

Abstract
The role of truncated androgen receptor splice variant-7 (AR-V7) in prostate cancer biology is an unresolved question. Is it simply a marker of resistance to 2nd generation androgen receptor signaling inhibitors (ARSi) like Abiraterone Acetate (Abi) and Enzalutamide (Enza) or a functional driver of lethal resistance via its ligand-independent transcriptional activity? To resolve this question, the correlation between resistance to ARSi and genetic chances and expression of full length AR (AR-FL) vs. AR-V7 were evaluated in a series of independent patient-derived xenografts (PDXs). While all PDXs lack PTEN expression, there is no consistent requirement for mutation in TP53, RB1, BRCA2, PIK3CA, or MSH2, or expression of SOX2 or ERG and ARSi-resistance. Elevated expression of AR-FL alone is sufficient for Abi- but not Enza-resistance, even if AR-FL is gain-of-function (GOF) mutated. Enza-resistance is consistently correlated with enhanced AR-V7 expression. In vitro and in vivo growth responses of Abi-/Enza-resistant LNCaP-95 cells in which CRISPR-Cas9 was used to knockout AR-FL or AR-V7 alone or in combination were evaluated. Combining these growth responses with RNAseq analysis demonstrates that both AR-FL and AR-V7 dependent transcriptional complementation are needed for Abi/Enza resistance.
Introduction
Androgen stimulates androgen receptor (AR)-dependent transcriptional regulation within prostate stromal cells activating secretion of a combination of paracrine growth and survival factors (e.g. IGF, EGF, FGFs) while simultaneously repressing secretion of paracrine death inducing factors (e.g. TGF-β ligands)18, 25. In the presence of physiologic androgen, prostate stromal cells secrete sufficient paracrine growth and survival factors to maintain homeostatic epithelial cell turnover, preventing gland regression without inducing neoplastic overgrowth25. Under these conditions, epithelial homeostasis is maintained and androgen-induced cell autonomous AR-dependent signaling within prostate epithelial cells induces their terminal differentiation [i.e. expression of prostate-specific differentiation marker genes such as prostate-specific antigen (PSA) and human kallikrein-2 (hK2)]15. This differentiation suppresses their proliferation, despite the chronic presence of high levels of stromal cell-derived paracrine growth factors15.
During prostatic carcinogenesis, there is conversion from AR-regulated stromal paracrine dependency by normal prostate epithelium to cancer cells acquiring autonomous stromal cell-independent AR-stimulated malignant growth25, 41. Such cell autonomous growth involves losing normal AR function as a growth suppressor and instead acquiring ability to act as an oncogenic GOF stimulator of malignant growth15, 25. These oncogenic acquisitions “addict” prostate cancer cells to cell autonomous AR signaling. This addiction can involve cancer cells acquiring cell autonomous ligand-dependent AR transcription preventing their apoptotic cell death while also inducing proliferation, making these cancers AR-dependent for their lethal growth19. Alternatively, prostate cancer cells can lose their dependence on AR survival signaling while retaining a sensitivity to AR signaling to enhance their rate of malignant proliferation25. This makes them AR-sensitive, but not absolutely dependent upon such continuous signaling. Regardless of whether AR signaling addiction results in dependency vs. sensitivity, it provides rationale for the use of androgen deprivation therapy (ADT) for metastatic prostate cancer25.
Eventual resistance to first-line (i.e. primary) ADT utilizing luteinizing hormone-releasing hormone (LHRH) analogs to suppress circulating testosterone (T) to a castrate level alone or in combination with Casodex (a 1st generation anti-androgen) is essentially universal. Such castration-resistant prostate cancer (CRPC) patients are subsequently given 2nd generation AR signaling inhibitors (ARSi) to suppress AR signaling using steroid synthesis inhibitors [e.g. Abiraterone Acetate, (Abi)] to eliminate non-testicular androgen ligands and/or next-generation ligand binding domain (LBD) antagonists [e.g. Enzalutamide, (Enza)] that target the full length AR (AR-FL) protein35. Subsequent resistance to these 2nd generation ARSi is also essentially universal and often associated with significantly elevated expression of both AR-FL and truncated AR splice variant-7 (AR-V7)2. AR-V7 originates from contiguous splicing of AR exons 1, 2, and 3 with the cryptic exon 3 (CE3) present within the canonical intron 3 of the AR gene. This generates a transcript which encodes for a truncated protein lacking C-terminal LBD, thus acquiring ligand-independent transcriptional activity7, 10, 12.
While expression of AR-V7 protein is rare in primary PC, nuclear AR-V7 expression is detectable in response to primary ADT alone in most patients, and further increases during Abi- or Enza- therapy2, 35. This raises the question of whether AR-V7 protein expression is simply associated with enhanced AR-FL expression as a marker of resistance to ARSi, or whether a critical level of AR-V7 is required for such ARSi-refractory lethal cancer growth. Consistent with this latter possibility is the observation that AR-V7 has cistromes and thus transcriptional outputs that are distinct from those directed by AR-FL and which are consistent with genomic features of disease progression in a low-androgen environment27. Thus, a series of PDXs in which the genetic and ph enotypic changes were followed before and after the development of ARSi resistance were used to determine the role of AR-V7 in this progression. In addition, in vitro and in vivo growth and transcriptional response of Abi- and Enza-resistant LNCaP-95 (LN-95) cells to CRISPR-Cas9 knockout (KO) of AR-FL vs. AR-V7 alone or in combination were evaluated.
Results
Response of Prostate Cancer PDX lacking AR-V7 to Abiraterone and Enzalutamide
CWR22 PDX is derived from a primary prostate cancer with an AR H875Y mutation from a hormone treatment-naïve European-American patient37. Its xenograft growth in adult male NSG mice is highly androgen sensitive as documented by its regression following castration with a subset (i.e. 40%) eventually relapsing14. Serial passage in castrated hosts of 1 of the relapses produced the CWR22-RH PDX so named because it is castration-Resistant and was produced at Hopkins14. CWR22-RH grows equally well in an intact or castrated NSG mouse with a doubling time (DT) of 10+/−2 days. Histologically, like the parental CWR22, it is a poorly differentiated adenocarcinoma (Fig. 1a), which expresses c-Myc and Ki67 in >80% of cells (Tbl. 1). Like the parental CWR22, CWR22RH cells express prostate specific HOXB13, and express luminal cell-specific, but not basal cell or NE specific, markers (Tbl. 1, Fig. 2). CWR22-RH secretes PSA (serum PSA of 249 +/− 41 ng/ml/gram tumor). Genetically, it retains the heterogeneous LOF mutation in BRCA2 (E984fs) and loss of homozygosity (LOH) and LOF TP53 [G154F] mutation from the parental CWR22 (Tbl 1).
