representing permeation of solute) was normalized to CD31-positive volume (representing the endothelium), and the value in En-IGF1R-KD aorta was expressed as relative to IGF1R-flox tissue (control).
Intravital fluorescence microscopy.
Intravital fluorescence microscopy was performed as described elsewhere (52). Mice were administered intravenously with Alexa Fluor 488-labeled anti-CD11b antibody (clone M1/70, ABLab, Vancouver, BC, Canada) via the jugular vein 1 h before assessment. Mice were placed on a Plexiglas board, and a section of the small intestine was exteriorized over a glass coverslip and superfused with a bicarbonate-buffered saline (37°C, pH 7.4). Body temperature was maintained between 36.5°C and 37.5°C by means of a thermostatically controlled heat lamp. The Plexiglas board was mounted on the stage of an inverted microscope (Eclipse TE2000, Nikon), and the intestinal microcirculation was observed through an ×20 objective lens. Fluorescent images (excitation = 420–490 nm, and emission = 520 nm) were detected with a charge-coupled device camera (Photometrics COOLSNAP ES). Images were projected onto a television monitor (PVM-1953MD, Sony) and recorded on a DVD recorder (DMR-E50, Panasonic). A time-date generator (WJ810, Panasonic) displayed this function on the monitor. The intestinal segment was scanned, and 10 single unbranched venules (diameter = 20–50 µm and length = 100 µm) were observed for at least 1 min. Monocyte-endothelial interactions (number of rolling monocytes and number of firmly attached monocytes) were quantified in each venule, followed by calculation of the mean for 10 venules. Monocytes were considered to be adherent if they did not move for at least 30 s. Rolling cells were defined to be those passing a cross line at a velocity significantly slower than the centerline velocity and are expressed as rolling cells per minute. Numbers of adherent cells were normalized in terms of millimeter squared surface area.
Vessel tension measurements using an organ bath.
Mice were fed a normal chow diet or a high-fat diet starting at 8 wk of age for 4 wk and then used for vessel tension experiments. Mice were euthanized by deep anesthesia with a ketamine (100 mg/kg) and xylazine (12 mg/kg) combination injected intraperitoneally based on body weight. Under a dissecting microscope, thoracic aortas were excised and fat and connective tissues were removed and cut transversely into rings (~5 mm in length) with extreme care to preserve the endothelium.
Aortic rings were mounted vertically by sliding them into two wire hooks. Aortic rings were immersed in 2 mL of KH buffer, consisting of118 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3, 11 mM glucose, and 2.5 mM CaCl2, pH 7.4 with 95% O2-5% CO2 at 37°C. Aortic rings were equilibrated for 90 min with a resting tension of 1 g. After baseline tension was determined, vessels were precontracted with phenylephrine (PE; 10−6 M), followed by administration of acetylcholine (ACh) at the indicated dose. Changes in vessel tension were time-dependently monitored (15 min each), and tension was recorded through the ADInstruments data acquisition system (Colorado Springs, CO).
Statistical analysis.
All numeric data and figures are expressed as means ± SE. Statistical analyses were performed with GraphPad PRISM software (version 8.3.0). Data sets were first assessed for residual distribution with the D’Agostino–Pearson omnibus normality test and for equal variances with the F test. Differences in outcomes were determined by unpaired Student’s t test with or without the Welch correction, or Mann–Whitney U test, accordingly with the normality of residuals distribution. Single-sample t test was used to compare ConA-positive volume per CD31-positive volume between En-IGF1R-KD and IGF1R-flox mice (Fig. 7). Differences were considered significant at P < 0.05.
Fig. 7.
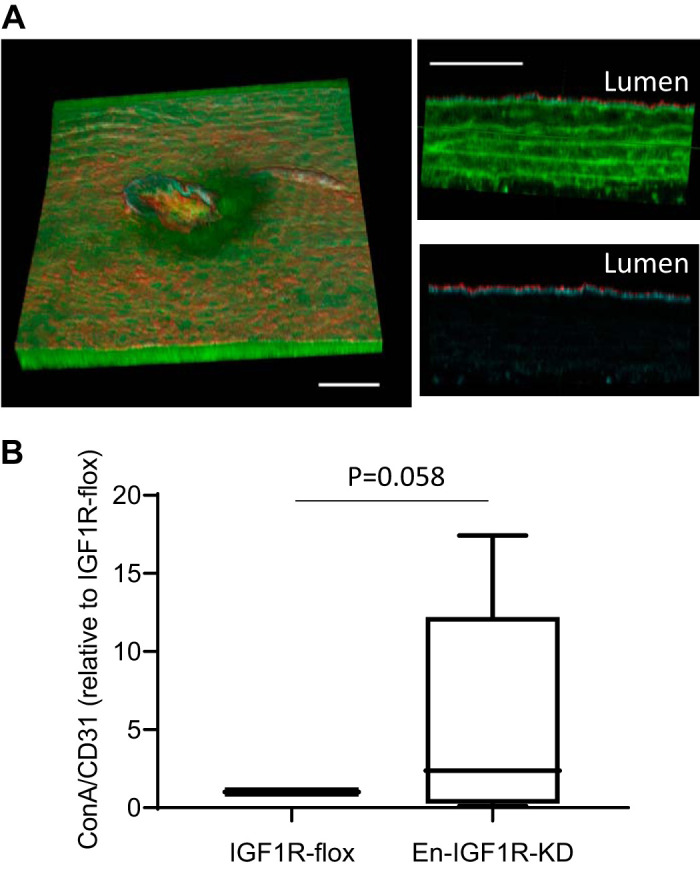
Endothelial IGF1R deficiency caused a strong trend of elevation of endothelial permeability in Apoe-deficient mice. Seven-week-old animals were fed on a high-fat diet for 4 wk and then perfused with biotin-labeled concanavalin A to assess solute permeability. A: after perfused fixation by 4% paraformaldehyde, tissues were dissected and stained with phalloidin (F-actin, green), anti-CD31 antibody [endothelial cells (ECs), red], and avidin-Atto 645 (concanavalin A, cyan). Left: 2-photon image showing a three-dimensional volume (582 µm × 536 µm lateral × 108 µm axial). Scale bar = 100 µm. Top, right: axial view of the aorta wall, where red = CD31-positive ECs, cyan = concanavalin A, and green = phalloidin, and autofluorescence of elastic laminar. Scale bar = 50 µm. Bottom, right: same view with top, right, but only showing CD31-positive ECs (red) and concanavalin A (cyan) to represent spatial localization of concanavalin A-positive part within the aorta wall. B: concanavalin A-positive stain was quantified and normalized to CD31-positive volume in the tissues. Single-sample t test was applied to evaluate statistical difference; n = 9 in each group.
