

Abstract
Histone deacetylases (HDACs) are validated drug targets for cancer treatment. Increased HDAC isozyme selectivity and novel strategies to inhibit HDAC activity could lead to safer and more effective drug candidates. Nonetheless, it is quite challenging to develop isozyme-specific HDACi due to the highly conserved catalytic domain. We discovered XZ9002, a first-in-class HDAC3-specific PROTAC that potently degraded HDAC3. Importantly, XZ9002 is more effective to inhibit cancer cell proliferation than its proteolysis-inactive counterpart, suggesting HDAC3 degradation is a novel and promising anticancer approach.
Graphical Abstract
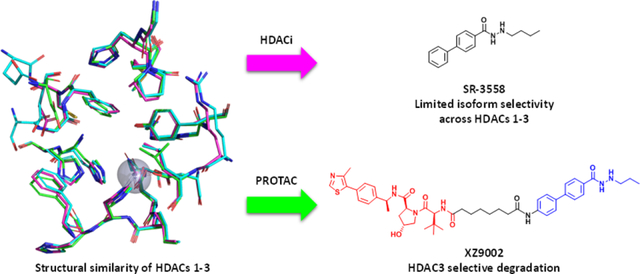
HDACs and histone acetyltransferases (HATs) are critical epigenetic regulators that impact transcription, replication, recombination, repair, metabolism, and other biological processes. In human cells, there are eleven isozymes of zinc-dependent HDACs which can be divided into four phylogenetic groups: class I (HDACs 1–3, and 8), class IIa (HDACs 4, 5, 7, and 9), class IIb (HDACs 6 and 10), and class IV (HDAC 11).1 HDACs are commonly overexpressed in various cancer types including hematologic and solid malignancies. Among them, class I HDACs are critical to activating oncogenes underlying tumorigenesis, disease progression and treatment resistance.2 Recent studies have shown that HDACs 1–3 are important for oncogene expression regulated by super enhancers in breast and other cancer types.3
To date, a number of HDAC inhibitors (HDACi) with different chemical scaffolds have been developed as anticancer agents, and four of them have been approved by USFDA for treating lymphomas, leukemias, and multiple myelomas. A typical HDACi consists of a zinc binding group (ZBG), a surface-recognition cap group, and an appropriate linker.4 Among those components, ZBG plays an important role in subtype selectivity. The majority of hydroxamate-derived HDACi are pan-HDACi with limited isozyme selectivity whereas benzamide-based HDACi prefer to bind to class I HDACs.5 Fine-tuned surface-recognition cap group can improve isozyme selectivity as well. Different types of HDACi have been tested clinically, but none of them have achieved clinical success for treating solid tumors as a single agent, which is probably due to their ineffectively low concentrations in tumor tissue. Importantly, dose-limiting adverse effects such as cardiac toxicity associated with hERG K+ channel activation hinder their application in the clinic.6 Increased HDAC isozyme selectivity and novel strategies to abolish HDAC activity could lead to more effective drug candidates to achieve clinical success.
Proteolysis targeting chimera (PROTAC) has emerged as a revolutionary technology in drug discovery.7 PROTACs possess several advantages over conventional inhibitors such as high potency, extended duration of action, and potential tissue/cell type selectivity.3b, 8 Moreover, due to their unique mechanism of action (MOA) via the formation of ternary complexes, specific proteasomal degradation of protein of interest (POI) may be achieved even when a promiscuous ligand of POI is used in a PROTAC. Recently, attempts have been made to degrade HDACs using the PROTAC approach. However, by conjugating either pan-HDACi or HDAC6 inhibitor as warheads, PROTACs 1–5 (Fig. S1, ESI†) with hydroxamate as ZBG can only degrade HDAC6.9 Of note, derivatives of 18β-glycyrrhetinic acid were reported to downregulate HDAC3 and HDAC6 at high concentrations, but the mechanism is unclear.10 By employing benzamide-based HDAC binder as the warhead, Smalley et al. developed PROTAC 6 (Fig. S1, ESI†) that degraded class I HDACs.11 More recently, a macrocyclic tetrapeptide HDACi was converted to a PROTAC that degraded HDACs 1–3.12 Nevertheless, weak and nonselective HDAC3 degradation was observed in those cases. Here we report the discovery of XZ9002, a PROTAC that potently and selectively degrades HDAC3 while sparing other HDAC isozymes in cancer cell lines.
