

Summary
Modelling cardiac disorders with human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes is a new paradigm for preclinical testing of candidate therapeutics. However, disease-relevant physiological assays can be complex, and the use of hiPSC-cardiomyocyte models of congenital disease phenotypes for guiding large-scale screening and medicinal chemistry have not been shown. We report chemical refinement of the antiarrhythmic drug mexiletine, via high-throughput screening of hiPSC-CMs derived from patients with the cardiac rhythm disorder long QT syndrome 3 (LTQ3) carrying SCN5A sodium channel variants. Using iterative cycles of medicinal chemistry synthesis and testing, we identified drug analogues with increased potency and selectivity for inhibiting late sodium current across a panel of 7 LTQ3 sodium channel variants and suppressing arrhythmic activity across multiple genetic and pharmacological hiPSC-CM models of LTQ3 with diverse backgrounds. These mexiletine analogues can be exploited as mechanistic probes and for clinical development.
Keywords: induced pluripotent stem cells, LQT3, mexiletine, arrhythmia, cardiomyocyte, drug development
eTOC Statement
McKeithan et al. used large-scale functional screening of hiPSC-cardiomyocytes carrying a mutation that causes an electrophysiological disorder (Long QT syndrome type 3) to direct the chemical optimization of mexiletine, an antiarrhythmic drug used to treat the disease. Four new analogues have greater potency and less proarrhythmic liability relative to mexiletine.
Graphical Abstract
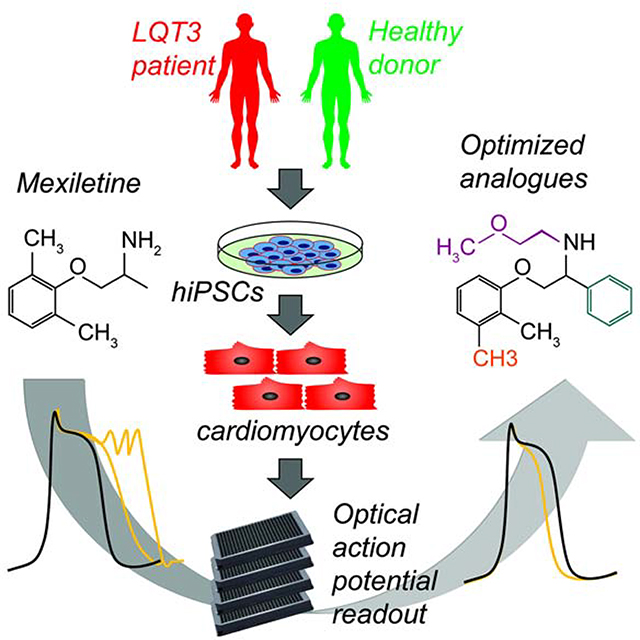
Introduction
Somatic cells derived from human induced pluripotent stem cells (hiPSCs) have been shown to recapitulate a wide range of disease phenotypes in vitro (Matsa et al., 2016; Tiscornia et al., 2011). By reproducing human pathophysiology, hiPSC-based models have been considered a new paradigm for the development of precision therapeutics that target specific disease mechanisms. Despite the immense promise of disease-specific hiPSC models, only a few large-scale drug development efforts have used hiPSCs and these studies were restricted to healthy donors (Hnatiuk and Mercola, 2019). Here, we describe the use of high throughput physiological screening for arrhythmic phenotypes in hiPSC-derived cardiomyocytes (hiPSC-CMs) from patients with long QT syndrome type 3 (LQT3) to facilitate the rapid medicinal chemical refinement of a small molecule drug, mexiletine.
Mexiletine is an orally available inhibitor of muscle and neuronal sodium channels and is a class 1B antiarrhythmic drug (Singh et al., 2020), and is used to suppress life-threatening ventricular arrhythmia and to shorten the heart-rate corrected QT interval (QTc) in LQT3 patients (Al-Khatib et al., 2018; Priori et al., 2015). The cardiac ventricular action potential (AP) is initiated by opening of Nav 1.5 channels that conduct a large inward Na+ current (peak Na+ current, INaP) that drives rapid conduction in the ventricles. The majority of Nav 1.5 channels rapidly inactivate with depolarization, but a small subset of channels fail to inactivate and mediate late Na+ current (INaL). LQT3 is caused by mutations in the pore-forming α-subunit of Nav 1.5 (encoded by SCN5A) that impair channel inactivation and accelerate recovery from the inactivated state increasing INaL to oppose repolarization and prolong the AP. These cellular effects prolong the QT interval on the surface electrocardiogram. LQT3 affects children and teenagers and is characterized by episodes of polymorphic ventricular tachycardia and carries a risk of sudden cardiac death. Mexiletine was shown to shorten the QTc in LQT3 patients (Schwartz et al., 1995) and subsequently to decrease the risk of ventricular tachycardia and ventricular fibrillation upon 35 month median follow-up (Mazzanti et al., 2016). Accordingly, mexiletine is included in current guidelines for treatment of LQT3 in addition to β-blockers and other antiarrhythmic drugs (Al-Khatib et al., 2018; Priori et al., 2015).In addition to LQT3, selective inhibition of INaL is considered a therapeutic target for electrical and contractile dysfunction in heart failure and cardiac ischemia (Horvath and Bers, 2014; Maier and Sossalla, 2013). Mexiletine is modestly selective for INaL over INaP, but also inhibits the repolarizing K+ current, IKr, prolongs the cardiac AP, and promotes early after depolarizations (EADs) in vitro at concentrations near the therapeutic plasma concentration and IC50 for inhibition of INaL (Gualdani et al., 2015; McKeithan et al., 2017). Currently, it is not known if the therapeutic and proarrhythmic properties of mexiletine are due to distinct determinants in the chemical structure from those that dictate inhibition of INaL. Studies of the skeletal muscle Na+ channel Nav 1.4 (encoded by SCN4A) and Nav 1.5 have shown that chemical substituents near the mexiletine center of chirality strongly modulate potency for inhibition of INa(De Bellis et al., 2013; De Luca et al., 2000; De Luca et al., 2003; Roselli et al., 2016). To explore a large number of chemical modifications, we took advantage of high throughput physiological screening in hiPSC-CMs. The human cardiomyocyte context is advantageous over simpler systems because it includes not only the Nav1.5 pore-forming α-subunit, but also ancillary proteins that regulate channel function. Furthermore, screening for a complex phenotype such as AP morphology in this context is unbaised with respect to target protein or molecular mechanism of action.
Based on the electrophysiologic responses of LQT3 and healthy hiPSC-CMs, we identified structural analogues of mexiletine with greater potency and selectivity for INaL that decreased AP prolongation and suppressed EADs. The analogues effectively suppressed arrhythmia in spontaneous genetic and pharmacologically-induced hiPSC-CM models of LQT3 from multiple genetic backgrounds. These studies illustrate the potential of using complex physiological models of patient-derived hiPSC-CMs for precision drug design.
Results
hiPSC-Cm generation and development of high throughput assays
hiPSC-CMs were generated from a previously characterized LQT3 patient harboring a de novo F1473C missense SCN5A mutation that presented with a QTc of 825 ms and 2:1 atrioventricular (AV) block (Bankston et al., 2007; Silver et al., 2009; Terrenoire et al., 2013). The patient tolerated a high dose (24 mg/kg/d) of mexiletine that provided a partial control of his arrhythmia. To conduct the high throughput screen, we used LQT3 SCN5A F1473C hiPSC-CMs derived from this individual prepared by Cellular Dynamics International (CDI)(MyCell Cardiomyocytes, see STAR Methods) and commercially available healthy donor hiPSC-CMs (iCell Cardiomyocytes, termed herein HD.CDI). Our previous studies showed that these hiPSC-CMs, generated by CDI as well as an independent hiPSC derivation, showed elevated INaL (relative to healthy controls) that was inhibitable by mexiletine (McKeithan et al., 2017; Terrenoire et al., 2013). In the high throughput screen, there was variation in APD75 and spontaneous beat rate that depended on the particular sample of hiPSC-CMs (Figure S1A–C). Despite this variation, LQT3 hiPSC-CMs showed characteristic AP prolongation relative to healthy donor controls when corrected for beat rate (Fridericia, 2003) (Figure 1B,D and Figure S1D), and a dose-dependent shortening of the APD in response to mexiletine (EC50= ~5 μM) (Figure 1A,B) that was highly reproducible across experiments (Figure S1E–E”). Interestingly, the LQT3 hiPSC-CMs showed only a modest APD prolongation (~100 ms) at high doses of mexiletine (Figure 1A,B and Figure S1E–E”) consistent with the patient’s tolerance of high dose mexiletine therapy (Bankston et al., 2007; Silver et al., 2009; Terrenoire et al., 2013). In contrast, HD.CDI hiPSC-CMs showed a proarrhythmic response to mexiletine (Figure 1C,D and Figure S1F–F”). hiPSC-CMs from three other unrelated healthy donors (HD.15S1, HD.113, HD.273) with normal SCN5A sequence also showed proarrhythmic responses (Figure 1C). None of the healthy donor lines exhibited APD shortening in response to mexiletine. Therefore, we used the LQT3 SCN5A F1473C hiPSC-CMs to screen for APD shortening and HD.CDI hiPSC-CMs to detect APD prolongation and EADs (Figure 1E). As a positive control for the proarrhythmic phenotype, experiments included hiPSC-CMs treated with dofetilide, a selective IKr inhibitor, that prolonged the AP and induced EADs in HD.CDI and LQT3 iPSC-CMs (EC50 = 4–7 nM, Figure S1G–G”, Figure S1H–H”) as well as the Na+ channel blocker tetrodotoxin (TTX) to confirm that the dependence on INa for AP generation of the SCN5A F1473C hiPSC-CMs (EC50 = 3.6 μM) (Figure S1I–I”). The effects of mexiletine, dofetilide and TTX on AP kinetics were highly reproducible across experiments (Figure S1E”–I”)
Figure 1: Phenotypic screening based on physiological response of hiPSC-CMs.

