

Abstract
Bone homeostasis is securely controlled by the dynamic well‐balanced actions among osteoclasts, osteoblasts and osteocytes. Osteoclasts are large multinucleated cells that degrade bone matrix and involve in the bone remodelling in conjunction with other bone cells, osteoblasts and osteocytes, the completely matured form of osteoblasts. Disruption of this controlling balance among these cells or any disparity in bone remodelling caused by a higher rate of resorption by osteoclasts over construction of bone by osteoblasts results in a reduction of bone matrix including bone mineral density (BMD) and bone marrow cells (BMCs). The dominating effect of osteoclasts results in advanced risk of bone crack and joint destruction in several diseases including osteoporosis and rheumatoid arthritis (RA). However, the boosted osteoblastic activity produces osteosclerotic phenotype and weakened its action primes to osteomalacia or rickets. On the other hand, senescent osteocytes predominately progress the senescence associated secretory phenotype (SASP) and may contribute to age related bone loss. Here, we discuss an advanced level work on newly identified cellular mechanisms controlling the remodelling of bone and crosstalk among bone cells as these relate to the therapeutic targeting of the skeleton.
Keywords: aging, bone remodelling, cellular senescence, osteocytes, osteoporosis, RANKL‐RANK pathway
1. INTRODUCTION
Bone homeostasis in the adult skeleton is complex processes. Human skeletal tissue is a constant state of remodelling. The three main bone cells involve in this remodelling process‐osteoblasts, osteoclasts and osteocytes via regulation of molecular signalling pathways. 1 In bone remodelling, the discrete zones of bone are resorbed by osteoclasts and substituted by fresh bone by osteoblasts, allowing for repair of bone micro‐injury and adapting of bone niche for control of mechanical strengths. Osteoblast cells are energetic in protein synthesis and matrix secretion to preserve and form new healthy bones. Following mineralization of bone matrix, fully differentiated and matured osteoblasts become osteocytes and are implanted in the bone matrix. 2 , 3 During bone remodelling process, mechanosensory cells, osteocytes act as bone orchestrators. This remodelling process is regulated by several local (e.g., growth factors, cytokines, chemokines) and systemic (e.g., oestrogens) factors that all together subscribe for bone homeostasis. 4 , 5 In bone modelling process, i.e., during bone development and bone resorption stages, osteocytes act autonomously to fine‐tune bone structure. Interestingly, in bone remodelling process, these cells act recycler to restore and keep skeletal health. 6 After osteoclast‐mediated bone resorption sequence, the eroded surface of trabecular bone is engaged by osteoblasts that make bone matrix and then undertake mineralization. 7 , 8 Under normal physiological conditions of bone homeostasis, osteoclastic action is closely associated with osteoblastic action in such a way that the eroded bone is completely exchanged by fresh bone. Definitively, fluctuating this homeostatic equilibrium in favour of excessive osteoclast activity turns to bone pathological conditions such as osteoporosis, Paget's disease, rheumatoid arthritis (RA), osteoarthritis and autoimmune arthritis. 7 However, imperfect osteoclast differentiation and/or function may cause osteopetrotic phenomena. On the other hand, the enhanced osteoblastic activity occurs osteosclerotic phenotype and diminished its action leads to osteomalacia or rickets. Thus, understanding the mechanism of bone homeostasis is important for the understanding of disease mechanisms and development of new therapeutics against bone diseases.
2. REGULATION OF BONE HOMEOSTASIS
2.1. Regulation of bone homeostasis by osteoclasts
The multinucleated giant cells, osteoclasts are differentiated from osteoclast precursor cells derived from haematopoietic stem cell (HSC) niche‐monocyte/ macrophage lineage cells in presence of two essential factors: the receptor activator of nuclear factor‐κB (NF‐κB) ligand (RANKL), a tumor necrosis factor (TNF) family cytokine and the macrophage/ monocyte colony‐stimulating factor (M‐CSF) (Figure 1). M‐CSF and RANKL are predominantly expressed in osteoblasts/stromal cells and essentially regulate osteoclast differentiation and function. 9 The M‐CSF signalling is important for osteoclast precursor cell growth and survival. 10 M‐CSF binds to its corresponding receptor, colony stimulating factor 1 receptor (c‐Fms), present on osteoclast precursors, providing the signals for supporting the survival of these progenitors. Binding of M‐CSF to its receptor c‐Fms recruits adapter proteins and cytosolic kinases, thereby activating a variety of intracellular signals. M‐CSF also induces the receptor RANK for RANKL and other RANK/NF‐κB (nuclear factor kappa‐light‐chain‐enhancer of activated B cells) pathway components such as PI3‐kinase, the indispensable regulators for osteoclastogenesis. 11 The RANK adopts a trimetric conformation by interacting with its extracellular signal factor RANKL. The intracellular domain of the RANK trimer is supposed to absence of signalling domains but instead, for signal transduction, recruits adaptor molecules such as TNF receptor‐associated factors (TRAFs) and Grb‐2‐associated binder (Gab) 2. 12 , 13 , 14 Of the several TRAF proteins that have been associated with RANKL, including TRAF1/2/3/5/6, TRAF6 is indeed an essential adaptor required for RANK‐associated signalling for osteoclastogenesis (Figure 2). Thus, intracellular RANK signalling by its interaction with RANKL induces recruitment and activation of its adaptor TRAF6, leading to the activation of multiple downstream signalling cascades such as mitogen activated protein kinases (MAPKs) including ERK, p38, and JNK (c‐Jun N‐terminal kinases) as well as TAK1 activate inhibitory κB (IκB) kinases (IKKs) NF‐κB, Rous sarcoma oncogene (Src) and AKT (also known as protein kinase B) [11]. In addition to these two positive regulators of osteoclast differentiation, osteoblasts also express a negative regulator of osteoclast differentiation, osteoprotegerin (OPG) a secreted member of the TNF receptor superfamily. 15 OPG inhibits osteoclastogenesis, osteoclast maturation and bone resorption by acting as a decoy receptor for both the RANK ligand (RANKL) in the RANK/RANKL/OPG axis 15 , 16 and tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL). 17 , 18 Overexpression of OPG causes osteoclast‐deficient osteopetrosis, while deletion of OPG leads to osteoporosis due to increased OC number and activity. 18 , 19 , 20 TRAIL stimulates osteoclastogenesis by interacting with specific TRAIL receptors on osteoclast precursor cell surfaces, activating TRAF6 signalling, inducing receptor activator of nuclear factor‐κB (NF‐κB) signalling and upregulating nuclear factor of activated T cells cytoplasmic 1 (NFATc1) expression. 16
Figure 1.