Previously, we synthesized a small library of HDACi with a novel benzoylhydrazide ZBG and identified SR-3558 as a potent and selective inhibitor of class I HDACs (Fig. 1A).13 We performed molecular docking studies using AutoDock Vina and found that the alkane tail in the ZBG occupies the bottom space of active-site pocket in class I HDACs (Fig. 1B). This binding mode well explained the unique selectivity of SR-3558 since other HDACs such as HDAC6 do not have this extra space. The docking studies also suggest that an n-propyl tail appears to adopt a better conformation fitting compared to the n-butyl group in SR-3558 (Fig. 1A and B). Moreover, the lowest binding energy pose of SR-3558 revealed that the terminal phenyl ring is exposed to solvent, providing a feasible linking site for E3 ligase ligand tethering. Based on these findings, we designed PROTACs 7a-c and 8a-d which recruit CRBN and VHL E3 ligases, respectively (Fig. 1C).
Fig. 1.
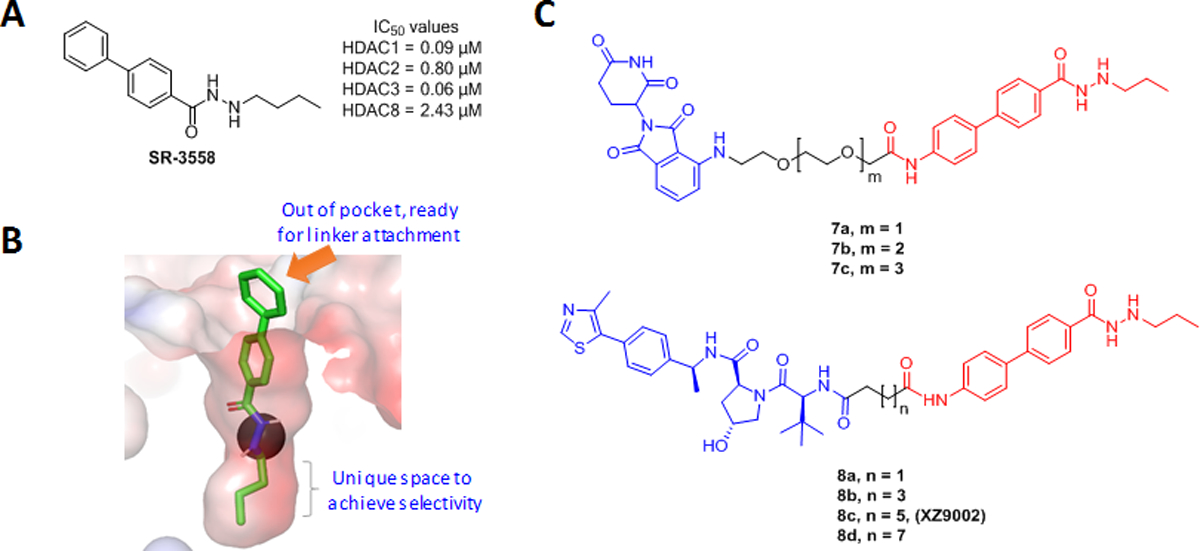
The rational design of HDAC3 specific degraders. (A) The chemical structure of lead compound SR-3558 that selectively binds to class I HDACs 1–3. (B) Molecular docking study revealed the lowest binding energy pose for SR-3558 with HDAC3 (PDB: 4A69). (C) The chemical structures of CRBN- and VHL-based PROTACs derived from SR-3558.
A multi-step procedure using advanced benzoic acid/ester and hydrazine as starting materials is reported for synthesizing benzoylhydrazide-containing HDACi.14 Due to the structural complexity of our designed PROTACs, it can be challenging to introduce ZBG in the late stage. We therefore developed a new strategy to build an advanced intermediate 13 in four steps with an excellent yield (Scheme 1). Briefly, 4-iodobenzohydrazide (9) was converted to 10 through a reductive amidation with propionaldehyde. The basic nitrogen in 10 was protected with Boc to give 11, which was coupled with 4-nitrophenylboronic acid through a Suzuki reaction followed by reduction of the nitro group in 12 via hydrogenolysis to afford precursor 13. The amidation between 13 and 14 in the presence of HATU, followed by cleavage of Boc group under acidic condition resulted in the synthesis of XZ9002. PROTACs 7a-c and 8a-d were synthesized using the same method (Scheme S1, ESI†).