Figure 1: Characterization of CWR22-RH.
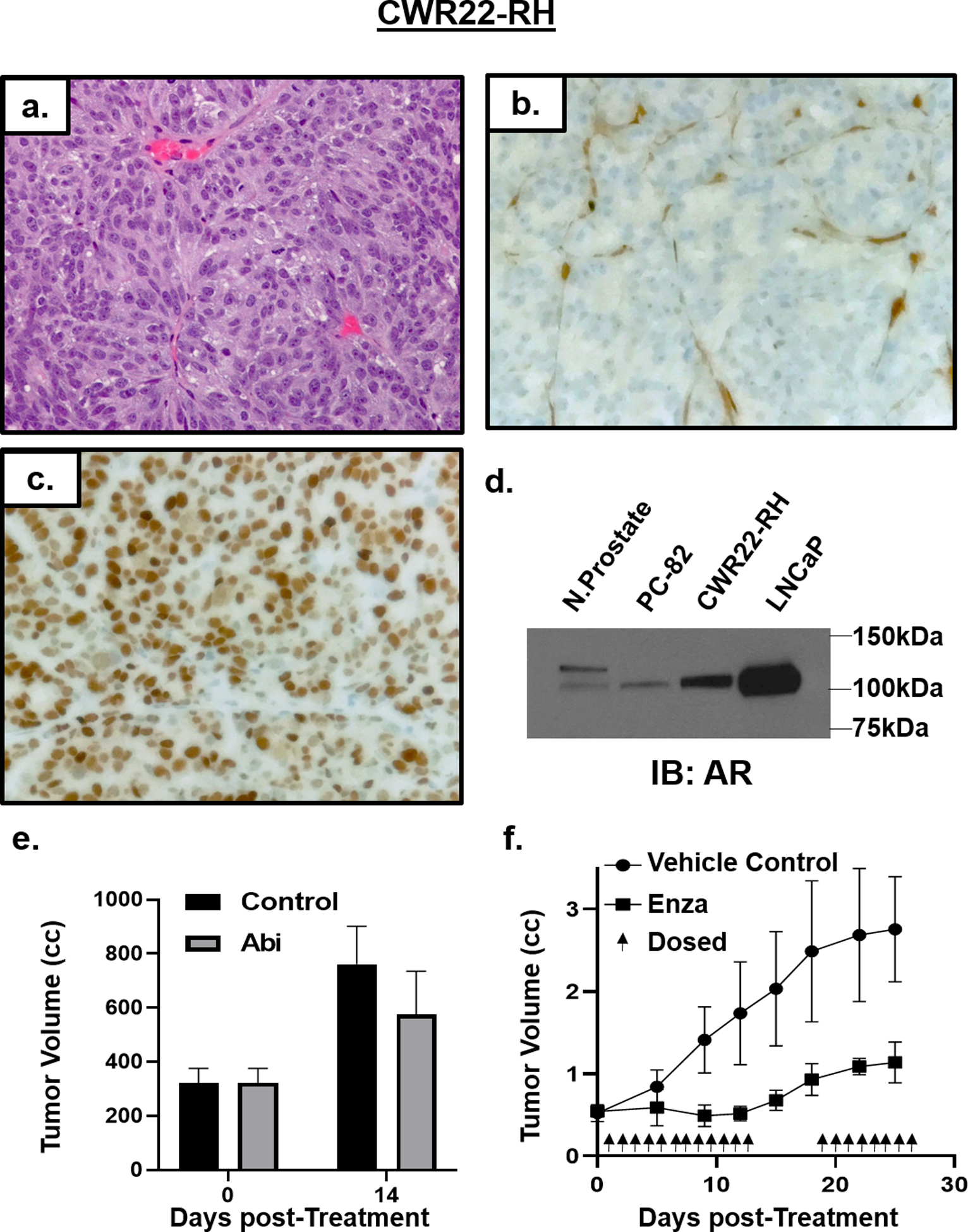
a) H & E histology (200x) of CWR22-RH xenografts. IHC staining (200x) for b) PTEN and c) AR. d) Western blot documenting AR expression in PC-82 relative to normal prostate, CWR22-RH, and LNCaP cells. e) m) Abi resistance of CWR22-RH xenografts in vivo (n = 3 each). f) Enzalutamide sensitivity of CWR22-RH xenografts in vivo (n = 5 each).
Table 1:
Phenotypic and Growth Characteristics of CWR22, CWR22-RH, LvCaP-2, LvCaP-2R, SkCaP-1, SkCaP-1R, LNCaP, and LN-95.
