

Abstract
PURPOSE
Low-grade serous ovarian carcinomas (LGSOCs) have historically low chemotherapy responses. Alterations affecting the MAPK pathway, most commonly KRAS/BRAF, are present in 30%-60% of LGSOCs. The purpose of this study was to evaluate binimetinib, a potent MEK1/2 inhibitor with demonstrated activity across multiple cancers, in LGSOC.
METHODS
This was a 2:1 randomized study of binimetinib (45 mg twice daily) versus physician’s choice chemotherapy (PCC). Eligible patients had recurrent measurable LGSOC after ≥ 1 prior platinum-based chemotherapy but ≤ 3 prior chemotherapy lines. The primary end point was progression-free survival (PFS) by blinded independent central review (BICR); additional assessments included overall survival (OS), overall response rate (ORR), duration of response (DOR), clinical-benefit rate, biomarkers, and safety.
RESULTS
A total of 303 patients were randomly assigned to an arm of the study at the time of interim analysis (January 20, 2016). Median PFS by BICR was 9.1 months (95% CI, 7.3 to 11.3) for binimetinib and 10.6 months (95% CI, 9.2 to 14.5) for PCC (hazard ratio,1.21; 95%CI, 0.79 to 1.86), resulting in early study closure according to a prespecified futility boundary after 341 patients had enrolled. Secondary efficacy end points were similar in the two groups: ORR 16% (complete response [CR]/partial responses[PRs], 32) versus 13% (CR/PRs, 13); median DOR, 8.1 months (range, 0.03 to ≥ 12.0 months) versus 6.7 months (0.03 to ≥ 9.7 months); and median OS, 25.3 versus 20.8 months for binimetinib and PCC, respectively. Safety results were consistent with the known safety profile of binimetinib; the most common grade ≥ 3 event was increased blood creatine kinase level (26%). Post hoc analysis suggests a possible association between KRAS mutation and response to binimetinib. Results from an updated analysis (n = 341; January 2019) were consistent.
CONCLUSION
Although the MEK Inhibitor in Low-Grade Serous Ovarian Cancer Study did not meet its primary end point, binimetinib showed activity in LGSOC across the efficacy end points evaluated. A higher response to chemotherapy than expected was observed and KRAS mutation might predict response to binimetinib.
INTRODUCTION
Serous carcinoma accounts for approximately 70%-80% of epithelial ovarian, tubal, and peritoneal cancers.1 Low-grade serous ovarian carcinoma (LGSOC) is a unique tumor that is distinguished from high-grade serous ovarian cancer not only by immunohistochemical profile but also by molecular characteristics, epidemiologic features and clinical behavior.2
CONTEXT
Key Objective
The objective of the MEK Inhibitor in Low-Grade Serous Ovarian Cancer (MILO)/ENGOT-ov11 study was to evaluate the MEK1/2 inhibitor binimetinib in patients with low-grade serous ovarian carcinomas (LGSOCs).
Knowledge Generated
This study did not meet its primary end point; however, binimetinib showed activity in LGSOC across the efficacy end points evaluated. Chemotherapy responses were higher than predicted. The safety results observed in this study are generally consistent with the known safety profile of binimetinib and with MEK inhibitor class effects.
Relevance
Currently, treatment options are limited for patients with LGSOC, and few offer objective decreases in disease burden or tumor-progression delays. Although this trial did not meet its primary end point, binimetinib did display a clinically meaningful progression-free survival and overall response rate and, therefore, should be considered a viable treatment option in this setting. Forthcoming biomarker analysis may ultimately identify a subset of patients who selectively benefit from binimetinib.
Aberrant signaling through the RAS/RAF/MEK/ERK pathway is a characteristic feature of many cancers, including LGSOC, with, 5%-16% and 16%-47% of LGSOCs having alterations in BRAF and RAS, respectively.3-7
Binimetinib is an oral, potent, selective, allosteric, small-molecule inhibitor of MEK1/2 and is approved in multiple countries in combination with encorafenib for the treatment of patients with unresectable or metastatic BRAF V600E or V600K mutation-positive melanoma.8,9 Inhibiting both basal and induced levels of ERK phosphorylation in numerous BRAF-mutated cancer cell lines (half maximal inhibitory concentration [IC50] values as low as 5 nM), binimetinib has nanomolar activity against purified MEK enzyme (IC50, 12 nM). Binimetinib has also demonstrated a decrease in pERK when tested in multiple cell lines, regardless of their mutational status and in vitro sensitivity.10 A prior single-arm, phase II study of the MEK inhibitor selumetinib showed promising activity in recurrent LGSOC.11
This phase III study was designed to evaluate the efficacy and safety of binimetinib in recurrent or persistent LGSOC. Patients were not selected on the basis of molecular profile; however, archival tumor tissue was collected at the time of enrollment for retrospective mutational analysis. Blinded independent central radiology review (BICR) was used to control for potential investigator variance in assessing response.
PATIENTS AND METHODS
Patients
Patients were > 18 years of age with a diagnosis of LGSOC, fallopian tube or primary peritoneum, confirmed histologically and verified by central pathology review. Archival tissue was also collected for biomarker testing using the FoundationOne Panel (Foundation Medicine, Cambridge, MA). Eligible patients had measurable recurrent or persistent disease (as defined by RECIST V1.1, per BICR) that had progressed (defined as radiologic and/or clinical progression; an increase in CA-125 alone was not sufficient) on or after last therapy, and was not amenable to potentially curative intent surgery, as determined by the investigator. Patients were required to have received ≥ 1 prior platinum-based chemotherapy regimen but ≤ 3 prior chemotherapy regimens in total, with no limit to the number of lines of prior hormonal therapy. Patients had an Eastern Cooperative Oncology Group performance status of 0 or 1. Patients were excluded if they had previous treatment with an MEK or BRAF inhibitor. Additional details regarding inclusion and exclusion criteria are provided in the Data Supplement.
The study was approved by the institutional review board for each site. All clinical work was conducted in compliance with current Good Clinical Practices as referenced in the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. All patients enrolled in the study provided written, informed consent prior to their participation.
Study Design and Treatments
The MEK Inhibitor in Low-Grade Serous Ovarian Cancer (MILO)/ARRAY-162-311/ENGOT-ov11 study was a multinational, randomized, two-arm, open-label, phase III study conducted at 102 sites in 20 countries (ClinicalTrials.gov identifier: NCT01849874; Appendix Table A1, online only). MILO was conducted in collaboration with European Network of Gynecologic Oncological Trial groups (ENGOT) according to the ENGOT Model C.12 Patients were stratified by their last platinum-free interval (≤ v > 182 days) and number of prior systemic regimens (1 to 2 v 3) and then randomly assigned 2:1 to receive binimetinib or physician’s choice chemotherapy (PCC; pegylated liposomal doxorubicin [PLD], paclitaxel, or topotecan). Patients randomly assigned to binimetinib received 45 mg orally twice daily with water irrespective of food, continuously, starting on day 1. Patients randomly assigned to PCC received one of the following: PLD (40 mg/m2 intravenously [IV] on day 1 of every 28-day cycle), paclitaxel (80 mg/m2 IV on days 1, 8, and 15 of every 28-day cycle), or topotecan (1.25 mg/m2 IV on days 1-5 of every 21-day cycle). Treatment continued until one of the following: locally determined progressive disease (PD) unacceptable toxicity, or inability to continue on protocol-directed therapy (additional information is provided in the Data Supplement). Patients randomly assigned to PCC who developed PD (by local and BICR assessment) were allowed to crossover to treatment with binimetinib provided they met the crossover eligibility requirements (Data Supplement).
