

Abstract
PURPOSE
BRCA1 or BRCA2 (BRCA) alterations are common in men with metastatic castration-resistant prostate cancer (mCRPC) and may confer sensitivity to poly(ADP-ribose) polymerase inhibitors. We present results from patients with mCRPC associated with a BRCA alteration treated with rucaparib 600 mg twice daily in the phase II TRITON2 study.
METHODS
We enrolled patients who progressed after one to two lines of next-generation androgen receptor–directed therapy and one taxane-based chemotherapy for mCRPC. Efficacy and safety populations included patients with a deleterious BRCA alteration who received ≥ 1 dose of rucaparib. Key efficacy end points were objective response rate (ORR; per RECIST/Prostate Cancer Clinical Trials Working Group 3 in patients with measurable disease as assessed by blinded, independent radiology review and by investigators) and locally assessed prostate-specific antigen (PSA) response (≥ 50% decrease from baseline) rate.
RESULTS
Efficacy and safety populations included 115 patients with a BRCA alteration with or without measurable disease. Confirmed ORRs per independent radiology review and investigator assessment were 43.5% (95% CI, 31.0% to 56.7%; 27 of 62 patients) and 50.8% (95% CI, 38.1% to 63.4%; 33 of 65 patients), respectively. The confirmed PSA response rate was 54.8% (95% CI, 45.2% to 64.1%; 63 of 115 patients). ORRs were similar for patients with a germline or somatic BRCA alteration and for patients with a BRCA1 or BRCA2 alteration, while a higher PSA response rate was observed in patients with a BRCA2 alteration. The most frequent grade ≥ 3 treatment-emergent adverse event was anemia (25.2%; 29 of 115 patients).
CONCLUSION
Rucaparib has antitumor activity in patients with mCRPC and a deleterious BRCA alteration, but with a manageable safety profile consistent with that reported in other solid tumor types.
INTRODUCTION
Therapies such as androgen receptor (AR)–directed therapy and taxane chemotherapy have led to improved outcomes for men with metastatic castration-resistant prostate cancer (mCRPC).1-4 However, patients will eventually progress, and subsequent treatment options are limited, highlighting the need for additional effective therapies.
CONTEXT
Key Objective
We evaluated rucaparib as treatment for men with metastatic castration-resistant prostate cancer (mCRPC) associated with a BRCA gene alteration who had received prior taxane and androgen receptor–directed therapy.
Knowledge Generated
A substantial proportion of patients achieved a confirmed radiographic response with rucaparib treatment in both blinded, central independent radiology review– and investigator-assessed analyses. Furthermore, we provide evidence of radiographic and prostate-specific antigen responses across subgroups based on baseline characteristics (eg, number of prior lines of therapy) and genomic characteristics (eg, gene, zygosity, and alteration types).
Relevance
Men with mCRPC and a BRCA alteration who receive a poly(ADP-ribose) polymerase inhibitor in this setting achieve higher objective and prostate-specific antigen response rates than those observed with previously approved therapies in an unselected population. These data illustrate the potential benefit of rucaparib in patients with mCRPC associated with a BRCA alteration.
Approximately 12% of men with mCRPC harbor a deleterious BRCA1 or BRCA2 (BRCA) alteration (BRCA1, 2%; BRCA2, 10%).5 Men with a germline BRCA alteration have an increased risk for prostate cancer and more commonly have nodal involvement and/or distant metastases.6,7 Poly(ADP-ribose) polymerase (PARP) inhibitors can induce cytotoxicity via synthetic lethality in tumor cells that are deficient in homologous recombination–directed DNA damage repair (DDR), including those carrying loss-of-function alterations in BRCA genes.8-10
The phase II TRITON2 study is evaluating the PARP inhibitor rucaparib for the treatment of men with mCRPC associated with a deleterious alteration in BRCA or other DDR gene who have progressed after next-generation AR-directed therapy and a taxane-based chemotherapy. Here, we present efficacy and safety data from TRITON2 for patients with mCRPC with a BRCA alteration.
METHODS
TRITON2 (ClinicalTrials.gov identifier: NCT02952534) is a fully enrolled, ongoing, international, open-label, phase II study evaluating rucaparib in patients with mCRPC associated with DDR deficiency. Men aged ≥ 18 years with histologically or cytologically confirmed mCRPC, Eastern Cooperative Oncology Group performance status of 0 or 1, and adequate organ function were enrolled. Eligible patients had a deleterious germline or somatic alteration in BRCA1, BRCA2, or another prespecified DDR gene that may confer sensitivity to PARP inhibition, as well as disease progression after one to two lines of next-generation AR-directed therapy for prostate cancer and one prior taxane-based chemotherapy for castration-resistant disease. Patients were required to receive a concomitant gonadotropin-releasing hormone analog or to have had prior bilateral orchiectomy. Patients who were previously treated with a PARP inhibitor, mitoxantrone, cyclophosphamide, or platinum-based chemotherapy or with an active secondary malignancy were excluded. Patients were enrolled irrespective of measurable disease status (Data Supplement). Full eligibility criteria are described in the protocol (Data Supplement).
Patients received a starting dose of 600 mg oral rucaparib twice daily. Dose reductions, in decrements of 100 mg, were permitted for grade ≥ 3 or persistent grade 2 treatment-emergent adverse events (TEAEs).
The study was approved by national or local institutional review boards and performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines of the International Council for Harmonisation. Patients provided written informed consent before participation.
Patient Populations
Efficacy and safety populations included patients with a deleterious BRCA alteration identified before enrollment who received ≥ 1 dose of rucaparib 600 mg. Patients with a non-BRCA DDR gene alteration (without a deleterious BRCA alteration) were not part of this analysis (data reported previously).11
Efficacy was assessed in the overall efficacy population (all evaluable patients regardless of measurable disease status), independent radiology review (IRR)–evaluable population (patients who had measurable disease at baseline per blinded, central IRR assessment), and investigator-evaluable population (patients who had measurable disease at baseline per investigator assessment).
Efficacy and safety analyses included all patients meeting the above criteria enrolled by May 8, 2019. The visit cutoff date for safety analyses was September 13, 2019. The visit cutoff date for efficacy analyses was December 23, 2019, to allow for more complete assessment (≥ 32 weeks of follow up) of the efficacy end points.