Scheme 1.
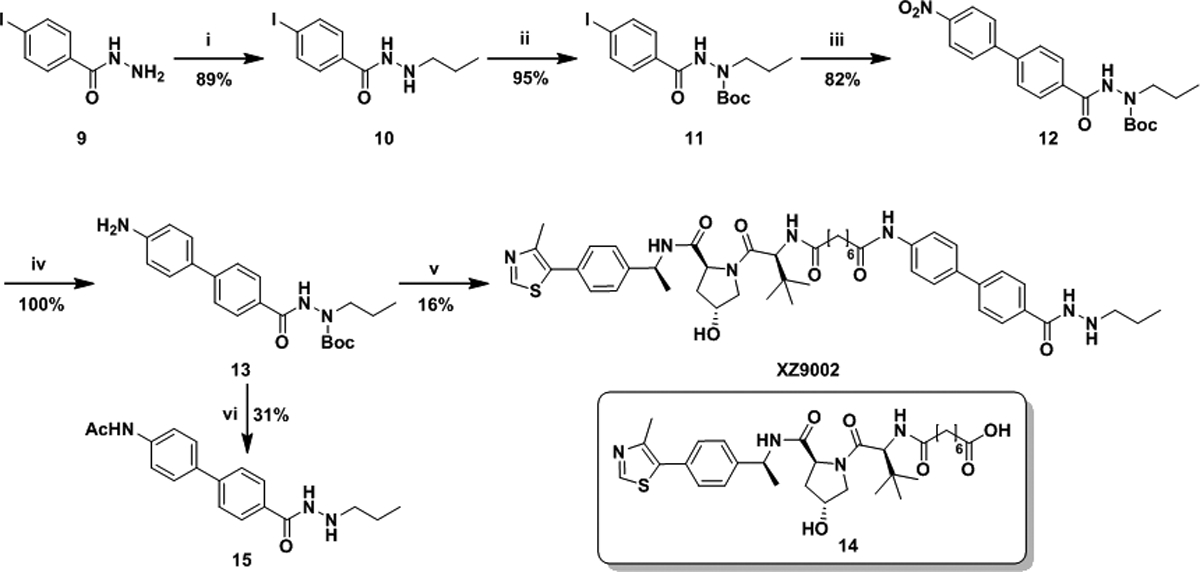
(i) a) Propionaldehyde, MeOH, THF; b) NaBH4, MeOH, THF. (ii) Boc2O, Et3N, CH2Cl2. (iii) 4-Nitrophenylboronic acid, Pd(PPh3)4, K2CO3, toluene, EtOH, water, 90 °C. (iv) Pd/C, H2, ethyl acetate. (v) a) 14, Et3N, HATU, CH2Cl2; b) TFA, CH2Cl2. (vi) a) Ac2O, Et3N, CH2Cl2; b) TFA, CH2Cl2.
To confirm if linker attachment is suitable at the proposed position, we synthesized an HDACi 15, in which the terminal phenyl ring in SR-3558 was replaced with an N-Ac-aniline (Scheme 1). The HDAC binding affinities were measured using the HDAC-Glo™ I/II Assays.13 Compound 15 exhibited good inhibitory activities against several HDACs with at least a 5.5-fold preference for HDAC3 (Table S1, ESI†), confirming the position is suitable for linker attachment. CRBN-based PROTACs 7a-c with PEG linkers maintained the in vitro HDAC binding affinities, whereas VHL-based PROTACs 8a-d with alkane linkers showed 10- to 20-fold decrease in HDAC inhibitory potencies. The HDAC binding activities slightly decreased when the linker length was increased in the 8a-d series.
The HDAC degradation ability of all designed PROTACs was examined in triple-negative breast cancer cell line MDA-MB-468 and the ER+ breast cancer cell line T47D. Western blot analysis (Fig. S2, ESI†) revealed that the VHL-based PROTACs 8a-d could induce HDAC3 degradation at 100 nM in MDA-MB-468 and T47D cells despite their compromised HDAC inhibitory activities, whereas these compounds had minimal or no effects on the protein levels of HDAC1 and HDAC2. In contrast, among PROTACs derived from CRBN, only 7c showed a small effect on HDAC3 degradation at 1.0 μM. Notably, most reported HDAC PROTACs only degrade HDAC6 or had a small effect on HDAC3 at high concentrations. The degradation of HDAC6 can even be achieved using pan-HDACi as warhead,9b which might be because HDAC6 is primarily localized in the cytoplasm and can be easily accessed by the ubiquitin proteasome system (UPS) compared with other HDACs mainly localized in the nucleus. Our PROTACs are derived from SR-3558, an HDACi with a unique benzoylhydrazide ZBG that preferentially binds to HDAC3, which may facilitate efficient degradation of this isozyme. In our case, VHL-recruiting degraders appeared to be more potent and selective in inducing HDAC3 degradation, and 8c (XZ9002) was the best candidate among its analogs. Thus, we decided to further characterize this compound.