| CWR22 | CWR22-RH | LvCaP-2 | LvCaP-2R | SkCaP-1 | SkCaP-1R | LNCaP | LNCaP-95 | |
|---|---|---|---|---|---|---|---|---|
| RNAseq Classification | AR+ PCa | AR+ PCa | AR+/NE+ PCa | AR+/NE+ PCa | AR+ PCa | AR+ PCa | AR+ PCa | AR+ PCa |
| Tissue of Origin | Primary | CWR22 | Liver Met | LvCaP-2 | Skin Met | SkCaP-1 | Lymph Node Met | LNCaP |
| Patient Treatment History | None | ADT, Abi, Carboplatin, and Enza | ADT, Taxane, Abi, Carboplatin, Enza | Castration | ||||
| Histology | Poorly Differentiated Adenocarcinoma | Poorly Differentiated Adenocarcinoma | Poorly Differentiated Amphicrine Carcinoma | Poorly Differentiated Amphicrine Carcinoma | Poorly Differentiated Adenocarcinoma | Poorly Differentiated Adenocarcinoma | Poorly Differentiated Adenocarcinoma | Poorly Differentiated Adenocarcinoma |
| in vivo Growth Response to ADT | Yes | No | Yes | No | Yes | No | Yes | No |
| Xenograft Doubling Time | 11 +/− 3 days (Intact Host) | 10 +/− 2 days (Intact or Castrate Host) | 10 +/− 3 days (Intact Host) | 9 +/− 2 days (Intact or Castrate Host) | 14 +/− 5 days (Intact Host) | 18 +/− 4 days (Intact or Castrate Host) | 12 +/− 5 days (Intact); 26 +/− 7 days (Castrate) | 6 +/− 3 days (Intact or Castrate Host) |
| AR | Homozygous GOF H878A mutation | Homozygous Double GOF H875Y & T878A mutation | Wild Type + low to no V7 | Wild Type + V7 | Wild Type | Wild Type + V7 | Homozygous T878A GOF mutation | Homozygous T878A GOF mutation + V7 |
| Normalized AR mRNA | 4 | 11 | 52 | 256 | 4 | 388 | 17 | 30 |
| Normalized AR protein | 6 | 25 | 11 | 50 | 7 | 80 | 33 | 59 |
| AR-FL/AR-V7 protein ratio | >100:1 | >100:1 | >100:1 | 6:1 | >100:1 | 12:1 | >100:1 | 8:1 |
| TP53 | Heterozygous LOF G154F mutation | Heterozygous LOF G154F mutation | LOF T211fs mutation | LOF T211fs mutation | Wild Type | Wild Type | Wild Type | Wild Type |
| PTEN | Wild Type | Heterozygous LOF T321fs mutation | LOH & Hemizygous Deleterious R130Q mutation | LOH & Hemizygous Deleterious R130Q mutation | Homozygous Deletion | Homozygous Deletion | LOH & Hemizygous p.K6fs Deleterious AA mutation | LOH & Hemizygous p.K6fs Deleterious AA mutation |
| ERG | No | No | No | No | Yes | Yes | No | No |
| c-Myc | >80% | >85% | >80% | >50% | >40% | >60% | >50% | >75% |
| Nkx3.1 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Ki67 | 72 +/− 27% | 83 +/− 6% | 80 +/− 6^ | 75 +/− 9% | 45 +/− 3% | 39 +/− 4% | 47 +/− 12% | 82 +/− 12% |
| PSA | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Serum PSA (ng/mL/g) | 462 +/− 67 (Intact Host) |
249 +/− 41 (Castrate Host) | 59 +/− 11 (Intact Host) |
25 +/− 6 (Castrate Host) |
284 +/− 51 (Intact Host) |
44 +/− 12 (Castrate Host) |
185 +/− 34 (Intact Host) |
50 +/− 10 (Castrate Host) |
| PSMA | Yes | Yes | Focal | Focal | >50% | >50% | Yes | Yes |
| CK5 | No | No | No | No | No | No | No | No |
| CK18 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| B-catenin | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type |
| RB | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type |
| ChgA | No | No | Yes | Yes | No | No | No | No |
| HoxB13 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| TP63 | No | No | No | No | No | No | No | No |
| Sox2 | No | No | No | No | No | No | No | No |
| BRCA2 | Heterozygous LOF E984fs mutation | Heterozygous LOF E984fs mutation | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type |
| PIK3CA | Heterozygous Q546R mutation | Heterozygous Q546R mutation | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type |
| MSH2 | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | Wild Type | MSI, Homozygous Deletion | MSI, Homozygous Deletion |
Figure 2:
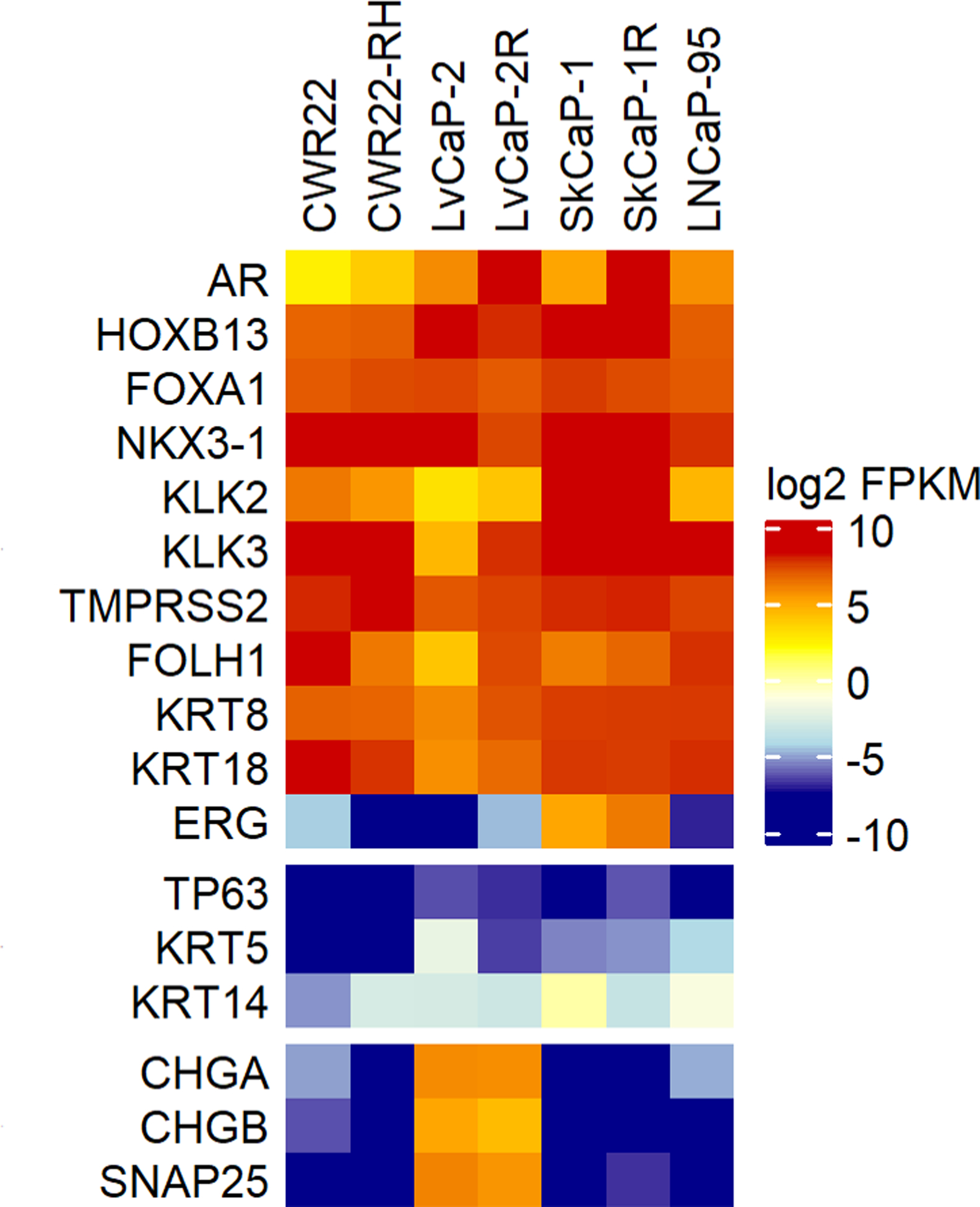
RNA-seq based expression analysis of a subset of genes across PDX models expressed as Log2 FPKM.