Analysis Outcomes
The primary end point was objective response rate (ORR) by blinded IRR per modified RECIST v1.1 and Prostate Cancer Clinical Trials Working Group 3 (PCWG3) criteria (IRR-evaluable population), supported by confirmed ORR by investigator assessment (investigator-evaluable population).
Secondary end points included duration of response (DOR) for radiographic response, rate of confirmed locally assessed prostate-specific antigen (PSA) response (≥ 50% decrease from baseline confirmed by a consecutive measurement ≥ 3 weeks later), time to PSA progression, radiographic progression-free survival (rPFS), overall survival (OS), and safety.
Exploratory subgroup analyses of confirmed ORR and confirmed PSA response rate were performed based on baseline disease characteristics (number of prior lines of therapy, measurable disease status, presence of hepatic metastases, and age) and genomic characteristics (BRCA1 or BRCA2, germline or somatic alteration, zygosity, and alteration types).
Safety was assessed by monitoring TEAEs, vital signs, laboratory testing, and physical examination. Dose intensity was calculated as the actual dose received divided by the first dose.
Procedures
Patients were screened for the presence of a deleterious somatic or germline alteration in BRCA1, BRCA2, or other DDR gene through central genomic testing of plasma or tumor tissue (archival or contemporaneous), or through local testing. Central testing was performed by Foundation Medicine.12,13 Germline testing was performed by Color Genomics.14,15 Additional details are provided in the Data Supplement.
Patients received rucaparib until radiographic disease progression (soft tissue or bone lesion) assessed by investigator per modified RECIST/PCWG3 criteria, unequivocal clinical progression, unacceptable toxicity or inability to tolerate additional treatment, loss to follow up, or withdrawal of consent. Tumor assessments by computed tomography or magnetic resonance imaging and bone scans were performed during screening, every 8 weeks for 24 weeks, then every 12 weeks thereafter. PSA assessments were conducted every 4 weeks.
TEAEs were monitored from the first dose of rucaparib until 28 days after the last dose. TEAEs were coded using Medical Dictionary for Drug Regulatory Activities v20.116 and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03.17 Additional details are provided in the Data Supplement.
Statistical Analyses
A Simon 2-stage design was used as a futility rule. Assuming a total sample size of 83 patients in the IRR-evaluable population, characteristics of the design included a null hypothesis of ORR = 20%, type I error rate of 5% (one sided), and 90% power when the true response rate is 35%. However, the final number of patients was to be defined based on regulatory considerations. Confirmed ORR and PSA responses were summarized descriptively with frequencies and 95% CIs (Clopper-Pearson).
DOR, time to PSA progression, and rPFS were summarized using Kaplan-Meier methodology. DOR was defined as the time from the date of the first confirmed response to the date progression was first documented plus 1 day. Time to PSA progression was defined as the time from the first rucaparib dose to the date of an increase ≥ 25% and absolute increase of ≥ 2 ng/mL above the nadir in PSA plus 1 day. PSA increases must have been confirmed by a consecutive assessment conducted ≥ 3 weeks later; early rises (< 12 weeks) were not considered in determining PSA progression.18 rPFS was defined as the time from first rucaparib dose to the date of first objective evidence of radiographic progression or death due to any cause, whichever occurred first.
Best change from baseline in the sum of the diameter of target lesions and PSA were summarized graphically. Statistical analyses were performed using SAS v9.4 (SAS Institute, Cary, NC).
RESULTS
Efficacy
The overall efficacy population included 115 patients who received ≥ 1 dose of rucaparib and had a deleterious BRCA alteration (BRCA1 [n = 13], BRCA2 [n = 102], germline [n = 44], and somatic [n = 71]; Data Supplement). Baseline patient demographics and disease characteristics are shown in Table 1 and the Data Supplement. At baseline, 62 patients had measurable disease per blinded IRR assessment (IRR-evaluable population) and 65 patients had measurable disease per investigator assessment (investigator-evaluable population); 57 patients had measurable disease by both IRR and investigator. Genomic characteristics (eg, germline/somatic status, zygosity, and alteration type) and co-occurring alterations are shown in the Data Supplement.
TABLE 1.
Baseline Patient and Disease Characteristics in TRITON2 Patients With a BRCA Alteration
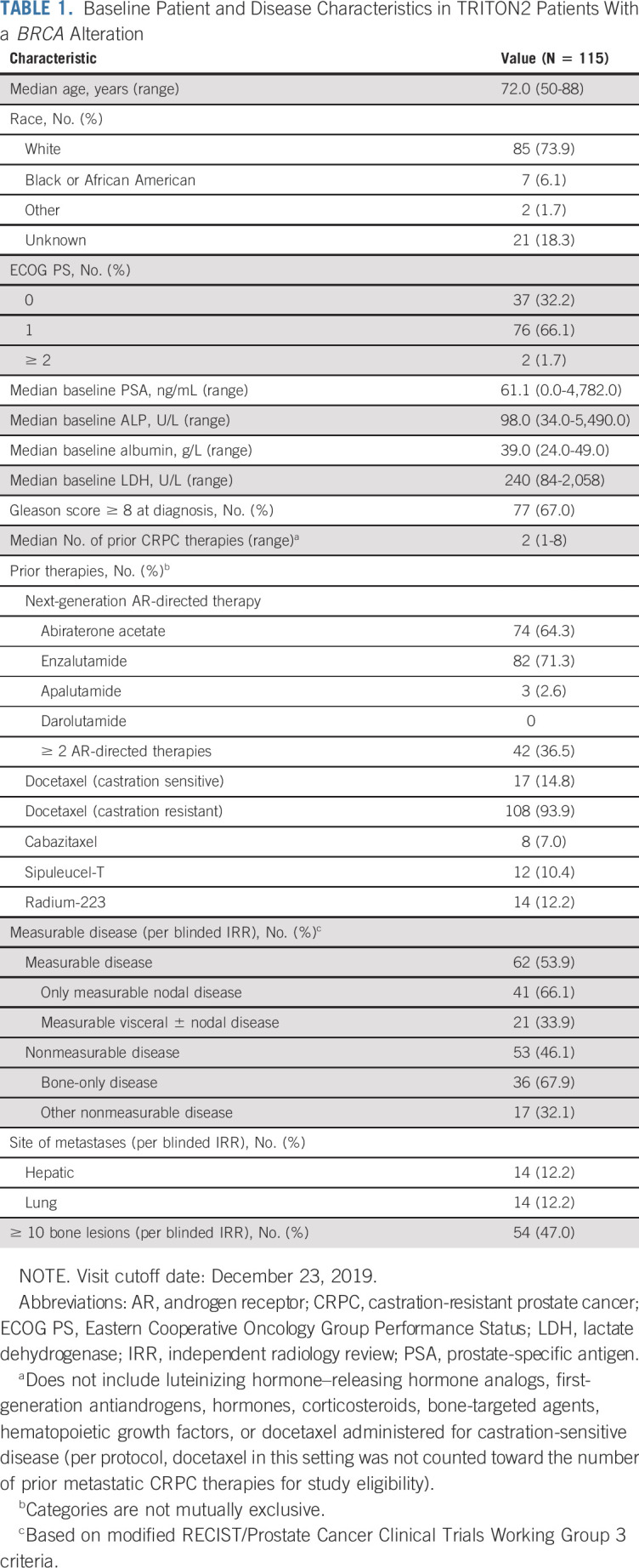
Median treatment duration for the overall efficacy population was 8.1 months (range, 0.5-30.3 months), and the median follow up was 17.1 months (range, 7.6-31.5 months). As of December 23, 2019, 29 patients (25.2%) remained on treatment.