RESULTS
Generation of endothelium IGF1R-deficient mice.
We cross-bred IGF-1 receptor floxed (IGF1R-flox) mice (25, 43), which had been bred onto an Apoe-deficient background, with transgenic mice that express Cre recombinase under the control of the Cdh5 promoter [B6;129-Tg(Cdh5-cre)1Spe/J, Jackson Laboratory]. The generated Cdh5-Cre+/Igf1rflox/flox/Apoe−/− mice have the floxed Igf1r gene selectively excised in vascular ECs. To confirm recombination of the floxed Igf1r gene, ECs were isolated enzymatically from lung tissues and tested for IGF1R expression (Fig. 1A). The endothelial IGF1R level determined by Western blot analysis was reduced by 60% (Fig. 1A), confirming that Cre recombination of the floxed Igf1r gene led to reduced expression levels of IGF1R (En-IGF1R-KD mice). IGF1R expression levels in aortic smooth muscle cells [Supplemental Fig. S1A (Supplemental Figures S1–S4, at Figshare repository: https://doi.org/10.6084/m9.figshare.12755861)] or Igf1r mRNA levels in circulating leukocytes or in the bone marrow (Supplemental Fig. S1, B and C) were not altered in En-IGF1R-KD mice.
Reduced expression of Igf1r in the endothelium did not affect animal growth (Fig. 1C). Circulating IGF-1 levels in En-IGF1R-KD mice were not significantly different from the levels in IGF1R-flox mice (Supplemental Fig. S2). Plasma total cholesterol levels were not different between En-IGF1R-KD and IGF1R-flox mice after 10 wk or 32 wk on a normal chow diet or after 12 wk on a high-fat diet (Supplemental Fig. S3). As it has been reported that reduced endothelial IGF-1 signaling alters vascular contractility (1, 3, 4, 23), we assessed whether endothelial function had been affected by IGF1R knockdown. There was no major difference in systolic blood pressure levels between En-IGF1R-KD and IGF1R-flox mice over 12 wk of feeding of a high-fat diet (Fig. 1D). We also assessed relaxation responses to acetylcholine in PE-constricted aortae (Fig. 1, E and F). Consistent with previous reports (1, 23), the relaxation response was enhanced in aortae from En-IGF1R-KD mice compared with those from IGF1R-flox mice on a normal chow diet (Fig. 1E). Feeding of a high-fat diet for 4 wk desensitized aortae to acetylcholine, and the IGF-1R knockdown did not influence the relaxation response (Fig. 1F). In fact, there was no difference in plasma nitrite/nitrate levels between En-IGF1R-KD and IGF1R-flox mice that were on a high-fat diet (Supplemental Fig. S4). We previously showed that IGF-1 increases antioxidant activity in vascular ECs (20). Hydroxyl radical antioxidant capacity was lower in the aortae of En-IGF1R-KD mice than in those of the control animals, but only when animals were fed on a high-fat diet (Fig. 1B).
Thus, Cdh5 promoter-driven Cre expression led to partial deficiency of IGF1R in the endothelium of IGF1R-floxed/Apoe-deficient mice and sensitized aortae to acetylcholine, resulting in improved relaxation responses following PE-induced vascular constriction. However, feeding of a high-fat diet compromised endothelial function, and IGF1R deficiency did not improve arterial relaxation response to acetylcholine. Endothelial IGF1R knockdown lowered aortic antioxidant capacity.
Atherosclerotic burden in CDH5-Cre/IGF1R-flox mice.
En-IGF1R-KD mice are on Apoe-deficient background; thus, they spontaneously develop atherosclerotic plaques in aortae at 20–40 wk of age on a normal chow diet, and a high-fat diet accelerates the development of atherosclerosis (34). After 12 wk on a high-fat diet, En-IGF1R-KD mice had a 30% increase in the plaque area compared with IGF1R-flox mice (P < 0.05, Fig. 2A). Moreover, plaque size at the aortic root was increased by 20% (P < 0.05, Fig. 2B). Relative cellular composition of the plaques (smooth muscle cells, Fig. 2C, and macrophages, Fig. 2D) was not altered by the reduced expression of IGF1R in ECs. We also assessed atherosclerosis development while animals were on a normal chow diet (Fig. 3). At 52 wk of age, En-IGF1R-KD mice had significantly higher atherosclerotic burden (66.1% increase, P < 0.05, Fig. 3). These observations indicate that endothelial IGF1R deficiency promoted atherosclerosis development.
Fig. 2.

Endothelial IGF1R deficiency promoted atherosclerotic burden in Apoe-deficient mice fed on a high-fat diet for 12 wk. Atherosclerotic lesion area was assessed on en face aorta (A) and cross-sections at aortic root (B) of En-IGF1R-KD or control mice fed on a high-fat diet for 12 wk. A: lesion surface area (% to total surface area) was assessed on entire aorta by en face Oil Red O staining. Horizontal bar indicates mean ± SE. *P < 0.05 by Student’s t test, n = 13 in IGF1R-flox and n = 27 in En-IGF1R-KD. B: lesion size (% to the tissue cross-sectional area) was assessed on cross-sections of aortic roots, which are stained with hematoxylin-eosin. *P < 0.05 by Student’s t test, n = 13 in IGF1R-flox, and n = 27 in En-IGF1R-KD. Smooth muscle (C) and macrophage (D) contents were assessed on the cross-sections of aortic root by immunologic staining against α-smooth muscle actin (smooth muscle, C) and Mac-3 (macrophage, D), respectively. Results were expressed as percentage of the plaque size (area), n = 13 in IGF1R-flox and n = 27 in En-IGF1R-KD. Scale bar = 250 µm.
Fig. 3.

Endothelial IGF1R deficiency promoted atherosclerotic burden in normal chow-fed Apoe-deficient mice at 12 mo of age. A: atherosclerotic burden was assessed by en face Oil Red O staining of aorta in En-IGF1R-KD mice or IGF1R-flox mice (control) after 12 mo of normal chow diet. B: En-IGF1R-KD mice have elevated levels of atherosclerotic burden by 66 ± 0.2% (P < 0.05, n = 5 in IGF1R-flox, and n = 6 in En-IGF1R-KD; horizontal bar indicates mean ± SE).
IGF-1 increases adherens or tight junction protein expression levels in endothelial cells and promotes endothelial barrier function.