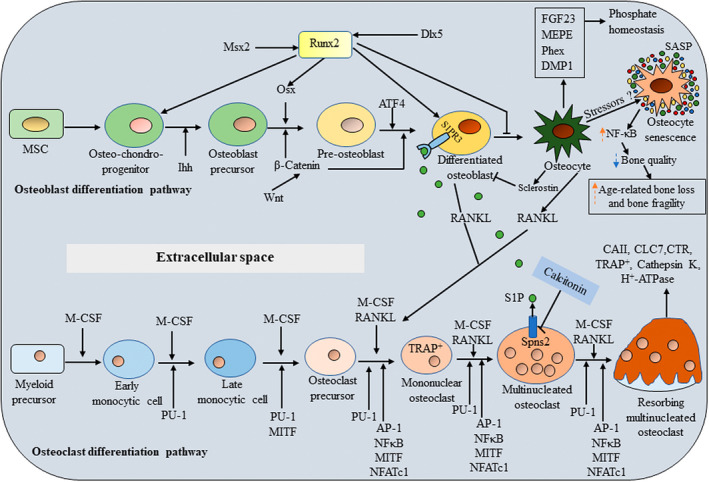
The regulation of bone homeostasis by cellular signalling molecules. Osteoblasts and chondrocytes originate from common MSC lineage precursors. Runx2, Osx, ATF4 are considered master genes for osteoblast differentiation. Ihh and Wnt/β‐catenin are also supporting signalling molecules in osteoblastogenesis. In response to various stressors such as oxidative stress, genomic instability and telomere shortening, osteocytes can be converted into osteocyte senescent cells which have SASP factor. The SASP upregulates NF‐κB and increases pro‐inflammatory factors such as IL‐1α. As a result, senescent cells and the SASP contribute to age‐related frailty and a number of age‐associated chronic diseases including osteoporosis. During osteoclast differentiation, osteoclasts derive from by the fusion of mononuclear progenitors of the monocyte/macrophage family, and osteocytes are non‐proliferative differentiated cells of the osteoblast lineage. M‐CSF and RANKL are essential external stimuli for osteoclastogenesis. PU.1, MITF, NF‐κB, AP‐1 and NFATc1 are essential for differentiation of functional osteoclasts. CT negatively regulates the osteoclast expression of spinster 2 (Spns2), which encodes a transporter for the signalling lipid sphingosine 1‐phosphate (S1P). CT suppresses Spns2 expression and reduces S1P release from osteoclasts. Sphingosine, derived from the membrane lipid sphingomyelin through intermediate ceramide, is phosphorylated to S1P by sphingosine kinases (Sphk1 and 2). S1P is either degraded by sphingosine lyase or secreted through obligatory interaction with Spns2 which is reduced by calcitonin. S1P acts through receptor S1PR3 in the osteoblast to increase osteoblast differentiation and bone formation. The osteocyte (stellate shaped blue cells on the right) expresses several membrane receptors including receptors including PTH and others and controls both osteoblast and osteoclast functions though sclerostin and RANKL. Osteocytes also secrete factors involved in phosphate homeostasis.  indicates upregulation and
indicates upregulation and  indicates down‐regulation.
indicates down‐regulation.
Figure 2.
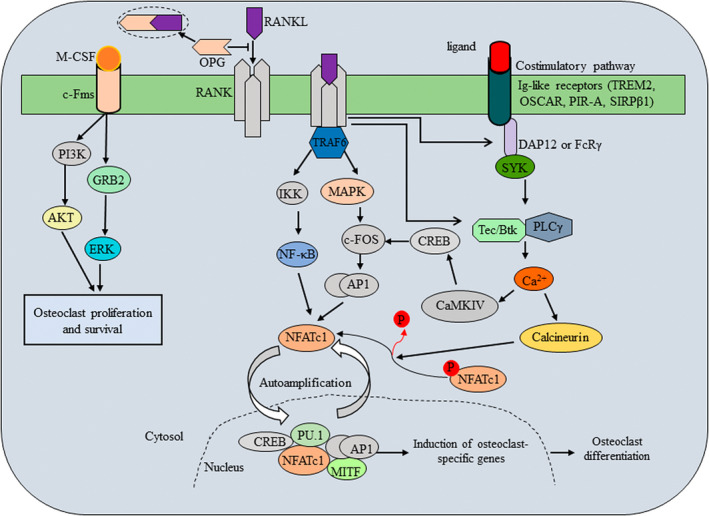
Major signaling pathways during the process of osteoclast differentiation. RANK and OPG are TNFR receptor‐related proteins, and RANKL is a TNF‐related cytokine that interacts specifically with either RANK or OPG. RANKL and M‐CSF regulate osteoclastogenesis through M‐CSF‐cFms, RANKL‐RANK as well as Ig‐like receptors associated with ITAM‐harbouring adaptor molecules such as DAP12 and Fc‐receptor common γ‐subunit (FcRγ). RANKL and its receptor RANK transduce a signal via the adaptor molecule TRAF6. TRAF6 recruits TAB2 and TAK1, which in turn activates the NF‐κB pathway and MAPK pathway. NF‐κB persuades c‐Fos expression via IKKs. Activation of MAPK pathway results in the activation of the Jun proteins. The c‐Fos and Jun proteins associate to form the complex AP‐1. The expression of the master regulator of osteoclastogenesis, NFATc1, is driven by AP‐1, NF‐κB and NFATc1 itself. The activation of NFATc1 is regulated by a co‐stimulatory signal pathway. FcRγ, DAP12 and their associating molecules activate Syk, which forms a complex with Btk/Tec and BLNK/SLP76. This complex further activates PLCγ, resulting in calcium signaling. The calcium signaling activates calcineurin, which dephosphorylates NFATc1, promoting its entry into the nucleus. Activated NFATc1 promotes its own expression making an autoamplification loop. The calcium signaling also induces c‐Fos expression via CAMKIV and CREB. NFATc1, together with other factors including PU.1 and MITF, promotes the expression of osteoclastogenic genes.
Based on osteoclast gene transcription, NF‐κB and NFATc1 are the key transcription factors. NF‐κB is inactive in the cytosol in the steady state and translocates to the nucleus upon activation, which is dependent on the classical or the alternative pathways. 21 In the classical pathway, the Iκ kinase (IKK) complex phosphorylates IκBs, the inactivators of NF‐κB. The phosphorylation of the IκB molecules leads to their ubiquitination and subsequent degradation, freeing NF‐κB. Activated NF‐κB dimers induce c‐Fos and NFATc1 expressions. 22 Since NF‐κB and MAPK signals are not RANKL‐specific pathways and RANKL almost exclusively induce osteoclastogenesis, there are some osteoclastogenesis specific transcription factors indicated to exist. In search for such factors, it has been reported that NFATc1 plays an essential and sufficient role in osteoclastogenesis. 23 In these cases, RANKL induces and activates NFATc1 through calcium signalling. Calcium oscillation activates calcineurin, which dephosphorylates NFATc1, enabling it to enter the nucleus, so as its expression becomes amplified. Calcium signalling is also reported inducing the c‐Fos via Ca2+/calmodulin‐dependent kinase (CAMK) IV‐cAMP response element‐binding protein (CREB) pathway. 24 Activation of downstream of NF‐κB and MAPK signalling, c‐Fos and Jun proteins are dimerized to form activator protein‐1 (AP‐1). NF‐κB, AP‐1 and other transcription factors are recruited to the promoter region of the NFATc1 after RANKL stimulation, together inducing NFATc1. 25 , 26 , 27 The expression of this transcription factor is greatly upregulated after RANKL stimulation by autoamplification machinery. 21 , 23 At the last stage of osteoclast differentiation, NFATc1 cooperates with Fos and Jun proteins to induce osteoclast‐specific genes such as DC‐STAMP (dendritic cell‐specific transmembrane protein); OC‐STAMP (osteoclast stimulatory transmembrane protein); TRAP (tartrate‐resistant acid phosphatase), calcitonin receptor, cathepsin K and integrin β3 integrin gene 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 together with other transcription factors such as PU.1 and microphthalmia transcription factor (MITF). 26 , 27 , 28 , 29 , 30