A,B) On-target phenotype: Representative optical action potential traces show a dose dependent shortening of action potential duration in response to mexiletine of LQT3 SCN5A F1473C hiPSC-CMs (A) and dose response curve for spontaneous beat rate corrected APD75 shortening in SCN5A F1473C hiPSC-CMs (B). Baseline APD75 of 482 ms shown in red (B).
C,D) Off-target phenotype: Representative optical action potential traces show a dose dependent prolongation of action potential duration in response to mexiletine of healthy donor hiPSC-CMs (C). Early after depolarizations (EADs) are indicated with an * (HD.113 trace) or delayed after depolarizations (DADs) are indicated with a † (HD.CDI trace). A dose response curve for spontaneous beat rate corrected APD75 prolongation in healthy donor hiPSCs (HD.CDI) (D). Baseline APD75 of 392 ms shown in green (D).
E) Drug reengineering strategy. hiPSC-CMs were generated from cells of an LQT3 patient and a healthy donor. Iterative cycles of chemical syntheses, optical high throughput screening and (in selected compounds) assessment of Na+ and K+ curren tinhibition using automate dplana rpatc hclam assay sresulte di n ase to flea dcandidates. Following confirmatio no fNa +current inhibition in LQT 3hiPSC-CMs,compound swere teste dfo refficac yagains t apane lo fLQT 3disease-causin gmutation sto asses sthe influence o fmutatio nan dindividua lhuma ngeneti cbackgroun do ncompound responsiveness.
Datapoints(B,D) indicat emea n ±SEM,n=3.
Physiological screening and mexiletine reengineering
We implemented a strategy (Figure 1E) using the LQT3 (SCN5A F1473C) patient and HD.CDI hiPSC-CMs to characterize the effect of mexiletine analogues on APD shortening and proarrhythmic phenotypes by high-throughput optical recording of APs. Analogues that shortened the APD in the SCN5A F1473C hiPSC-CMs were tested using automated planar patch clamp to directly measure inhibition of INa and IKr. Analogues were synthesized and analyzed iteratively (Figure 2A). Phenyl mexiletine (MexA1, Figure 2B), was tested because bulky substitutions at the alpha position to the amine of mexiletine had previously been shown to increase potency for INa inhibition in skeletal and cardiac muscle voltage-gated Na+ channels (De Bellis et al., 2013; De Luca et al., 2000; De Luca et al., 2003; Roselli et al., 2016). MexA1 increased the potency of APD shortening in SCN5A F1473C hiPSC-CMs with similar efficacy to mexiletine while also decreasing induction of EADs. The results are summarized in Figure 2C and representative AP recordings are shown in Figure S2A,C. Automated planar patch clamp studies confirmed that MexA1 had increased potency against INa and IKr relative to mexiletine, but also showed a loss of selectivity for INaL over INaP (Table S1).
Figure 2: Distinct chemical substituents discriminate the therapeutic and proarrhythmic effects of mexiletine on hiPSC-CMs action potential kinetics.

A) Compound triage. Analogues were screened through multiple cycles of synthesis and optical testing as in Figure 1E. Analogues that passed initial screen criteria were assessed for specific effects on Na+ (INa) and K+ (IK) currents, ultimately resulting in 4 lead candidates.
B) Chemical structures of mexiletine, phenyl mexiletine (MexA1) and lead candidates (MexA2–5).
C) Heat map representation of physiological responses of mexiletine and phenyl mexiletine analogues. Scale: green is improved and red is worse relative to mexiletine (black). See Figure S2 and Table S1 for numerical data. Note that phenyl mexiletine (MexA1) showed a favorable decrease in APD prolongation and EAD incidence (arrhythmia phenotypes) but worse selectivity ratios for INaL relative to mexiletine. Selectivity was restored by phenoxy substitutions (MexA2–5).
D) Summary of the phenotypic effects of chemical modifications to mexiletine.
E-I) Dose dependent effects on peak(INaP) and late(INaL) sodium current in LQT3 SCN5A F1473C hiPSC-CMs by whole cell patch clamp recording. Data are represented as mean ± SEM., n=4. See Table S2 for numerical summary.
Given that MexA1 was more potent but less selective than mexiletine, we sought to restore selectivity for INaL. Three modifications to the phenoxy region on the opposite side of the molecule, the ortho mono-substituted methyl (MexA2), 3,5-dimethyl (MexA3) and ortho trifluoromethyl-substituted (MexA4) analogues, potently shortened APD in SCN5A F1473C hiPSC-CMs (Figure 2B,C and Figure S2A,B). MexA2, MexA3 and MexA4 were not proarrhythmic in HD.CDI hiPSC-CMs at any dose examined (Figure 2C, and Figure S2C,D). MexA2, MexA3 and MexA4 were extremely potent against late sodium current (INaL) (IC50 ≤ 1 μM) and were more selective than mexiletine for inhibition of INaL over INaP (selectivity ratios were 53.4, 100.5, and 39.5 respectively, compared to 1.8 for MexA1 and 8.3 for mexiletine, see Table S1 and Figure S2E–I). MexA2, MexA3 and MexA4 were also more selective for INaL over IKr (ratios were >31 for MexA2–4, compared to 0.7 for MexA1 and 2.5 for mexiletine, Table S1 and Figure S2J–N). Less IKr block is desirable because it is a well-recognized mechanism of drug-induced proarrhythmia. The analogues, in particular MexA3 and MexA4, caused asystole at concentrations above 66 μM in both LQT3 and healthy donor hiPSC-CMs. Probing the chemical basis for asystole, we observed that N-ethyl methoxy substitution on the primary amine decreased this effect,exemplified by MexA5 (Figure 2B,C).
Validation of function in LQT3 patient hiPSC-CMs
To verify function in a cardiomyocyte context, mexiletine analogues were tested for their electrophysiological effects on INaL and INaP by whole cell patch clamp recording of LQT3SCN5A F1473C hiPSC-CMs (Figure 2E–I, Table S2). As in the heterologous expression system, MexA2–5 were more potent (28.3–134-fold) and selective (22.3–117-fold) than mexiletine for inhibiting INaL relative to INaP in LQT3 cardiomyocytes.
In summary (Figure 2D), introduction of a phenyl group alpha to the primary amine (MexA1) increased potency for APD shortening and decreased proarrhythmic liability, but also abrogated selectivity for inhibition of INaL over INaP. Introduction of substituents to the phenoxy moiety (i.e., MexA2–5) restored selectivity for INaL over INaP inhibition, while N-ethyl methoxy substitution (MexA5) decreased the asystolic effect observed at high concentrations of non-N-substituted phenyl mexiletine derivatives.
Evaluation of MexA2–5 against LQT3-causing SCN5A mutations
Given the diversity of SCN5A mutations causing LQT3, we tested INaP and INaL inhibition of MexA2–5 across a panel of 7 LQT3-causing mutant channels, including F1473C, expressed in HEK293T cells by automated planar patch clamp (Figure 3A). To compare efficacy against the panel of channels, each compound was tested at a single dose corresponding to the EC80 for the F1473C mutant channel (Figure 3B,C). Normalization of the dataset by analogue revealed differential inhibition profiles (Figure 3D). The compounds were generally effective against all mutant channels examined with the exception of MexA2 and MexA3 that did not block INaL in the L409P/R588 mutant (Figure 3B,C and Figure S2O,P). Among all mutants examined, MexA2–5 were most effective against INaL in F1473C and delKPQ (Figure 3D). Selectivity for F1473C might be expected because it was used in the primary screening.
Figure 3: Effects of SCN5A variants on responsiveness to mexiletine and phenyl mexiletine analogues.

A) Schematic of seven SCN5A variants transiently expressed in HEK293T cells and analyzed by planar patch clamp recording.
B,C) Mean % inhibition ± SEM of INaL and INaP inhibition. Each analogue was evaluated at a single dose (EC80 for SCN5A F1473C from Figure S2E–I, Table S1). See Figure S2O,P for individual data points.
D) Normalization of mean % inhibition by analogue (row normalization in the heatmap representation) portray compound-specific profiles of inhibition across the SCN5A mutants.
Effect of mexiletine analogues in hiPSC-CM models of LQT3
To gauge effectiveness in hiPSC-CMs from different individuals, we tested MexA2 in a second genetic LQT3 model with a distinct mutation in SCN5A (N406K)(Spencer et al., 2014) that is mexiletine responsive (Hu et al., 2018). In these and subsequent studies, the hiPSC-CMs were prepared using a metabolic maturation protocol (Feyen et al., 2020) (see STAR Methods). Because wildtype SCN5A is less responsive to mexiletine (Hu et al., 2018; Zhu et al., 2019), we also created pharmacological models of LQT3 by treating two healthy donor hiPSC-CMs (HD.15S1 and HD.113) with veratridine (0.37 μM) to cause persistent activation and ATX-II (0.5 μM) to inhibit inactivation of Nav 1.5 (Figure 4A). For comparison, we also tested INaL inhibitors GS945867 (GS-967) (Koltun et al., 2016) and ranolazine. Both are selective blockers of INaL over INaP but are structurally distinct from each other and mexiletine (Zablocki et al., 2016).