XZ9002 dose-dependently induced HDAC3 degradation in MDA-MB-468 cells, with a DC50 value (the concentration for 50% protein degradation) of 42 nM under 14 h treatment (Fig. 2A). In contrast, no significant changes in the protein levels of HDACs 1, 2, and 6 were observed under the same condition. Pre-incubation of MDA-MB-468 cells with an excess of the VHL ligand VH032 (10 μM) or a proteasome inhibitor MG132 blocked XZ9002-induced HDAC3 degradation (Fig. 2B), indicating that the PROTAC-mediated degradation depends on both the VHL E3 ligase and the UPS. To further confirm that the VHL E3 ligase is involved in XZ9002-induced HDAC3 degradation, we synthesized XZ9002-NC as a negative control compound (Scheme S1, ESI†), in which two chiral centers in the VHL binding motif of XZ9002 are reversed to abolish the interaction with VHL. As expected, XZ9002-NC did not induce HDAC3 degradation (Fig. 2B). Besides, the HDAC3 degradation induced by XZ9002 in MDA-MB-468 was time-dependent, starting within 2 h and after drug treatment for 8 h, 70% HDAC3 was degraded with 125 nM of XZ9002 (Fig. 2C). The effects of XZ9002 on HDAC3 protein levels in MDA-MB-468 were long-lasting and reversible. As indicated in the “washout” assay (Fig. 2D), it took more than 12 h for HDAC3 to rebound to the steady level. CRBN-based PROTACs 7a-c has minimal effect in degrading HDACs 1–3. However, treatment of 7a-c resulted in dramatically increased histone acetylation at H3K27 and H4K5 (Fig. S2, ESI†), which can be attributed to their potent HDAC inhibitory effect (Table S1). XZ9002 dose-dependently increased histone acetylation albeit with moderate effects compared to 15 and CRBN-based PROTACs (Fig. 2A and Fig. S2), which may reflect selective HDAC3 degradation rather than a broad inhibition of deacetylase activity of HDACs 1–3 by XZ9002.
Fig. 2.
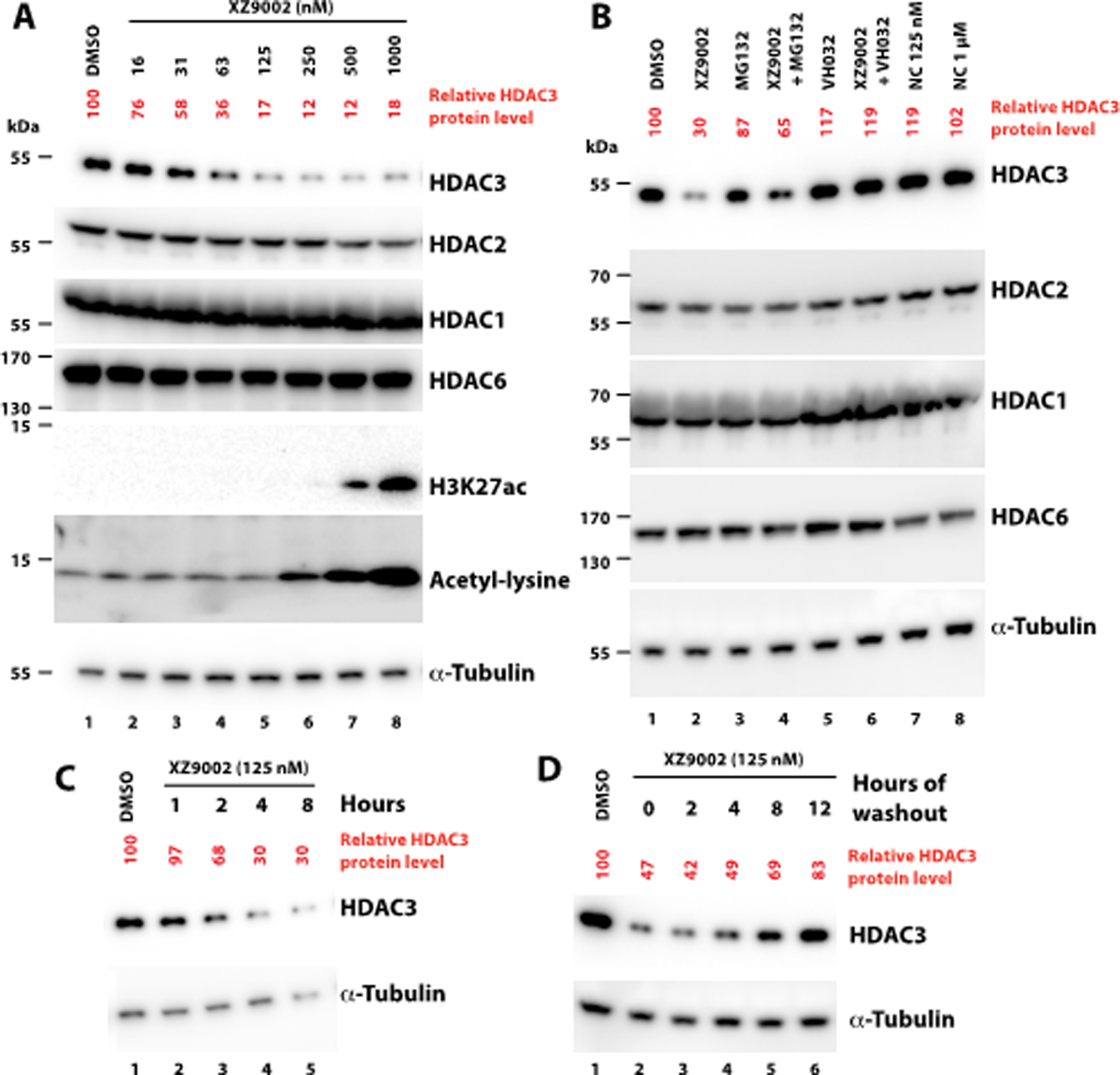
XZ9002 induces HDAC3 degradation. (A) Western blot showing HDAC protein levels in MDA-MB-468 cells treated with XZ9002 for 14 h. HDAC3 band intensity was normalized against that of α-tubulin in each sample. (B) Pretreatment with 1 μM MG132, or 10 μM VHL032 for 1 h blocked HDAC3 degradation by XZ9002 (125 nM). XZ9002-NC, the negative control of XZ9002 that does not bind VHL, cannot degrade HDAC3. NC: XZ9002-NC. (C) Time-course assay in MDA-MB-468 cells after treatment with 125 nM XZ9002. (D) MDA-MB-468 cells were incubated with 125 nM of XZ9002 for 14 h followed by drug washout, and incubation of the cells in drug-free medium for an additional time. Data are presented as representative figures of two independent experiments.