Assessments
The primary end point was BICR progression-free survival (PFS). Secondary end points included overall survival (OS), overall response rate (ORR; RECIST v1.1), duration of response (DOR), disease control rate (best response of complete response [CR] or partial response [PR], or stable disease [SD] documented ≥ week 24) and safety.
Tumors were assessed every 8 weeks for the first 72 weeks, then every 12 weeks until PD per BICR, irrespective of the days of study- drug administration. Safety was evaluated by ongoing monitoring, including ophthalmic examinations, dermatologic examinations, electrocardiograms, and cardiac scans of ejection fraction.
Statistical Methods
For efficacy, all randomly assigned patients were included in the analyses. For safety, all patients who received binimetinib or PCC were included. PFS was defined as the date of randomization to the date of first documented BICR PD or death due to any cause, whichever occurred first. If a patient had not experienced an event at the time of the analysis cutoff or at the start of any new therapy, PFS was censored at the date of last adequate tumor assessment. PFS and OS were summarized by treatment arm using the Kaplan-Meier method with 95% CIs for medians. The primary end point was compared between treatment arms using a stratified log-rank test, and a hazard ratio [HR] from the stratified Cox model was used to summarize the treatment effect estimate.
ORR was assessed and compared between arms using the Fisher exact test. Median DOR with 95% CIs was provided, with minimum, maximum, and the number still in response (censored) at the time of data cutoff.
A total of 195 events (PD or death) provided 90% power for testing the null hypothesis of no difference in PFS distribution functions between the two treatment arms assuming a true HR of 0.60 using a stratified log-rank test, a 1-tailed α of 0.025, and a 2:1 binimetinib arm to control arm randomization ratio. The HR required to achieve the final critical value was approximately 0.74. Historical evidence suggests that the median PFS in recurrent LGSOC is approximately 7 months.6,7 For exponential PFS, a HR of 0.60 translates to a median PFS of approximately 11.7 months in the binimetinib arm. A total of approximately 360 patients were planned. An interim analysis for early stopping for futility was planned at 40% information fraction (ie, n = 78 total progression events per BICR or deaths). The futility boundary was from the unified family of group sequential test designs with parameter P = 0.5.13 At 40% information fraction, this corresponds to an approximate boundary of 0.90 on the HR scale. A data cutoff date was set by the sponsor in advance of the occurrence of the 78th event. FoundationOne Panel genes that were prevalent in at least 5% of sequenced patients were tested for association with binary response (CR or PR v SD or PD) using two-sided Fisher exact tests.
RESULTS
Patient Characteristics and Drug Exposure
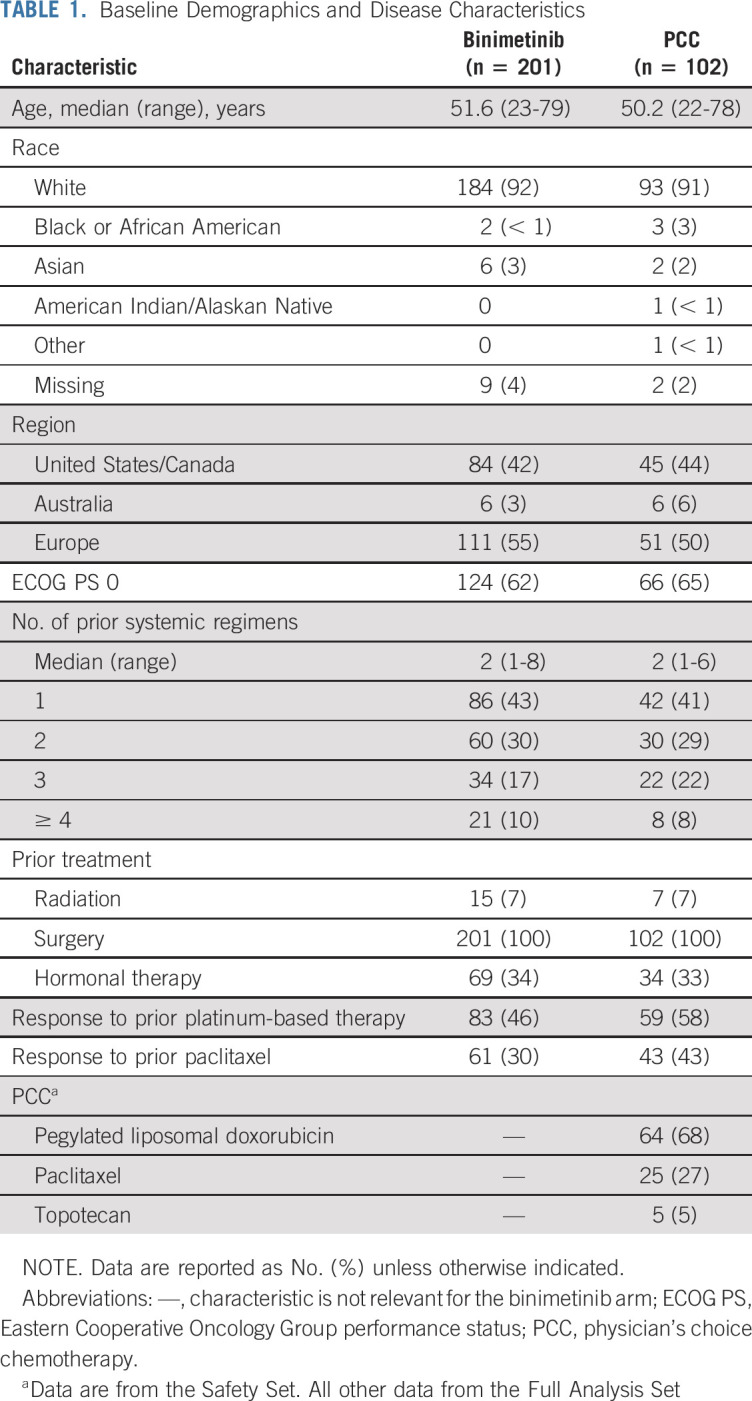
Patients were enrolled from June 28, 2013, to April 1, 2016. Per recommendation of the data monitoring committee, enrollment was discontinued after the planned interim analysis showed the HR for PFS crossed the predefined futility boundary. The interim analysis was conducted with 303 patients and then, at the time of the decision to discontinue enrollment for the study, 341 patients. Results presented here include an assessment of end points during the randomized period, up to the data cutoff date for the interim analysis of January 20, 2016, for a total of 303 patients (n = 201 patients receiving binimetinib; n = 102 receiving PCC) in the full analysis set and 294 patients (n = 200 receiving binimetinib; n = 94 receiving PCC) in the safety population (Fig 1). At the time of data cutoff, (January 20, 2016), 107 patients (53%) and 48 patients (47%) had discontinued treatment of binimetinib and PCC, respectively. The most common reasons for discontinuing initial treatment were disease progression (binimetinib, 24%; PCC, 20%) and adverse events (binimetinib, 20%; PCC, 11%; Fig 1). Patient baseline demographics and disease characteristics were generally well balanced between the two groups (Table 1).