The RANKL signalling pathway is also coordinated co‑stimulatory signalling and calcium signalling in osteoclastogenesis. Immunoglobulin (Ig)‐like receptors are a group of proteins consisted of an extracellular region, which has an Ig‐like domain and an intracellular region, which associates with ITAM (immunoreceptor tyrosine‐based activation motif)‐ or ITIM (immunoreceptor tyrosine‐based inhibitory motif)‐harbouring adaptor proteins. (Ig)‐like receptors and their adaptors are expressed on osteoclasts as well as immune cells. FcRγ (Fc receptor common γ subunit) and DAP (DNAX‐activating protein) 12 are ITAM‐harbouring adaptors expressed in osteoclast cells that have been found to have an important role in osteoclastogenesis through NFATc1 induction. 35 PIR‐A (paired Ig‐like receptor‐A), OSCAR (osteoclast‐associated receptor) and FcγRs (Fcγ receptors) are described to associate with FcRγ; whereas triggering receptor expressed in myeloid cells (TREM)‐2, signal‐regulatory protein (SIRP) β1, and myeloid DAP12‐associating lectin (MDL)‐1 associate with DAP12. 35 , 36 , 37 , 38 Lastly, the integrated activation of RANK‐RANKL and Ig‐like receptor signalling pathways in osteoclast differentiation are regulated by the master transcription factor, NFATc1. 21 The activated NFATc1 expression upregulates cathepsin K, MMP9, the H+‐ATPase subunits and carbonic anhydrase II expressions, the vital molecules for the bone resorbing action of osteoclasts [9]. During the advanced stages of osteoclast differentiation, the several useful markers such as αvβ3 integrin and TRAP are also upregulated in associated with osteoclast maturation. 39 These markers have been identified in osteoclasts and their precursors for facilitating the focal bone erosion in osteoporosis and rheumatoid arthritis. However, it has also been found that TRAP activity of macrophages has not substantial capability to resorb mineralized bone matrix. 40
Calcitonin (CT), an endogenous calcium regulatory hormone, directly inhibits bone‐resorbing activity of mature osteoclasts by binding the CT receptor (CTR). 41 It has been found that CT stops vesicular trafficking to and from the ruffled border and that it alters the cytoskeleton of the cells. 42 CT has also been demonstrated to interfere with osteoclast differentiation from precursor cells and fusion of mononucleated precursors to form multinucleated osteoclasts in bone marrow cultures. 41 , 43 Indeed, the presence of the CTR is thought to be a later marker of osteoclast differentiation, and its presence is often taken as marker of a mature bone‐resorbing osteoclast. 44 , 45 As a pharmacologically inhibitor of bone resorption, 46 CT is widely used therapeutic to treat metabolic bone disorders such as osteoporosis Paget's disease and malignancy‐associated hypercalcemia. 41 , 43 , 47 However, it is well‐recognized that continuous CT treatment eventually causes a loss of its suppressive effects on osteoclastic bone resorption. 41 This process is referred to as the ‘escape’ phenomenon. The mechanism of this escape phenomenon has been studied by using mouse and rat osteoclasts and the results suggest that escape or desensitization to CT is closely associated with the down‐regulation of the CT receptor (CTR) 41 by its internalization as well as by reduced its expression of cell surface through inhibition of de novo CTR synthesis. 43 The intracellular signalling responsible for CT‐induced CTR down‐regulation in mouse osteoclasts is activation of the protein kinase A pathway through cell surface CTR. 43 However, the physiological role of CT is uncertain as mice lacking CT and CT gene‐related peptide (CGRP) have a high bone mass phenotype due to an increase in bone formation parameters. 48 This is surprising as the CTR is expressed on osteoclasts but not on osteoblasts. 48 To clarify the cellular and molecular mechanisms of CT action in bone remodelling, Keller and colleagues identified a novel mediator of the inhibitory effect of CT on bone formation. 49 CT negatively regulates bone formation by inhibiting the release of the anabolic bone factor sphingosine‐1‐phosphate (S1P) 50 a lysophospholipid from osteoclasts. 51 CT decreases expression of Spns2 (which encodes spinster 2, a transmembrane exporter protein for S1P) and reduce S1P levels in wild‐type but not in CTR knock‐out osteoclasts. S1P acts through its receptor S1P receptor 3 (S1PR3) in the osteoblast to increase osteoblast differentiation and bone formation. 49 , 51 , 52 Thus, Spns2 acts as an osteoclast‐secreted coupling factor that stimulates bone formation 51 and is found to be negatively regulated by CT in wild‐type osteoclasts.
2.2. Regulation of bone homeostasis by osteoblasts
Adult bone marrow is known to contain mesenchymal lineage progenitors which are capable to differentiate into a range of cell lineages that form different types of tissues such as bone, cartilage and muscle. 53 , 54 Bone formation occurs through two developmental processes‐intramembranous and endochondral ossification. Intramembranous ossification which mainly forms craniofacial bone is mediated by the differentiation of the condensed mesenchymal stem cells (MSCs) into osteoblasts, the fundamental players for bone formation. In endochondral ossification, MSC condensation differentiates into chondrocytes, forming a cartilage template that is later replaced by bone. 55 There are three major stages of osteoblastogenesis: proliferation, matrix maturation and mineralization. Commitment of MSCs to tissue‐specific cell types is orchestrated by several transcriptional regulators that serve as “master switches.” In skeletogenesis, the key transcription factors‐ Runx2 (Runt‐related transcription factor 2), osterix (Osx), ATF4 (activating transcription factor 4) and Dlx5 are known to play important roles in the cell‐fate decision process by which MSCs become osteoblasts through activation of cell type‐specific genes. Runt‐domain containing Runx2, a central regulator of bone formation is expressed in the condensed MSCs. Runx2 fulfils its role as a master regulatory switch through segregation of Runx2+cells from osteochondro‐progenitors to form precursor osteoblasts. 56 Runx2 is also reported to have two main functions‐induction of osteogenesis and suppression of adipocyte differentiation. 2 Although Runx2 is crucial for osteoblast differentiation, multiple genes regulate Runx2 activity and the effectiveness of Runx2 in stimulating osteoblast formation. Osx, a zinc finger‐containing transcription factor is one of the regulators of this differentiation program and acts as a down‐stream of Runx2. 2 Osx also involves in osteogenesis, by promoting primary bone matrix formation and the commitment of MSCs to the osteogenic lineage. Thus, it is evident that Runx2 is essential to main the prechondrogenic MSCs segregate into the osteoblast precursors, whereas Osx is subsequently obligatory to complete the osteoblast differentiation pathway (Figure 1). Thus, Runx2 is called the master regulator/platform protein of osteoblast differentiation and is indispensable to bone formation.