Class I HDACs are frequently overexpressed in various cancer types. HDAC3 exhibits biological functions distinct from HDAC1 and HDAC2. HDAC3 requires a cofactor such as NCOR2 for its catalytic activity.15 It is involved in G2/M progression in the cell cycle,16 DNA replication and DNA damage responses.17 Inhibition of HDAC3 induces DNA replication stress in cutaneous T cell lymphomas.18 Several studies have suggested that HDAC3 may be a critical factor for breast cancer metastatic progression. In an analysis of clinical breast cancer samples, it has been shown that HDAC3 is highly expressed in tumours with features of aggressive subtypes and that HDAC3 expression levels inversely correlate with patient survival rate.2a, 2c Inhibition of HDAC3 attenuates breast cancer cell proliferation2b, 19 and suppresses the expression of genes underlying cancer stem cell phenotypes.3a Encouragingly, class I-selective HDACi provided survival benefits when combined with the aromatase inhibitor exemestane for patients with hormone receptor-positive breast cancers.20 Notably, HDAC3 also has deacetylase activity-independent function,21 which could not be blocked by conventional HDACi. These studies suggest that HDAC3 inhibition can induce distinct cellular response and likely exerts underappreciated anticancer effects. We next evaluated the effects of XZ9002 on the survival and proliferation of breast cell lines using colony formation assays. As shown in Fig. 3, compared to XZ9002-NC, XZ9002 was markedly more potent to suppress clonogenic growth of T47D, HCC1143, and BT549 breast cancer cells. XZ9002 was also more effective than XZ9002-NC to inhibit clonogenic growth of MDA-MB-468 cells, although the difference was moderate (Fig. 3). Considering that the major difference between XZ9002 and XZ9002-NC is the HDAC3 degradation activity, these data indicate that HDAC3 degradation may be a more effective anticancer strategy than the traditional enzymatic inhibition approach, highlighting translational potential of HDAC3 degraders in cancer treatment.