The confirmed ORR for the IRR-evaluable population was 43.5% (95% CI, 31.0% to 56.7%; 27 of 62), and the confirmed ORR for the investigator-evaluable population was 50.8% (95% CI, 38.1% to 63.4%; 33 of 65; Table 2). Most patients had a best response of stable disease or better (88.7% in the IRR-evaluable population and 89.2% in the investigator-evaluable population). Eight patients had a confirmed complete response in soft-tissue disease per blinded IRR and/or investigator assessment. Among these patients, one had hepatic metastases, one had a non-nodal pelvic mass, and 6 had nodal-only disease at baseline per IRR; one had a BRCA1 alteration and seven had a BRCA2 alteration. Among responders in the IRR-evaluable population, 19 of 27 patients (70.4%) demonstrated a response by the first tumor assessment (week 8; Data Supplement). Median DOR in the IRR-evaluable population was not reached (NR; 95% CI, 6.4 months to NR; Data Supplement) and was 6.4 months in the investigator-evaluable population (95% CI, 5.5 to 11.7 months; Data Supplement). Within the IRR-evaluable population, 15 of 27 patients with a confirmed objective response had a DOR ≥ 6 months; three patients with ongoing responses were followed for < 6 months from the onset of response. In the IRR- and investigator-evaluable populations, 64.5% and 63.1% of patients demonstrated a ≥ 30% reduction in target lesion size from baseline (Fig 1A and Data Supplement).
TABLE 2.
Rate of Response to Rucaparib Treatment

FIG 1.

Best change from baseline in (A) sum of target lesion(s) in the independent radiology review–evaluable population and in (B) prostate-specific antigen (PSA) in the overall efficacy population. Visit cutoff date: December 23, 2019. In (A), the upper dotted line indicates the threshold for progressive disease, a 20% increase in the sum of the longest diameter of the target lesions, whereas the lower dotted line indicates the threshold for partial response, a 30% decrease in the sum of the longest diameter of the target lesions. In (B), the upper dotted line indicates the threshold for PSA progression, a 25% increase from baseline (accompanied by an absolute increase of ≥ 2 ng/mL above the nadir), whereas the lower dotted line indicates the threshold for PSA response, a 50% decrease from baseline. Bars were capped at 100% for visual clarity. PSA increases for the 4 leftmost patients were 689%, 231%, 183%, and 133%. In both panels, patients with 0% change from baseline are shown as 0.5% for visual clarity.
In the overall efficacy population, 63 of 115 patients had a confirmed PSA response (54.8%; 95% CI, 45.2% to 64.1%; Table 2), and median time to PSA response was 1.9 months (95% CI, 1.3 to 1.9 months). Median time to PSA progression was 6.5 months (95% CI, 5.9 to 7.8 months; Data Supplement). The majority of patients (60.0%) demonstrated a single best PSA reduction ≥ 50% from baseline (Fig 1B).
Although ORRs were similar across subgroups based on baseline clinical or genomic characteristics, some differences were observed with PSA response rates (Fig 2 and Data Supplement). PSA responses were observed among patients with BRCA1 (15.4%; 2 of 13 patients) or monoallelic alterations (11.1%; 1 of 9 patients), albeit at a lower rate than the overall population; however, the number of patients in these subgroups was low. Conversely, a higher proportion of patients with biallelic alterations (75.0%; 27 of 36 patients) and/or homozygous loss (81.0%; 17 of 21 patients) experienced a PSA response compared with the overall population.
FIG 2.