A wide range of cytokines modulate osteoblast differentiation, including bone matrix‐derived TGF‐β (transforming growth factor beta), bone morphogenic protein 2 (BMP‐2), BMP‐4, and BMP‐7 and their inhibitors noggin, chordin and gremlin. BMP does not directly stimulate the Runx2 expression in MSCs 57 but it helps the expression of Dlx5 (distal‐less homeobox 5) in osteoblasts 58 , 59 and Dlx5 then persuades expression of Runx2 in osteoprogenitor cells. Dlx5 has been found to augment Runx2 promoter activity. 58 , 60 Furthermore, there is cumulative evidence that Dlx5 endorses activation of osteocalcin (OCN) by establishing heterodimers with Runx2 upstream protein, Msx2 (Msh homeobox 2), a homeobox‐containing transcription factor expressed in osteoblasts during development. 61 , 62 ATF4 is identified by Yang et al. as a critical substrate of ribosomal protein S6 kinase polypeptide 3 (RSK2) that is obligatory for the timely commencement of osteoblast differentiation, for terminal differentiation of osteoblasts, for bone sialoprotein (BSP), and for OCN expression. 52 Additionally, RSK2 and ATF4 post‐transcriptionally control the synthesis of type I collagen, the main constituent of the bone matrix. The study indicates that ATF4 is a critical regulator of osteoblast differentiation and function, and suggests that absence of ATF4 phosphorylation by RSK2 may part to the bone phenotype of Coffin‐Lowry syndrome. 63
These transcription factors regulate multiple signalling pathways involved in MSC differentiation, including Ihh (Indian hedgehog), AKT, BMP, IGF and WNT/β‐catenin. 64 Ihh and WNT/β‐catenin are key signalling molecules in osteoblastogenesis. Ihh acts in differentiation of osteoblast progenitors into Runx2+ osteoblast precursors. WNT/β‐catenin signalling acts later in the differentiation pathway to Osx+ osteoblast precursors and then to bone‐secreting osteoblasts (Figure 1). 65 BMP2 and IGF‐1 are shown to work synergistically in the Osx upregulation. 66 IGF‐1 is shown to promote early stages of differentiation, acting through the AKT/MAP kinase pathway, with its concentration observed to be very low in later osteogenic phenotypes. 65 In contrast, BMP2 is observed to promote osteogenesis in a Runx2‐dependent manner, with its expression not requiring all three MAPK components (ERK, p38, and JNK), relying only on p38 and JNK. Moreover, the combined expression of BMP2 and IGF‐1 further promotes osteoblast differentiation, working through the MAPK and PKD (protein kinase D) pathways. 65 , 66 , 67 In addition, alkaline phosphatase (ALP), bone sialoprotein (BSP), and collagen type 1 alpha 1 (Col1a1) are early markers of osteoblast differentiation, while parathyroid hormone (PTH)/PTH‐related peptide (PTH/PTHrP) receptor and OCN appear late, concomitantly with mineralization. Osteopontin (OPN) peaks twice, during proliferation and then again in the later stages of differentiation. 68 In intermediate stage of osteoblast development, preosteoblasts express STRO‐1, ALP and type I collagen, and is committed to the osteoblast lineage with extensive replicative capacity, but no self‐renewal capacity. 69 However, in the final stage of osteoblast development, the mature osteoblasts express ALP, OPN, BSP, and OCN, and lies adjacent to newly synthesized osteoid. This stage, which is responsible for the laying down of bone, has limited replicative potential. 70 The fibroblast growth factors (FGFs) and BMP signal synergistically promote osteoblast differentiation. BMP signalling is essential for FGF‐induced osteoblast differentiation 71 and the osteogenic effect of BMP2 is also repressed in the absence of FGF2. 72 FGF2 is responsible for BMP‐induced nuclei translocation and accumulation of Runx2 and P‐Smad (mothers against decapentaplegic homolog)1/5/8. 73
2.3. Regulation of bone homeostasis by osteocytes
Last decade has viewed a renewed curiosity in the role and biology of matrix‐embedded osteocytes and these cells have appeared as master regulators of bone homeostasis. At present, osteocytes are known to directly involve bone remodelling through the regulation of sclerostin (the product of the SOST gene), a WNT‐inhibitor signalling pathway that suppresses bone formation via osteoblasts 74 and RANKL signalling pathways a cytokine required for osteoclastogenesis (Figure 1).
Canonical WNT/β‐catenin signalling is critical to regulate bone‐mass homeostasis 75 , 76 and occurs in an autocrine or paracrine fashion. 77 Canonical WNT signalling is triggered by binding of extracellular WNT ligand to the low‐density lipoprotein receptor‐related protein 5/6 (LRP5/6), the transmembrane proteins, established as WNT co‐receptors and a frizzled (FZD) family cell‐surface receptor to promote formation of a ternary complex, leading to phosphorylation of the cytoplasmic domain of LRP5/6 (called activated ‘WNT on’ state). 77 The intracellular abundance and location of β‐catenin provides a central switch in the canonical WNT signalling pathway. The LRP5/6‐WNT‐FZD ternary complex leads to translocate the destruction complex to the cell membrane, phosphorylates the intracellular region of LRP5/6, recruits AXIN protein which disrupts destruction complex activity and allows free β‐catenin to accumulate in the cytoplasm. Non‐phosphorylated β‐catenin translocates from the cytoplasm to the nucleus to regulate gene expression. 77
Osteocytes are key players in the regulation of the canonical WNT signalling pathway as producers and targets of WNT ligands and as secretors of molecules that modulate WNT actions. 75 , 76 A potent antagonist of WNT signalling secreted by osteocytes is sclerostin, primarily expressed by mature osteocytes but not by early osteocytes or osteoblasts. 78 In the WNT off (sclerostin inhibited) state, sclerostin interacts with the WNT co‐receptors LRP5/6 79 , 80 (via the first two YWTD‐EGF repeat domains) antagonizing downstream signalling. 81 , 82 LRP5 and LRP6 are type I transmembrane receptors (C‐terminus in cytosol) and structurally related proteins sharing around 71% homology at the nucleotide level. 83 The structural organization of LRP4, another member of the LRP family proteins is markedly different from LRP5/6. 83 , 84 LRP4 is a type II transmembrane receptor (N‐terminus in cytosol) and it belongs to the LRP subfamily III along with LRP5 and LRP6. 83 , 85 Sclerostin also interacts with LRP4 (via the extracellular domain) which acts as a chaperone and is required for the inhibitory action of sclerostin on WNT/βcatenin signalling. 82 , 86 , 87 In such ways, sclerostin prevents formation of the LRP4/5/6‐WNT‐FZD ternary complex and hence inhibits canonical WNT signalling. In the WNT off state (basal/inhibited), cytosolic β‐catenin is phosphorylated and targeted for ubiquitination by the destruction complex and degraded in the proteasome.
It has been found that calcitonin treatment significantly increases sclerostin expression in bone as well as down‐regulates two other osteocyte gene products, DMP1 (dentin matrix protein 1), MEPE (matrix extracellular phosphoglycoprotein). 88 , 89 , 90 Because freshly isolated osteocytes from calvariae and long bones express CTR mRNA and immunohistochemistry studies reveal co‐localization of CTR and sclerostin in osteocytes in calvarial sections. 88 , 89 , 90 Moreover, CTR and sclerostin expression are both lost in long‐term culture of osteocytes as well as in osteocytes declined as mice aged. 88 , 89 These data indicate that osteocyte‐mediated responses to CT are most likely to be of physiological relevance in young rodents. However, the mechanism by which CT might stimulate sclerostin remains undefined.
Osteocytes adjacent the endosteal surface can interconnect directly with bone marrow cells by expanding their dendrites to the marrow space and by secreting a wide variety of molecules such as sclerostin, PTHrP, prostaglandin E2 (PGE2), fibroblast growth factor‐23 (FGF23) and RANKL capable of acting locally or on distant organs. Osteocytes express also a plethora of genes required for proper matrix mineralization and phosphate homeostasis such as PHEX (phosphate‐regulating neutral endopeptidase), DMP1, MEPE as well as FGF23. 91 Finally, these cells also synthesize a phosphoprotein, osteopontin (OPN) involved in mineralization and hematopoiesis. 92 OPN is an HSC niche component that negatively regulates stem cell pool size. Sclerostin is highly regulated by mechanical stimuli, 93 PTH and several other factors in humans and animals. 94 , 95 Osteocytes are also key regulators of bone resorption by secreting both pro‐ and anti‐osteoclastogenic factors, such as RANKL, 96 M‐CSF and OPG. 97 , 98
Osteocytes also appear to be centrally involved in calcium phosphate homeostasis through vitamin D signalling pathway. 99 Active form of vitamin D, 1α,25(OH)2D3 (also known as calcitriol) plays in bone mineralization role through binding to the vitamin D receptor (VDR) present mainly in intestine, bone, kidney, and parathyroid gland. 100 Systemic VDR knockout mice are phenotypically very similar to those with vitamin D deficiency. 101 In support of this notion, there is also evidence for presence of VDR transcripts in osteocytes and direct activation of 25‐hydroxyvitamin D (25(OH)D3) in osteocytes. 102 Interestingly calcium, PTH and calcitriol increase circulating FGF23 produced primarily by osteocytes and by matured osteoblasts. 103 FGF23 level is regulated by locally supplied bone‐derived factors including PHEX and DMP‐1 through activation of complex Klotho‐FGF‐receptor 1 and by systemic factors including calcitriol and PTH. 104 , 105 , 106 Excess FGF23 in both humans and mouse models causes hypophosphatemia, suppression of calcitriol levels and rickets (in childhood) or osteomalacia (during adulthood). 103 Elevated levels of FGF23 also cause acquired hypophosphatemic disorders, such as in tumor‐induced osteomalacia (TIO) and is an adaptive response in chronic kidney disease (CKD). In addition, increased FGF23 level affects calcitriol synthesis and degradation, thus hindering its ability to counterbalance hypophosphatemia. FGF23 impairs the production of renal calcitriol by inhibiting the expression of extra‐renal 25(OH)D3‐1α‐hydroxylase (encoded by CYP27B1), the enzyme that converts 25(OH)D3 to its active metabolite, calcitriol. FGF23 also upregulates the expression of cytochrome P450 enzyme 24‐hydroxylase (encoded by CYP24A1), a mitochondrial enzyme responsible for the inactivation of 25(OH)D3 and its hormonal form, calcitriol into 24‐hydroxylated products for excretion. 103 Both of these effects of FGF23 lower circulating concentrations of calcitriol which consequently decreases the intestinal absorption of calcium phosphate. The over‐expression of CYP24A1 leads to dysfunction of the VDR as it over metabolized the 25(OH)D3 and calcitriol. In the parathyroid, FGF23 also directly suppresses the synthesis and secretion of PTH, which indirectly contributes to suppression of calcitriol, since PTH is the primary stimulus of CYP27B1. Thus, CKD patients ought to experience vitamin D deficiency and subsequent osteoporosis.