Fig. 3.
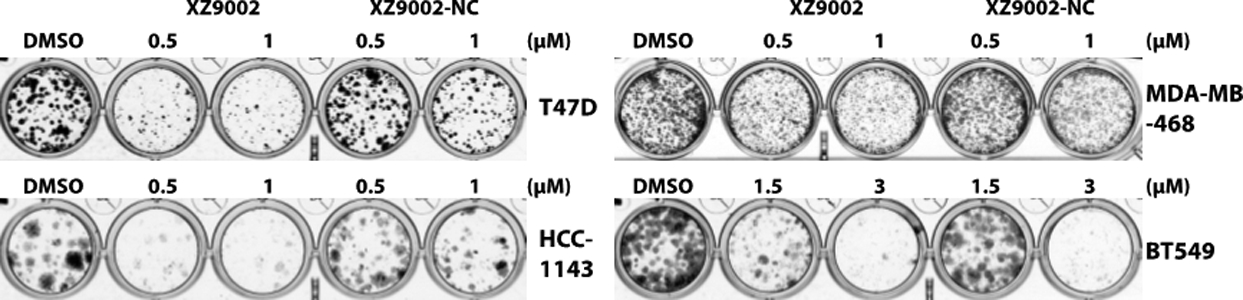
XZ9002 is more potent than XZ9002-NC to inhibit clonogenic growth of cancer cells. The indicated cancer cell lines were exposed to DMSO, XZ9002, or XZ9002-NC at the indicated concentrations. Medium with freshly added compound was changed every five days until colonies grew to appropriate sizes. Colonies were stained. Shown are representative images of four biological replicates.
XZ9002 appears to bind HDAC3 more strongly than other HDACs (Table S1). However, this alone may be insufficient to achieve selective HDAC3 degradation. Other factors likely contribute to this unique selectivity. Notably, when an inhibitor is converted to a PROTAC, additional selectivity can be achieved, which may be attributed to the isoform-specific cooperativity of ternary complexes bridged by a PROTAC22 and/or the availability of appropriately placed and surface-exposed lysine residues as ubiquitin acceptor sites for ubiquitination of a target protein.23 Additionally, HDAC3, HDAC1, and HDAC2 exist in distinct protein complexes: HDAC3 is a component of the NCOR complex, while HDAC1 and HDAC2 are both found in NuRD, CoREST, and SIN3 complexes.1 The structural contexts of these complexes could influence PROTAC-induced formation of substrate and E3 ligase complex. Further work is required to determine how XZ9002 preferentially targets HDAC3 but not HDAC1 and HDAC2 for degradation.
Compared to 15, XZ9002 was less potent to induce histone hyperacetylation (Fig. S2) and cytotoxicity (Fig. S3). However, the level of histone acetylation induced by an HDACi does not directly correlate with its potency to provoke cytotoxicity.24 In general, pan HDACi induce a higher level of histone acetylation irrespectively of acetylation sites. Lauffer et al. attributed this to inhibition of multiple HDAC isozymes by pan HDACi. Additionally, increased acetylation of non-histone proteins such as p53 induced by HDACi also contributes to cytotoxicity. Notably, XZ9002 and XZ9002-NC induced a similar level of histone acetylation (data not shown). Thus, it is plausible that specific HDAC3 degradation triggered by XZ9002 rather than its ability to increase histone acetylation per se impairs cell growth.