FIG 1.
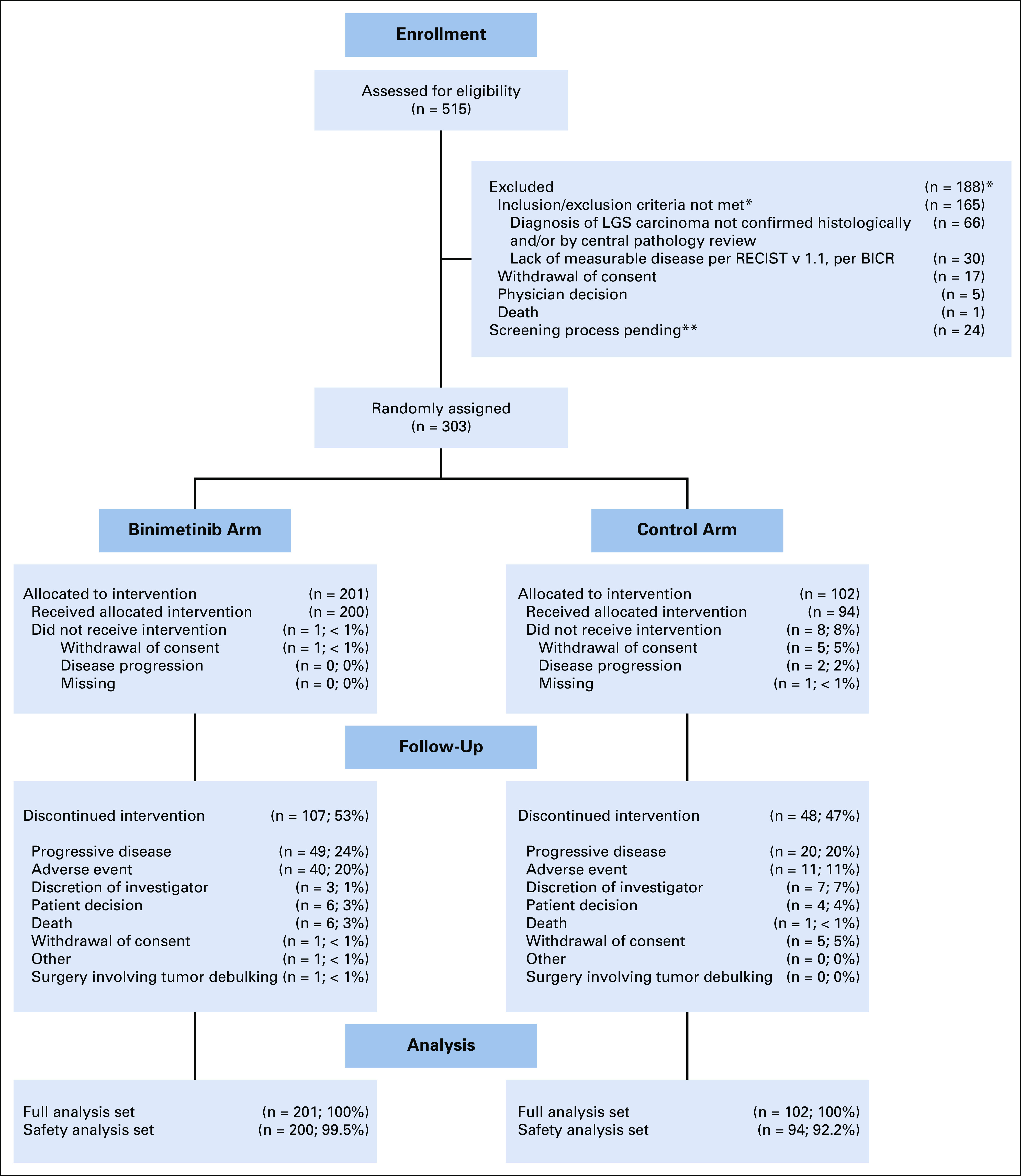
CONSORT diagram (data cutoff date: January 20, 2016). BICR, blinded independent central review; LGS, low-grade serous. (*) Patients may be counted as not meeting > 1 criterion; most common reasons provided. (**) These patients had signed ICF prior to the data cutoff date but the outcome of their screening process was still pending as of the cutoff date.
TABLE 1.
Baseline Demographics and Disease Characteristics
The median duration of exposure to binimetinib was 4.1 months (range, 0-24 months) and the median relative dose intensity was 67.6% (range, 6%-100%). The median duration of exposure to any of the PCC was 4.1 months (range, 0-18 months). Patients in the PCC group received PLD (n = 64 patients; 68%); paclitaxel (n = 25 patients; 27%), or topotecan (n = 5 patients; 5%). The median (range) relative dose intensity was 71.3% (40%-100%) for topotecan, 95.9% (0%-116%) for PLD, and 89.4% (33%-102%) for paclitaxel.
Efficacy
The primary end point of PFS by BICR is shown in Figure 2. The median PFS was 9.1 months (95% CI, 7.3 to 11.3) in the binimetinib group and 10.6 months (95% CI, 9.2 to 14.5) in the PCC group. The HR from the stratified Cox model was 1.21 (95% CI, 0.79 to 1.86). Based on a point-estimate futility boundary of HR > 0.84 for the 103 events observed in the interim analysis, the futility boundary was crossed, indicating a low probability of reaching statistical significance in favor of binimetinib with continued follow-up. In the local investigator assessment, patients in the binimetinib arm had a median PFS of 12.5 months (95% CI, 9.1 to 17.7) compared with 11.6 months (95% CI, 10.0 to 16.1) in the PCC group. The stratified HR was 0.87 (95% CI, 0.56 to 1.34).
FIG 2.