There are several unique genetic changes associated with castration resistance of the CWR22-RH. These include a loss of homozygosity (LOH) and a LOF truncating PTEN [T321fs] mutation (Tbl. 1) resulting in these cells being null for PTEN protein expression (Fig 1b). In addition, during relapse to castration, CWR22-RH acquired an additional AR T878A mutation and is thus hemizygous for H875Y/T878A double mutation (Tbl. 1). This double mutated AR is highly expressed in nuclei of CWR22-RH cells (Fig 1c) at a 25-fold higher level of AR-FL protein compared to normal prostate luminal cells. This is consistent with such elevation in AR-FL protein being the most common molecular determinant of resistance to first-line ADT in CRPC patients5, 6. This elevation in AR-FL, however, is not accompanied by detectable AR-V7 protein expression (Fig. 1d). Double AR mutations in codons 875 and 878 result in a GOF, because such ARs are strongly stimulated by progesterone binding, which is only a very weak agonist for wild type AR6. This is consistent with CWR22-RH growing equally well in intact vs. castrated hosts, (Tbl. 1), since castration does not lower serum progesterone in mice (1–2 ng/ml)30. Daily oral treatment of castrated adult male NSG mice bearing CWR22-RH tumors with a therapeutically effective dose of Abi (i.e. 0.5 mmol/kg20) no growth inhibitory effect (Fig. 1e). This resistance is predictable because Abi inhibits steroid metabolism downstream from progesterone and castration does not lower serum progesterone levels in mice6, 30.
Despite its Abi-resistance, growth of the CWR22-RH PDX in castrated adult male NSG mice is profoundly inhibited by daily oral treatment with a therapeutically effective dose of Enza (10 mg/kg40), even though it has high expression of double mutated (i.e. H875Y/T878A) AR-FL protein (Fig. 1f). This is not unexpected since Enz blocks progesterone binding to AR and thus can inhibit progesterone induced growth of PCA cells28.
AR-FL vs. AR-V7 expression in Prostate Cancer PDXs resistant to Abiraterone and Enzalutamide
LvCaP-2 PDX is derived from a liver metastasis obtained at rapid autopsy from a 75-year old European-American who following a prostate biopsy (Gleason Sum 9) was treated over a 3-year period with ADT, then Abi, docetaxel plus carboplatin, and finally Enza (Suppl. Fig. 1a). Histologically, CRPC PDX is a poorly differentiated adenocarcinoma (Fig. 3a). LvCaP-2 has wild type AR, which it expresses at a 52-fold higher mRNA level (Tbl. 1) and an 11-fold higher AR-FL protein level compared to normal prostate luminal cells with a low level of AR-V7 protein that it loses with serial passaging in intact hosts (Fig. 3b, inset). Essentially all LvCaP-2 cells exhibit nuclear localization of AR protein (Fig. 3b). At the transcriptome and protein level (Tbl. 1), a high proportion (>80%) of the parental LvCaP-2 cells express c-Myc (Fig. 3c) and Ki67 (Fig. 3d). Besides expressing prostate specific HOXB13 (Fig. 3e), it expresses luminal cell-specific (Fig. 3f–h), but not basal cell, markers (Tbl. 1, Fig. 2). It does express NE markers, however and is thus an “amphicrine” carcinoma3. Genetically, it has a hemizygous LOF truncating mutation in TP53 (T211fs) and hemizygous deleterious mutation (R130Q) in PTEN31 with loss of PTEN protein expression. While LvCaP-2 has wild type RB1, there is only limited focal expression of RB1 protein. It secrets PSA [serum PSA of 59 +/− 11 ng/ml/gram tumor, (Tbl. 1)].
Figure 3: Characterization of LvCaP-2 and LvCaP-2R.
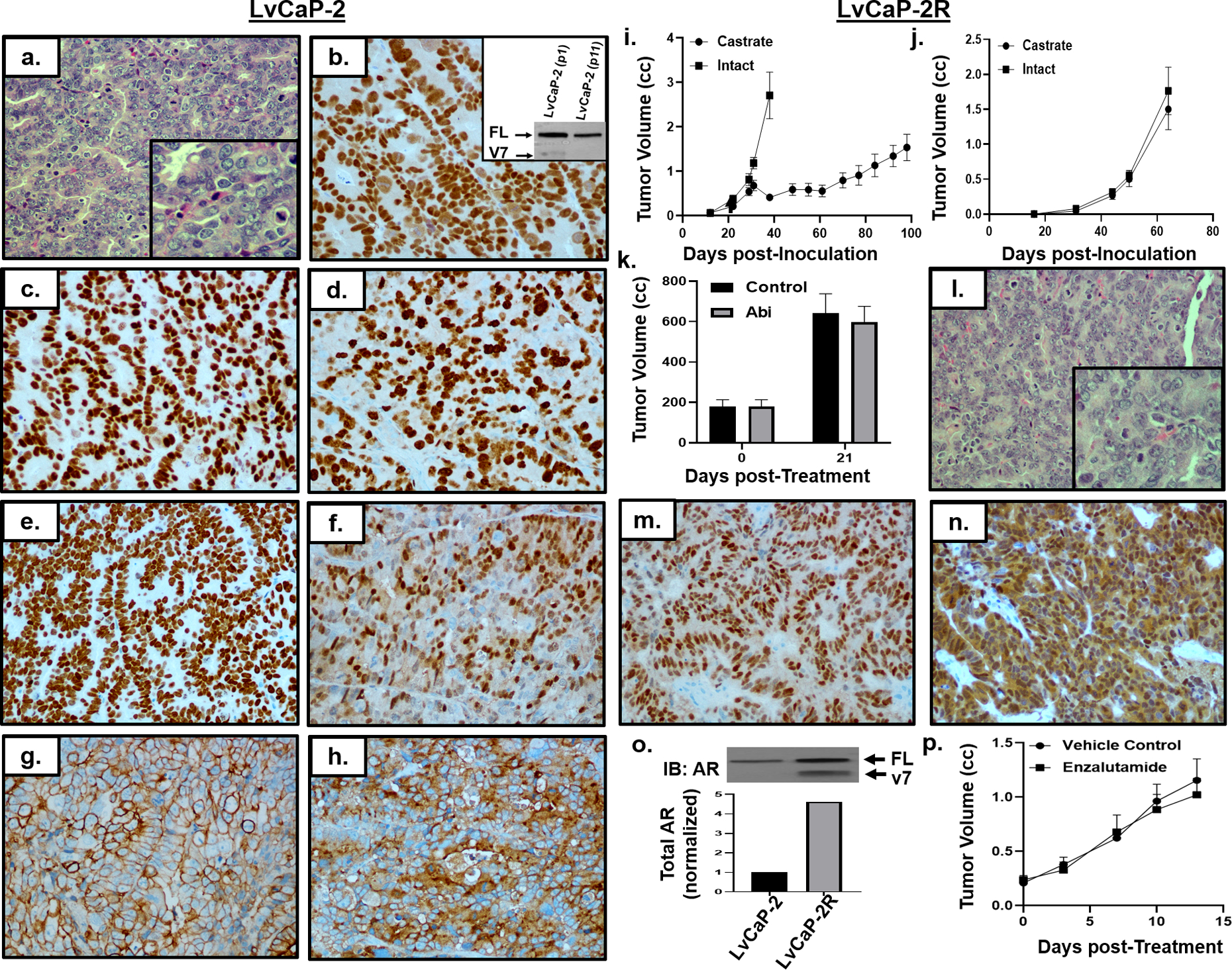