2.4. Cellular Senescence in bone remodelling with aging
Advanced age is one of the major risk factors for osteoporosis. 107 Cellular senescence is a common hallmark of aged tissues and is generally defined as a dynamic process whereby a cell loses its proliferative potential, undergoing an essentially irreversible growth arrest in response to various stressors such as oxidative stress, DNA damage, genomic instability and telomere shortening. 108 Senescent cells develop a complex, altered gene expression profile that is characterized by upregulation of senescent cell antiapoptotic pathways (SCAPs). 109 A further common feature of senescent cells is the frequent development of a distinctive pro‐inflammatory secretome, termed the senescence‐associated secretory phenotype (SASP) which comprises of cytokines, chemokines, interleukins, growth factors/regulators, and matrix‐degrading proteases. 109 Indeed, even a relatively low abundance of senescent cells (e.g., ~10–15% in aged primates) 110 is sufficient to cause tissue dysfunction as well as aberrant remodelling and disruption of the normal function of neighbouring tissues via secretion of their SASP. 108 , 109 Thus, senescent cells and the SASP are known as contributors to age‐related frailty 108 , 109 and a number of age‐associated chronic diseases including osteoporosis. 111
Recently, there have been several reports describing the role of cellular senescence in the pathophysiology of osteoporosis via altered expression of SASP factors. 111 , 112 Senescent cells accumulate in the bone microenvironment with aging and clearance of these cells using genetic and pharmaceutical interventions improves bone quality in aged mice. 113 , 114 In advanced aging, multiple cell types such as containing B cells, T cells, myeloid cells, osteoprogenitors, osteoblasts and osteocytes in the bone microenvironment become senescent, 115 , 116 although senescent myeloid cells and senescent osteocytes predominantly develop the SASP, a pro‐inflammatory environment. 117 The SASP has been shown to be initiated by NF‐κB 118 and maintained in an autocrine loop, at least in part, by IL‐1α. 117 , 119
Given the accumulating evidence that osteocyte regulates myeloid lineage cells (eg, osteocyte RANKL production leading to osteoclast development from myeloid progenitors. 96 , 97 Thus, it is speculated that with aging, a subset of osteocytes become senescent and produce a SASP signal that is communicated to neighbouring myeloid lineage cells. These myeloid lineage cells may, in turn, become senescent and amplify this signal, resulting in excessive production and secretion of SASP factor, thereby creating a toxic local microenvironment that may not only lead to senescence of neighbouring cells but also contribute to age‐related bone loss. From these findings, it is clear that senescence cells act as a causal mediator of age‐related bone loss like osteoporosis. 112 , 113 , 117 , 120 Given the critical role of osteocytes in regulating bone remodelling, 117 targeted elimination of dysfunctional senescent osteocytes can potentially reverse the SASP, enhance bone formation, and prevent age‐related bone loss. Because osteocytes are long‐lived cells that constitute more than 95% of all bone cells. 117 For example, a Janus kinase inhibitor previously discovered to inhibit production of multiple SASP factors, 121 ruxolitinib administration is found to induce improvements in overall bone strength in old mice. 113 Thus, targeting senescent cells (so‐called “senotherapeutics”) may potentially aid regeneration of dysfunctional aged tissues and attractive therapeutic potential for the treatment of bone diseases like osteoporosis.
3. AN OVERVIEW OF EXISTING THERAPIES FOR THE TREATMENT OF BONE DISEASES SUCH AS OSTEOPOROSIS
Osteoporosis is a skeletal disease and one of the major health problems worldwide. It is characterized by degradation of bone tissue and increase in the fragility of the bones. 122 The loss of bone mineral density (BMD) is due to the action of osteoclast cells, which are associated with modified hormone levels and factors such as ageing. Thus, rheumatic and musculoskeletal diseases like osteoporosis markedly increases the risk of skeletal fractures as well as the prevalence of comorbidities. Effective fracture prevention by reducing the loss of bone mass is the primary treatment goal for people with osteoporosis. Osteoporosis can be prevented and improved musculoskeletal health by using numerous pharmacotherapies such as biphosphonates; selective oestrogen receptor modulators (SERMs); hormone therapies; strontium ranelate; denosumab (a human monoclonal antibody with specificity for RANKL); romosozumab (a monoclonal antibody that binds to and inhibits sclerostin) or stimulating bone formation called anabolic medications e.g., PTH preparations and calcitonin therapy have been verified the effects of increased BMD and decreased risk of skeletal fractures. 123 , 124 , 125 However, these treatments have some side effects, such as oily skin, fluid retention, nausea, long‐term toxicity, and even prostate cancer in males and thus natural therapies that incur better therapeutic activities and fewer side effects are hunted. Therefore, searching small molecules that precisely suppress osteoclastic action is a favourable approach of the drug discovery for the treatment and management of bone‐related diseases including osteoporosis. 126
4. SUMMARY
Bone homeostasis is a dynamic equilibrium by the regulatory actions of three key bone cells, osteoclasts, osteoblasts and osteocytes. Bone homeostasis remains intact as long as the activities of these cells are well‐adjusted, and thus net bone mass is maintained. This balance implies the existence of molecular mechanisms that tightly coordinate the differentiation of osteoblasts, osteocytes and osteoclasts, as well as their migration to locations where they function. Targeting senescent cells may attractive therapeutic potential for the treatment of bone diseases like osteoporosis in the near future.
CONFLICTS OF INTEREST
All of the authors clearly declare that they have no competing and commercial interests.
AUTHORS’ CONTRIBUTIONS
MAAB designed the study and revised the manuscript. MAAM assisted to revise the manuscript.
TRANSPARENCY DECLARATIONS
None to declare.
ACKNOWLEDGEMENTS
All of the authors thank to Faculty of Science, University of Rajshahi for financial support and sponsorship.