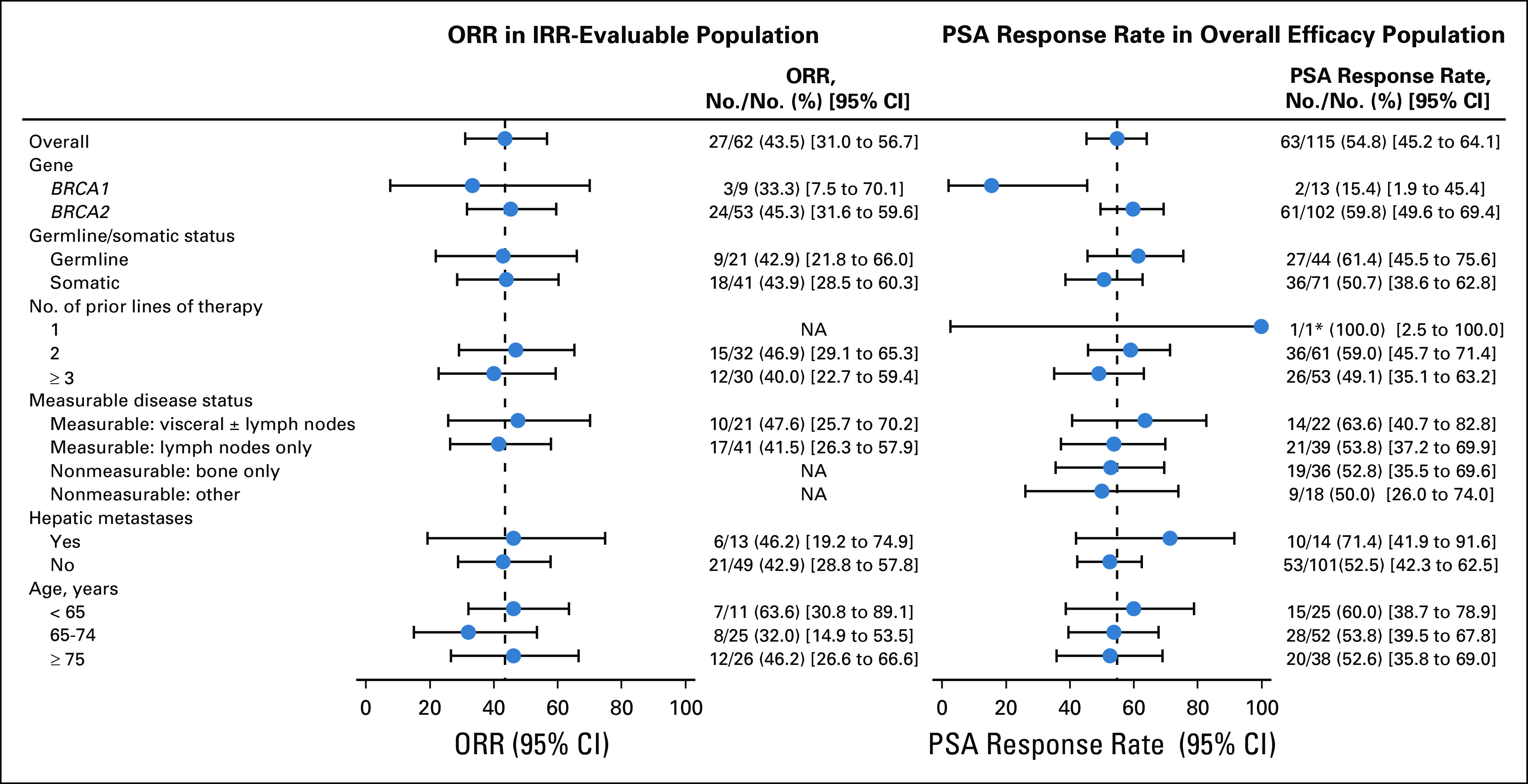
Subgroup analysis of objective response rate (ORR) in independent radiology review (IRR)–evaluable population and prostate-specific antigen (PSA) response rate in overall efficacy population by baseline characteristics. Visit cutoff date: December 23, 2019. The vertical dotted line corresponds to the overall ORR or PSA response. (*)One patient received taxane in the hormone-sensitive setting only, which per protocol was not counted as a line of therapy for eligibility; not receiving taxane for castration-resistant prostate cancer was considered a protocol deviation. NA, not applicable.
Median rPFS was 9.0 months (95% CI, 8.3 to 13.5 months) per blinded IRR assessment (Fig 3) and 8.5 months (95% CI, 8.1 to 11.2 months) per investigator assessment (Data Supplement). Although OS data were not yet mature at the time of the analysis (41% of events reported), the Kaplan-Meier estimate of 12-month OS was 73.0% (95% CI, 62.9% to 80.7%).
FIG 3.

Radiographic progression-free survival by blinded independent radiology review assessment. Visit cutoff date: December 23, 2019. Progression was assessed per modified RECIST/PWCG3 criteria. Details on reasons for censoring are provided in the Data Supplement. IRR, independent radiology review; PCWG3, Prostate Cancer Clinical Trials Working Group 3; RECIST, Response Evaluation Criteria In Solid Tumors version 1.1.
Safety
The safety population included 115 patients who received one or more dose of rucaparib. As of the visit cutoff, median treatment duration was 6.5 months (range, 0.5-26.7 months), and median follow up was 13.7 months (range, 4.2-28.2 months). Mean dose intensity was 0.88 (standard deviation, 0.15).
A TEAE of any grade occurred in 114 patients (99.1%), and a grade ≥ 3 TEAE was reported in 70 patients (60.9%; Table 3 and Data Supplement). The most frequent TEAEs (any grade) were asthenia/fatigue (61.7%), nausea (52.2%), and anemia/decreased hemoglobin (43.5%). The most frequent grade ≥ 3 TEAE was anemia/decreased hemoglobin (25.2%). Overall, 32 patients (27.8%) received ≥ 1 transfusion of packed RBCs. Other TEAEs of interest are presented in the Data Supplement.
TABLE 3.
Most Commonly Reported TEAEs (N = 115)

ALT/AST elevations (any grade) were observed in 38 patients (33.0%)—the majority were grade 1 or 2. These typically occurred within the first 4 weeks of rucaparib treatment, normalized over time with continued treatment, and were not associated with abnormal increases in bilirubin or other criteria for drug-induced hepatotoxicity (Data Supplement). Increases in creatinine—predominantly grade 1 or 2—were also observed with the initiation of rucaparib and typically stabilized by the third week (Data Supplement) without being accompanied by a change in blood urea nitrogen. Decreased phosphate was observed in 75 (67.6%) of 111 patients (Data Supplement); however, most patients had low baseline phosphate and/or were receiving concomitant medications associated with decreased phosphate (eg, antacids or bone-targeting agents).
Treatment interruption due to a TEAE occurred in 65 patients (56.5%), most commonly because of anemia/decreased hemoglobin (21.7%), thrombocytopenia/decreased platelets (13.9%), and asthenia/fatigue (9.6%). Dose reduction due to a TEAE occurred in 47 patients (40.9%), most commonly due to anemia/decreased hemoglobin (13.0%), asthenia/fatigue (9.6%), and thrombocytopenia/decreased platelets (7.0%). Overall, 73 patients (63.5%) had either a treatment interruption or dose reduction due to a TEAE.
Nine patients (7.8%) discontinued because of a TEAE, one patient each due to acute respiratory distress syndrome; ALT/AST increased; anemia; balance disorder; cardiac failure; decreased appetite, fatigue, and weight decreased; leukopenia and neutropenia; pneumonia; and prolonged QT. There were three deaths as a result of TEAEs, including one each from pneumonia and prolonged QT, both considered unrelated to rucaparib, and one from acute respiratory distress syndrome, considered related to rucaparib by the investigator (additional details are in the Data Supplement).