To obtain insights into mechanisms underlying the increase in plaque burden that ensued from a reduction in endothelial IGF1R, we assessed endothelial barrier function, the loss of which makes the endothelium leaky and contributes to development or progression of atherosclerosis (33). Intercellular junctions (tight or adherens junctions) between ECs form a tight barrier and thus are integral to regulating traffic of molecular solutes. We screened potential IGF-1 effects on expression levels of intercellular junction proteins in cultured human AoECs (Fig. 4A). IGF-1 elevated expression levels of adherens junction proteins (CD31, VE-cadherin) and tight junction proteins (occludin, claudin 5, JamA, but not JamC). Intriguingly, IGF-1 downregulated VCAM1 and ICAM1, which are involved in leukocyte adhesion to the endothelium. To further confirm IGF-1’s effect on these proteins, we determined if a loss of IGF-1-dependent intracellular signaling alters expression levels of these proteins (Fig. 4, B–D). PPP (PubChem CID 72435) is a receptor kinase inhibitor specific to IGF1R. ECs were cultured in a medium supplemented with 2% FBS and growth factors, including IGF-1, and then exposed to 1 µM PPP. As expected, PPP exerted effects opposite to IGF-1’s effects. PPP downregulated adherens junction proteins (CD31, VE-cadherin) and tight junction proteins (occludin, claudin 5), whereas it upregulated VCAM1 and ICAM1 (Fig. 4B). We further confirmed these results by using other methods to block IGF-1 signal transduction (Fig. 4, C and D). Anti-IGF1R monoclonal antibody clone αIR3 is a human IGF1R-specific neutralizing antibody, and it downregulated CD31, VE-cadherin, occludin, claudin5, and JamA, while upregulating VCAM1 and ICAM1 (Fig. 4C). Likewise, RNA silencing of IGF1R produced identical results (Fig. 4D). Consistently, IGF-1 elevated levels of claudin 5 (Fig. 5A) and VE-cadherin (Fig. 5C), which colocalized with ZO-1 (Fig. 5A) and β-catenin (Fig. 5C), respectively, at the interface between neighboring cells. Conversely, inhibition of IGF1R by PPP decreased the colocalized detection of claudin 5 and ZO-1 (Fig. 5B) and of VE-cadherin and β-catenin (Fig. 5D) at the cell-cell interface.
Fig. 4.

IGF-1 increases intercellular junction protein expression levels in cultured human aortic endothelial cells. A: human aortic endothelial cells (hAoECs) were exposed to 100 ng/mL IGF-1 for 24 h and assessed for tight junction, adherens junction, and adhesion protein expression levels by Western blot analysis. Data representative of 3 independent experiments are shown. B–D: hAoECs were cultured in a complete medium (EGM-2, Lonza), which contains 2% FBS and IGF-1. IGF-1-dependent signaling was inhibited by 1 μM PPP (B), by anti-IGF1R neutralizing antibody (clone αIR3; C) for 24 h, or by siRNA to IGF1R (D, 36 h after siRNA transfection), and intercellular junction protein expression levels were assessed by Western blot analysis. Quantification from 3 to 4 independent blots (A and B; *P < 0.05, **P < 0.01) or 2 independent blots (C and D) is indicated as a fold difference to the control. Experiments were performed 3 times to confirm reproducibility.
Fig. 5.

IGF-1 and IGF1R signaling pathway positively regulate intercellular junctions in cultured human aortic endothelial cells (hAoECs). The hAoECs were exposed to 100 ng/mL IGF-1 (A and C; in serum- and IGF-1-free EGM-2 medium) or 1 µM PPP (B and D; in EGM-2 medium containing FBS and IGF-1) for 24 h. A and B: claudin 5 (green) and ZO-1 (red) were visualized by immunofluorescence staining. C and D: VE-cadherin (green) and β-catenin (red) were visualized by immunofluorescence staining. Merged detection of yellow color was quantified as described in materials and methods. *P < 0.05, **P < 0.01, n = 6–8. Data representative of 3 independent experiments are shown. Scale bar = 50 µm.
To determine if a loss of IGF-1 action results in a disruption of endothelial barrier function, ECs were exposed to 1 µM PPP for 24 h and then tested for dextran-FITC permeability over 20 min (Fig. 6A). The cell layer exposed to DMSO only (solvent for PPP) had negligible levels of leakage of dextran-FITC (1.2 ± 0.1% of “no monolayer” control), whereas exposure to PPP caused an approximate fivefold increase in leakage (5.9 ± 0.6% of “no monolayer” control). Thus, the loss of endothelial IGF-1 signaling increased permeability, consistent with the downregulation of junction proteins. We also assessed if IGF-1 regulated leukocyte adhesion on an EC monolayer. THP-1, human monocytic leukemia cells, adhered less on to a confluent monolayer of ECs after exposure to IGF-1 (Fig. 6B left). Vice versa, PPP inhibition of IGF1R-dependent signaling significantly increased THP-1 adhesion onto ECs (Fig. 6B, right). These results are consistent with the negative regulation of ICAM1 and VCAM1 by IGF-1 (Fig. 4).
Fig. 6.
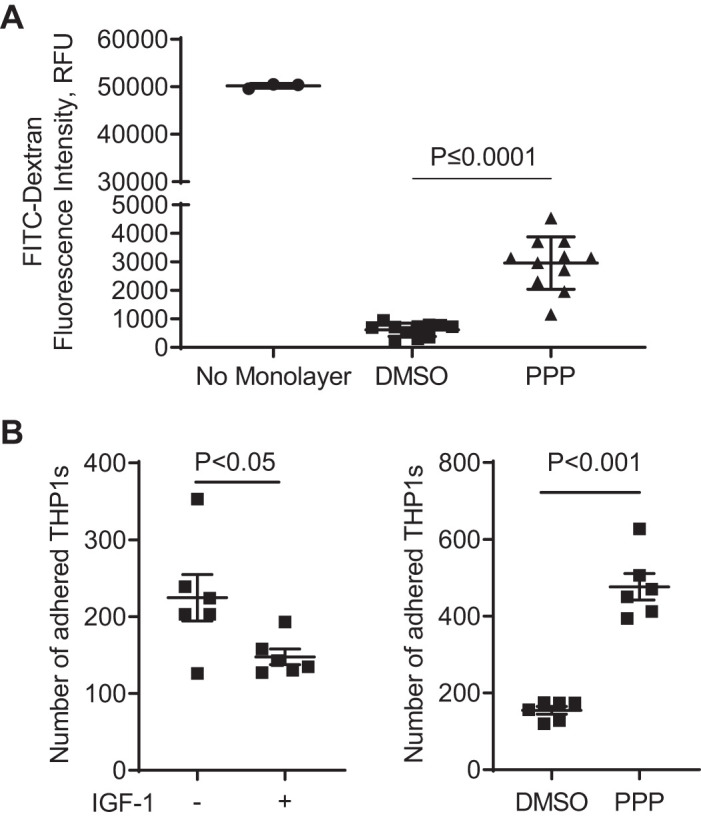
Inhibition of IGF-1 signaling elevates solute permeability and THP-1 monocyte adhesion on monolayer of human aortic endothelial cells (hAoECs). A: transwell permeability assay testing FITC-labeled dextran permeation through a monolayer culture of hAoECs (expressed as relative fluorescence units). Confluent hAoECs were exposed to 1 µM PPP (or 0.1% DMSO as a solvent control) for 24 h and assessed for solute permeability in 20 min. “No Monolayer” column represents the solute permeation through the porous membrane without cells. B: IGF-1 decreased and IGF-1R inhibition increased adhesion of THP-1 monocytes to hAoECs. Confluent hAoECs were exposed to 100 ng/mL IGF-1 or 1 µM PPP (or 0.1% DMSO as a solvent control) for 24 h and tested for THP-1 adherence. THP-1 cells were labeled with 2 µM CellTracker Green CMFDA, added into hAoEC culture at 5 × 105 cells/mL and incubated for 40 min at 37°C. After removal of nonadherent cells, adherent green THP-1 cells on the EC monolayer were photographed (viewing area = 696 × 524 nm) and counted. The average cell count of 3 random views represents 1 sample, and the sample size is 6 in each group.