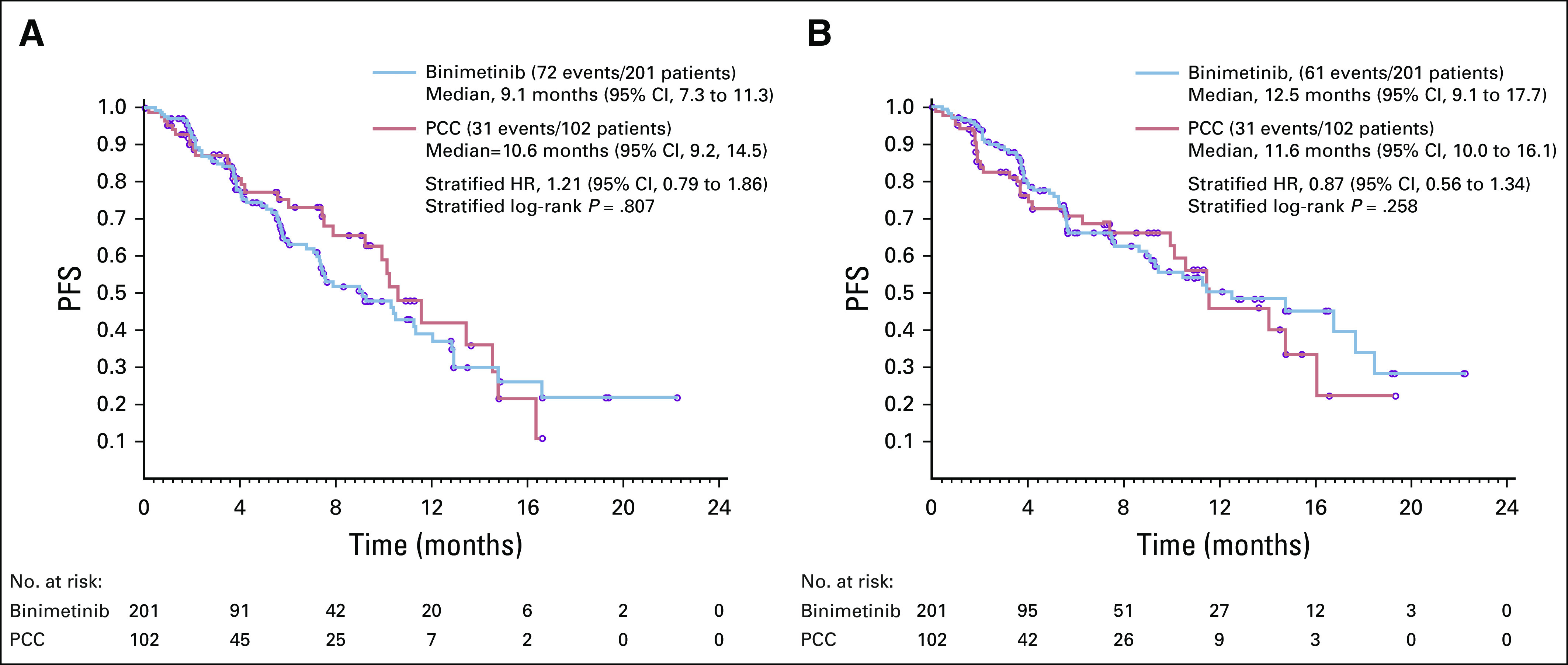
Kaplan-Meier plot of progression-free survival per (A) blinded independent central review and (B) local assessment. HR, hazard ratio; PCC, physician’s choice chemotherapy; PFS, progression free survival.
The OS results are depicted in the Data Supplement. They were similar between groups, with 164 patients (82%) in the binimetinib group alive at the time of data cutoff for interim analysis compared with 82 patients (80%) in the PCC group. The median OS was 25.33 months (95% CI, 18.46 to not reached [NR]) in the binimetinib group and 20.83 months (95% CI, 17.45 to NR) in the PCC group. The HR from the stratified Cox model was 0.85 (95% CI, 0.49 to 1.48).
The response analysis is shown in Table 2. The ORR by BICR was 16% in the binimetinib group and 13% in the PCC group. The median DOR in the binimetinib group was 8.05 months (95% CI, 5.55 to NR) compared with 6.67 months (95% CI, 3.71 to NR) in the PCC group; 23 patients in the binimetinib group and 8 patients in the PCC group had responses ongoing at the data cutoff date. For the response assessment by local investigator, the ORR was 18% in the binimetinib group and 13% in the PCC group. Median DOR was 15.84 months (95% CI, 10.41 to NR) in the binimetinib group and 9.89 months (95% CI, 6.41 to 9.89) in the PCC group. A waterfall plot displaying percent change in sum of longest diameters per BICR is displayed in the Data Supplement.
TABLE 2.
Summary of Best Overall Response and Duration of Response

At the time enrollment to the study ended in April 2016, patients being treated with binimetinib or PCC were notified of the interim results, but if desired, they were allowed to continue receiving treatment until treatment discontinuation criteria were met. Crossover was stopped at that time. An updated analysis was conducted when the remaining data were collected after the discontinuation of enrollment, with a data cutoff of January 2019 (n = 341). In this analysis, median PFS by BICR was 10.4 months (95% CI, 7.5 to 12.9) in the binimetinib group and 11.5 months (95% CI, 9.9 to 14.8) in the PCC group (HR, 1.15;95% CI, 0.76 to 1.74; Data Supplement). Median OS was 34.6 months (95% CI, 28.0 to NR) and 34.2 months (95% CI, 21.6 to NR) for the binimetinib and PCC groups, respectively (HR, 0.93; 95% CI, 0.65 to 1.33; Data Supplement). Updated ORR by local investigator assessment was 24% in both groups (Data Supplement). It is important to note the median OS estimates in both arms increased at the follow-up analysis, possibly as a result of the instability of the median estimates at the time of the initial analysis, when the potential follow-up was substantially (3 years) shorter.
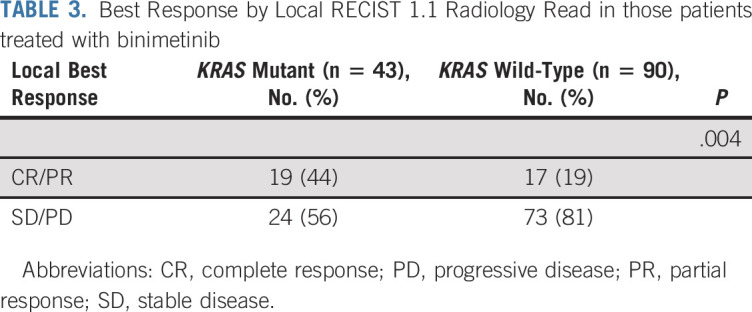
Molecular testing was performed on all consenting patients with adequate archival tissue. At the time of the January 2019 data cutoff, 215 patients had tumor tests available. There were 47 mutations detected in at least 5% of patients, most commonly KRAS, which was found in 33% of patients. The frequency of KRAS mutation was evenly distributed between the two groups and was found in 46 patients (32%) treated with binimetinib and 24 patients (34%) treated with PCC. Unbiased univariate analyses evaluating best ORR to therapy as a binary response showed KRAS mutation was significantly associated with response to treatment with binimetinib (odds ratio [OR], 3.4; 95% CI, 1.53 to 7.66; unadjusted P = .003; Fig 3A) but not PCC (OR, 2.13; 95% CI, 0.67 to 6.81; P = .2; Fig 3B). KRAS mutation was also associated with prolonged PFS in patients treated with binimetinib (median PFS: KRAS mutant: 17.7 months [95% CI, 12 to NR]; KRAS wild-type (WT): 10.8 months [95% CI, 5.5 to 16.7]; P = .006), but not PCC (median PFS: KRAS mutant: 14.6 months [95% CI, 9.4 to NA]; KRAS WT: 11.5 months [95% CI, 5.7 to 26.6]; P = .502).Among those patients treated with binimetinib for whom updated local RECIST 1.1 response data and molecular data were available (n = 133), KRAS mutation status was significantly associated with local best response (P = .004); 44% of patients with KRAS mutation versus 19% of patients with KRAS WT had CR or PR (Table 3). Mutations identified by Foundation Medicine FoundationOne Panel in ≥ 1 tumor sample are listed in the Data Supplement.
FIG 3.