Figure 4: Relative therapeutic efficacy of MexA2 and INaL inhibitors in a cohort of LQT3 hiPSC-CM models reflecting different human genetic backgrounds and disease manifestations.

A) Experimental setup to test the therapeutic efficacy and toxicity of MexA2 and other INaL blockers in genetic and pharmacological LQT3 hiPSC-CM models.
B-G) Genetic models of LQT3 in hiPSC-CMs: Representative optical voltage recordings of action potentials from individual compounds at the lowest dose with maximum APD75 shortening effect(B,E). Each trace is an average normalized ΔF/F0 vs. time plot (33Hz) from multiple peaks in a well (n=3–5peaks). The grey lines represent AP recording from respective control wells. Maximum APD75 shortening (C,F) and prolongation (D,G) attained by each compound in individual models of LQT3. APD75 shortening and prolongation times (ms) are represented as change from their respective control wells (n = 3–6 wells). Data are represented as mean ± SEM.
H-P) Pharmacological models of LQT3 in hiPSC-CMs: Representative optical voltage recordings of action potentials from individual compounds at the lowest dose with maximum APD75 shortening effect (H,K,N). Each trace is an average normalized ΔF/F0 vs. time plot (33Hz) from multiple peaks in each well (n= 3–5 peaks). The grey lines represent AP recording from respective control wells. Maximum APD75 shortening (I,L,O) and prolongation (J,M,P) attained by each compound in each model of LQT3. APD75 shortening and prolongation times (ms) are represented as change from their respective control wells (n = 3–6 wells).
Q) Experimental setup for evaluating arrythmia suppression by the small molecules. Genetic LQT3 SCN5A N406K hiPSC-CMs led to spontaneous arrythmias and high dose of ATX-II (1μM) induced triggered activity in healthy donor (HD.113) hiPSC-CMs.
R) Representative optical voltage recordings of action potential in LQT3 SCN5A N406K hiPSC-CMs with spontaneous arrhythmia. An individual AP trace is shown for each compound at the lowest dose that attained shortest arrhythmic period and lowest number of arrhythmic peaks (more information about metrics in Figure S4A).
S,T) The graph indicates the shortest arrhythmic period (S) and lowest number of arrhythmic peaks (T) attained by individual drugs(n=3 wells).
U) Representative optical voltage recording of the ATX-II (1 μM) induced arrythmia in HD.113 hiPSC-CMs for each compound at the lowest dose that achieved maximal reversion of APD75 instability and beat-to-beat instability (instability= standard deviation/ mean).
V,W) Quantification of ATX-II induced arrythmia and the lowest dose which achieved maximal suppression of APD75 instability (V) and beat-to-beat instability (W) for each compound (n=3 wells).
Data are presented as mean ± SEM. Statistical significance was calculated by unpaired T-test comparing either MexA2 and mexiletine groups (*) or the individual compound vs untreated control (†) for B-P. One-way ANOVA with Dunnett’s method for multiple comparisons was used in S-T and V-W (vs DMSO control group)(*). ††† and ***, P<0.001; †† and **, P<0.01; † and *, P<0.05. Mexiletine is denoted ‘mex’, ranolazine as ‘ran’ and GS-967 as ‘GS.’
Mexiletine shortened APD at intermediate doses in LQT3 SCN5A F1473C and weakly in LQT3 SCN5A N406K (Figure 4B–G, Figure S3A,B) but not in all the pharmacological models (Figure 4H–P, Figure S3C–E). At higher doses, mexiletine substantially prolonged the AP in all hiPSC-CMs examined, often inducing EADs, except in LQT3 SCN5A F1473C. This finding was consistent with earlier results (Figure 1A). In contrast, MexA2 shortened APD across both genetic and both pharmacologic LQT3 models without prolongation (Figure 4B–P, Figure S3A– E). MexA5 showed similar efficacy to MexA2 in shortening the APD of genetic and pharmacological LQT3 models (Figure S4B–I). The other INaL blockers showed mutation-specific efficacy. For instance, ranolazine shortened APD without prolongation at high doses in LQT3 SCN5A F1473C hiPSC-CMs, but did not shorten APD and induced EADs at high doses in LQT3 SCN5A N406K hiPSC-CMs (Figure 4B–G, Figure S3A,B). In general, MexA2, MexA5 and GS-967 showed the most pronounced shortening at low doses while mexiletine and ranolazine prolonged the APD at high doses (Figure 4B–P, Figure S3A–E, Figure S4B–I).
Effect of MexA2 on suppression of arrhythmia
We tested the ability of MexA2, MexA5 and other INaL inhibitors to suppress spontaneous arrhythmias that arise in about half of SCN5A N406K hiPSC-CM differentiations as well as a high dose ATX-II induced Journ arrhythmia in healthy donor hiPSC-CMs (Figure 4Q, Figure S3F,G, Figure S4J–M). Both MexA2 and MexA5 terminated spontaneous arrhythmias in SCN5A N406K hiPSC-CMs. MexA2 terminated arrhythmia at 3-fold lower doses than mexiletine in the SCN5A N406K and ATX-II models. GS-967 was effective, but ranolazine blocked arrhythmia only in the genetic LQT3 model (SCN5A N406K). In summary, MexA2 and MexA5 potently reverted LQT3 phenotypes in hiPSC-CMs of different genotypes and LQT3-causing SCN5A mutations.
Discussion
We described the iterative chemical refinement of mexiletine analogues based on their effects on desirable AP shortening, unwanted AP prolongation and EAD phenotypes in hiPSC-CMs. Our study demonstrates that a disease phenotype of patient-derived hiPSC-CMs can guide the improvement and optimization of a drug. We succeeded in identifying four mexiletine analogues (MexA2–5) that did not induce AP prolongation or EADs and yet retained the ability to suppress arrhythmia in hiPSC-CM models of LQT3.
MexA2–5 were 28–134-fold more potent and 22–117-fold more selective for inhibiting INaL relative to INaP compared to mexiletine in SCN5A F1473C hiPSC-CMs. These compounds also showed greatly improved selectivity for INaL relative to IKr inhibition (> 31-fold) compared to mexiletine. However, it is not able that they retained comparable IKr inhibition to mexiletine. This finding implies that another mechanism is responsible for the absence of AP prolongation and EADs. Thus, conventional planar patch screening for INa and IKr block (Table S1) as logical determinants of the AP prolongation and EAD propensity would not have predicted the absence of AP prolongation and EADs upon treatment with high doses of MexA2–5. AP waveforms depend on a complex interplay of multiple ion channels. Our results illustrate the advantages of screening for alterations in hiPSC-CMs AP waveform morphology directly when investigating the chemical basis for complex electrophysiological phenotypes such as EADs, rather than relying on effects on individual channel types in non-CM over expression systems.
More than 750 SCN5A mutations have been reported, of which 227 have been associated with LQT3 (Stenson et al., 2017). Individual mutant channels respond differently to mexiletine (Ruan et al., 2010; Ruan et al., 2007). In addition to F1473C, MexA2–5 were tested against six different LQT3-causing mutant channels expressed in HEK293 cells to eliminate the influence of genetic background (Figure 3). MexA2–5 blocked five of the seven SCN5A mutant channels, demonstrating broad efficacy against multiple mutations albeit with subtle mutation-specific effects (Figure 3D). MexA2–5 were uniformly effective at inhibiting INaL mediated by SCN5A F1473C mutant channels (Figure 3D), suggesting that reliance on a single variant in primary screening might bias the development of therapeutics in favor of individuals carrying that variant. A channel with a missense mutation (i.e., L409P) combined with a common genetic variant (i.e., R588) was inhibited by mexiletine and two analogues (MexA4 and MexA5) but not by two others (MexA2 and MexA3). This channel is responsible for a highly malignant LQT3 phenotype that was originally detected in utero and is also resistant to block by lidocaine that binds to the same site as mexiletine in Nav 1.5 (Murphy et al., 2012). The mechanism of resistance is unclear but might reflect alterations in biophysical properties of Nav 1.5 (e.g., ultra-rapid recovery from inactivation and shift of steady state inactivation to depolarized voltage potentials)(Murphy et al., 2012). Interestingly, MexA2 and MexA3 preferentially blocked INaP over INaL that may point to a unique interaction between these compounds and the channel mutants. More generally, the structural basis for variable effectiveness of mexiletine is of considerable research and clinical interest. Current models hold that mexiletine preferentially binds with greatest affinity when the channel is in its inactivated state (Desaphy et al., 2001). LQT3 mutations that increase the persistence of the inactivated state (that occurs when the channel’s inactivation gate blocks access to the pore) correlate with efficacy of QT interval shortening in patients (Ruan et al., 2007). Other mutation-induced conformational changes in the channel modulate drug responsiveness. For instance, Zhu et al. (Zhu et al., 2019) recently proposed, based on mutational analysis, that the activated conformation of domain III voltage sensing domain of the α-subunit (DIII-VSD) enhances mexiletine binding and inhibition. Mexiletine docking to the channel pore maintains the DIII-VSD in an activated conformation (Zhu et al., 2019). The mexiletine analogues described herein that show variant-selectivity might be useful tools to probe how mutations affect channel pharmacology.