REFERENCES
- 1. Chotiyarnwong P, McCloskey EV. Pathogenesis of glucocorticoid‐induced osteoporosis and options for treatment. Nat Rev Endocrinol. 2020;16(8):437–447. [DOI] [PubMed] [Google Scholar]
- 2. Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2(4):389–406. [DOI] [PubMed] [Google Scholar]
- 3. Takayanagi H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7(4):292–304. [DOI] [PubMed] [Google Scholar]
- 4. Phan TCA, Xu J, Zheng MH. Interaction between osteoblast and osteoclast: Impact in bone disease. Histol Histopathol. 2004;19(4):1325–1344. [DOI] [PubMed] [Google Scholar]
- 5. Crockett JC, Mellis DJ, Scott DI, Helfrich MH. New knowledge on critical osteoclast formation and activation pathways from study of rare genetic diseases of osteoclasts: focus on the RANK/RANKL axis. Osteoporos Int. 2011;22(1):1–20. [DOI] [PubMed] [Google Scholar]
- 6. Brylka LJ, Schinke T. Chemokines in physiological and pathological bone remodeling. Front Immunol. 2019;10:2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khosla S, Riggs BL. Pathophysiology of age‐related bone loss and osteoporosis. Endocrinol Metab Clin North Am. 2005;34(4):1015–1030. [DOI] [PubMed] [Google Scholar]
- 8. Theill LE, Boyle WB, Penninger JM. RANK‐L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795–823. [DOI] [PubMed] [Google Scholar]
- 9. Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009;5(12):667–676. [DOI] [PubMed] [Google Scholar]
- 10. Nakashima T, Hayashi M, Takayanagi H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol Metab. 2012;23(11):582–590. [DOI] [PubMed] [Google Scholar]
- 11. Okamoto K, Nakashima T, Shinohara M, et al. Osteoimmunology: The conceptual framework unifying the immune and skeletal systems. Physiol Rev. 2017;97(4):1295–1349. 10.1152/physrev.00036.2016 [DOI] [PubMed] [Google Scholar]
- 12. Bai S, Kitaura H, Zhao H, et al. FHL2 inhibits the activated osteoclast in a TRAF6‐dependent manner. J Clin Invest. 2005;115(10):2742–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lamothe B, Webster WK, Gopinathan A, Besse A, Campos AD, Darnay BG. TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochem Biophys Res Commun. 2007;359(4):1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naito A, Azuma S, Tanaka S, et al. Severe osteopetrosis, defective interleukin‐1 signalling and lymph node organogenesis in TRAF6‐deficient mice. Genes Cells. 1999;4(6):353–362. [DOI] [PubMed] [Google Scholar]
- 15. Glass DA, Bialek P, Ahn JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8(5):751–764. [DOI] [PubMed] [Google Scholar]
- 16. Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89(2):309–319. [DOI] [PubMed] [Google Scholar]
- 17. Chamoux E, Houde N, L'Eriger K, Roux S. Osteoprotegerin decreases human osteoclast apoptosis by inhibiting the TRAIL pathway. J Cell Physiol. 2008;216(2):536–542. [DOI] [PubMed] [Google Scholar]
- 18. Lacey DL, Tan HL, Lu J, et al. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo . Am J Pathol. 2000;157(2):435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis‐inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95(7):3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamashita T, Yao Z, Li F, et al. NF‐kappaB p50 and p52 regulate receptor activator of NF‐kappaB ligand (RANKL) and tumor necrosis factor‐induced osteoclast precursor differentiation by activating c‐Fos and NFATc1. J Biol Chem. 2007;282(25):18245–18253. [DOI] [PubMed] [Google Scholar]
- 21. Takayanagi H, Kim S, Koga T, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3(6):889–901. [DOI] [PubMed] [Google Scholar]
- 22. Sato K, Suematsu A, Nakashima T, et al. Regulation of osteoclast differentiation and function by the CaMK‐CREB pathway. Nat Med. 2006;12(12):1410–1416. [DOI] [PubMed] [Google Scholar]
- 23. Asagiri M, Sato K, Usami T, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202(9):1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cao H, Yu S, Yao Z, et al. Activating transcription factor 4 regulates osteoclast differentiation in mice. J Clin Invest. 2010;120(8):2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maruyama K, Fukasaka M, Vandenbon A, et al. The transcription factor Jdp2 controls bone homeostasis and antibacterial immunity by regulating osteoclast and neutrophil differentiation. Immunity. 2012;37(6):1024–1036. [DOI] [PubMed] [Google Scholar]
- 26. Matsuo K, Galson DL, Zhao C, et al. Nuclear factor of activated T‐cells (NFAT) rescues osteoclastogenesis in precursors lacking c‐Fos. J Biol Chem. 2004;279(25):26475–26480. [DOI] [PubMed] [Google Scholar]
- 27. Kim K, Lee S‐H, Kim JH, Choi Y, Kim N. NFATc1 induces osteoclast fusion via up‐regulation of Atp6v0d2 and the dendritic cell‐specific transmembrane protein (DC‐STAMP). Mol Endocrinol. 2008;22(1):176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsumoto M, Kogawa M, Wada S, et al. Essential role of p38 mitogen‐activated protein kinase in cathepsin k gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem. 2004;279(44):45969–45979. [DOI] [PubMed] [Google Scholar]
- 29. Ikeda F, Nishimura R, Matsubara T, et al. Critical roles of c‐Jun signaling in regulation of NFAT family and RANKL‐regulated osteoclast differentiation. J Clin Invest. 2004;114(4):475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miyamoto H, Suzuki T, Miyauchi Y, et al. Osteoclast stimulatory transmembrane protein and dendritic cell–specific transmembrane protein cooperatively modulate cell–cell fusion to form osteoclasts and foreign body giant cells. J Bone Miner Res. 2012;27(6):1289–1297. [DOI] [PubMed] [Google Scholar]
- 31. Crotti TN, Flannery M, Walsh NC, Fleming JD, Goldring SR, McHugh KP. NFATc1 directly induces the human beta3 integrin gene in osteoclast differentiation. J Musculoskelet Neuronal Interact. 2005;5(4):335–337. [PMC free article] [PubMed] [Google Scholar]
- 32. Kim K, Kim JH, Lee J, et al. Nuclear factor of activated t cells c1 induces osteoclast‐associated receptor gene expression during tumor necrosis factor‐related activation‐induced cytokine‐mediated osteoclastogenesis. J Biol Chem. 2005;280(42):35209–35216. [DOI] [PubMed] [Google Scholar]
- 33. Kim Y, Sato K, Asagiri M, Morita I, Soma K, Takayanagi H. Contribution of nuclear factor of activated t cells c1 to the transcriptional control of immunoreceptor osteoclast‐associated receptor but not triggering receptor expressed by myeloid cells‐2 during osteoclastogenesis. J Biol Chem. 2005;280(38):32905–32913. [DOI] [PubMed] [Google Scholar]
- 34. McHugh KP, Hodivala‐Dilke K, Zheng MH, et al. Mice lacking beta3 integrins are osteosclerotic because of dysfunctional osteoclasts. J Clin Invest. 2000;105(4):433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koga T, Inui M, Inoue K, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428(6984):758–763. [DOI] [PubMed] [Google Scholar]
- 36. Joyce‐Shaikh B, Bigler ME, Chao C‐C, et al. Myeloid DAP12‐associating Lectin (MDL)‐1 regulates synovial inflammation and bone erosion associated with autoimmune arthritis. J Exp Med. 2010;207(3):579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kameda Y, Takahata M, Komatsu M, et al. Siglec‐15 regulates osteoclast differentiation by modulating RANKL‐induced phosphatidylinositol 3‐kinase/AKT and Erk pathways in association with signaling adaptor DAP12. J Bone Miner Res. 2013;28(12):2463–2475. [DOI] [PubMed] [Google Scholar]
- 38. Negishi‐Koga T, Gober H‐J, Sumiya E, et al. Immune complexes regulate bone metabolism through FcRγ signalling. Nat Commun. 2015;6:6637. [DOI] [PubMed] [Google Scholar]
- 39. Ross FP, Teitelbaum SL. Alphavbeta3 and macrophage colony‐stimulating factor: partners in osteoclast biology. Immunol Rev. 2005;208:88–105. [DOI] [PubMed] [Google Scholar]