a) H & E histology (200x) of LvCaP-2 (inset, 400x). IHC (200x) for b) AR (inset, AR immunoblot), c) c-Myc, d) Ki67, e) HoxB13, f) Nkx3.1, g) cytokeratin-18, and h) PSA. i) Growth rate of LvCaP-2 in intact (i.e. ADT-equivalent) mice with subsequent regression and relapse in castrate (i.e. ARSi-equivalent) male NSG mice (n = 5 each). j) Growth rate of LvCaP-2R in intact vs. castrate hosts (n = 5 each). k) Abi resistance of LvCaP-2R xenografts in vivo (n = 3). i) H & E histology (200x) of LvCaP-2R (inset, 400x). IHC (200x) for m) Nkx3.1 and n) AR in LvCaP-2R PDX in castrate hosts. o) AR immunoblot of LvCaP-2 vs. LvCaP-2R and quantification based on densitometry. p) LvCaP-2R resistance to daily oral Enzalutamide treatment.
When adult male hosts bearing the LvCaP-2 PDX are castrated, the cancer stops growing for ~1 month before relapsing (Fig. 3i). Passage of a relapsing tumor in castrated hosts results in a variant, termed LvCaP-2R, that grows equally well in intact vs. castrated hosts [DT of 10 +/− 3 days vs. 9 +/− 2 days, respectively (Fig. 3j)]. Growth of LvCaP-2R in a castrated mouse is resistant to daily oral treatment with 0.5 mmol/kg of Abi (Fig. 3k)
Histologically (Fig. 3l) and phenotypically (Tbl. 1, Fig 2), this Abi-resistant LvCaP-2R remains a poorly differentiated amphicrine adenocarcinoma with retained expression of NKX3.1 (Fig. 3m), PSA, HOXB13 and PSMA (Tbl. 1). LvCaP-2R has a 50% decrease in RB1 mRNA with undetectable expression of RB1 protein, and an additional 5-fold increase in AR mRNA compared to the parental LvCaP-2, raising the level to 256-fold higher than in normal prostate luminal cells (Tbl. 1, Fig. 2). This results in a 4.7-fold increase in total AR protein in LvCaP-2R in castrated hosts vs. parental LvCaP-2 in intact mice (Fig. 3n), which is 50-fold higher total AR protein than in normal prostatic luminal cells (Tbl. 1).
Importantly, progression of LvCaP-2 to the Abi-resistant LvCaP-2R variant is associated with the gain of AR-V7 protein expression at a ratio of 6 to 1 [AR-FL: AR-V7] (Fig. 3o). This translates to an 8-fold higher level of AR-V7 protein in LvCaP-2R than the level of AR-FL expression in normal prostate luminal cells. AR is located not only in the cytoplasm, but also strongly present in nuclei of essentially all LvCaP-2R cells despite being in castrated hosts (Fig. 3n). Growth of the Abi-resistant LvCaP-2R PDX in castrated NSG adult male mice is not inhibited by daily oral treatment with 10 mg/kg of Enza (Fig. 3p). Likewise, Enza-treatment had no effect on serum PSA expressed as ng/ml/gram of tumor (i.e. 25 +/− 4 vs. 22 +/− 6 for controls vs. Enza-treated, respectively).
To address the generalizability of coordinated AR-FL and AR-V7 expression in the development of Abi- and Enza-resistance, an additional PDX, termed SkCaP-1, was evaluated. The SkCaP-1 PDX is derived from a biopsy of a CRPC skin metastasis obtained from a 52-year old European-American who underwent a radical prostatectomy (Gleason Sum 7), and subsequently progressed over a 12-year period to sequential treatment with salvage XRT/ADT/Taxane/Abi/Carboplatin/ Enza treatment before rapid autopsy (Suppl. Fig. 1b). Histologically, it is a poorly differentiated adenocarcinoma (Fig. 4a). In addition to expressing prostate-specific HOXB13, it expresses luminal cell-specific including AR (Fig. 4b), Nkx3.1 (Fig. 4c), and PSMA (Fig. 4d); but not basal cell or NE specific, markers (Tbl. 1, Fig. 2). SkCaP-1 expresses wild type AR at a 4- and 7-fold higher level on a mRNA and AR-FL protein basis, respectively, compared to normal prostate luminal cells (Tbl. 1), but has very low detectable AR-V7 expression (Fig. 4e, inset). Essentially, all SkCaP-1 cells have nuclear localization of AR protein in an intact male NSG mouse (Fig. 4b). This growth is associated with SkCaP-1 cells expressing c-Myc and RB1 (Tbl. 1) and Ki67 (Fig. 4f). The major genetic characteristic of SkCaP-1 cells is homozygous deletion of PTEN and thus they are null for PTEN protein (Tbl. 1). It secretes PSA [serum PSA of 284 +/− 51 ng/ml/gram tumor, (Tbl. 1)].