In summary, we have developed novel PROTACs that can specifically degrade HDAC3 among different HDAC isozymes. XZ9002, the best PROTAC among its analogues, selectively and potently induced HDAC3 degradation in a dose- and time-dependent manner. MOA studies validated that PROTAC-induced degradation is mediated by both the VHL E3 ligase and the UPS. Furthermore, the HDAC3 degrader XZ9002 has potent antiproliferative activity against cancer cells. Due to the catalytical mechanism of action and isozyme selectivity, this class of novel HDAC3-specific degraders may overcome dose-limiting toxicity associated with conventional HDACi. This may be crucial to fulfilling the considerable therapeutic potential of HDAC inhibition in the clinic. Also, considering the complicated functions of different HDACs, isozyme-specific degraders like XZ9002 is a useful toolkit to dissect the functions of HDACs.
Supplementary Material
Acknowledgments
This work was supported by a UFHCC pilot grant (P0157383), and grants (to D.L.) from James and Esther King Biomedical Research Program (6JK03 and 20K07), Florida Department of Health, and Florida Breast Cancer Foundation. HRMS studies were supported in part by NIH grant S10OD021758. Jia Wang was supported by a scholarship from China Scholarship Council.
Footnotes
Conflicts of interest
X.Z., Y.X., and D.L. are inventors of a provisional patent application for the use of HDAC PROTACs as anticancer agents.
Electronic supplementary information (ESI) available. Full protocols, assays and 1H NMR, 13C NMR, LC-MS, and HRMS data. See DOI: 10.1039/x0xx00000x
Notes and References
- 1.Yang X-J and Seto E, Nat. Rev. Mol. Cell Biol, 2008, 9, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Muller BM, Jana L, Kasajima A, Lehmann A, Prinzler J, Budczies J, Winzer KJ, Dietel M, Weichert W and Denkert C, BMC Cancer, 2013, 13, 215; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kwak SM, Seo J, Hwang JT, Sung GJ, Song JH, Jeong JH, Lee SH, Yoon HG, Choi HK and Choi KC, Cells, 2019, 8, 930; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cui Z, Xie M, Wu Z and Shi Y, Med. Sci. Monit, 2018, 24, 2456–2464; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tang X, Li G, Su F, Cai Y, Shi L, Meng Y, Liu Z, Sun J, Wang M, Qian M, Wang Z, Xu X, Cheng YX, Zhu WG and Liu B, Nucleic Acids Res, 2020, 48, 2912–2923; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) West AC and Johnstone RW, J. Clin. Invest, 2014, 124, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Caslini C, Hong S, Ban YJ, Chen XS and Ince TA, Oncogene, 2019, 38, 6599–6614; [DOI] [PubMed] [Google Scholar]; (b) Gryder BE, Wu L, Woldemichael GM, Pomella S, Quinn TR, Park PMC, Cleveland A, Stanton BZ, Song Y, Rota R, Wiest O, Yohe ME, Shern JF, Qi J and Khan J, Nat. Commun, 2019, 10, 3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner FF, Weiwer M, Lewis MC and Holson EB, Neurotherapeutics, 2013, 10, 589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L, Han Y, Jiang Q, Wang C, Chen X, Li X, Xu F, Jiang Y, Wang Q and Xu W, Med. Res. Rev, 2015, 35, 63–84. [DOI] [PubMed] [Google Scholar]
- 6.(a) Guha M, Nat. Rev. Drug Discov, 2015, 14, 225–226; [DOI] [PubMed] [Google Scholar]; (b) Gryder BE, Sodji QH and Oyelere AK, Future Med. Chem, 2012, 4, 505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Zou Y, Ma D and Wang Y, Cell Biochem. Funct, 2019, 37, 21–30; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paiva S-L and Crews CM, Curr. Opin. Chem. Biol, 2019, 50, 111–119; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Khan S, He Y, Zhang X, Yuan Y, Pu S, Kong Q, Zheng G and Zhou D, Oncogene, 2020, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Thummuri D, He Y, Liu X, Zhang P, Zhou D and Zheng G, Chem. Commun, 2019, 55, 14765–14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Yang H, Lv W, He M, Deng H, Li H, Wu W and Rao Y, Chem. Commun, 2019, 55, 