- 40. Athanasou NA, Quinn J. Immunophenotypic differences between osteoclasts and macrophage polykaryons: Immunohistological distinction and implications for osteoclast ontogeny and function. J Clin Pathol. 1990;43(12):997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pondel M. Calcitonin and calcitonin receptors: bone and beyond. Int J Exp Pathol. 2000;81(6):405–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baron R, Neff L, Brown W, Louvard D, Courtoy PJ. Selective internalization of the apical plasma membrane and rapid redistribution of lysosomal enzymes and mannose 6‐phosphate receptors during osteoclast inactivation by calcitonin. J Cell Sci. 1990;97(Pt 3):439–447. [DOI] [PubMed] [Google Scholar]
- 43. Samura A, Wada S, Suda S, Iitaka M, Katayama S. Calcitonin receptor regulation and responsiveness to calcitonin in human osteoclast‐like cells prepared in vitro using receptor activator of nuclear factor‐kappaB ligand and macrophage colony‐stimulating factor. Endocrinol. 2000;141(10):3774–3782. [DOI] [PubMed] [Google Scholar]
- 44. Kim MS, Day CJ, Selinger CI, Magno CL, Stephens SRJ, Morrison NA. MCP‐1‐induced human osteoclast‐like cells are tartrate‐resistant acid phosphatase, NFATc1, and calcitonin receptor‐positive but require receptor activator of NFκB ligand for bone resorption. J Biol Chem. 2006;281:1274–1285. [DOI] [PubMed] [Google Scholar]
- 45. Kurihara N, Gluck S, Roodman GD. Sequential expression of phenotype markers for osteoclasts during differentiation of precursors for multinucleated cells formed in long‐term human marrow cultures. Endocrinol. 1990;127(6):3215–3221. [DOI] [PubMed] [Google Scholar]
- 46. Karsdal MA, Henriksen K, Arnold M, Christiansen C. Calcitonin: a drug of the past or for the future? Physiologic inhibition of bone resorption while sustaining osteoclast numbers improves bone quality. BioDrugs. 2008;22(3):137–144. [DOI] [PubMed] [Google Scholar]
- 47. Camozzi V, Luisetto G, Basso SMM, Cappelletti P, Tozzoli R, Lumachi F. Treatment of chronic hypercalcemia. Med Chem. 2012;8(4):556–563. [DOI] [PubMed] [Google Scholar]
- 48. Dacquin R, Davey RA, Laplace C, et al. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol. 2004;164(4):509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Keller J, Catala‐Lehnen P, Huebner AK, et al. Calcitonin controls bone formation by inhibiting the release of sphingosine 1‐phosphate from osteoclasts. Nat Commun. 2014;5(5215). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sartawi Z, Schipani E, Ryan KB, Waeber C. Sphingosine 1‐phosphate (S1P) signalling: Role in bone biology and potential therapeutic target for bone repair. Pharmacol Res. 2017;125(Pt B):232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin TJ, Sims NA. Calcitonin physiology, saved by a lysophospholipid. J Bone Miner Res. 2015;30(2):212–215. [DOI] [PubMed] [Google Scholar]
- 52. Holmes D. Bone: Key role for S1P in bone remodelling. Nat Rev Endocrinol. 2015;11(1):3. [DOI] [PubMed] [Google Scholar]
- 53. Karsenty G, Ferron M. The contribution of bone to whole‐organism physiology. Nature. 2012;481(7381):314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fakhry M, Hamade E, Badran B, Buchet R, Magne D. Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J Stem Cells. 2013;5(4):136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. [DOI] [PubMed] [Google Scholar]
- 56. Lian JB, Javed A, Zaidi SK, et al. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit Rev Eukaryot Gene Expr. 2004;14(1–2):1–41. [PubMed] [Google Scholar]
- 57. Lee KS, Kim HJ, Li QL, et al. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast‐specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol. 2000;20(23):8783–8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee M‐H, Kim Y‐J, Kim H‐J, et al. BMP‐2‐induced Runx2 expression is mediated by Dlx5, and TGF‐beta 1 opposes the BMP‐2‐induced osteoblast differentiation by suppression of Dlx5 expression. J Biol Chem. 2003;278(36):34387–34394. [DOI] [PubMed] [Google Scholar]
- 59. Miyama K, Yamada G, Yamamoto TS, et al. A BMP‐inducible gene, dlx5, regulates osteoblast differentiation and mesoderm induction. Dev Biol. 1999;208(1):123–133. [DOI] [PubMed] [Google Scholar]
- 60. Lee M‐H, Kim Y‐J, Yoon W‐J, et al. Dlx5 specifically regulates Runx2 type II expression by binding to homeodomain‐response elements in the Runx2 distal promoter. J Biol Chem. 2005;280(42):35579–35587. [DOI] [PubMed] [Google Scholar]
- 61. Newberry EP, Latifi T, Towler DA. Reciprocal regulation of osteocalcin transcription by the homeodomain proteins Msx2 and Dlx5. Biochemistry. 1998;37(46):16360–16368. [DOI] [PubMed] [Google Scholar]
- 62. Shirakabe K, Terasawa K, Miyama K, Shibuya H, Nishida E. Regulation of the activity of the transcription factor Runx2 by two homeobox proteins, Msx2 and Dlx5. Genes Cells. 2001;6(10):851–856. [DOI] [PubMed] [Google Scholar]
- 63. Yang X, Matsuda K, Bialek P, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin‐Lowry Syndrome. Cell. 2004;117(3):387–398. [DOI] [PubMed] [Google Scholar]
- 64. Rodda SJ, McMahon AP. Distinct roles for hedgehog and canonical Wnt signalling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–3244. [DOI] [PubMed] [Google Scholar]
- 65. Celil AB, Campbell PG. BMP‐2 and insulin‐like growth factor‐I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J Biol Chem. 2005;280(36):31353–31359. [DOI] [PubMed] [Google Scholar]
- 66. Celil AB, Hollinger JO, Campbell PG. Osx transcriptional regulation is mediated by additional pathways to BMP2/Smad signaling. J Cell Biochem. 2005;95(3):518–528. [DOI] [PubMed] [Google Scholar]
- 67. Lemonnier J, Ghayor C, Guicheux J, Caverzasio J. Protein kinase C‐independent activation of protein kinase D is involved in BMP‐2‐induced activation of stress mitogen‐activated protein kinases JNK and p38 and osteoblastic cell differentiation. J Biol Chem. 2004;279(1):259–264. [DOI] [PubMed] [Google Scholar]
- 68. Soltanoff CS, Yang S, Chen W, Li Y‐P. Signaling networks that control the lineage commitment and differentiation of bone cells. Crit Rev Eukaryot Gene Expr. 2009;19(1):1–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gronthos S, Zannettino AC, Graves SE, Ohta S, Hay SJ, Simmons PJ. Differential cell surface expression of the STRO‐1 and alkaline phosphatase antigens on discrete developmental stages in primary cultures of human bone cells. J Bone Miner Res. 1999;14(1):47–56. [DOI] [PubMed] [Google Scholar]
- 70. Stein GS, Lian JB, Stein JL, Wijnen AJV, Montecino M. Transcriptional control of osteoblast growth and differentiation. Physiol Rev. 1996;76(2):593–629. [DOI] [PubMed] [Google Scholar]
- 71. Jiang T, Ge S, Shim YH, Zhang C, Cao D. Bone morphogenetic protein is required for fibroblast growth factor 2‐dependent later‐stage osteoblastic differentiation in cranial suture cells. Int J Clin Exp Pathol. 2015;8(3):2946–2954. [PMC free article] [PubMed] [Google Scholar]
- 72. Naganawa T, Xiao L, Coffin JD, et al. Reduced expression and function of bone morphogenetic protein‐2 in bones of Fgf2 null mice. J Cell Biochem. 2008;103(6):1975–1988. [DOI] [PubMed] [Google Scholar]
- 73. Agas D, Sabbieti MG, Marchetti L, Xiao L, Hurley MM. FGF‐2 enhances Runx‐2/Smads nuclear localization in BMP‐2 canonical signaling in osteoblasts. J Cell Physiol. 2013;228(11):2149–2158. [DOI] [PubMed] [Google Scholar]
- 74. Winkler DG, Sutherland MK, Geoghegan JC, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003;22(23):6267–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. [DOI] [PubMed] [Google Scholar]
- 76. Gori F, Lerner U, Ohlsson C, Baron R. A new WNT on the bone: WNT16, cortical bone thickness, porosity and fractures. Bonekey Rep. 2015;4:669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Holdsworth G, Roberts SJ, Ke HZ. Novel actions of sclerostin on bone. J Mol Endocrinol. 2019;62(2):R167–R185. [DOI] [PubMed] [Google Scholar]
- 78. Poole KES, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19(13):1842–1844. [DOI] [PubMed] [Google Scholar]