Figure 4: Characterization of SkCaP-1 and SkCaP-1R.
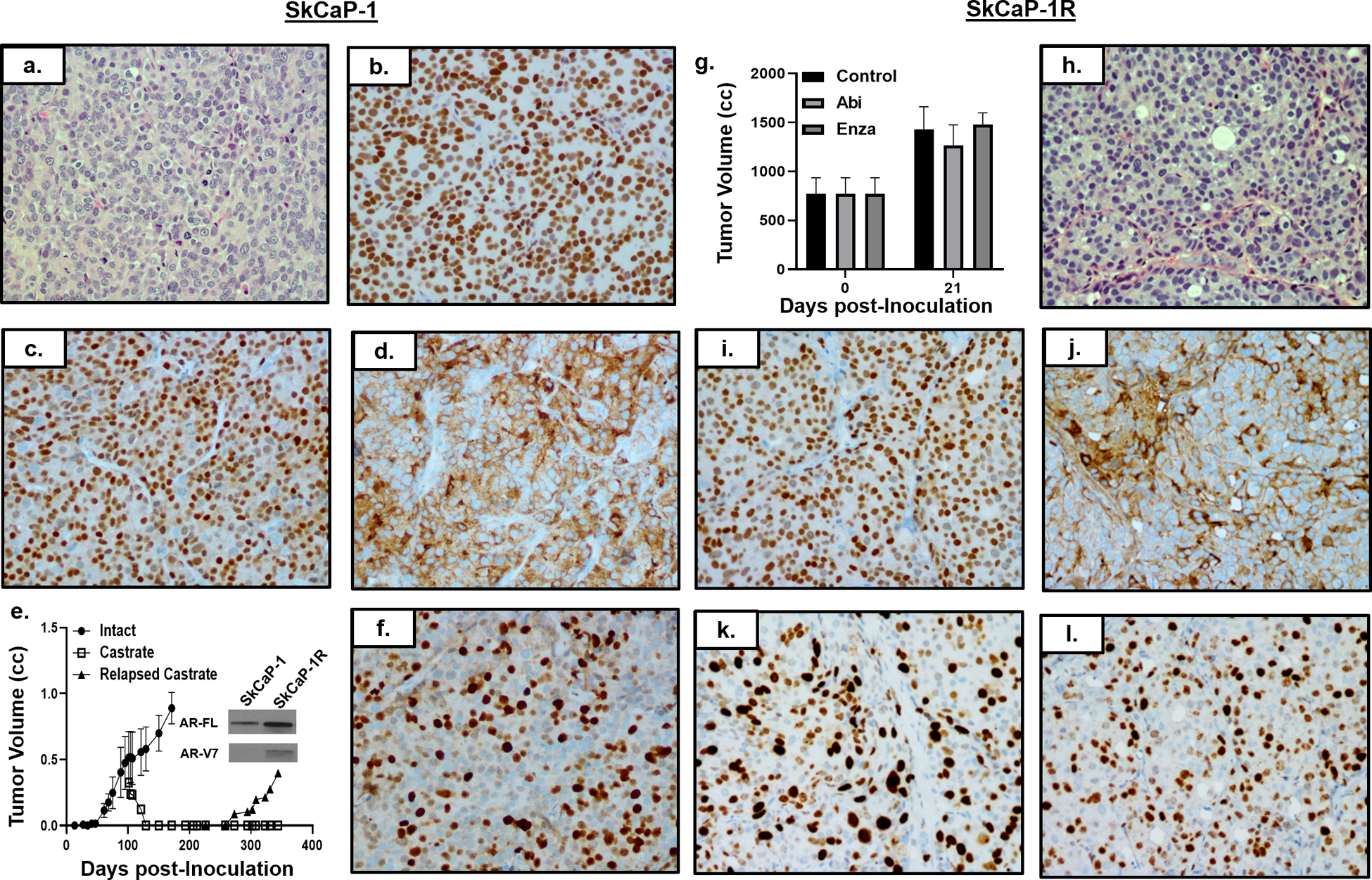
a) H & E histology (200x) of SkCaP-1. IHC (200x) of SKCaP-1 for b) AR, c) Nkx3.1, and d) PSMA. e) Growth rate of SkCaP-1 in intact (i.e. ADT-equivalent) mice with subsequent regression and relapse in castrate (i.e. ARSi-equivalent) male NSG mice (n = 5 each). AR-FL and AR-V7 immunoblots of SkCaP-1 vs. SkCaP-1R (inset). f) IHC (200x) of SkCaP-1 for Ki67. g) Abi and Enza resistance of SkCaP-1R in vivo (n = 3 each). h) H & E histology (200x) of SkCaP-1R. IHC (200x) of i) AR, j) PSA, k) c-Myc, and i) Ki67 in SkCaP-1R PDX.
When adult male mice bearing established SkCaP PDXs are castrated, cancers regress over a 40-day period to a non-palpable size before relapsing (Fig. 4e). Passage of such a relapsing cancer results in a variant, SkCaP-1R, that grows equally well in intact vs. castrated hosts [DT of 18 +/− 4 days] (Tbl. 1). Growth of the SkCaP-1R in castrated adult male NSG mice is not inhibited by daily oral treatment with Abi- or Enza- over a 3-week period (Fig. 4g). Neither Abi- nor Enza- treatment has an effect on serum PSA expressed as ng/ml/gram of tumor (i.e. 50 +/− 8 for controls vs. 54+/−12 for Abi vs. 44 +/− 12 for Enza).
Histologically, Abi/Enza-resistant SkCaP-1R remains a poorly differentiated adenocarcinoma (Fig. 4h). It retains expression of prostate specific HOXB13 and luminal cell-specific including AR (Fig. 4i) and PSA (Fig. j); but not basal cell or NE specific, markers (Tbl. 1, Fig. 2). A major transcriptional difference between SkCaP-1R growing in castrated hosts is an additional 12-fold increase in AR mRNA compared to the SkCaP-1 growing in intact hosts (Fig. 2), raising the level to 388-fold higher than in normal prostate luminal cells (Tbl. 1). This results in an 11-fold increase in total AR protein in SkCaP-1R in castrated hosts vs. parental SkCaP-1 in intact mice, which is 80-fold higher total AR protein than in normal prostatic luminal cells. Progression to the Abi/Enza-resistant SkCaP-1R variant is associated with an enhanced expression of AR-V7 protein at a ratio of AR-FL to AR-V7 of 12:1 (Fig. 4e, inset). This translates to a 6-fold higher level of AR-V7 protein in SkCaP-1R than the level of AR-FL in normal prostate luminal cells (Tbl. 1). AR is located in nuclei of essentially all SkCaP-1R cells despite being in a castrated host (Fig. 4i). This is consistent with their retained expression of c-Myc (Fig. 4k) and Ki67 (Fig. 4l).