DISCUSSION
Rucaparib treatment demonstrated significant clinical activity in men with mCRPC associated with a BRCA alteration, resulting in meaningful radiographic and PSA responses, including complete responses in soft-tissue disease, with a manageable safety and tolerability profile. Based on these results, rucaparib was granted accelerated approval by the US Food and Drug Administration for the treatment of men with a deleterious BRCA mutation (germline and/or somatic) associated mCRPC who have been treated with AR-directed therapy and a taxane-based chemotherapy.19
Baseline characteristics in the efficacy population were as expected for a population of patients with mCRPC receiving third-line or later treatment. More than one third of patients were treated with ≥ 2 next-generation AR-directed therapies.
For men with mCRPC with disease progression after next-generation AR-directed therapy and taxane-based chemotherapy, response to previously approved therapies has been historically poor, with confirmed ORRs ranging from 8%-15% and PSA response (≥ 50% decrease) rates ranging from 8%-39%.20-22 Of note, these data come from studies of men with mCRPC who were not selected based on a potential predictive biomarker (eg, BRCA alteration).
Data from TRITON2 illustrate the importance of genomic screening to identify men who may benefit from treatment with a PARP inhibitor.2,4,23 TRITON2 patients with mCRPC associated with a BRCA alteration who received rucaparib had substantially higher RECIST and PSA response rates than those typically observed with other treatments in an unselected population, including responses in clinically relevant subgroups. For example, although the numbers are small, the confirmed ORR of 46.2% (6 of 13) among patients with hepatic metastases (Fig 2) is potentially clinically important as hepatic metastases are known to be an indicator of poor prognosis in mCRPC.24
RECIST and PSA responses were observed in patients with BRCA2 and BRCA1 alterations, germline and somatic alterations, and all categories of alteration zygosity. Although the rates of confirmed PSA response were higher in BRCA2 versus BRCA1 and biallelic versus monoallelic subgroups, the numbers of patients in the BRCA1 and monoallelic subgroups were small. Due to the limited number of tissue samples received (66 of 115 patients [57.4%]), alteration zygosity could only be determined in 45 patients (39.1%) based on the availability of sufficiently high-quality next-generation sequencing data from tissue for copy-number analysis. Importantly, despite the relatively small numbers, confirmed PSA responses were observed in all molecular and clinical subgroups examined.
Our results are consistent with those from other studies demonstrating the clinical activity of PARP inhibitors (olaparib, niraparib, and talazoparib) in patients with mCRPC and a BRCA alteration who received prior AR-directed therapy.25-29 For example, in the PROfound study, patients with a BRCA alteration and prior AR-directed therapy showed improved rPFS with olaparib versus abiraterone acetate or enzalutamide (median, 9.8 v 3.0 months; hazard ratio, 0.22; 95% CI, 0.15 to 0.32).29 Although there are important differences in the study designs for these trials (eg, differences in the method of determining genomic alteration, type of alterations eligible for enrollment, assessment of response by RECIST, PSA decrease, or a composite that includes changes in circulating tumor cell count), these studies reinforce the potential benefit of PARP inhibitors in patients with mCRPC associated with a BRCA alteration.
The safety profile of rucaparib in patients with mCRPC was consistent with that observed in prior studies conducted in patients with ovarian cancer and other solid tumor types,19,30,31 as well as in studies of men with mCRPC who received other PARP inhibitors, with asthenia/fatigue, GI adverse effects, and myelosuppression among the most common TEAEs reported.25-27,29 Similar to studies of rucaparib in ovarian cancer, elevations in ALT, AST, and creatinine were commonly reported; however, these laboratory abnormalities were not associated with liver or kidney toxicity. Elevated creatinine has been observed with multiple PARP inhibitors and is thought to be due to inhibition of renal transporters (eg, MATE-1, MATE2-K, OCT2) rather than a direct impact on renal function.19,32-35 Although there have been reports of fatal pneumonitis with other PARP inhibitors,34 interstitial lung disease has not been identified as a potential risk from rucaparib treatment when evaluated across studies in multiple tumor types; the majority of cases had an alternative etiology, and most resolved with continued rucaparib treatment or after dose interruption with negative rechallenge.
A strength of our analysis is the inclusion of both IRR- and investigator-assessed end points; a substantial proportion of patients achieved a confirmed radiographic response with rucaparib treatment via both assessment methods. Discordance in assessment by independent reviewers and investigators may be influenced by variations in lesion selection or by historical/clinical information available to investigators but not the blinded IRR.36 One limitation of our analysis was that the DOR and rPFS results for the IRR-evaluable population were more heavily impacted by censoring. Per the protocol, radiographic assessments were to be continued until disease progression was observed by the investigator. If the investigator reported disease progression on a scan but the blinded IRR did not, the patient became censored in the IRR analysis. Additional limitations include the lack of a control arm and the immaturity of OS data. The phase III TRITON3 study (ClinicalTrials.gov identifier: NCT02975934) is ongoing to define the clinical benefit (eg, rPFS and OS) of rucaparib in an earlier disease setting among patients with mCRPC associated with a BRCA or ATM alteration who have progressed after one next-generation AR-directed therapy and who have not received taxane-based chemotherapy in the mCRPC setting. Rucaparib is being compared with physician’s choice of next-generation AR-directed therapy or docetaxel and will provide additional evidence of the effects of rucaparib treatment in men with mCRPC.
Altogether, results from the TRITON2 study demonstrate that rucaparib has meaningful antitumor activity and a manageable safety profile in patients with mCRPC, as well as a deleterious germline or somatic BRCA alteration, and support the use of rucaparib in this patient population.
ACKNOWLEDGMENT
The authors thank Cheryl Chun and Vivian Chen of Clovis Oncology for assistance in manuscript preparation. Medical writing and editorial support funded by Clovis Oncology were provided by Nathan Yardley and Frederique H. Evans of Ashfield Healthcare Communications.
PRIOR PRESENTATION
Presented at the European Society for Medical Oncology Annual Congress, Munich, Germany, October 19-23, 2018, and European Society for Medical Oncology Annual Congress, Munich, Barcelona, Spain, September 27-October 1, 2019; the American Society of Clinical Oncology ASCO Annual Meeting, Chicago, IL, May 31-June 4, 2019; and the ASCO Genitourinary Cancers Symposium, San Francisco, CA, February 13-15, 2020.