Selective INaL inhibitors ranolazine and a clinical candidate related to GS-967 (GS-6615, eleclazine) were previously under clinical evaluation for LQT3 (Priori et al., 2018). MexA2 and MexA5 compared favorably to these structurally distinct compounds for the ability to shorten APD and suppress spontaneous arrhythmia in both hiPSC-CM LQT3 genetic (SCN5A N406K) and pharmacologic (ATX-II) models that reflect different mechanisms of impaired channel inactivation and patient backgrounds (Figure 4).
hiPSC modeling of genetic heart disease is an enabling technology for drug development because it introduces aspects of the clinical presentation of disease into the earliest stages of the discovery and development pipeline. Precedents include predicting proarrhythmic and cardiotoxic responses to drugs and the discovery of small molecule probes of diabetic cardiomyopathy and ischemic heart disease (Blinova et al., 2018; Drawnel et al., 2014; Fiedler et al., 2019; Sharma et al., 2017). We believe that the use of congenital disease models in large scale screening or to guide medicinal chemistry is the next step for implementation of hiPSC-CMs in the drug discovery pipeline, and our study demonstrates the feasibility of this approach. There are several steps to translate these findings to drug development, including functional testing in a murine (Fabritz et al., 2010; Nuyens et al., 2001; Schroder et al., 2014) or pharmacological (e.g., ATX-II treated ex vivo rabbit heart) models (Belardinelliet al.,2013). These would be followed by pharmacokinetic, pharmacodynamic and toxicity studies in large animals in anticipation of clinical testing.
In summary, we showed that patient-derived and pharmacological hiPSC-CM models can recapitulate the beneficial and adverse pharmacological effects of mexiletine. Through medicinal chemistry refinement, four phenyl mexiletine analogues (MexA2–5) were found to have greater on-target potency and selectivity in addition to decreased proarrhythmic liability relative to mexiletine. These compounds were generally effective against different SCN5A mutations and suppressed arrhythmia in a cohort of hiPSC-CM LQT3 models reflective of distinct human genetic backgrounds. This study highlights the use of disease-specific hiPSCs in drug discovery. In particular, the results illustrate the use of hiPSC-derived models to develop small molecule drugs based on the quantitative assessment of therapeutic potential and liabilities directly in a disease-relevant human cellular context.
Limitations of the Study
Limitations of this study include the use of hiPSC-CMs that are relatively immature compared to adult CMs. CM immaturity might bias the development of compounds towards those that target proteins and processes that operate in early fetal rather than adult CMs. It would not have been possible to conduct a largescalescreenusing adult humancardiomyocytes,thereforetohelp mitigate the limitations caused by physiological immaturity, we employed a metabolism-based maturation protocol that yielded hiPSC-CMs with Na+-dependent APs (STAR Methods) (Feyen et al., 2020). Key modifications to the media were Pre-prooflowglucoseandadditionofoxidativeenergy substrates that resulted in a low resting membrane potential and fast depolarization (>200V/s) due to large inwardly rectifying K+ current, IK1, that sets the resting membrane potential as well as increases the number of activatable Na+ channels. Although the Na+ channel electrophysiology and cellular basis for arrhythmia are suitable for screening and initial characterization of compounds, the hiPSC-CM model does not fully replicate complex multicellular arrhythmia substrates. Therefore, additional evaluation of lead compounds for safety and efficacy for LQT3 or suppression of ventricular arrhythmia should be conducted in animal models (e.g., canine models) that have a similar electrophysiological basis for AP morphology and arrhythmia susceptibility as humans.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mark Mercola (mmercola@stanford.edu).
Materials availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The published article includes all datasets generated or analyzed during this study. This study did not generate code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
iCell Cardiomyocyte Culture (corresponds to Figure 2)
MyCell Cardiomyocytes (LQT3)referred to as (SCN5A F1473C) and iCell Cardiomyocytes (healthy donor) referred to as (HD.CDI) (Cellular Dynamics International, Wisconsin, USA) were used for optical assessment of AP kinetics and whole cell patch clamp recording (Figure 2). The LQT3 cells were from a male. The HD.CDI iCell Cardiomyocytes were from a female. Purity of MyCell Cardiomyocytes was ≥97% hiPSC-CMs and iCell Cardiomyocytes was ≥99% hiPSC-CMs (as per CDI data sheets). Cryopreserved vials were thawed according to the manufacturer’s specifications. The vial was transferred to a 37°C water bath for 4min. Once completely thawed, the contents were slowly transferred to a 50 mL conical vial and diluted dropwise in iCell Cardiomyocyte Plating Media (iCCPM) to a concentration of 2.5 × 105 cells/mL. hiPSC-CMs were transferred into 384 well plates pre-coated (Greiner Bio-One) with 0.1% (w/v) gelatin (Stem Cell Technologies). The plates were then placed in a 37°C incubator with 5% CO2. After 24 hours, the media was exchanged with iCell Cardiomyocyte Maintenance Media (iCCMM), supplemented with 5mM D-glucose. This process was repeated two additional times, ultimately yielding a final volume of 100 μL/well. Media was exchanged every other day for 14 days prior to imaging by removing 50 μL of media and adding 50 μL of fresh iCCMM (supplemented with 5 mM D-glucose).
Differentiation of hiPSCs to cardiomyocytes (corresponds to Figure 4)
Healthy donor cells (HD.113, HD.15S1, HD.273) were obtained from the Stanford CVI Biobank and originally derived under a Stanford University IRB approved protocol. Healthy donor [HD.113 (male), HD.15S1 (male), HD.273 (female)] and LQT3 [SCN5A F1473C (male) and SCN5A N406K (female)] hiPSCs were differentiated and used for tertiary assays shown in Figure 4. Briefly, hiPSCs were dissociated using 0.5 mM EDTA (ThermoFisher Scientific, Waltham, MA, USA) in PBS without CaCl2 or MgCl2 (Corning, New York, USA) for 7 minutes at room temperature. The dissociated hiPSCs were plated in growth factor reduced matrigel-coated dishes in E8 culture media (ThermoFisher Scientific, Waltham, MA, USA) supplemented with 1 μM Y-27632 (Tocris, Bristol, UK). After 24 hours, the media was replaced with E8 without Y-27632 and was replenished daily for 3–5 days until the cells reached ≥ 90% confluence to begin differentiation. Cardiomyocytes were differentiated by methods previously described (Feyen et al., 2020). Briefly, hiPSCs were treated with 4–8 μM CHIR99021 (Tocris, Bristol, UK), depending on the hiPSC line used for differentiation, for 3 days in RPMI 1640 supplemented with B27 without insulin (RPMI/B27-). Subsequently, the cells were treated with 2 μM Wnt inhibitor C59 (Tocris, Bristol, UK) in RPMI/B27- for another 2 days. Between 5–11 days of differentiation, RPMI/B27- media was used and changed every other day until beating cells were observed when the media was switched to RPMI 1640 supplemented with B27 with insulin (RPMI/B27+). To improve the CM purity, cells were cultured in RPMI/B27+ without glucose, but supplemented with 5 mM sodium L-lactate for 3 days. On day 14, the hiPSC-CMs were dissociated with TrypLE 10x for 10 minutes (or until the cells lifted from the plate) and seeded in 6-well Matrigel coated plates at a density of 3x106 per well in RPMI/B27+ containing 10% Knockout Serum Replacement (KOSR) and ROCK inhibitor Y-27632 (Tocris, Bristol, UK). hiPSC-CMs were cultured in RPMI/B27+ with glucose for 3–5 days prior to switching to 3 mL of maturation media in each well of a 6 well plate (Feyen et al., 2020). Maturation media was composed as follows: DMEM without glucose (Thermo Fisher Scientific, 11966025) supplemented with 3 mM glucose (Sigma Aldrich, G7021), 10 mM L-lactate (Sigma Aldrich, 71718), 5 μg/ml Vitamin B12 (Sigma Aldrich, V6629), 0.82 μM Biotin (Sigma Aldrich, B4639), 5 mM Creatine monohydrate (Sigma Aldrich, C3630), 2 mM Taurine (Sigma Aldrich, T0625), 2 mM L-carnitine (Sigma Aldrich, C0283), 0.5 mM Ascorbic acid (Sigma Aldrich, A8960), 1x NEAA (Thermo Fisher Scientific, 11140),0.5%(w/v) Albumax (ThermoFisher Scientific, 11020021), 1x B27 and 1% KOSR (Thermo Fisher Scientific, 10828028). Cells were kept in maturation media culture for 3–4 weeks with media changes occurring every 4 days. For the optical voltage assay, cells were dissociated similarly to day 14 and replated in maturation media with 10% KOSR and Y-27632 and plated onto Matrigel-coated 384-well tissue culture plates (Greiner Bio-One, Kremsmünster, Austria) at a density of 20,000 cells/well.
METHOD DETAILS
High throughput optical action potential recording
All dye loading and compound administration methods were described and validated in detail in McKeithan et al. (McKeithan et al., 2017). 384 well plates containing hiPSC-CMs were placed on a 37°C heated surface for all manipulations to prevent temperature fluctuation. Each well was washed to remove the tissue culture media and replaced with Tyrode’s solution. VF2.1.Cl (Miller et al., 2012) loading solution was prepared as described in McKeithan et al. (McKeithan et al., 2017). Each plate was incubated at 37°C in a 5% CO2 incubator for 50 min. After incubation with VF2.1.Cl solution, each well was washed with Tyrode’s solution. Each test compound was loaded and incubated for 5 minutes prior to imaging. Time series images were acquired at 100Hz using an IC200 KIC instrument (Vala Sciences, California, USA) with excitation wavelength of 485/20 nm and emission filter 525/30 nm using a 0.75 NA 20x Nikon Apo VC objective. Image analysis was conducted using Cyteseer (Vala Sciences, California, USA) as previously described (Cerignoli et al., 2012; Lu et al., 2015; McKeithan et al., 2017). The output images from the IC200 KIC were loaded into Cyteseer and a whole well cardiac time series algorithm was executedontheimagefiles.Physiologicalparameters (beat rate, normalized area under the peak trace (normalized peak integral), and APD25, APD50, APD75 and APD90) were automatically calculated for each time series. EADs were quantified automatically by identifying peaks following a local minimum above a user defined threshold above the diastolic interval minimum. Data tables were analyzed with Microsoft Excel and dose responses and heat maps were generated with GraphPad Prism 7 software.