LN-95 variant as a prototypic model of ARSi-resistance
These results document that elevated expression of AR-FL alone is sufficient for Abi-, but not Enza-resistance, even if AR-FL has a GOF mutation and that Enza-resistance is correlated with a critical level of AR-V7 expression. To test the role of AR-FL vs. AR-V7 in CRPC resistance to Enza directly requires an Enza-resistant cell line amenable to CRISPR-Cas9 gene KO that expresses both AR-FL and AR-V7. These conditions are met by a variant of the LNCaP cell line known as the LNCaP-95 (a.k.a. LN-95). LNCaP is derived from a supraclavicular lymph node metastasis from a CRPC patient11. It expresses wild type RB1, but has a 2-bp deletion in codon 6 in PTEN leading to a LOF frame shift mutation22, and has a GOF AR T878A mutation42, and methylation of the GSTP1 and TGFβR2 promoters resulting in a loss of expression of these latter 2 proteins9, 24, 46. Thus, LN-95 are not sensitive to ADT-induced cell death4, 8, 9, 39, 46. This is significant because LNCaP cells are passaged in phenol red-containing RPMI-1640 media supplemented with 10% fetal bovine serum (i.e. FBS media)11. FBS media contains a castrate serum level of testosterone [i.e. 22.0 +/− 6.1 pg/ml (55.1–97.5 pM)]34. LNCaP cells have microsatellite instability (MSI) due to homozygous deletion of exons 9 to 16 in the mismatch repair gene hMSH2, resulting in truncation and LOF of the protein21. Thus, LNCaP is genetically unstable and accumulates mutations during serial in vitro passaging. This provides a mechanism for why LNCaP acquires a faster growth rate coupled with a decrease in PSA expression and acquisition of resistance during serial in vitro culture17.
In low androgen FBS media, LNCaP cells express a high level of mutated AR-FL (T878A) protein [i.e. 33-fold higher than normal prostate luminal epithelial cells, (Fig. 1d)]40, but no detectable level of AR-V7 (Fig. 5a). In this low androgen media, AR-signaling is functional as documented by its secretion of 70 ng of PSA/ml of media/106 cells per day. Functional AR-signaling is also confirmed by the fact that addition of Enza (10 μM) to the FBS media inhibits LNCaP growth by ~75% (Fig. 5b) due to the inhibition of AR-dependent cell autonomous autocrine signaling41. Similarly, in vitro growth of LNCaP cells is inhibited by ~90% when cultured in phenol red-free RPMI-1640 media supplemented with 10% charcoal-stripped FBS [C/S media, (Fig. 5b)], containing and even more depleted testosterone level [i.e. 5.0 +/− 0.49 pg/ml (15.6 – 19.0 pM)] equivalent to that of the serum of patients treated with LHRH analogs plus abi34. This growth inhibition is associated with a similar 95% reduction in PSA secretion (i.e. only 3 ng/ml/106 cells per day). These results document that LNCaP cells are stimulated, but not absolutely dependent upon AR signaling (i.e. cells are CR, but still AR-signaling sensitive). Thus, LNCaP is tumorigenic when xenografted in immune-deficient male mice, but their growth is faster in intact compared to castrated hosts (Fig. 5c).
Figure 5: Characterization of LNCaP variant under long-term ARSi-equivalent conditions (i.e. LN-95 cells).
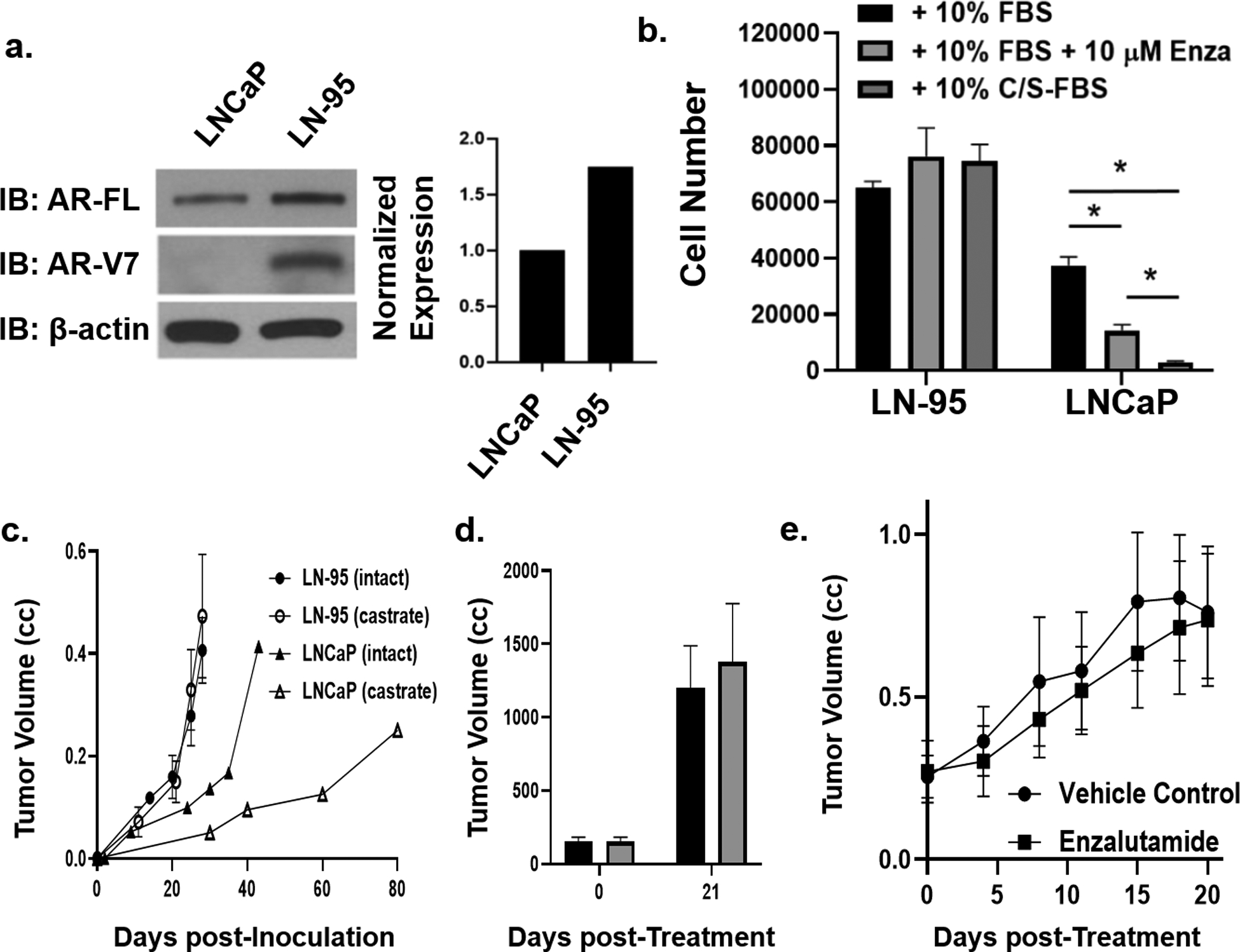
a) AR-FL and AR-V7 immunoblot of LNCaP vs. LN-95 variant and quantification via densitometry. b) Cell number after 5 days of in vitro growth of LN-95 in 10% FBS media, 10% FBS media containing 10 μM enzalutamide, or 10% CS-FBS media vs. LNCaP growth under the same conditions with asterisks denoting significant difference at p < 0.05. c) Growth rate of LN-95 in castrated (i.e. ARSi-equivalent) vs. LNCaP in intact (i.e. ADT-equivalent) mice. d) Abi resistance of LN-95 xenografts in vivo (n = 3 each). e) In vivo growth response of LN-95 growing in castrated (i.e. ARSi-equivalent) male NSG mice given daily oral dosing with 25 mg of enzalutamide/kg/d vs. vehicle controls (n = 5 each).