- 79. Semënov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J. Biol. Chem. 2005;280(29):26770–26775. [DOI] [PubMed] [Google Scholar]
- 80. Holdsworth G, Slocombe P, Doyle C, et al. Characterization of the interaction of sclerostin with the low density lipoprotein receptor‐related protein (LRP) family of Wnt co‐receptors. J Biol Chem. 2012;287(32):26464–26477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280(20):19883–19887. [DOI] [PubMed] [Google Scholar]
- 82. Delgado‐Calle J, Sato AY, Bellido T. Role and mechanism of action of sclerostin in bone. Bone. 2017;96:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lara‐Castillo N, Johnson ML. LRP receptor family member associated bone disease. Rev Endocr Metab Disord. 2015;16(2):141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rey J‐P, Ellies DL. Wnt modulators in the biotech pipeline. Dev Dyn. 2010;239(1):102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Li Y, Cam J, Bu G. Low‐density lipoprotein receptor family: endocytosis and signal transduction. Mol Neurobiol. 2001;23(1):53–67. [DOI] [PubMed] [Google Scholar]
- 86. Leupin O, Piters E, Halleux C, et al. Bone overgrowth‐associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489–19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bullock WA, Hoggatt AM, Horan DJ, et al. Lrp4 mediates bone homeostasis and mechanotransduction through interaction with sclerostin in vivo. iScience.. 2019;20:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gooi JH, Pompolo S, Karsdal MA, et al. Calcitonin impairs the anabolic effect of PTH in young rats and stimulates expression of sclerostin by osteocytes. Bone. 2010;46(6):1486–1497. [DOI] [PubMed] [Google Scholar]
- 89. Gooi JH, Chia LY, Walsh NC, et al. Decline in calcitonin receptor expression in osteocytes with age. J Endocrinol. 2014;221(2):181–191. [DOI] [PubMed] [Google Scholar]
- 90. Paic F, Igwe JC, Nori R, et al. Identification of differentially expressed genes between osteoblasts and osteocytes. Bone. 2009;45(4):682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pajevic PD, Krause DS. Osteocyte regulation of bone and blood. Bone. 2019;119:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stier S, Ko Y, Forkert R, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med. 2005;201(11):1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Spatz JM, Wein MN, Gooi JH, et al. The Wnt inhibitor Sclerostin is up‐regulated by mechanical unloading in osteocytes in vitro . J Biol Chem. 2015;290(27):16744–16758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bellido T, Ali AA, Gubrij I, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146(11):4577–4583. [DOI] [PubMed] [Google Scholar]
- 95. Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37(2):148–158. [DOI] [PubMed] [Google Scholar]
- 96. Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–1234. [DOI] [PubMed] [Google Scholar]
- 97. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix‐embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Harris SE, MacDougall M, Horn D, et al. Meox2Cre‐mediated disruption of CSF‐1 leads to osteopetrosis and osteocyte defects. Bone. 2012;50(1):42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bonewald LF. The amazing osteocyte. J. Bone Miner. Res. 2011;26(2):229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lieben L, Carmeliet G. Vitamin D signaling in osteocytes: effects on bone and mineral homeostasis. Bone. 2013;54(2):237–243. [DOI] [PubMed] [Google Scholar]
- 101. Quarles LD. Skeletal secretion of FGF‐23 regulates phosphate and vitamin D metabolism. Nat Rev Endocrinol. 2012;8(5):276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chen H, Senda T, Kubo K‐Y. The osteocyte plays multiple roles in bone remodeling and mineral homeostasis. Med Mol Morphol. 2015;48(2):61–68. [DOI] [PubMed] [Google Scholar]
- 103. Edmonston D, Wolf M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat Rev Nephrol. 2020;16(1):7–19. [DOI] [PubMed] [Google Scholar]
- 104. Sapir‐Koren R, Livshits G. Bone mineralization is regulated by signaling cross talk between molecular factors of local and systemic origin: the role of fibroblast growth factor 23. BioFactors. 2014;40(6):555–568. [DOI] [PubMed] [Google Scholar]
- 105. Pereira RC, Juppner H, Azucena‐Serrano CE, Yadin O, Salusky IB, Wesseling‐Perry K. Patterns of FGF‐23, DMP1, and MEPE expression in patients with chronic kidney disease. Bone. 2009;45(6):1161–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hu MC, Shiizaki K, Kuro‐o M, Moe OW. Fibroblast growth factor 23 and Klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75:503–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Drake MT, Clarke BL, Lewiecki EM. The Pathophysiology and treatment of osteoporosis. Clin Ther. 2015;37(8):1837–1850. [DOI] [PubMed] [Google Scholar]
- 108. Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol. 2020;16(5):263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311(5765):1257. [DOI] [PubMed] [Google Scholar]
- 111. Farr JN, Khosla S. Cellular senescence in bone. Bone. 2019;121:121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Khosla S, Farr JN, Kirkland JL. Inhibiting cellular senescence: A new therapeutic paradigm for age‐related osteoporosis. J Clin Endocrinol Metab. 2018;103(4):1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Farr JN, Xu M, Weivoda MM, et al. Targeting cellular senescence prevents age‐related bone loss in mice. Nat Med. 2017;23(9):1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chandra A, Lagnado AB, Farr JN, et al. Targeted reduction of senescent cell burden alleviates focal radiotherapy‐related bone loss. J Bone Miner Res. 2020;35(6):1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Piemontese M, Almeida M, Robling AG, et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight. 2017;2(17):e93771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Farr JN, Fraser DG, Wang H, et al. Identification of senescent cells in the bone microenvironment. J Bone Miner Res. 2016;31(11):1920–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Salminen A, Kauppinen A, Kaarniranta K. Emerging role of NF‐kappaB signaling in the induction of senescence‐associated secretory phenotype (SASP). Cell Signal. 2012;24(4):835–845. [DOI] [PubMed] [Google Scholar]
- 119. Nelson G, Wordsworth J, Wang C, et al. A senescent cell bystander effect: senescence‐induced senescence. Aging Cell. 2012;11(2):345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Farr JN, Rowsey JL, Eckhardt BA, et al. Independent roles of estrogen deficiency and cellular senescence in the pathogenesis of osteoporosis: Evidence in young adult mice and older humans. J Bone Miner Res. 2019;34(8):1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Xu M, Tchkonia T, Ding H, et al. JAK inhibition alleviates the cellular senescence‐associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. 2015;112:E6301–E6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int. 2006;17(12):1726–1733. [DOI] [PubMed] [Google Scholar]
- 123. Gupta T, Das N, Imran S. The prevention and therapy of osteoporosis: a review on emerging trends from hormonal therapy to synthetic drugs to plant‐based bioactives. J Diet Suppl. 2019;16(6):699–713. [DOI] [PubMed] [Google Scholar]
- 124. Reid IR. A broader strategy for osteoporosis interventions. Nat Rev Endocrinol. 2020;16(6):333–339. [DOI] [PubMed] [Google Scholar]
- 125. Shoback D, Rosen CJ, Black DM, Cheung AM, Murad MH, Eastell R. Pharmacological management of osteoporosis in postmenopausal women: an endocrine society guideline update. J Clin Endocrinol Metab. 2020;105(3):dgaa048. [DOI] [PubMed] [Google Scholar]
- 126. Chen L‐R, Ko N‐Y, Chen K‐H. Medical treatment for osteoporosis: from molecular to clinical opinions. Int J Mol Sci. 2019;20(9):2213. [DOI] [PMC free article] [PubMed] [Google Scholar]