Ion channel modulators were purchased: Mexiletine (Toronto Research Chemical, Toronto, Canada), dofetilide (Tocris, Bristol, UK), tetrodotoxin (Tocris Bristol, UK), GS-967 (Medchem Express, Monmouth Junction, NJ, USA), ranolazine (Tocris, Bristol, UK), veratridine (Tocris, Bristol, UK) and ATX-II (Alomone Labs, Jerusalem, Israel).
Automated planar patch clamp
Whole cell planar patch clamp recordings were used to directly study the effects of test compounds on ion channel currents: INaP, INaL, and IKr (hERG) channels (Farre and Fertig, 2012). In all experiments the number of cells studied (n) for each drug concentration cited was indicated.
For dose response studies (Figure 2E–I), at least 6 increasing concentrations were included. For Na+ channel experiments, we used a stable HEK cell line expressing a long QT mutant, F1473C (Bankston et al., 2007), and for IKr we used a commercially available CHO stable cell line, hERG Duo (Bsys, Switzerland). Normalized blocking ratio data, for each of IKr, INaP and INaL, were fitted by a 4-parameter logistic fit. The following solutions were used in these experiments: Internal solution for K+ channel experiments: 50 mM KCl, 10 mM NaCl, 60 mM K-Fluoride, 20 mM EGTA, and 10 mM HEPES/KOH, pH 7.2. Internal solution for Na+ channel experiments: 50 mM CsCl, 10 mM NaCl, 60 mM Cs-Fluoride, 20 mM EGTA, 10 mM HEPES/CsOH, pH 7.2 External solutions for K+ and Na+ channel experiments: 140 mM NaCl, 4 mM KCl, 1mM MgCl2, 5 mM D-glucose monohydrate, and10 mM HEPES/NaOH pH 7.4. Control experiments contained 0.01% DMSO as did all drug containing solutions. Reagents were obtained from Sigma-Aldrich (St Louis, MO, USA). Patch clamp data were acquired with PatchMaster (HEKA, Germany) and PatchLiner Data acquisition (Nanion, Munich, Germany) and analyzed with Origin 7.0 (OriginLab, Northampton, MA, USA) and IgorPro (WaveMetrics, Portland, OR, USA) and MATLAB (Mathworks, Natick, MA, USA). Data were shown as mean ± SEM. Statistical data analyses was assessed with Student’s t test. Differences at P <0.05 were considered as statistically significant.
For the study of LQT3-causing SCN5A mutations, plasmids encoding WT and LQT3-mutations were transiently transfected Jouinto HEK-293Tcells byelectroporationusingtheMaxcyteSTX system (MaxCyte Inc., Gaithersburg, MD, USA). HEK-293T cells were grown to 70–80% confluency and then harvested using 5% trypsin, then a 500 μl aliquot of cell suspension was used to determine cell number and viability on an automated cell counter (ViCell, Beckman Coulter, Brea, CA, USA). Remaining cells were collected by gentle centrifugation (160 × g, 4 minutes), then the cell pellet was washed with 3 ml electroporation buffer (EBR100, MaxCyte Inc., Gaithersburg, MD, USA) and re-suspended in electroporation buffer at a density of 108 viable cells/ml. For each electroporation, plasmids encoding WT or LQT3-causing SCN5A mutations (40 μg) were added to 100 μl cell suspension (108 cells/ml). The DNA-cell suspension mix was then transferred to an OC-100 processing assembly (MaxCyte Inc., Gaithersburg, MD, USA) and electroporated using the optimization-5 preset protocol. Immediately after electroporation, 10 μl of DNase I (Sigma-Aldrich, St. Louis, MO, USA) was added to the DNA-cell suspension and the entire mixture was transferred to a 60 mm tissue culture dish and incubated for 30 min at 37°C in 5% CO2. Following incubation, cells were gently re-suspended in culture media, transferred to a T75 tissue culture flask and grown for 48 hours at 37°C in 5% CO2. Following incubation, cells were harvested, counted and re-suspended in external solution at 180,000 cells/ml. Cells were allowed to recover for at least 30 min at 15 °C while shaking on a rotating platform. Following equilibration, 10 μl of cell suspension was added to each well of a 384-well, single-hole, low resistance(2 MΘ) ‘chip’(Nanion Technologies, München, Germany). Whole-cell currents were recorded at room temperature in the whole-cell configuration. The external solution contained (in mM) the following: NaCl 140, KCl 4, CaCl2 2, MgCl2 1, HEPES 10, glucose 5, pH = 7.4). The internal solution contained (in mM) the following: CsF 110, CsCl 10, NaCl 10, HEPES 10, EGTA 20, pH = 7.2. Automated patch clamp recording was conducted using a Syncropatch 768PE (Nanion Technologies, Munich, Germany) using single hole low resistance 384-well patch plates. Pulse generation and data collection were done with PatchController384 V.1.3.0 and DataController384 V1.2.1(Nanion Technologies). Whole-cell currents were filtered at 3 kHz and acquired at 10 kHz. The access resistance and apparent membrane capacitance were estimated using built-in protocols. Series resistance was compensated 95% and leak and capacitance artifacts were subtracted out using the P/4 method. Cells were excluded from analysis if the maximum peak current was less than 300 pA. Test compounds were diluted in the external solution and prepared in a separate 24 wells plate. DMSO concentration was the same for each compound.
hiPSC-CM electrophysiology
Sodium currents were recorded from single hiPSC-CMs using the whole-cell patch-clamp technique. 5mm coverslips were coated with a 0.1% gelatin solution. hiPSC-CMs were plated and cultured for at least 2 weeks prior to recording. Each coverslip was transferred to a recording chamber (RC-25-F, Warner Instruments, Hamden, CT) perfused with Tyrode’s solution that was mounted on the stage of an inverted Olympus microscope. Patch pipettes were pulled glass capillaries (CORNING 7740, 1.65mm) with a P-2000 laser pipette puller (Sutter Instruments, Novato, CA, USA). Pipettes had electrode tip resistance of 1.5 MΘ to 5.5 MΘ and access resistance of < 8MΘ. In some voltage recordings, series resistance and cell capacitance were compensated 30 to 60%. The intracellular solution consisted of 120 mM CsCl, 20 mM tetraethylammonium chloride (TEA-Cl), 10 mM HEPES, 2.25 mM EGTA, 1 mM CaCl2, 2 mM MgCl2, pH 7.4. All recordings were conducted at room temperature. Current response traces were acquired using the Axon 200B amplifier and were digitally sampled at 10 kHz using Digidata 1322A digitizer hardware and pClamp 10.2 software (Molecular Devices, SanJose, CA, USA). Current responses were filtered with an 8-pole Bessel analog low-pass filter at 1–2 kHz cutoff frequency. Current amplitude at the end of the depolarizing step was corrected by the leak current and the corrected current amplitude was averaged for n=10 cells. After the leak-correction, the average value of the recorded current amplitude was computed over the last 50 ms of the 150 ms depolarizing pulse of each 250 ms sweep. The resulting value was averaged over 3 consecutive sweeps.
Chemistry
Chemical names were generated with ChemDraw Ultra version 11 (PerkinElmer, Waltham, MA, USA). Experiments were carried out under inert atmosphere when oxygen- or moisture-sensitive reagents or intermediates were employed. Commercial solvents and reagents were used without further purification. Mexiletine was purchased from Toronto Research Chemicals (Ontario, Canada). 2-(2,6-dimethylphenoxy)-1-phenylethan-1-amine(phenylmexiletine, MexA1) was purchased from Millipore Sigma (Burlington, MA, USA).
Microwave reactions were conducted using a Biotage Initiator microwave synthesizer (Biotage, Uppsala, Sweden). Reaction products were purified, when necessary, using an Isco Combiflash Rf flash chromatography system (Teledyne-Isco, Lincoln, NE, USA) with the solvent systems indicated. Nuclear magnetic resonance (NMR) data were recorded on a Varian Mercury 300 MHz Spectrometer (Agilent, Santa Clara, CA, USA) or with a Bruker 500 MHz instrument at NuMega Resonance Laboratories (San Diego, CA, USA) using CDCl3 as a solvent except where indicated. Chemical shifts for nuclear magnetic resonance (NMR) data were expressed in parts per million (ppm, d) referenced to residual peaks from the deuterated solvents. Mass spectrometry (MS) data was reported from liquid chromatography-mass spectrometry (LCMS) instrumentation. Electrospray ionization (ESI) mass spectral data was obtained using an Agilent 1100 LC/MS (Agilent, Santa Clara, CA, USA). Final test compounds had a purity greater than 95% based on LCMS analysis using UV-Vis detection at 275 nM and 220 nM.