14848–14851; [DOI] [PubMed] [Google Scholar]; (b) Yang K, Song Y, Xie H, Wu H, Wu Y-T, Leisten ED and Tang W, Bioorg. Med. Chem. Lett, 2018, 28, 2493–2497; [DOI] [PubMed] [Google Scholar]; (c) Wu H, Yang K, Zhang Z, Leisten ED, Li Z, Xie H, Liu J, Smith KA, Novakova Z and Barinka C, J. Med. Chem, 2019, 62, 7042–7057; [DOI] [PubMed] [Google Scholar]; (d) An Z, Lv W, Su S, Wu W and Rao Y, Protein Cell, 2019, 10, 606–609; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yang K, Wu H, Zhang Z, Leisten ED, Nie X, Liu B, Wen Z, Zhang J, Cunningham MD and Tang W, ACS Med. Chem. Lett, 2020, 11, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang M, Xie X, Gong P, Wei Y, Du H, Xu Y, Xu Q, Jing Y and Zhao L, Eur. J. Med. Chem, 2020, 188, 111991. [DOI] [PubMed] [Google Scholar]
- 11.Smalley JP, Adams GE, Millard CJ, Song Y, Norris JKS, Schwabe JWR, Cowley S and Hodgkinson JT, Chem. Commun, 2020, 56, 4476–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roatsch M, Vogelmann A, Herp D, Jung M and Olsen CA, ChemRxiv, 2020, Preprint. [Google Scholar]
- 13.Wang Y, Stowe RL, Pinello CE, Tian G, Madoux F, Li D, Zhao LY, Li JL, Wang Y, Wang Y, Ma H, Hodder P, Roush WR and Liao D, Chem. Biol, 2015, 22, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) McClure JJ, Zhang C, Inks ES, Peterson YK, Li J and Chou CJ, J. Med. Chem, 2016, 59, 9942–9959; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li X, Peterson YK, Inks ES, Himes RA, Li J, Zhang Y, Kong X and Chou CJ, J. Med. Chem, 2018, 61, 2589–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watson PJ, Fairall L, Santos GM and Schwabe JW, Nature, 2012, 481, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Y and Hsieh J, Proc. Natl. Acad. Sci. U.S.A, 2014, 111, 13541–13546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, Yenamandra A, Locke K, Yuan JL, Bonine-Summers AR, Wells CE, Kaiser JF, Washington MK, Zhao Z, Wagner FF, Sun ZW, Xia F, Holson EB, Khabele D and Hiebert SW, Cancer Cell, 2010, 18, 436–447; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Summers AR, Fischer MA, Stengel KR, Zhao Y, Kaiser JF, Wells CE, Hunt A, Bhaskara S, Luzwick JW, Sampathi S, Chen X, Thompson MA, Cortez D and Hiebert SW, J. Clin. Invest, 2013, 123, 3112–3123; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, Aladjem MI and Pommier Y, Cancer Res, 2010, 70, 4470–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wells CE, Bhaskara S, Stengel KR, Zhao Y, Sirbu B, Chagot B, Cortez D, Khabele D, Chazin WJ, Cooper A, Jacques V, Rusche J, Eischen CM, McGirt LY and Hiebert SW, PLoS One, 2013, 8, e68915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Ridolfi E, Matteucci E, Maroni P and Desiderio MA, Br. J. Cancer, 2008, 99, 1623–1634; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Matteucci E, Ridolfi E, Maroni P, Bendinelli P and Desiderio MA, Mol. Cancer Res, 2007, 5, 833–845. [DOI] [PubMed] [Google Scholar]
- 20.(a) Jiang Z, Li W, Hu X, Zhang Q, Sun T, Cui S, Wang S, Ouyang Q, Yin Y, Geng C, Tong Z, Cheng Y, Pan Y, Sun Y, Wang H, Ouyang T, Gu K, Feng J, Wang X, Wang S, Liu T, Gao J, Cristofanilli M, Ning Z and Lu X, Lancet Oncol, 2019, 20, 806–815; [DOI] [PubMed] [Google Scholar]; (b) Yardley DA, Ismail-Khan RR, Melichar B, Lichinitser M, Munster PN, Klein PM, Cruickshank S, Miller KD, Lee MJ and Trepel JB, J. Clin. Oncol, 2013, 31, 2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, Everett LJ, Nabel CS, Li Y, Selvakumaran V, Won KJ and Lazar MA, Mol. Cell, 2013, 52, 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M and Ciulli A, Nat. Chem. Biol, 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robb CM, Contreras JI, Kour S, Taylor MA, Abid M, Sonawane YA, Zahid M, Murry DJ, Natarajan A and Rana S, Chem. Commun, 2017, 53, 7577–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, Kaminker JS, Heise CE and Steiner P, J. Biol. Chem, 2013, 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.