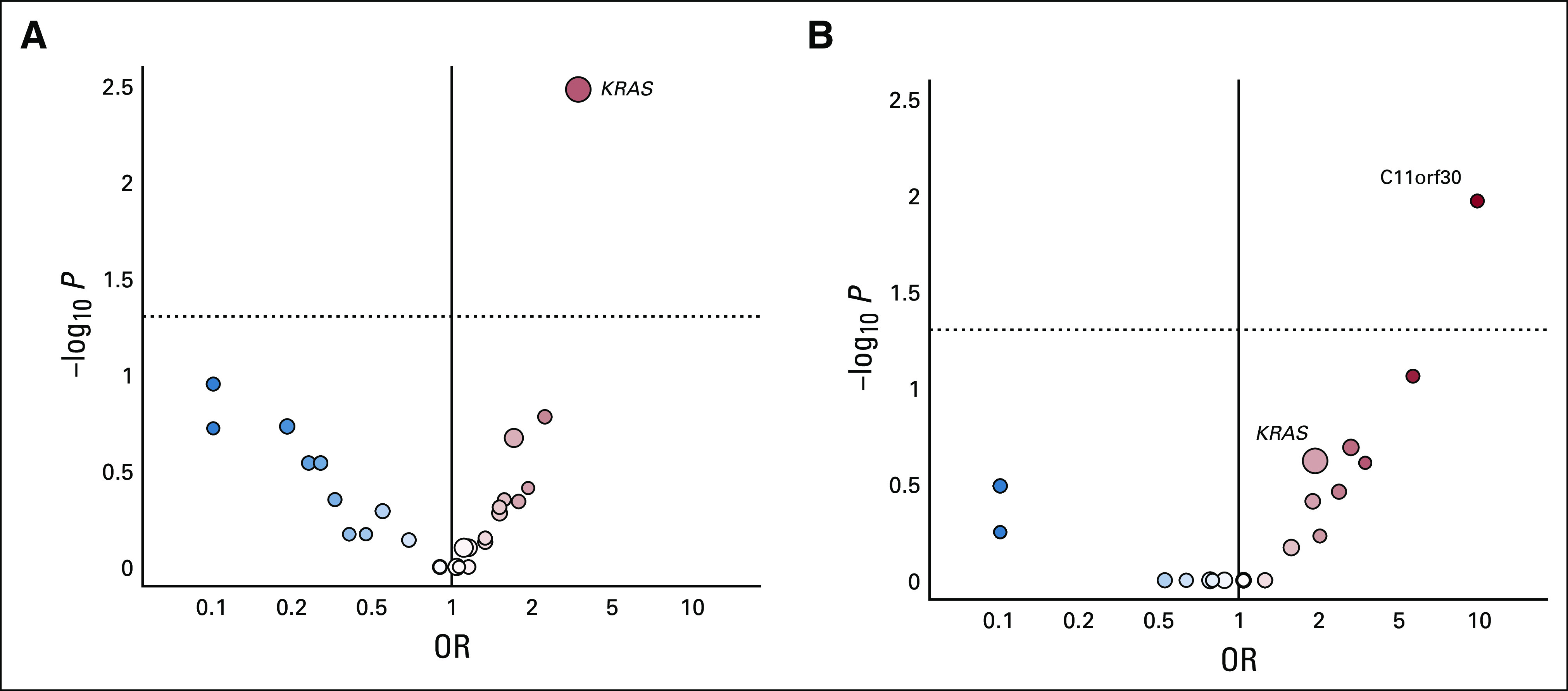
(A) Binimetinib treatment group: univariate analysis of molecular alterations and response to therapy. (B) Physician’s choice chemotherapy group: univariate analysis of molecular alterations and response to therapy. OR, odds ratio.
TABLE 3.
Best Response by Local RECIST 1.1 Radiology Read in those patients treated with binimetinib
Safety
Grade ≥ 3 adverse events were reported in 76% and 44% of patients for binimetinib and PCC, respectively (Table 4). Adverse events that led to permanent discontinuation of study drug were reported by 62 patients (31%) for binimetinib and 16 patients (17%) in the PCC group. Adverse events leading to binimetinib discontinuation in ≥ 5 patients were decreased ejection fraction (n = 8 patients; 4%), vomiting (n = 6 patients; 3%), intestinal obstruction and retinal vein occlusion (n = 5 patients; 2% each). The adverse event leading to discontinuation of PCC in ≥ 5 patients was palmar-plantar erythrodysesthesia syndrome (n = 5 patients; 5%). A total of six patients (3%) in the binimetinib group experienced a retinal vein occlusion event, all of which resulted in treatment discontinuation. All events were considered resolved or resolving, two with sequelae. No permanent blindness or permanent loss of vision was observed.
TABLE 4.
Adverse Events Reported in > 20% of Treated Patients in Either Arm

DISCUSSION
Binimetinib did not demonstrate a significant difference in the primary end point of PFS versus PCC in patients with recurrent or persistent LGSOC. In addition, the proportion of patients achieving an objective response and the median DOR appeared similar between arms. Of note, the responses to chemotherapy in this study were greater than anticipated on the basis of previously reported, single-institution retrospective case series.
Although the MILO/ENGOT-ov11 trial did not meet its primary end point, binimetinib did display a clinically meaningful PFS and ORR and, therefore, should be considered a viable treatment option in this setting. The median OS for patients with advanced LGSOC approaches 10 years, with patients often experiencing significant morbidity from their disease during that time.2 Currently, treatment options are limited for patients with this disease and few offer objective decreases in disease burden or delays in tumor progression. Recent results from another phase II/III trial in 260 patients with recurrent LGSOC showed trametinib was associated with significantly improved PFS (median, 13.0 v 7.2 months; HR, 0.48; 95% CI, 0.36 to 0.64; P < .0001) and ORR (trametinib: 26.2% v control:6.2%; OR, 5.4; 95% CI, 2.39 to 12.21; P < .0001) compared with physician’s choice standard of care, also indicating the potential of MEK inhibition in this patient population.14 Of note, the control arm in that study did not appear to perform as well as in the current study, possibly because of differences in inclusion criteria. The trametinib study allowed for an unlimited number of prior chemotherapies, whereas the binimetinib study was limited to patients who had received a maximum of three prior lines of chemotherapy. Differences in study design and inclusion criteria likely selected for a more chemotherapy-resistant population in the trametinib study, explaining the similar activity of MEK inhibitors between the two studies (response rate of 24% on updated analysis of binimetinib study; 26.2% in the trametinib study) but difference in activity within the control arms. Safety results from this study show that patients treated with binimetinib had higher rates of nonserious and serious adverse events overall, as well as grade ≥ 3 adverse events compared with the PCC group, and there were more frequent dose reductions, dose interruptions, and permanent discontinuations due to adverse events experienced by patients in the binimetinib group, resulting in a lower relative dose intensity for the binimetinib group compared with any of the drugs in the PCC group. The majority of adverse events assessed as related to binimetinib were reversible with or without drug interruption. The safety profile observed in this study is consistent with the known binimetinib profile and consistent with those for the class of MEK inhibitors.15
There are several limitations of the study. First, the lack of suitable, validated biomarkers led to a design with an unselected patient population. Post hoc analysis suggests a possible association between KRAS mutation and response to binimetinib. Additional exploration is warranted to determine if patients with KRAS mutation may derive greater benefit from binimetinib. Although KRAS has been an elusive target across multiple cancer types, prior early-phase studies have found promising response rates to MEK inhibitors and MEK inhibitor combinations in those patients with KRAS-mutant LGSOC.16-18 This has led to considerable interest in the use of mutation status when weighing the expected adverse effects versus benefits of MEK inhibitor therapy. Adverse events in the binimetinib group led to study discontinuations and a low dose intensity. The safety events noted in this study were resolved with conservative supportive care and could potentially be mitigated in future protocols with more proactive management.