To corroborate these results in vivo, we evaluated vascular permeability in En-IGF1R-KD mice (Fig. 7). Animals were fed on a high-fat diet for 4 wk and then perfused with saline containing biotinylated ConA (ConA-biotin), which binds to the basement membrane but not the luminal surface of ECs (6, 50). Perfused aortas were dissected out, stained with streptavidin (detecting ConA-biotin) and anti-CD31 antibody, and prepared for multiphoton microscopy imaging. As a site for assessment, we chose thoracic aorta intercostal artery branch sites, as they are an atherosclerosis-prone area and we could obtain images at a consistent location on each tissue. We measured ConA-stained tissue volume (representing permeation) normalized to CD31-stained tissue volume (representing the endothelium) within 0.29 ± 0.01 mm2 (a square of ~0.54 × 0.54 mm; Fig. 7A). There was a relatively large intragroup variance in measured ConA-positive volume, with a strong trend toward increased permeability in aortae of En-IGF1R-KD mice (Fig. 7B; P = 0.058, n = 9). Leukocyte adhesion on the endothelium assessed in vivo was not different between En-IGF1R-KD and IGF1R-flox mice, whereas feeding of a high-fat diet significantly elevated leukocyte binding by threefold (Fig. 8).
Fig. 8.
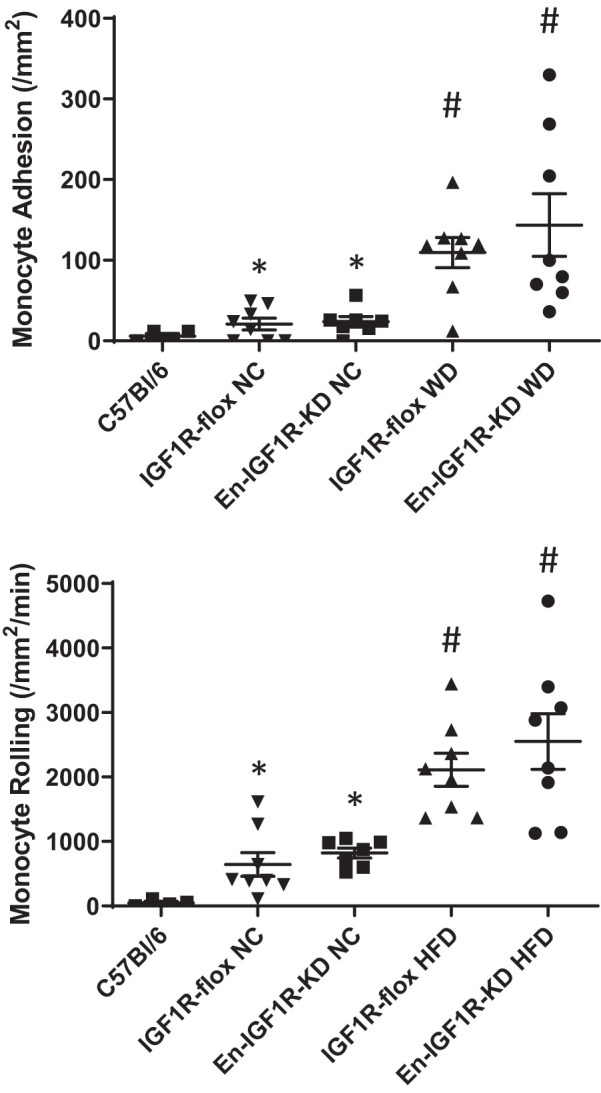
CD11+ leukocyte adhesion on endothelium was enhanced by feeding of a high-fat diet but not altered by endothelial IGF1R deficiency. Eight-week-old En-IGF1R-KD and IGF1R-flox mice were on a normal chow diet or a high-fat diet for 4 wk. Adhesion or rolling of CD11+ leukocytes was assessed by intravital microscopy. Age-matched C57Bl/6 mice were on a normal chow diet for a negative control for hyperlipidemia. Apoe deficiency increased the number of leukocyte adhesion and rolling, and feeding of a high-fat diet further increased leukocyte adhesion and rolling on the endothelium. NC, normal chow diet; HFD, high-fat diet. *P < 0.01 vs. C57Bl/6, n = 8; #P < 0.01 vs. IGF1R-flox NC, n = 8, assessed by the Mann–Whitney test.
DISCUSSION
Our investigation revealed that endothelial IGF1R deficiency promoted atherosclerosis development, as assessed by measurement of the whole aorta en face lesion area and the aortic root plaque cross-sectional area. IGF-1 positively regulated intercellular junction protein expression levels in ECs, and loss of IGF-1 signaling downregulated junction protein expression and markedly increased endothelial permeability, indicating that IGF-1 plays an important role in maintaining endothelial barrier function. Thus, there was a strong trend toward increased permeability in aortae of En-IGF1R-KD mice. Endothelial IGF1R deficiency also lowered antioxidant capacity in aortae of animals on a high-fat diet, suggesting that elevated oxidative stress could potentially contribute to the elevation of vascular permeability.