Illustration of methods for the synthesis of compounds MexA2–5
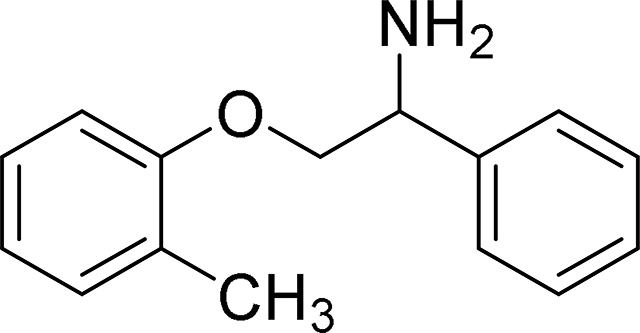
1-Phenyl-2-(o-tolyloxy)ethan-1-amine (MexA2)
Step 1: 1-phenyl-2-(o-tolyloxy)ethan-1-one
To a solution of α-bromoacetophenone (1.0 g, 5.0 mmol) and 2-methylphenyl (0.65 g, 6.0 mmol) in 5 mL of DMF added potassium carbonate (0.83 g, 6.0 mmol). The mixture was stirred at 21 ºC for 16 h and then diluted with water. The product was extracted with ether. The organic phase was washed (1M sodium hydroxide, water, brine), dried (magnesium sulfate) and subjected to chromatography on silica gel using a gradient of ethyl acetate in hexanes to afford 0.61g (45%) of the product as an off white solid.
ESI/MS: calculated C15H14O2 m/z = 226.1, found m/z = 227.0 [M+H]+.
1H NMR (CDCl3): 2.30 (s, 3H), 5.26 (s, 2H), 6.75 (d, J =8.0 Hz, 1H), 6.89 (t, J = 7.5 Hz, 1H), 7.09 – 7.17 (m, 2H), 7.46 – 7.51 (m, 2H), 7.58 – 7.64 (m, 1H), 7.99 – 8.02 (m, 2H).
Step 2: 1-phenyl-2-(o-trolley)ethanone O-benzyl oxime
To 0.2 g (0.9 mmol) of 1-phenyl-2-(o-tolyloxy)ethan-1-one in 5 mL of ethanol was added pyridine (0.35 mL, 4.4 mmol) and O-benzylhydroxylamine (0.51 mL, 4.4 mmol). The mixture was stirred at 40 ºC for 16 h, treated with additional pyridine (0.17 mL, 2.2 mmol) and O-benzylhydroxylamine (0.25 mL, 2.2 mmol) and stirred at 55 ºC for additional 16 h. The volatiles were removed under reduced pressure and the residue was partitioned between aqueous 1M hydrochloric acid and ether. The organic phase was washed (water, brine) and dried over magnesium sulfate. The oxime was purified by chromatography on silica gel using a mixture of ether and hexanes to afford the product as a mixture of E/Z isomers (64 mg, 21% yield).
ESI/MS: calculated C22H21NO2 m/z = 331.2, found m/z = 332.0[M+H]+.
1H NMR (CDCl3): 2.03 (s,3H), 5.25 (s, 2H), 5.29 (s, 2H), 6.80 – 6.89 (m, 2H), 7.03 – 7.09 (m, 2H), 7.31 – 7.45 (m, 9H), 7.63 – 7.66 (m, 1H).
1-Phenyl-2-(o-tolyloxy)ethan-1-amine (MexA2)
A solution of 30 mg (0.09 mmol) of the above oxime in 0.4 mL of THF was treated with 0.4 mL of 1M borane-THF (0.4 mmol) and stirred at 21 ºC for 16 h. The mixture was treated with 1M aqueous hydrochloric acid and extracted with ether. The combined organic phases were dried over sodium sulfate. The crude product was purified by preparative thin layer chromatography and the product was treated with an excess of hydrogen chloride in ether. The precipitated solid was filtered, rinsed with ether and vacuum dried to afford the product as a hydrochloride salt (16.4 mg, 68% yield).
ESI/MS: calculated C15H17NO m/z = 227.1, found m/z = 228.0 [M+H]+.
1H NMR (CDCl3): 2.24 (s, 3H), 2.88 (bs, 2H), 3.98 – 4.05 (m, 1H), 4.13 (dd, J = 4.1 Hz and 9.1 Hz, 1H), 4.45 – 4.49 (m, 1H), 6.75 – 6.78 (m, 1H), 6.83 – 6.88 (m, 1H), 7.08 – 7.13 (m, 2H), 7.29 – 7.37 (m, 4H), 7.44 – 7.47 (m, 1H).
The following compounds were generated by the sequence described above with appropriate modifications:
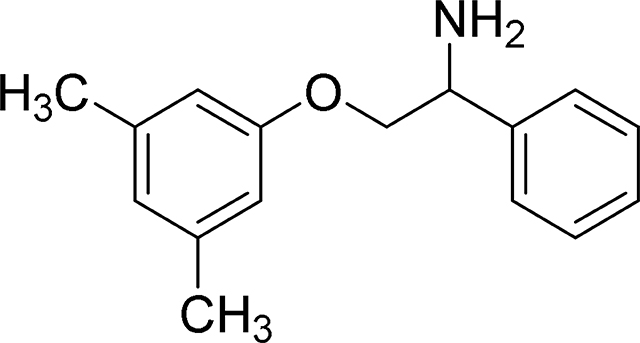
2-(3,5-Dimethylphenoxy-1-phenylethan-1-amine (MexA3)
ESI/MS: calculated m/z for C16H19NO: 241.1 Found m/z: 242.1 [M+H]+.
1H NMR (300 MHz, Methanol-d4) δ 7.42 – 7.58 (m, 4H), 7.42 – 7.58 (m, 4H), 6.64 (s, 3H), 4.73 (dd, J = 4.1, 8.4 Hz, 1H), 4.18 – 4.38 (m, 2H), 2.27 (d, J = 0.7 Hz, 6H).
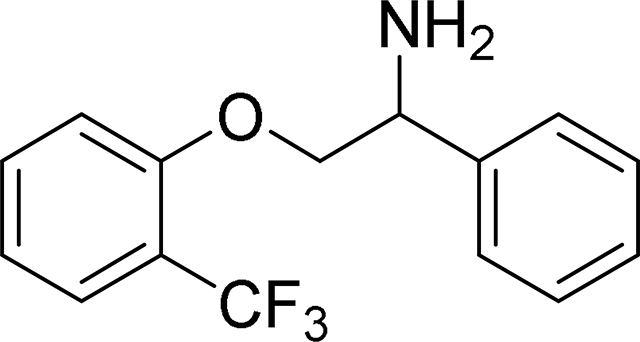
1-Phenyl-2-(2-(trifluoromethyl)phenoxy)ethan-1-amine (MexA4)
ESI/MS: calculated C15H14F3NO m/z = 281.1, found m/z = 282.0 [M+H]+.
1H NMR (CDCl3): 2.38 (bs, 2H), 4.02 – 4.11 (m, 1H), 4.19 – 4.27 (m, 1H), 4.48 – 4.58 (m, 1H), 6.91 – 6.94 (m, 1H), 6.98 – 7.03 (m, 1H), 7.29 – 7.42 (m, 4H), 7.44 – 7.49 (m, 2H), 7.54 – 7.58 (m, 1H).
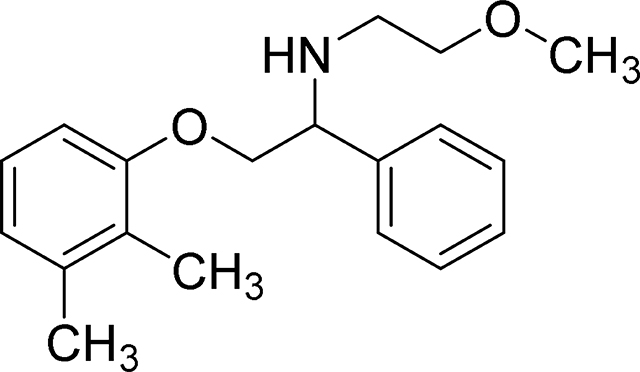
2-(2,3-dimethylphenoxy)-N-(2-methoxyethyl)-1-phenylethan-1-amine (MexA5)
This compound was synthesized in two steps from the precursor 2-(2,3-dimethylphenoxy)-1-phenylethan-1-amine, which in turn was generated by the method described above.
Properties for precursor precursor 2-(2,3-dimethylphenoxy)-1-phenylethan-1-amine:
ESI/MS: calculated m/z for C16H19NO: 241.1 Found m/z: 242.0 [M+H]+.
1H NMR (300 MHz, CDCl3) δ 7.47 (d, J = 7.43 Hz, 1H), 7.28 – 7.43 (m, 4H), 7.02 (t, J = 7.84 Hz, 1H), 6.79 (d, J = 7.70 Hz, 1H), 6.68 (d, J = 7.98 Hz, 1H), 4.47 (dd, J = 3.85, 8.25 Hz, 1H), 4.10 (dd, J = 3.85, 9.08 Hz, 1H), 3.87 – 4.04 (m, 1H), 2.23 – 2.35 (m, 3H), 2.11 – 2.22 (m, 3H)
Step 1: N-(2-(2,3-dimethylphenoxy)-1-phenylethyl)-2-methoxyacetamide.
To a solution of 2-methoxyacetic acid (0.068 mL, 0.89 mmol) and trimethylamine (0.126 mL, 0.9 mmol) in dichloromethane (1 mL) added 400 mg (0.9 mmol) of (Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP). After 5 min a solution of 198 mg (0.82 mmol) of 2-(2,3-dimethylphenoxy)-1-phenylethan-1-amine was added. After stirring overnight at room temperature, the mixture was diluted with ethyl acetate. The organic phase was washed (water, saturated aqueous sodium bicarbonate, water and brine) and dried over magnesium sulfate. The crude product was purified on silica gel using a methanol:dichloromethane gradient. 156 mg (61%) of the target compound was obtained.