In conclusion, although this study did not meet its primary end point, binimetinib showed activity in LGSOC across the efficacy end points evaluated. Chemotherapy responses were greater than predicted. The safety results observed in this study are generally consistent with the known safety profile of binimetinib and with MEK inhibitor class effects. Forthcoming biomarker analysis may ultimately identify a subset of patients who selectively benefit from binimetinib, and additional clinical evaluation is warranted.
ACKNOWLEDGMENT
We thank patients, their families, and the sites that participated in this study. For editorial support, we thank J. D. Cox, PhD, and Mayville Medical Communications.
Appendix
TABLE A1.
MILO-ENGOT-ov11 Study Investigators: List of Investigators Who Consented a Study Participant

SUPPORT
This study was sponsored by Array BioPharma, which was acquired by Pfizer in July 2019 (B.J.M.); the National Institutes of Health, Memorial Sloan Kettering Cancer Center Support Grant No. P30 CA008748 (R.N.G. and C.A.); Cycle for Survival (R.N.G.); and Ovarian Cancer Research Fund Alliance (R.N.G.). Editorial support was funded by Pfizer.
CLINICAL TRIAL INFORMATION
NCT01849874 (PROSPER)
See accompanying editorial on page 3731
AUTHOR CONTRIBUTIONS
Conception and design: Bradley J. Monk, Rachel N. Grisham, Susana Banerjee, Mansoor Raza Mirza, Robert L. Coleman, Amit M. Oza, Carol Aghajanian, Janna Christy-Bittel, Regina Berger, Adam P. Boyd, Ignace Vergote
Administrative support: Robert L. Coleman
Provision of study material or patients: Rachel N. Grisham, Elsa Kalbacher, Ignacio Romero, Peter Vuylsteke, Robert L. Coleman, Felix Hilpert, Amit M. Oza, Martin K. Oehler, Sandro Pignata, Carol Aghajanian, Christian Marth, Jalid Sehouli, Gunnar Kristensen, Andrew Clamp, Isabelle Ray-Coquard, Ignace Vergote
Collection and assembly of data: Bradley J. Monk, Rachel N. Grisham, Elsa Kalbacher, Mansoor Raza Mirza, Peter Vuylsteke, Robert L. Coleman, Felix Hilpert, Amit M. Oza, Anneke Westermann, Martin K. Oehler, Carol Aghajanian, Kathleen N. Moore, Janna Christy-Bittel, Josep M. del Campo, Regina Berger, Christian Marth, Jalid Sehouli, David M. O’Malley, Cristina Churruca, Adam P. Boyd, Gunnar Kristensen, Andrew Clamp, Isabelle Ray-Coquard, Ignace Vergote
Data analysis and interpretation: Bradley J. Monk, Rachel N. Grisham, Susana Banerjee, Elsa Kalbacher, Mansoor Raza Mirza, Ignacio Romero, Peter Vuylsteke, Robert L. Coleman, Felix Hilpert, Amit M. Oza, Sandro Pignata, Carol Aghajanian, Nicoletta Colombo, Esther Drill, David Cibula, Kathleen N. Moore, Janna Christy-Bittel, Josep M. del Campo, Christian Marth, Jalid Sehouli, Adam P. Boyd, Andrew Clamp, Isabelle Ray-Coquard, Ignace Vergote
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
MILO/ENGOT-ov11: Binimetinib versus Physician’s Choice Chemotherapy in Recurrent or Persistent Low-Grade Serous Carcinomas of the Ovary, Fallopian Tube, or Primary Peritoneum
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Bradley J. Monk
Leadership: US Oncology
Honoraria: AbbVie, Advaxis, Agenus, Akeso Biopharma, Amgen, Aravive, AstraZeneca, Asymmetric Therapeutics, Boston Biomedical, ChemoCare, ChemoID, Clovis Oncology, Dicepheria, Easai, Geistlich, Genmab/Seattle Genetics, ImmunoGen, Immunomedics, Incyte, Iovance, Laekna Health Care, Merck, Mersana, Myriad, Nucana, Oncomed, Oncoquest, Oncosec, Perthera, Pfizer, Puma, Regeneron, Roche, Senti Biosciences, Takeda, Tarveda, Tesaro/GlaxoSmithKline, Vavotar Liofe Sciences, VBL, Vigeo, GOG Foundation
Consulting or Advisory Role: AbbVie, Advaxis, Agenus, Akeso Biopharma, Amgen, Aravive, AstraZeneca, Asymmetric Therapeutics, Boston Biomedical, ChemoCare, ChemoID, Clovis Oncology, Dicepheria, Easai, Geistlich, Genmab/Seattle Genetics, GOG Foundation, ImmunoGen, Immunomedics, Incyte, Iovance, Laekna Health Care, Merck, Mersana, Myriad, Nucana, Oncomed, Oncoquest, Oncosec, Perthera, Pfizer, Puma, Regeneron, Roche, Senti Biosciences, Takeda, Tarveda, Tesaro/GlaxoSmithKline, Vavotar Liofe Sciences, VBL, Vigeo
Speakers' Bureau: Roche, AstraZeneca, Clovis Oncology, Easai, Tesaro/GlaxoSmithKline, Merck, Novartis (Inst), Amgen (Inst), Genentech (Inst), Eli Lilly (Inst), Janssen (Inst), Array BioPharma (Inst), Tesaro (Inst), Morphotek (Inst), Pfizer (Inst), Advaxis (Inst), AstraZeneca (Inst), Immunogen (Inst), Regeneron (Inst), Nucana (Inst)
Rachel N. Grisham
Consulting or Advisory Role: Mateon Therapeutics, Clovis Oncology, Regeneron
Research Funding: Context Therapeutics (Inst)
Travel, Accommodations, Expenses: EMD Serono
Other Relationship: Prime Oncology, MCM Education, OncLive
Susana Banerjee
Honoraria: Roche, AstraZeneca, GlaxoSmithKline, Clovis
Consulting or Advisory Role: AstraZeneca/MedImmune, Tesaro, Clovis Oncology, Merck, Seattle Genetics, GenMab, Carrick Therapeutics (Inst), Amgen, Roche, GlaxoSmithKline, MSD Oncology, Mersana
Research Funding: AstraZeneca (Inst), Janssen-Cilag (Inst), GlaxoSmithKline (Inst)
Travel, Accommodations, Expenses: NuCana BioMed, AstraZeneca
Elsa Kalbacher
Consulting or Advisory Role: AstraZeneca, GlaxoSmithKline/Tesaro
Travel, Accommodations, Expenses: AstraZeneca, Tesaro, Sanofi, GlaxoSmithKline/Novartis, LEO Pharma
Mansoor Raza Mirza
Leadership: Karyopharm Therapeutics, Sera Prognostics
Stock and Other Ownership Interests: Karyopharm Therapeutics, Sera Prognostics
Honoraria: Roche, Advaxis, AstraZeneca, Cerulean Pharma, Clovis Oncology, Novocure, Pfizer, Tesaro
Consulting or Advisory Role: AstraZeneca, Cerulean Pharma, Clovis Oncology, Genmab, Karyopharm Therapeutics, Novocure, Pfizer, Tesaro, BioCad, Sotio