To our knowledge, this is the first report assessing endothelial IGF1R-mediated effects on atherosclerosis. Cre recombinase expression driven by the Cdh5 promoter in Igf1Rflox/flox mice achieved partial deletion of IGF1R expression (~60% reduction, Fig. 1A), which is comparable with a similar model reported elsewhere, in which IGF1R deletion was achieved by Tie2 promoter-driven Cre expression (1). Abbas et al. (1) showed that IGF1R is a negative regulator of nitric oxide bioavailability via inhibition of insulin receptor-dependent signaling pathways, thereby attenuating endothelium-dependent vasorelaxation responses. In line with their report, we observed that the relaxation response to acetylcholine was improved in aortas of En-IGF1R-KD mice compared with those of IGF1R-flox mice (Fig. 1E) fed on normal chow. Hyperlipidemia is known to induce endothelial dysfunction and elevate constrictive vascular tone (11, 17, 31), via downregulation of endothelial nitric oxide synthase (eNOS) and induction of vascular hypersensitivity to endothelin-1 (28, 31). In fact, feeding of a high-fat diet blunted the vascular relaxation response to acetylcholine in both IGF1R-flox and En-IGF1R-KD aortas; therefore, there was no difference between the groups (Fig. 1F). Overall, our results imply that although the IGF1R may be a negative regulator of the vascular relaxation response, this effect is lost in the setting of prolonged hyperlipidemia, which results in severely attenuated aortic relaxation responses (Fig. 1F). Of note, there was no difference in serum nitrates and nitrites levels between En-IGF1R-KD and IGF1R-flox mice under feeding of a high-fat diet (Supplemental Fig. S4), suggesting no difference in NO bioavailability between the groups. Thus, changes in nitric oxide bioavailability do not explain the enhanced atheroma formation in En-IGF1R-KD mice. This finding is consistent with our previous findings indicating that the atheroprotective effect of an increase in circulating IGF-1 is nitric oxide independent (44).
We used Apoe-deficient mice, a widely used murine model of atherosclerosis. The Apoe gene product, apolipoprotein E (APOE), functions as a lipid transport protein mediating clearance of circulating lipoproteins and also mediating cholesterol efflux from peripheral tissues and cells for reverse cholesterol transport (32). Therefore, Apoe deficiency promotes atherosclerosis via hyperlipidemia and via cholesterol accumulation in macrophages and smooth muscle cells in plaques. Our data show that there was no difference in levels of hyperlipidemia between IGF1R-flox and En-IGF1R-KD mice (Supplemental Fig. S2). The lipid efflux pathways are active in ECs and support eNOS-mediated nitric oxide production (2, 49, 53); therefore, Apoe deficiency may result in lowered eNOS activity. Our data show that En-IGF1R-KD improved vasorelaxation (Fig. 1E), consistent with elevated eNOS activity (23), suggesting that En-IGF1R-KD counteracted the effect of Apoe deficiency at least in animals fed normal chow.
We found that IGF-1 positively regulated a variety of intercellular junction protein expression levels (Fig. 4A), and vice versa, a loss of IGF-1 signaling caused downregulation of junction proteins (Fig. 4, B and C), along with elevated permeability of a monolayer of ECs (Fig. 6). Consistent with these findings in cell culture, there was a strong trend toward elevated vascular leak in aortae of En-IGF1R-KD mice (Fig. 7). We also found that IGF-1 decreased the expression of the cell adhesion molecules ICAM-1 and VCAM-1 and reduced adhesion of THP-1 monocytes to an EC monolayer. Conversely, inhibition of IGF-1 signaling resulted in an increase of THP-1 adhesion to ECs (Fig. 6). Our in vivo data, however, showed that endothelial IGF1R deficiency did not increase leukocyte adhesion to the endothelium (Fig. 8). The reasons for this discrepancy between in vitro and in vivo observations are unclear and await future investigation. More importantly, our results suggest that changes in leukocyte adhesion are not a likely reason for elevated atherosclerosis in En-IGF1R-KD mice. Rather, reduced endothelial barrier function and elevated solute permeability are more likely to explain the increase in atherosclerosis present in En-IGF1R-KD mice. Previous reports provide conflicting observations about IGF-1’s effects on endothelial permeability. Intraocular IGF-1 disrupted the blood-retinal barrier (14, 18), an effect mediated by vascular endothelial growth factor (13, 36). Ribozyme-mediated targeting of IGF1R prevented high glucose-induced downregulation of occludin, and thus disruption of tight junctions in retinal ECs (42). It has been shown that young En-IGF1R-KD mice had an intact blood-brain barrier (BBB) without elevation of leakage (27). However, in middle-aged rats with cerebral ischemia, IGF-1 improved BBB function (5). In a unilateral ureteral obstruction-induced renal fibrosis model, IGF1R deficiency was associated with endothelial barrier dysfunction (29). These conflicting observations may be due to a difference in ECs from different vascular beds or may be due to differences in the primary insults causing the endothelial dysfunction, e.g., high glucose, high lipid, or ischemia and/or reperfusion. It is also possible that acute IGF-1 exposure may downregulate endothelial barrier function (13, 14, 18, 36, 42), whereas sustained increase in IGF-1 signaling leads to enhanced barrier function (5, 29).
Our results indicate that IGF-1 coordinately regulates a variety of intercellular junction proteins and cell adhesion proteins in aortic ECs. It is possible that IGF-1 regulates them individually or regulates a master regulatory event that changes expression levels of these proteins. It had been described that reactive oxygen species (ROS) disrupt endothelial tight and adherens junctions and thus disrupt endothelial barrier function leading to elevated paracellular permeability (30, 37). ROS cause dissociation of tight and adherens junctions (9) and downregulation of junction proteins (38, 54). We and others have previously shown antioxidant effects of IGF-1 in arterial ECs or arterial tissue. For instance, IGF-1 upregulated glutathione peroxidase-1 expression levels and antioxidant activity in aortic ECs (20). Liver-specific knockdown of IGF-1, which causes a decrease of circulating IGF-1 levels by 50%, caused dysregulation of Nrf2-dependent antioxidant responses in the vasculature (3). Our current data (Fig. 1B) indicate that endothelium IGF1R knockdown lowered antioxidant capacity in aorta, which is consistent with antioxidant effects of IGF-1 in ECs and suggests a redox-mediated mechanism underlying IGF-1 regulation of intercellular junctions and endothelial permeability.
Aging is the most significant independent risk factor for atherosclerotic cardiovascular diseases, and it is noteworthy that circulating IGF-1 levels decline with age. Thus, our experimental results may support the hypothesis that age-dependent decline of circulating IGF-1 causes diminished IGF-1 signaling in the endothelium, leading to an elevated risk for atherosclerosis. Our previous study using congenic mice with low-serum IGF-I levels (B6.C3H-6T mice) (39) showed that these mice had higher atherosclerotic burden, which is consistent with a significant contribution of the age-dependent decline of serum IGF-1 levels to atherosclerosis (41). Not only circulating (i.e., endocrine) IGF-1 but also vascular IGF-1 production and signaling (i.e., autocrine or paracrine effects) may decline with age. For instance, a recent publication reported significant changes in miRNA expression profiles in aged mouse aorta (24). MiR-29a, MiR-30, MiR-195, MiR-365, and let-7i are miRNAs that increased with age, and they were shown to target IGF-1 and IGF-1R mRNAs. Therefore, these miRNAs potentially downregulate IGF-1 signaling in aged aorta. On the other hand, it was recently shown that developmental IGF-1 deficiency elicits persisting late-life changes in miRNA expression in the vasculature, potentially leading to perturbation of extracellular matrix homeostasis and loss of structural integrity of vessels (48). Further investigation will be required to assess potential links between low IGF-1 availability and changes in miRNA expression profile in the endothelium, which may lead to loss of endothelial function. Interestingly, novel evidence (15, 35) suggests that IGF-1 protects against features of cognitive loss, vascular rarefaction, and sensorimotor decline with aging. These observations suggest that IGF-1 may have a protective effect in the cerebrovasculature.