Rf = 0.7 (19:1 dichloromethane:methanol)
1H NMR (300 MHz, CDCl3) δ 7.22 – 7.49 (m, 5H), 7.04 (t, J = 7.98 Hz, 1H), 6.80 (d, J = 7.70 Hz, 1H), 6.68 (d, J = 8.25 Hz, 1H), 5.39 – 5.59 (m, 1H), 4.18 – 4.39 (m, 2H), 3.97 (s, 2H), 3.39 – 3.51 (m, 3H), 2.28 (s, 3H), 2.10 – 2.18 (m, 3H)
Step 2: 2-(2,3-dimethylphenoxy)-N-(2-methoxyethyl)-1-phenylethan-1-amine.
A solution of 156 mg (0.5 mmol) of N-(2-(2,3-dimethylphenoxy)-1-phenylethyl)-2-methoxyacetamide in 3.0 mL of THF was added dropwise at room temperature to a 1M solution of lithium aluminum hydride in THF (2.4 mL, 2.4 mmol). The mixture was then heated at 75 oC overnight. The mixture was cooled in an ice/brine bath and treated with water(0.087 mL) followed by 1M aq NaOH (0.087 mL). The precipitate was removed by filtration and rinsed with ethyl acetate. The filtrate was dried over magnesium sulfate, concentrated and purified by chromatography using a methanol dichloromethane gradient to afford the amine free base as a colorless oil.
1H NMR (300 MHz, CDCl3) δ 7.18 – 7.54 (m, 5H), 6.91 – 7.07 (m, 1H), 6.77 (d, J = 7.43 Hz, 1H), 6.65 (d, J = 7.98 Hz, 1H), 4.11 – 4.22 (m, 1H), 3.92 – 4.11 (m, 2H), 3.43 – 3.60 (m, 2H), 3.38 (s, 3H), 2.59 – 2.87 (m,2H), 2.29 (s, 3H), 2.18 (s, 3H).
The oil was dissolved in ether (9 mL) and treated with 4M HCl in dioxane (0.5 mL, 2.0 mmol). Hexanes and toluene were added and the mixture was evaporated under reduced pressure. The residue was treated with hexane:ether and the solid obtained was filtered, rinsed with hexanes and vacuum dried. 56 mg (33%) of the hydrochloride salt was obtained.
ESI/MS: calculated m/z for C16H25NO2: 299.1 Found m/z: 300.1 [M+H]+.
1H NMR (300 MHz, DMSO-d6) δ 9.89 – 9.27 (br.d., 2H), 7.64 (br. s., 1H), 7.16 – 7.60 (m, 3H), 7.02 (br. s., 1H), 6.77 (br. s., 1H), 4.77 (br. s., 1H), 4.51 (br. s., 1H), 4.34 (br. s., 1H), 3.74 – 3.46 (m., 2H), 3.33 – 3.43 (m, 2H), 3.30 (s, 3H), 2.18 (s, 3H), 1.99 (s, 3H)
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± standard error of the mean (SEM) as indicated. Statistical analysis was conducted using GraphPad Prism 7(GraphPad Software, Inc., San Diego, CA). Concentration-dependent IC50 curves were fitted using a log(inhibitor) versus response -variable slope (four parameters). Significance was calculated using the two-tailed unpaired Student’s t tests. Data that were not normally distributed were tested using the Mann–Whitney-Wilcoxon test. One-way ANOVA with Dunnett’s method was used for multiple comparisons. P value < 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Y-27632 dihydrochloride | Tocris | 1254 |
| CHIR99021 | Tocris | 4423 |
| Wnt-C59 | Tocris | 5148 |
| VF2.1.Cl | (Miller et al., 2012) | N/A |
| Mexiletine hydrochloride | Toronto Research Chemical | M340800 |
| Dofetilide | Tocris | 3757 |
| Tetrodotoxin citrate | Tocris | 1069 |
| GS-967 | Medchem Express | HY-12593 |
| Ranolazine dihydrochloride | Tocris | 3118 |
| Veratridine | Tocris | 2918 |
| ATX-II | Alomone Labs | STA-700 |
| MexA1-A5 | This study | MexA1-MexA5 |
| Experimental Models: Cell Lines | ||
| Human iPSC line (healthy donor) | SCVI Biobank | SCVI-273 |
| Human iPSC line (healthy donor) | SCVI Biobank | SCVI-15S1 |
| Human iPSC line (healthy donor) | SCVI Biobank | SCVI-113 |
| Human iPSC line SCN5A F1473C | (McKeithan et al., 2017) | N/A |
| Human iPSC line SCN5A N406K | (Spencer et al., 2014) | N/A |
| MyCell Cardiomyocytes (SCN5A F1472C) | Cellular Dynamics International | Lot #: 1583.763.CM001 |
| iCell Cardiomyocytes (healthy donor) | Cellular Dynamics International | Lot #s: 1097546, 1291715, 1093711, 1031999 |
| CHO hERG-Duo Cell Line | Bsys | https://www.bsys.ch/ |
| Software and Algorithms | ||
| Cyteseer | Vala Sciences | http://www.valasciences.com/ |
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| PatchMaster | HEKA | http://www.heka.com/ |
| PatchLiner Data acquisition | Nanion | https://www.nanion.de/en/ |
| Origin 7.0 | OriginLab | https://www.originlab.com/ |
| IgorPro | WaveMetrics | https://www.wavemetrics.com/ |
| MATLAB | Mathworks | https://www.mathworks.com/ |
| PatchController384 V.1.3.0 | Nanion | https://www.nanion.de/en/ |
| DataController384 V1.2.1 | Nanion | https://www.nanion.de/en/ |
| pClamp 10.2 | Molecular Devices | https://www.moleculardevices.com/ |
| ChemDraw Ultra version 11 | PerkinElmer | https://www.perkinelmer.com/ |
Highlights.
LQT3 hiPSC-cardiomyocytes recapitulate disease phenotypes and drug responses
Large-scale functional screens using LTQ3 hiPSC-CMs enable drug optimization
Optimized mexiletine analogues decrease proarrhythmic liability and improve potency
Acknowledgements
We would like to thank J. Wu (Stanford Cardiovascular Institute, Stanford, CA) and the Stanford Biobank for the healthy donor hiPSC lines. We would also like to thank B. Conklin (Gladstone Institute of Cardiovascular Disease) for the LQT3 SCN5A N406K hiPSC line, and Tatiana Abramova (Northwestern University) for engineering and expressing mutant Nav 1.5 plasmid constructs. This research was made possible by grants from the National Institutes of Health (NIH R01HL113601, R01HL130840, R01HL141358, P01HL141084 and R21HL141019 to MM; U01 HL131419 (ALG); R01HL123483 to RSK), a grant from the American Heart Association (9SFRN34820006; ALG), and a grant from the California Institute for Regenerative Medicine (CIRM Grant Number TR4-06857 to JRC and MM). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official view of CIRM or any other agency of the State of California. Flow cytometry and high throughput screening services were supported by NIH P30CA030199 at the Sanford-Burnham-Prebys Medical Discovery Institute. This research was also supported by Stanford Cardiovascular Institute and Stanford School of Medicine funds to MM. DAMF was funded by the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie grant agreement No. 708459.