Research Funding: AstraZeneca (Inst), Boehringer Ingelheim (Inst), Pfizer (Inst), Tesaro (Inst), Clovis Oncology (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Karyopharm Therapeutics, Pfizer, Roche, Tesaro, SeraCare
Ignacio Romero
Consulting or Advisory Role: Clovis Oncology, GlaxoSmithKline, AstraZeneca Spain, Roche
Speakers' Bureau: Roche, AstraZeneca, PharmaMar, GlaxoSmithKline
Research Funding: Roche (Inst)
Travel, Accommodations, Expenses: PharmaMar, AstraZeneca Spain, GlaxoSmithKline, Clovis Oncology, Roche
Peter Vuylsteke
Honoraria: Roche, Novartis, MSD Oncology, Bristol Myers Squibb, Pfizer
Consulting or Advisory Role: Pfizer
Research Funding: MSD Oncology (Inst)
Travel, Accommodations, Expenses: Pfizer, Mundifarma, AstraZeneca
Robert L. Coleman
Consulting or Advisory Role: Clovis Oncology, Roche, Esperance Pharmaceuticals, AstraZeneca/MedImmune, Genmab, Tesaro, OncoMed, Sotio, Oncolytics, AbbVie/Stemcentrx, Immunogen, AbbVie, Agenus, OncoSec, Novocure
Research Funding: AstraZeneca/MedImmune, Esperance Pharmaceuticals, OncoMed, Array BioPharma, Clovis Oncology, Johnson & Johnson, Merck, Roche/Genentech, Abbott/AbbVie
Travel, Accommodations, Expenses: Merck, AstraZeneca/MedImmune, Array BioPharma, Clovis Oncology, Roche/Genentech, Research to Practice, GOG, Clovis Oncology, Sotio, Vaniam Group
Felix Hilpert
Honoraria: Roche, AstraZeneca, Tesaro, Clovis Oncology, PharmaMar, MSD Oncology
Consulting or Advisory Role: Tesaro, AstraZeneca, PharmaMar, Roche
Travel, Accommodations, Expenses: Tesaro, AstraZeneca, PharmaMar
Amit M. Oza
Uncompensated Relationships: Ozmosis Research
Martin K. Oehler
Honoraria: AstraZeneca
Consulting or Advisory Role: AstraZeneca
Patents, Royalties, Other Intellectual Property: Autoantibody Biomarkers for Ovarian Cancer (PCT application no. PCT/AU2014/000925), Plasma Biomarkers for Ovarian Cancer (PCT application no. PCT/AU2016/050040)
Sandro Pignata
Honoraria: AstraZeneca, Roche, Pharmamar, Tesaro, Pfizer, MSD Oncology
Consulting or Advisory Role: AstraZeneca, Roche, PharmaMar, Pfizer, Tesaro
Research Funding: Roche (Inst), AstraZeneca (Inst), MSD Oncology (Inst), Pfizer (Inst)
Carol Aghajanian
Consulting or Advisory Role: Tesaro, Mersana, Eisai, Roche, AbbVie
Research Funding: Roche (Inst), AbbVie (Inst), Clovis Oncology (Inst), AstraZeneca (Inst), Clovis Oncology (Inst), AstraZeneca (Inst)
Nicoletta Colombo
Honoraria: Roche, AstraZeneca, Tesaro, PharmaMar, GlaxoSmithKline, MSD Oncology, Clovis, Pfizer, Amgen, Immunogen
Consulting or Advisory Role: Roche, PharmaMar, AstraZeneca, Clovis Oncology, Pfizer, MSD Oncology, Tesaro, BioCad, GlaxoSmithKline, Immunogen, Immunogen
David Cibula
Consulting or Advisory Role: Astra Zeneca, Sotio, Roche
Kathleen N. Moore
Honoraria: Research To Practice, Prime Oncology
Consulting or Advisory Role: Roche, Immunogen, AstraZeneca (Inst), Clovis Oncology, Tesaro (Inst), VBL Therapeutics, Janssen Oncology, Merck, Aravive, Samumed, OncoMed, Pfizer/EMD Serono, Eisai, AbbVie, Vavotar, Mersana (Inst)
Research Funding: PTC Therapeutics (Inst), Eli Lilly (Inst), Merck (Inst), Tesaro (Inst), Genentech (Inst), Clovis Oncology (Inst), Lilly Foundation (Inst), Regeneron (Inst), Advaxis (Inst), Bristol Myers Squibb, Verastem (Inst), Novartis Pharmaceuticals UK (Inst), AstraZeneca (Inst), Agenus (Inst), Takeda (Inst), Forty Seven (Inst), Stem CentRx (Inst), Immunogen (Inst), Bayer (Inst), Novogen (Inst), AbbVie/Stemcentrx (Inst)
Janna Christy-Bittel
Employment: Array Biopharma, Pfizer
Stock and Other Ownership Interests: Gilead Sciences, Array BioPharma
Regina Berger
Travel, Accommodations, Expenses: Roche (Inst), Merck Sharp & Dohme (Inst), Clovis Oncology (Inst), BioCad (Inst)
Christian Marth
Honoraria: AstraZeneca, Pfizer, Roche, GlaxoSmithKline
Consulting or Advisory Role: AstraZeneca, Roche, MSD Oncology, Pfizer, GlaxoSmithKline
Travel, Accommodations, Expenses: Roche, AstraZeneca, GlaxoSmithKline
Jalid Sehouli
Honoraria: AstraZeneca, Eisai, Clovis Oncol, Olympus Medical Systems, Johnson & Johnson, PharmaMar, Pfizer, Teva, Tesaro, MSD Oncology, GlaxoSmithKline, Bayer
Consulting or Advisory Role: AstraZeneca, Clovis Oncology, PharmaMar, Merck, Pfizer, Tesaro, MSD Oncology, Eli Lilly, Novocure, Johnson & Johnson
Research Funding: AstraZeneca (Inst), Clovis Oncology (Inst), Merck (Inst), Bayer (Inst), PharmaMar (Inst), Pfizer (Inst), Tesaro (Inst), MSD Oncology (Inst), Roche (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Clovis Oncology, PharmaMar, Roche Pharma AG, Tesaro, Tesaro, MSD Oncology, Olympus
David M. O'Malley
Consulting or Advisory Role: Janssen Oncology, AstraZeneca, Clovis Oncology, Tesaro, Novocure, AbbVie, Roche, OncoQuest, Immunogen, GOG Foundation, Translational Genomics/Cordgenics, Agenus, Marker Therapeutics, Eisai, Genelux, Iovance Biotherapeutics, Ambry Genetics, Tarveda Therapeutics, Leap Therapeutics, Myriad Genetics, GlaxoSmithKline, Regeneron
Research Funding: Amgen (Inst), AstraZeneca (Inst), Roche (Inst), Regeneron (Inst), Immunogen (Inst), Janssen Research & Development (Inst), Clovis Oncology (Inst), EMD Serono (Inst), Ergomed (Inst), Ajinomoto (Inst), Immunogen (Inst), Cerulean Pharma (Inst), PharmaMar (Inst), Array BioPharma (Inst), Bristol Myers Squibb (Inst), Agenus (Inst), Tesaro (Inst), TRACON Pharma (Inst), Genmab (Inst), Seattle Genetics (Inst), Iovance Biotherapeutics (Inst), Leap Therapeutics (Inst), Merck (Inst), AbbVie/Stemcentrx (Inst), AbbVie (Inst)
Adam P. Boyd
Employment: Pfizer, Array BioPharma