In summary, our findings revealed a novel role for IGF-1 in supporting endothelial barrier function via upregulation of junction proteins. Endothelial IGF1R deficiency significantly elevated atherosclerotic burden after long-term normal chow feeding or when animals were fed on a high-fat diet. The endothelial IGF1R deficiency did not alter leukocyte adhesion or rolling on the endothelium in vivo and did not change nitric oxide levels in animals on a high-fat diet. Thus, the loss of endothelial barrier function and elevated endothelial permeability are likely, at least in part, to be responsible for elevated atherosclerotic burden in En-IGF1R-KD mice. Potential mechanisms for IGF-1 regulation of junction proteins are unclear but could include a redox-sensitive mechanism, as suggested by the finding that aortic hydroxyl radical antioxidant capacity was reduced in En-IGF1R-KD mice on a high-fat diet. Further investigation into IGF-1 regulation of endothelial barrier function could lead to novel therapeutic approaches for atherosclerotic vascular disease.
GRANTS
This work was supported by American Heart Association Grant 1-13-BS-210 (to T.C.W.) and National Institute of Health Grants R01-HL070241 (to P.D.), R01-HL142796 (to S.S.), R01-HL127092 (to T.C.W.), and R01-AA022108 (to R.J.K.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.H. conceived and designed research; Y.H., S.S., S.-Y.S., S.D., P.S., Z.L., X.H., T.C.W., M.W., D.W., and H.Y. performed experiments; Y.H., S.S., S.-Y.S., S.D., P.S., X.H., T.C.W., and H.Y. analyzed data; Y.H., S.S., S.D., M.H.H., T.C.W., R.J.K., T.Y., and P.D. interpreted results of experiments; Y.H., S.-Y.S., and S.D. prepared figures; Y.H. drafted manuscript; P.D. edited and revised manuscript; Y.H. and P.D. approved final version of manuscript.
ACKNOWLEDGMENTS
All the confocal images published in this article were obtained at the Molecular Cytology Core, University of Missouri. We thank Drs. Baker and Jurkevich (University of Missouri) for expert advice for obtaining confocal images and quantitative analysis. We thank Daniel J. Lightell, Jr., for technical help and support for the project.
REFERENCES
- 1.Abbas A, Imrie H, Viswambharan H, Sukumar P, Rajwani A, Cubbon RM, Gage M, Smith J, Galloway S, Yuldeshava N, Kahn M, Xuan S, Grant PJ, Channon KM, Beech DJ, Wheatcroft SB, Kearney MT. The insulin-like growth factor-1 receptor is a negative regulator of nitric oxide bioavailability and insulin sensitivity in the endothelium. Diabetes 60: 2169–2178, 2011. doi: 10.2337/db11-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Assanasen C, Mineo C, Seetharam D, Yuhanna IS, Marcel YL, Connelly MA, Williams DL, de la Llera-Moya M, Shaul PW, Silver DL. Cholesterol binding, efflux, and a PDZ-interacting domain of scavenger receptor-BI mediate HDL-initiated signaling. J Clin Invest 115: 969–977, 2005. doi: 10.1172/JCI23858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bailey-Downs LC, Mitschelen M, Sosnowska D, Toth P, Pinto JT, Ballabh P, Valcarcel-Ares MN, Farley J, Koller A, Henthorn JC, Bass C, Sonntag WE, Ungvari Z, Csiszar A. Liver-specific knockdown of IGF-1 decreases vascular oxidative stress resistance by impairing the Nrf2-dependent antioxidant response: a novel model of vascular aging. J Gerontol A Biol Sci Med Sci 67A: 313–329, 2012. doi: 10.1093/gerona/glr164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey-Downs LC, Sosnowska D, Toth P, Mitschelen M, Gautam T, Henthorn JC, Ballabh P, Koller A, Farley JA, Sonntag WE, Csiszar A, Ungvari Z. Growth hormone and IGF-1 deficiency exacerbate high-fat diet-induced endothelial impairment in obese Lewis dwarf rats: implications for vascular aging. J Gerontol A Biol Sci Med Sci 67A: 553–564, 2012. doi: 10.1093/gerona/glr197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bake S, Okoreeh AK, Alaniz RC, Sohrabji F. Insulin-Like Growth Factor (IGF)-I Modulates Endothelial Blood-Brain Barrier Function in Ischemic Middle-Aged Female Rats. Endocrinology 157: 61–69, 2016. doi: 10.1210/en.2015-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barber AJ, Antonetti DA. Mapping the blood vessels with paracellular permeability in the retinas of diabetic rats. Invest Ophthalmol Vis Sci 44: 5410–5416, 2003. doi: 10.1167/iovs.03-0244. [DOI] [PubMed] [Google Scholar]
- 7.Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol 23: 168–175, 2003. doi: 10.1161/01.ATV.0000051384.43104.FC. [DOI] [PubMed] [Google Scholar]
- 8.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87: 840–844, 2000. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- 9.Chattopadhyay R, Raghavan S, Rao GN. Resolvin D1 via prevention of ROS-mediated SHP2 inactivation protects endothelial adherens junction integrity and barrier function. Redox Biol 12: 438–455, 2017. doi: 10.1016/j.redox.2017.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.d’Uscio LV, Smith LA, Katusic ZS. Hypercholesterolemia impairs endothelium-dependent relaxations in common carotid arteries of apolipoprotein e-deficient mice. Stroke 32: 2658–2664, 2001. doi: 10.1161/hs1101.097393. [DOI] [PubMed] [Google Scholar]
- 12.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation 109, Suppl 1: III27–III32, 2004. doi: 10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 13.Deissler HL, Deissler H, Lang GE. Inhibition of vascular endothelial growth factor (VEGF) is sufficient to completely restore barrier malfunction induced by growth factors in microvascular retinal endothelial cells. Br J Ophthalmol 95: 1151–1156, 2011. doi: 10.1136/bjo.2010.192229. [DOI] [PubMed] [Google Scholar]