SUPPORT
Funded by Clovis Oncology; supported in part by the National Cancer Institute (NCI) Cancer Center Support Grant No. P30-CA008748, NCI Prostate Specialized Program of Research Excellence (SPORE) Grant No. P50-CA092629-16, Department of Defense Prostate Cancer Research Program Grant No. W81XWH-17-1-0124, and a Prostate Cancer Foundation Young Investigator Award (W.A.); and supported in part by a Prostate Cancer Foundation Challenge Award and NCI Prostate SPORE Grant No. P50-CA180995 (A.P.).
Written on behalf of the TRITON2 investigators.
See accompanying editorial on page 3735
AUTHOR CONTRIBUTIONS
Conception and design: Wassim Abida, Simon P. Watkins, Tony Golsorkhi, and Simon Chowdhury
Provision of study materials or patients: Wassim Abida, Akash Patnaik, David Campbell, Jeremy Shapiro, Alan H. Bryce, Ray McDermott, Brieuc Sautois, Nicholas J. Vogelzang, Richard M. Bambury, Eric Voog, Jingsong Zhang, Josep M. Piulats, Charles J. Ryan, Axel S. Merseburger, Gedske Daugaard, Axel Heidenreich, Karim Fizazi, Celestia S. Higano, Laurence E. Krieger, Cora N. Sternberg, Simon Chowdhury
Collection and assembly of data: Wassim Abida, Akash Patnaik, David Campbell, Jeremy Shapiro, Alan H. Bryce, Ray McDermott, Brieuc Sautois, Nicholas J. Vogelzang, Richard M. Bambury, Eric Voog, Jingsong Zhang, Josep M. Piulats, Charles J. Ryan, Axel S. Merseburger, Gedske Daugaard, Axel Heidenreich, Karim Fizazi, Celestia S. Higano, Laurence E. Krieger, Cora N. Sternberg, Simon Chowdhury
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Wassim Abida
Honoraria: CARET
Consulting or Advisory Role: Clovis Oncology, Janssen, MORE Health, ORIC Pharmaceuticals, Daiichi Sankyo
Research Funding: AstraZeneca (Inst), Zenith Epigenetics (Inst), Clovis Oncology (Inst), GlaxoSmithKline (Inst)
Travel, Accommodations, Expenses: GlaxoSmithKline, Clovis Oncology, ORIC Pharmaceuticals
Akash Patnaik
Stock and Other Ownership Interests: Gilead Sciences, Genentech
Honoraria: Merck, Prime Oncology, Jounce Therapeutics, Exelixis
Consulting or Advisory Role: Jounce Therapeutics, Exelixis, Guidepoint, Gerson Lehrman Group
Research Funding: Bristol Myers Squibb (Inst), Progenics (Inst), Clovis Oncology (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb, Prime Oncology, Exelixis
Alan H. Bryce
Honoraria: Astellas Pharma, Bayer
Travel, Accommodations, Expenses: Clovis Oncology (Inst), Phosplatin Therapeutics (Inst)
Ray McDermott
Honoraria: Bayer, Sanofi, Janssen, Astellas Pharma, Bristol Myers Squibb, Merck Sharp & Dohme, Pfizer, Novartis, Clovis Oncology
Speakers' Bureau: MSD Oncology
Research Funding: Sanofi (Inst), Janssen (Inst), Bayer (Inst), Astellas Pharma (Inst)
Travel, Accommodations, Expenses: Pfizer, Janssen-Cilag, Roche, Ipsen
Brieuc Sautois
Consulting or Advisory Role: Clovis Oncology, Astellas Pharma, Janssen, Sanofi
Travel, Accommodations, Expenses: Janssen
Nicholas J. Vogelzang
Employment: US Oncology
Stock and Other Ownership Interests: Caris Life Sciences
Honoraria: UpToDate, Pfizer, Novartis
Consulting or Advisory Role: Pfizer, Bayer, Genentech, AstraZeneca, Caris Life Sciences, Tolero Pharmaceuticals, Merck, Astellas Pharma, Boehringer Ingelheim, Corvus, Modra, Clovis Oncology, Janssen Oncology, Eisai
Speakers' Bureau: Bayer, Sanofi, Genentech, Bristol Myers Squibb, Seattle Genetics, Astellas Pharma, Clovis Oncology
Research Funding: US Oncology (Inst), Endocyte (Inst), Merck (Inst), Suzhou Kintor Pharmaceuticals (Inst)
Expert Testimony: Novartis
Travel, Accommodations, Expenses: Genentech, US Oncology, Pfizer, Bayer, Onyx, Exelixis, AstraZeneca, MedImmune, Sanofi
Richard M. Bambury
Leadership: Portable Medical Technology
Stock and Other Ownership Interests: Portable Medical Technology
Consulting or Advisory Role: Bayer, Janssen, Roche
Travel, Accommodations, Expenses: Pfizer, AbbVie, Ipsen
Eric Voog
Consulting or Advisory Role: Pfizer, Ipsen, Sanofi, LEO Pharma
Jingsong Zhang
Honoraria: AstraZeneca, MedImmune, Sanofi, Merck
Consulting or Advisory Role: AstraZeneca, MedImmune, Bayer, Seattle Genetics, Clovis Oncology, Janssen Oncology, Dendreon
Speakers' Bureau: Sanofi, Merck Sharp & Dohme, AstraZeneca
Research Funding: AstraZeneca (Inst), MedImmune (Inst)
Josep M. Piulats
Consulting or Advisory Role: Janssen Oncology, Astellas Pharma, VCN Biosciences, Clovis Oncology, Genentech, Bristol Myers Squibb, Merck Sharp & Dohme, BeiGene
Research Funding: Bristol Myers Squibb, AstraZeneca, MedImmune, Merck Sharp & Dohme, Pfizer, EMD Serono, Incyte, Janssen Oncology
Travel, Accommodations, Expenses: Janssen Oncology, Roche, Bristol Myers Squibb
Charles J. Ryan
Honoraria: Janssen Oncology, Bayer
Consulting or Advisory Role: Bayer, Dendreon, AAA, Myovant Sciences, Clovis Oncology (Inst), Roivant
Research Funding: Clovis Oncology (Inst), Sanofi (Inst)
Axel S. Merseburger
Honoraria: Janssen-Cilag, Astellas Pharma, Ipsen, Roche, Bristol Myers Squibb, Eisai, Takeda, Pfizer, Novartis
Consulting or Advisory Role: MSD Oncology, Bristol Myers Squibb, Janssen-Cilag, Astellas Pharma, Ipsen, Clovis Oncology
Speakers' Bureau: Ipsen
Research Funding: Novartis (Inst), AstraZeneca (Inst), Janssen-Cilag (Inst), Bristol Myers Squibb (Inst), Clovis Oncology (Inst)
Travel, Accommodations, Expenses: Janssen-Cilag, Astellas Pharma, Ipsen
Gedske Daugaard