Stock and Other Ownership Interests: Array BioPharma
Travel, Accommodations, Expenses: Array BioPharma
Andrew Clamp
Consulting or Advisory Role: AstraZeneca, GlaxoSmithKline
Speakers' Bureau: Clovis, AstraZeneca (Inst), Clovis Oncology (Inst), Millennium (Inst), Pfizer (Inst), Immunogen (Inst), Eli Lilly (Inst)
Travel, Accommodations, Expenses: AstraZeneca, Tesaro
Isabelle Ray-Coquard
Honoraria: Roche, PharmaMar, AstraZeneca, Clovis Oncology, Tesaro, MSD Oncology, Genmab, AbbVie, Pfizer, Bristol Myers Squibb
Consulting or Advisory Role: Pfizer, AbbVie, Genmab, Roche, AstraZeneca, Tesaro, Clovis Oncology, PharmaMar, MSD Oncology, Bristol Myers Squibb, Deciphera, Mersana, Mersana, GlaxoSmithKline
Research Funding: MSD Oncology, Bristol Myers Squibb
Travel, Accommodations, Expenses: Roche, AstraZeneca, Tesaro, PharmaMar, GlaxoSmithKline, Clovis, Clovis, Bristol Myers Squibb, Advaxis
Uncompensated Relationships: GINECO Group, Inca National Cancer Institute France
Ignace Vergote
Consulting or Advisory Role: PharmaMar (Inst), Roche (Inst), MSD Belgium (Inst), Genmab (Inst), Millennium (Inst), Clovis Oncology (Inst), Immunogen (Inst), Tesaro (Inst), Oncoinvent (Inst), Sotio (Inst), Amgen Europe (Inst), AstraZeneca (Inst), Carrick Therapeutics (Inst), Debiopharm Group (Inst), F. Hoffmann La Roche (Inst), GlaxoSmithKline Pharmaceuticals (Inst), Medical University of Vienna (Inst), Octimet Oncology (Inst), Deciphera (Inst), Verastem (Inst)
Research Funding: Roche (Inst), Genmab (Inst), Amgen (Inst), Oncoinvent (Inst)
Travel, Accommodations, Expenses: Roche, AstraZeneca, Tesaro, Amgen, MSD/Merck Zurich + USA
No other potential conflicts of interest were reported.
REFERENCES
- 1.Colombo N, Peiretti M, Parma G, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v23–v30. doi: 10.1093/annonc/mdq244. [DOI] [PubMed] [Google Scholar]
- 2.Diaz-Padilla I, Malpica AL, Minig L, et al. Ovarian low-grade serous carcinoma: A comprehensive update. Gynecol Oncol. 2012;126:279–285. doi: 10.1016/j.ygyno.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 3.Grisham RN, Iyer G, Garg K, et al. BRAF mutation is associated with early stage disease and improved outcome in patients with low-grade serous ovarian cancer. Cancer. 2013;119:548–554. doi: 10.1002/cncr.27782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grisham RN, Sylvester BE, Won H, et al. Extreme outlier analysis identifies occult mitogen-activated protein kinase pathway mutations in patients with low-grade serous ovarian cancer. J Clin Oncol. 2015;33:4099–4105. doi: 10.1200/JCO.2015.62.4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunter SM, Anglesio MS, Ryland GL, et al. Molecular profiling of low grade serous ovarian tumours identifies novel candidate driver genes. Oncotarget. 2015;6:37663–37677. doi: 10.18632/oncotarget.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vang R, Shih IeM, Kurman RJ. Ovarian low-grade and high-grade serous carcinoma: Pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv Anat Pathol. 2009;16:267–282. doi: 10.1097/PAP.0b013e3181b4fffa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong KK, Tsang YT, Deavers MT, et al. BRAF mutation is rare in advanced-stage low-grade ovarian serous carcinomas. Am J Pathol. 2010;177:1611–1617. doi: 10.2353/ajpath.2010.100212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon S, Seger R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 9.Scholl FA, Dumesic PA, Khavari PA. Effects of active MEK1 expression in vivo. Cancer Lett. 2005;230:1–5. doi: 10.1016/j.canlet.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 10.Winski S, Anderson D, Bouhana K, et al. MEK162 (ARRY-162), a novel MEK 1/2 inhibitor, inhibits tumor growth regardless of KRas/Raf pathway mutations. Eur J Cancer. 2010;8:56. [Google Scholar]
- 11.Farley J, Brady WE, Vathipadiekal V, et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: An open-label, single-arm, phase 2 study. Lancet Oncol. 2013;14:134–140. doi: 10.1016/S1470-2045(12)70572-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vergote I, Pujade-Lauraine E, Pignata S, et al. European Network of Gynaecological Oncological Trial Groups’ requirements for trials between academic groups and pharmaceutical companies. Int J Gynecol Cancer. 2010;20:476–478. doi: 10.1111/IGC.0b013e3181d3caa8. [DOI] [PubMed] [Google Scholar]
- 13.Kittelson JM, Emerson SS. A unifying family of group sequential test designs. Biometrics. 1999;55:874–882. doi: 10.1111/j.0006-341x.1999.00874.x. [DOI] [PubMed] [Google Scholar]
- 14. Gershenson DM, Miller A, Brady W, et al: A randomized phase II/III study to assess the efficacy of trametinib in patients with recurrent or progressive low-grade serous ovarian or peritoneal cancer. Ann Oncol 30: v851-v934, 2019 (suppl 5) [Google Scholar]
- 15.Mektovi: US Prescribing Information. Pfizer Inchttp://www.arraybiopharma.com/documents/Mektovi_Prescribing_information.pdf
- 16.Bedard PL, Tabernero J, Janku F, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21:730–738. doi: 10.1158/1078-0432.CCR-14-1814. [DOI] [PubMed] [Google Scholar]
- 17.Said R, Ye Y, Falchook GS, et al. Outcomes of patients with advanced cancer and KRAS mutations in phase I clinical trials. Oncotarget. 2014;5:8937–8946. doi: 10.18632/oncotarget.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spreafico A, Oza AM, Clarke BA, et al. Genotype-matched treatment for patients with advanced type I epithelial ovarian cancer (EOC) Gynecol Oncol. 2017;144:250–255. doi: 10.1016/j.ygyno.2016.12.002. [DOI] [PubMed] [Google Scholar]