- 14.Devi TS, Singh LP, Hosoya K-I, Terasaki T. GSK-3β/CREB axis mediates IGF-1-induced ECM/adhesion molecule expression, cell cycle progression and monolayer permeability in retinal capillary endothelial cells: Implications for diabetic retinopathy. Biochim Biophys Acta 1812: 1080–1088, 2011. doi: 10.1016/j.bbadis.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Farias Quipildor GE, Mao K, Hu Z, Novaj A, Cui M-H, Gulinello M, Branch CA, Gubbi S, Patel K, Moellering DR, Tarantini S, Kiss T, Yabluchanskiy A, Ungvari Z, Sonntag WE, Huffman DM. Central IGF-1 protects against features of cognitive and sensorimotor decline with aging in male mice. Geroscience 41: 185–208, 2019. doi: 10.1007/s11357-019-00065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hama H, Hioki H, Namiki K, Hoshida T, Kurokawa H, Ishidate F, Kaneko T, Akagi T, Saito T, Saido T, Miyawaki A. ScaleS: an optical clearing palette for biological imaging. Nat Neurosci 18: 1518–1529, 2015. doi: 10.1038/nn.4107. [DOI] [PubMed] [Google Scholar]
- 17.Hasdai D, Nielsen MF, Rizza RA, Holmes DR Jr, Richardson DM, Cohen P, Lerman A. Attenuated in vitro coronary arteriolar vasorelaxation to insulin-like growth factor I in experimental hypercholesterolemia. Hypertension 34: 89–95, 1999. doi: 10.1161/01.HYP.34.1.89. [DOI] [PubMed] [Google Scholar]
- 18.Haurigot V, Villacampa P, Ribera A, Llombart C, Bosch A, Nacher V, Ramos D, Ayuso E, Segovia JC, Bueren JA, Ruberte J, Bosch F. Increased intraocular insulin-like growth factor-I triggers blood-retinal barrier breakdown. J Biol Chem 284: 22961–22969, 2009. doi: 10.1074/jbc.M109.014787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heitzer T, Schlinzig T, Krohn K, Meinertz T, Münzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104: 2673–2678, 2001. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- 20.Higashi Y, Pandey A, Goodwin B, Delafontaine P. Insulin-like growth factor-1 regulates glutathione peroxidase expression and activity in vascular endothelial cells: Implications for atheroprotective actions of insulin-like growth factor-1. Biochim Biophys Acta 1832: 391–399, 2013. doi: 10.1016/j.bbadis.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higashi Y, Sukhanov S, Anwar A, Shai SY, Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci 67A: 626–639, 2012. doi: 10.1093/gerona/gls102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higashi Y, Sukhanov S, Parthasarathy S, Delafontaine P. The ubiquitin ligase Nedd4 mediates oxidized low-density lipoprotein-induced downregulation of insulin-like growth factor-1 receptor. Am J Physiol Heart Circ Physiol 295: H1684–H1689, 2008. doi: 10.1152/ajpheart.00548.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imrie H, Viswambharan H, Sukumar P, Abbas A, Cubbon RM, Yuldasheva N, Gage M, Smith J, Galloway S, Skromna A, Rashid ST, Futers TS, Xuan S, Gatenby VK, Grant PJ, Channon KM, Beech DJ, Wheatcroft SB, Kearney MT. Novel role of the IGF-1 receptor in endothelial function and repair: studies in endothelium-targeted IGF-1 receptor transgenic mice. Diabetes 61: 2359–2368, 2012. doi: 10.2337/db11-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiss T, Giles CB, Tarantini S, Yabluchanskiy A, Balasubramanian P, Gautam T, Csipo T, Nyúl-Tóth Á, Lipecz A, Szabo C, Farkas E, Wren JD, Csiszar A, Ungvari Z. Nicotinamide mononucleotide (NMN) supplementation promotes anti-aging miRNA expression profile in the aorta of aged mice, predicting epigenetic rejuvenation and anti-atherogenic effects. Geroscience 41: 419–439, 2019. doi: 10.1007/s11357-019-00095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klöting N, Koch L, Wunderlich T, Kern M, Ruschke K, Krone W, Brüning JC, Blüher M. Autocrine IGF-1 action in adipocytes controls systemic IGF-1 concentrations and growth. Diabetes 57: 2074–2082, 2008. doi: 10.2337/db07-1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T. A simple method of isolating mouse aortic endothelial cells. J Atheroscler Thromb 12: 138–142, 2005. doi: 10.5551/jat.12.138. [DOI] [PubMed] [Google Scholar]
- 27.Kondo T, Hafezi-Moghadam A, Thomas K, Wagner DD, Kahn CR. Mice lacking insulin or insulin-like growth factor 1 receptors in vascular endothelial cells maintain normal blood-brain barrier. Biochem Biophys Res Commun 317: 315–320, 2004. doi: 10.1016/j.bbrc.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 28.Lerman A, Webster MW, Chesebro JH, Edwards WD, Wei CM, Fuster V, Burnett JC Jr. Circulating and tissue endothelin immunoreactivity in hypercholesterolemic pigs. Circulation 88: 2923–2928, 1993. doi: 10.1161/01.CIR.88.6.2923. [DOI] [PubMed] [Google Scholar]
- 29.Liang M, Woodard LE, Liang A, Luo J, Wilson MH, Mitch WE, Cheng J. Protective role of insulin-like growth factor-1 receptor in endothelial cells against unilateral ureteral obstruction-induced renal fibrosis. Am J Pathol 185: 1234–1250, 2015. doi: 10.1016/j.ajpath.2015.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol Cell Physiol 280: C719–C741, 2001. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- 31.Mathew V, Cannan CR, Miller VM, Barber DA, Hasdai D, Schwartz RS, Holmes DR Jr, Lerman A. Enhanced endothelin-mediated coronary vasoconstriction and attenuated basal nitric oxide activity in experimental hypercholesterolemia. Circulation 96: 1930–1936, 1997. doi: 10.1161/01.CIR.96.6.1930. [DOI] [PubMed] [Google Scholar]
- 32.Matsuura F, Wang N, Chen W, Jiang XC, Tall AR. HDL from CETP-deficient subjects shows enhanced ability to promote cholesterol efflux from macrophages in an apoE- and ABCG1-dependent pathway. J Clin Invest 116: 1435–1442, 2006. doi: 10.1172/JCI27602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mundi S, Massaro M, Scoditti E, Carluccio MA, van Hinsbergh VW, Iruela-Arispe ML, De Caterina R. Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review. Cardiovasc Res 114: 35–52, 2018. doi: 10.1093/cvr/cvx226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 14: 133–140, 1994. doi: 10.1161/01.ATV.14.1.133. [DOI] [PubMed] [Google Scholar]