Consulting or Advisory Role: Sanofi, Astellas Pharma, Bayer
Travel, Accommodations, Expenses: Astellas Pharma
Axel Heidenreich
Honoraria: Amgen, Astellas Pharma, Bayer, Ferring, Ipsen, Janssen-Cilag, Sanofi, Takeda
Consulting or Advisory Role: Astellas Pharma, Bayer, Janssen-Cilag, Clovis Oncology
Speakers' Bureau: Amgen, Astellas Pharma, Bayer, Ipsen, Johnson & Johnson, Sanofi, Takeda, Pfizer
Research Funding: Astellas Pharma, Bayer, Sanofi
Karim Fizazi
Honoraria: Janssen, Sanofi, Astellas Pharma, Bayer
Consulting or Advisory Role: Janssen Oncology (Inst), Bayer, Astellas Pharma, Sanofi (Inst), Orion Pharma, Curevac, AstraZeneca (Inst), ESSA (Inst), Amgen (Inst)
Travel, Accommodations, Expenses: Janssen, MSD Oncology
Celestia S. Higano
Honoraria: Astellas Pharma
Consulting or Advisory Role: Bayer, Ferring, Clovis Oncology, Blue Earth Diagnostics, Janssen, Hinova, Pfizer, AstraZeneca, Carrick Therapeutics, Novartis, Merck Sharp & Dohme
Research Funding: Aragon Pharmaceuticals (Inst), AstraZeneca (Inst), Medivation (Inst), Emergent BioSolutions (Inst), Bayer (Inst), Pfizer (Inst), Roche (Inst), Astellas Pharma (Inst), Clovis Oncology (Inst), Ferring Pharmaceuticals (Inst), eFFECTOR Therapeutics (Inst)
Travel, Accommodations, Expenses: Bayer, Clovis Oncology, Blue Earth Diagnostics, Ferring, Pfizer, Hinova Pharmaceuticals, Janssen Oncology, Novartis, Merck Sharp & Dohme, Carrick Therapeutics
Laurence E. Krieger
Honoraria: Roche, Janssen-Cilag, Ipsen, MSD Oncology, Ferring, Astellas Pharma, Pfizer
Consulting or Advisory Role: Roche, Sanofi, Novartis, Ipsen, Astellas Pharma, Bristol Myers Squibb, Merck Sharp & Dohme, Bayer, Clovis Oncology, Pfizer, Amgen, AstraZeneca, Ferring
Speakers' Bureau: Roche, Janssen-Cilag, Merck Sharp & Dohme, AbbVie, Janssen
Research Funding: Janssen-Cilag (Inst), Roche (Inst), Bristol Myers Squibb (Inst), Astellas Pharma (Inst)
Expert Testimony: Novartis, Astellas Pharma, Ipsen
Travel, Accommodations, Expenses: Novartis, Bayer, Ipsen, Astellas Pharma, AstraZeneca, Janssen-Cilag, MSD Oncology, Pfizer, Janssen
Cora N. Sternberg
Consulting or Advisory Role: Bayer, MSD Oncology, Pfizer, Roche, Incyte, AstraZeneca, Merck, Medscape, UroToday, Astellas Pharma, Genentech, Sanofi
Simon P. Watkins
Employment: Clovis Oncology
Stock and Other Ownership Interests: Clovis Oncology, United Health Group
Darrin Despain
Employment: Clovis Oncology
Stock and Other Ownership Interests: Clovis Oncology
Andrew D. Simmons
Employment: Clovis Oncology
Stock and Other Ownership Interests: Clovis Oncology
Consulting or Advisory Role: Redwood Bioscience
Andrea Loehr
Employment: Clovis Oncology
Stock and Other Ownership Interests: Clovis Oncology
Melanie Dowson
Employment: Clovis Oncology
Stock and Other Ownership Interests: Clovis Oncology
Travel, Accommodations, Expenses: Clovis Oncology
Tony Golsorkhi
Employment: Clovis Oncology
Stock and Other Ownership Interests: Clovis Oncology
Simon Chowdhury
Honoraria: Clovis Oncology, Novartis
Consulting or Advisory Role: Clovis Oncology, Astellas Pharma, Bayer, Pfizer, Janssen-Cilag, BeiGene, Novartis
Speakers' Bureau: Pfizer, Janssen-Cilag
Research Funding: Sanofi (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.National Comprehensive Cancer Network NCCN Clinical Practice Guidelines in Oncology: Prostate cancer (version 2.2020) https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf
- 2.Parker C, Gillessen S, Heidenreich A, et al. Cancer of the prostate: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26:v69–v77. doi: 10.1093/annonc/mdv222. [DOI] [PubMed] [Google Scholar]
- 3.European Association of Urology Guidelines: Prostate cancer. http://uroweb.org/guideline/prostate-cancer/
- 4.Gillessen S, Attard G, Beer TM, et al. Management of patients with advanced prostate cancer: Report of the Advanced Prostate Cancer Consensus Conference 2019. Eur Urol. 2020;77:508–547. doi: 10.1016/j.eururo.2020.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Abida W, Armenia J, Gopalan A, et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol. 2017;2017:1–16. doi: 10.1200/PO.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castro E, Goh C, Olmos D, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31:1748–1757. doi: 10.1200/JCO.2012.43.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bancroft EK, Page EC, Castro E, et al. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: Results from the initial screening round of the IMPACT study. Eur Urol. 2014;66:489–499. doi: 10.1016/j.eururo.2014.01.003. [Erratum: Eur Urol 67:e126, 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drew Y, Mulligan EA, Vong WT, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst. 2011;103:334–346. doi: 10.1093/jnci/djq509. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen M, Simmons AD, Harding TC. Preclinical assessment of the PARP inhibitor rucaparib in homologous recombination deficient prostate cancer models. Cancer Res. 2017;77(abstr 2476) [Google Scholar]
- 10.Robillard L, Nguyen M, Harding TC, et al. In vitro and in vivo assessment of the mechanism of action of the PARP inhibitor rucaparib. Cancer Res. 2017;77(abstr 2475) [Google Scholar]