Footnotes
Declaration of Interests
Human BioMolecular Research Institute has filed a patent application on mexiletine analogues, in which J.R.C., K.J.O. and D.A.R. are co-inventors. M.M. serves on the scientific advisory board of Vala Sciences, which manufactures a high content instrument used in these studies. A.L.G. serves on a scientific advisory board for cardiometabolic diseases at Amgen, Inc. The other authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, Deal BJ, Dickfeld T, Field ME, Fonarow GC, et al. (2018). 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Circulation 138, e272–e391. [DOI] [PubMed] [Google Scholar]
- Bankston JR, Yue M, Chung W, Spyres M, Pass RH, Silver E, Sampson KJ, and Kass RS (2007). A novel and lethal de novo LQT-3 mutation in a newborn with distinct molecular pharmacology and therapeutic response. PLoS One 2, e1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L, Liu G, Smith-Maxwell C, Wang WQ, El-Bizri N, Hirakawa R, Karpinski S, Li CH, Hu L, Li XJ, et al. (2013). A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J Pharmacol Exp Ther 344, 23–32. [DOI] [PubMed] [Google Scholar]
- Blinova K, Dang Q, Millard D, Smith G, Pre-proofPierson J, Guo L,Brock M,Lu HR, Kraushaar U, Zeng H, et al. (2018). International Multisite Study of Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Drug Proarrhythmic Potential Assessment. Cell Rep 24, 3582–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerignoli F, Charlot D, Whittaker R, Ingermanson R, Gehalot P, Savchenko A, Gallacher DJ, Towart R, Price JH, McDonough PM, et al. (2012). High throughput measurement of Ca(2)(+) dynamics for drug risk assessment in human stem cell-derived cardiomyocytes by kinetic image cytometry. J Pharmacol Toxicol Methods 66, 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bellis M, De Luca A, Desaphy JF, Carbonara R, Heiny JA, Kennedy A, Carocci A, Cavalluzzi MM,Lentini G,Franchini C, et al. (2013). Combined Modifications of Mexiletine Pharmacophores for New Lead Blockers of Na(v)1.4 Channels. Biophys J 104, 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Natuzzi F, Desaphy JF, Loni G, Lentini G, Franchini C, Tortorella V, and Camerino DC (2000). Molecular determinants of mexiletine structure for potent and use-dependent block of skeletal muscle sodium channels. Mol Pharmacol 57, 268–277. [PubMed] [Google Scholar]
- De Luca A, Talon S, De Bellis M, Desaphy JF, Franchini C, Lentini G, Catalano A, Corbo F, Tortorella V, and Conte-Camerino D (2003). Inhibition of skeletal muscle sodium currents by mexiletine analogues: specific hydrophobic interactions rather than lipophilia per se account for drug therapeutic profile. N-S Arch Pharmacol 367, 318–327. [DOI] [PubMed] [Google Scholar]
- Desaphy JF, De Luca A, Tortorella P, De Vito D, George AL Jr., and Conte Camerino D (2001). Gating of myotonic Na channel mutants defines the response to mexiletine and a potent derivative. Neurology 57, 1849–1857. [DOI] [PubMed] [Google Scholar]
- Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, Gerard R, Badi L, Kam-Thong T, Bu L, et al. (2014). Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep 9, 810–821. [DOI] [PubMed] [Google Scholar]
- Fabritz L, Damke D, Emmerich M, Kaufmann SG, Theis K, Blana A, Fortmuller L, Laakmann S, Hermann S, Aleynichenko E, et al. (2010). Autonomic modulation and antiarrhythmic therapy in a model of long QT syndrome type 3. Cardiovasc Res 87, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre C, and Fertig N (2012). HTS techniques for patch clamp-based ion channel screening -advances and economy. Expert Opin Drug Discov 7, 515–524. [DOI] [PubMed] [Google Scholar]
- Feyen DAM, McKeithan WL, Bruyneel AAN, Spiering S, Hormann L, Ulmer B, Zhang H, Briganti F, Schweizer M, Hegyi B, et al. (2020). Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep 32, 107925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler LR, Chapman K, Xie M, Maifoshie E, Jenkins M, Golforoush PA, Bellahcene M, Noseda M, Faust D, Jarvis A, et al. (2019). MAP4K4 Inhibition Promotes Survival of Human Stem Cell-Derived Cardiomyocytes and Reduces Infarct Size In Vivo. Cell Stem Cell 24, 579–591 e512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridericia LS (2003). The duration of systole in an electrocardiogram in normal humans and in patients with heart disease. 1920. Ann Noninvasive Electrocardiol 8, 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualdani R, Tadini-Buoninsegni F, Roselli M, Defrenza I, Contino M, Colabufo NA, and Lentini G (2015). Inhibition of hERG potassium channel by the antiarrhythmic agent mexiletine and its metabolite m-hydroxymexiletine. Pharmacol Res Perspect 3, e00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnatiuk A, and Mercola M (2019). Stars in the Night Sky: iPSC-Cardiomyocytes Return the Patient Context to Drug Screening. Cell Stem Pre-proofCell 24,506–507. [DOI] [PubMed] [Google Scholar]
- Horvath B, and Bers DM (2014). The late sodium current in heart failure: pathophysiology and clinical relevance. ESC Heart Fail 1, 26–40. [DOI] [PubMed] [Google Scholar]
- Hu RM, Tester DJ, Li R, Sun T, Peterson BZ, Ackerman MJ, Makielski JC, and Tan BH (2018). Mexiletine rescues a mixed biophysical phenotype of the cardiac sodium channel arising from the SCN5A mutation, N406K, found in LQT3 patients. Channels (Austin) 12, 176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltun DO, Parkhill EQ, Elzein E, Kobayashi T, Notte GT, Kalla R, Jiang RH, Li X, Perry TD, Avila B, et al. (2016). Discovery of triazolopyridine GS-458967, a late sodium current inhibitor (Late INai) of the cardiac NaV 1.5 channel with improved efficacy and potency relative to ranolazine. Bioorg Med Chem Lett 26, 3202–3206. [DOI] [PubMed] [Google Scholar]
- Lu HR, Whittaker R, Price JH, Vega R, Pfeiffer ER, Cerignoli F, Towart R, and Gallacher DJ (2015). High Throughput Measurement of Ca++ Dynamics in Human Stem Cell-Derived Cardiomyocytes by Kinetic Image Cytometery: A Cardiac Risk Assessment Characterization Using a Large Panel of Cardioactive and Inactive Compounds. Toxicol Sci 148, 503–516. [DOI] [PubMed] [Google Scholar]
- Maier LS, and Sossalla S (2013). The late Na current as a therapeutic target: where are we? J Mol Cell Cardiol 61, 44–50. [DOI] [PubMed] [Google Scholar]
- Matsa E, Ahrens JH, and Wu JC (2016). Human Induced Pluripotent Stem Cells as a Platform for Personalized and Precision Cardiovascular Medicine. Physiol Rev 96, 1093–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzanti A, Maragna R, Faragli A, Monteforte N, Bloise R, Memmi M, Novelli V, Baiardi P, Bagnardi V, Etheridge SP, et al. (2016). Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3. J Am Coll Cardiol 67, 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeithan WL, Savchenko A, Yu MS, Cerignoli F, Bruyneel AAN, Price JH, Colas AR, Miller EW, Cashman JR, and Mercola M (2017). An Automated Platform for Assessment of Congenital and Drug-Induced Arrhythmia with hiPSC-Derived Cardiomyocytes. Front Physiol 8, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EW, Lin JY, Frady EP, Steinbach PA, Kristan WB, and Tsien RY (2012). Optically monitoring voltage in neurons by photo-induced electron transfer through molecular wires. Proc Natl Acad Sci U S A 109, 2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy LL, Moon-Grady AJ, Cuneo BF, Wakai RT, Yu S, Kunic JD, Benson DW, and George AL Jr. (2012). Developmentally regulated SCN5A splice variant potentiates dysfunction of a novel mutation associated with severe fetal arrhythmia. Heart Rhythm 9, 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuyens D, Stengl M, Dugarmaa S,Rossenbacker T,Compernolle V,Rudy Y,Smits JF, Flameng W, Clancy CE, Moons L, et al. (2001). Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med 7, 1021–1027. [DOI] [PubMed] [Google Scholar]
- Priori S, Denjoy I, Blair C, Hellawell J, Satler CA, and Ackerman MJ (2018). A Phase 3 Single-Blind Study of Eleclazine in Subjects With Type 3 Long QT Syndrome. Circulation 136. [Google Scholar]
- Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G,et al. (2015).2015ESCGuidelinesforthe management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 36, 2793–2867. [DOI] [PubMed] [Google Scholar]
- Roselli M, Carocci A, Budriesi R, Micucci M, Toma M, Mannelli LD, Lovece A, Catalano A, Cavalluzzi MM, Bruno C, et al. (2016). Synthesis, antiarrhythmic activity, and toxicological evaluation of mexiletine analogues. Eur J Med Chem 121, 300–307. [DOI] [PubMed] [Google Scholar]
- Ruan Y, Denegri M, Liu N, Bachetti T, Seregni M, Morotti S, Severi S, Napolitano C, and Priori SG (2010). Trafficking defects and gating abnormalities of a novel SCN5A mutation question gene-specific therapy in long QT syndrome type 3. Circ Res 106, 1374–1383. [DOI] [PubMed] [Google Scholar]
- Ruan Y, Liu N, Bloise R, Napolitano C, and Priori SG (2007). Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation 116, 1137–1144. [DOI] [PubMed] [Google Scholar]
- Schroder EA, Burgess DE, Manning CL, Zhao Y, Moss AJ, Patwardhan A, Elayi CS, Esser KA, and Delisle BP (2014). Light phase-restricted feeding slows basal heart rate to exaggerate the type-3 long QT syndrome phenotype in mice. Am J Physiol Heart Circ Physiol 307, H1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen LS, et al. (1995). Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 92, 3381–3386. [DOI] [PubMed] [Google Scholar]
- Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, Churko JM, Kitani T, Wu H, Holmstrom A, et al. (2017). High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver ES, Liberman L, Chung WK, Spotnitz HM, Chen JM, Ackerman MJ, Moir C, Hordof AJ, and Pass RH (2009). Long QT syndrome due to a novel mutation in SCN5A: treatment with ICD placement at 1 month and left cardiac sympathetic denervation at 3 months of age. J Interv Card Electrophysiol 26, 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Kerndt C, and Zeltser R (2020). Mexiletine. In StatPearls (Treasure Island (FL)). [Google Scholar]
- Spencer CI, Baba S, Nakamura K, Hua EA, Sears MA, Fu CC, Zhang J, Balijepalli S, Tomoda K, Hayashi Y, et al. (2014). Calcium transients closely reflect prolonged action potentials in iPSC models of inherited cardiac arrhythmia. Stem Cell Reports 3, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, and Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 136, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrenoire C, Wang K, Tung KW, Chung WK, Pass RH, Lu JT, Jean JC, Omari A, Sampson KJ, Kotton DN, et al. (2013). Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol 141, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Vivas EL, and Izpisua Belmonte JC (2011). Diseases in a dish: modeling human genetic disorders using induced pluripotent cells. Nat Med 17, 1570–1576. [DOI] [PubMed] [Google Scholar]
- Zablocki JA, Elzein E, Li X, Koltun DO, Parkhill EQ, Kobayashi T, Martinez R, Corkey B, Jiang H, Perry T, et al. (2016). Discovery of Dihydrobenzoxazepinone (GS-6615) Late Sodium Current Inhibitor (Late INai), a Phase II Agent with Demonstrated Preclinical Anti-Ischemic and Antiarrhythmic Properties. J Med Chem 59, 9005–9017. [DOI] [PubMed] [Google Scholar]
- Zhu W, Mazzanti A, Voelker TL, Hou P, Moreno JD, Angsutararux P, Naegle KM, Priori SG, and Silva JR (2019). Predicting Patient Response to the Antiarrhythmic Mexiletine Based on Genetic Variation. Circ Res 124, 539–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study. This study did not generate code.