- 35.Norling AM, Gerstenecker AT, Buford TW, Khan B, Oparil S, Lazar RM. The role of exercise in the reversal of IGF-1 deficiencies in microvascular rarefaction and hypertension. Geroscience 42: 141–158, 2020. doi: 10.1007/s11357-019-00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poulaki V, Joussen AM, Mitsiades N, Mitsiades CS, Iliaki EF, Adamis AP. Insulin-like growth factor-I plays a pathogenetic role in diabetic retinopathy. Am J Pathol 165: 457–469, 2004. doi: 10.1016/S0002-9440(10)63311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci 13: 7210–7226, 2008. doi: 10.2741/3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rochfort KD, Collins LE, Murphy RP, Cummins PM. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: consequences for interendothelial adherens and tight junctions. PLoS One 9: e101815, 2014. doi: 10.1371/journal.pone.0101815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosen CJ, Ackert-Bicknell CL, Adamo ML, Shultz KL, Rubin J, Donahue LR, Horton LG, Delahunty KM, Beamer WG, Sipos J, Clemmons D, Nelson T, Bouxsein ML, Horowitz M. Congenic mice with low serum IGF-I have increased body fat, reduced bone mineral density, and an altered osteoblast differentiation program. Bone 35: 1046–1058, 2004. doi: 10.1016/j.bone.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 40.Shai SY, Sukhanov S, Higashi Y, Vaughn C, Kelly J, Delafontaine P. Smooth muscle cell-specific insulin-like growth factor-1 overexpression in Apoe-/- mice does not alter atherosclerotic plaque burden but increases features of plaque stability. Arterioscler Thromb Vasc Biol 30: 1916–1924, 2010. doi: 10.1161/ATVBAHA.110.210831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shai SY, Sukhanov S, Higashi Y, Vaughn C, Rosen CJ, Delafontaine P. Low circulating insulin-like growth factor I increases atherosclerosis in ApoE-deficient mice. Am J Physiol Heart Circ Physiol 300: H1898–H1906, 2011. doi: 10.1152/ajpheart.01081.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spoerri PE, Afzal A, Li Calzi S, Shaw LC, Cai J, Pan H, Boulton M, Grant MB. Effects of VEGFR-1, VEGFR-2, and IGF-IR hammerhead ribozymes on glucose-mediated tight junction expression in cultured human retinal endothelial cells. Mol Vis 12: 32–42, 2006. [PubMed] [Google Scholar]
- 43.Stachelscheid H, Ibrahim H, Koch L, Schmitz A, Tscharntke M, Wunderlich FT, Scott J, Michels C, Wickenhauser C, Haase I, Brüning JC, Niessen CM. Epidermal insulin/IGF-1 signalling control interfollicular morphogenesis and proliferative potential through Rac activation. EMBO J 27: 2091–2101, 2008. doi: 10.1038/emboj.2008.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sukhanov S, Higashi Y, Shai SY, Blackstock C, Galvez S, Vaughn C, Titterington J, Delafontaine P. Differential requirement for nitric oxide in IGF-1-induced anti-apoptotic, anti-oxidant and anti-atherosclerotic effects. FEBS Lett 585: 3065–3072, 2011. doi: 10.1016/j.febslet.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sukhanov S, Higashi Y, Shai SY, Itabe H, Ono K, Parthasarathy S, Delafontaine P. Novel effect of oxidized low-density lipoprotein: cellular ATP depletion via downregulation of glyceraldehyde-3-phosphate dehydrogenase. Circ Res 99: 191–200, 2006. doi: 10.1161/01.RES.0000232319.02303.8c. [DOI] [PubMed] [Google Scholar]
- 46.Sukhanov S, Higashi Y, Shai SY, Snarski P, Danchuk S, D’Ambra V, Tabony M, Woods TC, Hou X, Li Z, Ozoe A, Chandrasekar B, Takahashi SI, Delafontaine P. SM22α (Smooth Muscle Protein 22-α) Promoter-Driven IGF1R (Insulin-Like Growth Factor 1 Receptor) Deficiency Promotes Atherosclerosis. Arterioscler Thromb Vasc Biol 38: 2306–2317, 2018. doi: 10.1161/ATVBAHA.118.311134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sukhanov S, Higashi Y, Shai SY, Vaughn C, Mohler J, Li Y, Song YH, Titterington J, Delafontaine P. IGF-1 reduces inflammatory responses, suppresses oxidative stress, and decreases atherosclerosis progression in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 27: 2684–2690, 2007. doi: 10.1161/ATVBAHA.107.156257. [DOI] [PubMed] [Google Scholar]
- 48.Tarantini S, Giles CB, Wren JD, Ashpole NM, Valcarcel-Ares MN, Wei JY, Sonntag WE, Ungvari Z, Csiszar A. IGF-1 deficiency in a critical period early in life influences the vascular aging phenotype in mice by altering miRNA-mediated post-transcriptional gene regulation: implications for the developmental origins of health and disease hypothesis. Age (Dordr) 38: 239–258, 2016. doi: 10.1007/s11357-016-9943-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terasaka N, Yu S, Yvan-Charvet L, Wang N, Mzhavia N, Langlois R, Pagler T, Li R, Welch CL, Goldberg IJ, Tall AR. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J Clin Invest 118: 3701–3713, 2008. doi: 10.1172/JCI35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thurston G, Baluk P, Hirata A, McDonald DM. Permeability-related changes revealed at endothelial cell borders in inflamed venules by lectin binding. Am J Physiol 271: H2547–H2562, 1996. doi: 10.1152/ajpheart.1996.271.6.H2547. [DOI] [PubMed] [Google Scholar]
- 51.van Hinsbergh VW. Endothelium—role in regulation of coagulation and inflammation. Semin Immunopathol 34: 93–106, 2012. doi: 10.1007/s00281-011-0285-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang WZ, Jones AW, Wang M, Durante W, Korthuis RJ. Preconditioning with soluble guanylate cyclase activation prevents postischemic inflammation and reduces nitrate tolerance in heme oxygenase-1 knockout mice. Am J Physiol Heart Circ Physiol 305: H521–H532, 2013. doi: 10.1152/ajpheart.00810.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westerterp M, Tsuchiya K, Tattersall IW, Fotakis P, Bochem AE, Molusky MM, Ntonga V, Abramowicz S, Parks JS, Welch CL, Kitajewski J, Accili D, Tall AR. Deficiency of ATP-Binding Cassette Transporters A1 and G1 in Endothelial Cells Accelerates Atherosclerosis in Mice. Arterioscler Thromb Vasc Biol 36: 1328–1337, 2016. doi: 10.1161/ATVBAHA.115.306670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, Meda P, Imhof BA, Nourshargh S. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol 12: 761–769, 2011. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]