- 11.Abida W, Campbell D, Patnaik A, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: Analysis from the phase 2 TRITON2 study. Clin Cancer Res. 2020;26:2487–2496. doi: 10.1158/1078-0432.CCR-20-0394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clark TA, Chung JH, Kennedy M, et al. Analytical validation of a hybrid capture–based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J Mol Diagn. 2018;20:686–702. doi: 10.1016/j.jmoldx.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford B, Adams SB, Sittler T, et al. Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast Cancer Res Treat. 2017;163:383–390. doi: 10.1007/s10549-017-4181-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neben CL, Zimmer AD, Stedden W, et al. Multi-gene panel testing of 23,179 individuals for hereditary cancer risk identifies pathogenic variant carriers missed by current genetic testing guidelines. J Mol Diagn. 2019;21:646–657. doi: 10.1016/j.jmoldx.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Brown EG, Wood L, Wood S. The medical dictionary for regulatory activities (MedDRA) Drug Saf. 1999;20:109–117. doi: 10.2165/00002018-199920020-00002. [DOI] [PubMed] [Google Scholar]
- 17.National Cancer Institute NCI term browser: CTCAE. https://nciterms.nci.nih.gov/ncitbrowser/pages/multiple_search.jsf?nav_type=terminologies
- 18.Scher HI, Morris MJ, Stadler WM, et al. Trial design and objectives for castration-resistant prostate cancer: Updated recommendations from the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016;34:1402–1418. doi: 10.1200/JCO.2015.64.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clovis Oncology . Rubraca (rucaparib) tablets [prescribing information] Boulder, CO: Clovis Oncology; 2020. [Google Scholar]
- 20.Loriot Y, Bianchini D, Ileana E, et al. Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100) Ann Oncol. 2013;24:1807–1812. doi: 10.1093/annonc/mdt136. [DOI] [PubMed] [Google Scholar]
- 21.Pezaro CJ, Omlin AG, Altavilla A, et al. Activity of cabazitaxel in castration-resistant prostate cancer progressing after docetaxel and next-generation endocrine agents. Eur Urol. 2014;66:459–465. doi: 10.1016/j.eururo.2013.11.044. [DOI] [PubMed] [Google Scholar]
- 22.Al Nakouzi N, Le Moulec S, Albigès L, et al. Cabazitaxel remains active in patients progressing after docetaxel followed by novel androgen receptor pathway targeted therapies. Eur Urol. 2015;68:228–235. doi: 10.1016/j.eururo.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Nuhn P, De Bono JS, Fizazi K, et al. Update on systemic prostate cancer therapies: Management of metastatic castration-resistant prostate cancer in the era of precision oncology. Eur Urol. 2019;75:88–99. doi: 10.1016/j.eururo.2018.03.028. [DOI] [PubMed] [Google Scholar]
- 24.Halabi S, Lin C-Y, Kelly WK, et al. Updated prognostic model for predicting overall survival in first-line chemotherapy for patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2014;32:671–677. doi: 10.1200/JCO.2013.52.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mateo J, Porta N, Bianchini D, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:162–174. doi: 10.1016/S1470-2045(19)30684-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith MR, Sandhu SK, Kelly WK, et al. Pre-specified interim analysis of GALAHAD: A phase II study of niraparib in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) and biallelic DNA-repair gene defects (DRD) Ann Oncol. 2019;30(abstr 3405):v851–v934. [Google Scholar]
- 28.De Bono JS, Mehra N, Higano CS, et al. TALAPRO-1: A phase II study of talazoparib (TALA) in men with DNA damage repair mutations (DDRmut) and metastatic castration-resistant prostate cancer (mCRPC)—First interim analysis (IA) J Clin Oncol. 2020;38(suppl 6; abstr 119) [Google Scholar]
- 29.de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382:2091–2102. doi: 10.1056/NEJMoa1911440. [DOI] [PubMed] [Google Scholar]
- 30.Clovis Oncology . Rubraca (rucaparib) tablets [summary of product characteristics] Swords, Ireland: Clovis Oncology; 2019. [Google Scholar]
- 31.Kristeleit R, Shapiro GI, Burris HA, et al. A phase I-II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin Cancer Res. 2017;23:4095–4106. doi: 10.1158/1078-0432.CCR-16-2796. [DOI] [PubMed] [Google Scholar]
- 32.McCormick A, Swaisland H. In vitro assessment of the roles of drug transporters in the disposition and drug-drug interaction potential of olaparib. Xenobiotica. 2017;47:903–915. doi: 10.1080/00498254.2016.1241449. [DOI] [PubMed] [Google Scholar]
- 33.Kikuchi R, Lao Y, Bow DA, et al. Prediction of clinical drug-drug interactions of veliparib (ABT-888) with human renal transporters (OAT1, OAT3, OCT2, MATE1, and MATE2K) J Pharm Sci. 2013;102:4426–4432. doi: 10.1002/jps.23737. [DOI] [PubMed] [Google Scholar]
- 34.AstraZeneca . Lynparza (olaparib) tablets [prescribing information] Wilmington, DE: AstraZeneca; 2020. [Google Scholar]
- 35.Zibetti Dal Molin G, Westin SN, Msaouel P, et al. Discrepancy in calculated and measured glomerular filtration rates in patients treated with PARP inhibitors. Int J Gynecol Cancer. 2020;30:89–93. doi: 10.1136/ijgc-2019-000714. [DOI] [PubMed] [Google Scholar]
- 36.Tang PA, Pond GR, Chen EX. Influence of an independent review committee on assessment of response rate and progression-free survival in phase III clinical trials. Ann Oncol. 2010;21:19–26. doi: 10.1093/annonc/mdp478. [DOI] [PubMed] [Google Scholar]