Pflug et al. serially passaged LNCaP cells in C/S media to induce “adaptation” to an Abi-equivalent androgen deprivation state over a period of several months32, producing the LN-95 variant. LN-95 cells cultured in C/S media retain the major genetic alterations of parental LNCaP [i.e. PTEN loss, hMSH2 and AR mutations, plus methylation and loss of GSTP1 and TGFβR2, (Tbl. 1)]. Associated with LN-95 adaption to growth in C/S media is a 1.8-fold increase in AR mRNA (Fig. 2) and protein compared to the parental LNCaP in FBS media [i.e. 58-fold higher AR protein than in normal prostate luminal cells, (Fig. 5a)]13. LN-95 cells in C/S media not only express an elevated level of mutated AR-FL protein, but also AR-V7 at a ratio of 8:1 (Fig. 5a). These changes in the AR axis are associated with LN-95 cells growing faster than parental LNCaP cells, growing equally well in C/S vs. FBS media, and their growth not inhibited by Enza (Fig. 5b).
Unlike LNCaP that grows much faster in an intact (DT of 12+/−5 days) vs. castrated male mouse (DT of 26+/−7days), LN-95 xenografts grow equally well in intact vs. castrated male mice at a 2-times faster rate (DT of 6+/−2 days in either host) than LNCaP cells in intact mice (Fig. 5c). LN-95 growing in castrated hosts express PSA [i.e. 50 +/− 10 ng PSA/ml per gram of xenograft tumor, (Tbl. 1)], and neither their PSA secretion nor growth is inhibited by daily oral treatment with Abi (Fig. 5d). This is consistent with tissue levels of both T and DHT in castrated mice bearing LN-95 xenografts being >50 pg/gram of tumor45, which is equivalent to levels in prostate cancer tissue in Abi-treated patients29. Like LvCaP-2R, SkCaP-1R expresses both AR-FL and AR-v7 and its growth in castrated NSG mice is not inhibited by daily oral treatment with either Abi- or Enza (Fig. 5e). These results validate that LN-95 is an appropriate prototypic model for evaluating the role of AR-V7 in ARSi-resistance.
Role of AR-FL vs. AR-V7 in resistance of LN-95 cells to enzalutamide
To address the role of AR-FL vs. AR-V7 in Enza-resistance, the growth response to Enza was compared between LN-95 cells in which CRISPR-Cas9 was used to KO either AR-FL or AR-V7 alone or in combination (Fig. 6A). Clones of LN-95 cells were obtained in androgen-depleted C/S media in which AR-FL or AR-V7 alone or in combination were knocked out as validated by sequence analysis (Suppl. Fig. 2), Western blotting (Fig. 6b), and IHC (Fig. 6c). IHC documents that in single KOs, the remaining AR-FL or AR-V7 is localized in the nuclei despite being in androgen-depleted C/S media (Fig. 6c). This is significant for the AR-FL KO cells that only express AR-v7, because this documents that AR-V7, which contains the classical nuclear localization domain (i.e. AA608–628)12, 16 translocates to the nuclei even though it lacks the LBD. This ability of the AR-V7 protein to nuclear translocate without co-expression of AR-FL is confirmed by cell fractionation and Western blot analysis (Fig. 6d). In these AR-FL KO cells, nuclear AR-V7 is transcriptionally active even without AR-FL as demonstrated by its ability to increase transcription of a subset of AR target genes (Fig. 6e). These target genes are defined by their transcriptional down regulation in total AR (i.e. AR-FL/AR-V7) double KO cells and transcriptional upregulation by the addition of synthetic androgen (i.e. R1881) to parental LN-95 cells and AR-v7 KO cells that only express AR-FL (Fig. 6f). Significantly, when R1881 is added to the media, the LN-95 cells decrease their AR RNA expression by 40% (p < 0.05) and stop expressing a detectable level of AR-V7 protein, and this loss of AR-V7 protein does not occur in AR-FL KO cells (Fig. 6b). This supports that there is an autoregulatory negative feedback loop between level of ligand dependent AR-FL signaling and AR-V7 expression as described previously13, 26.
Figure 6: Characterization of AR-FL, AR-V7, vs. Total AR Knockout in LN-95 cells in vitro.
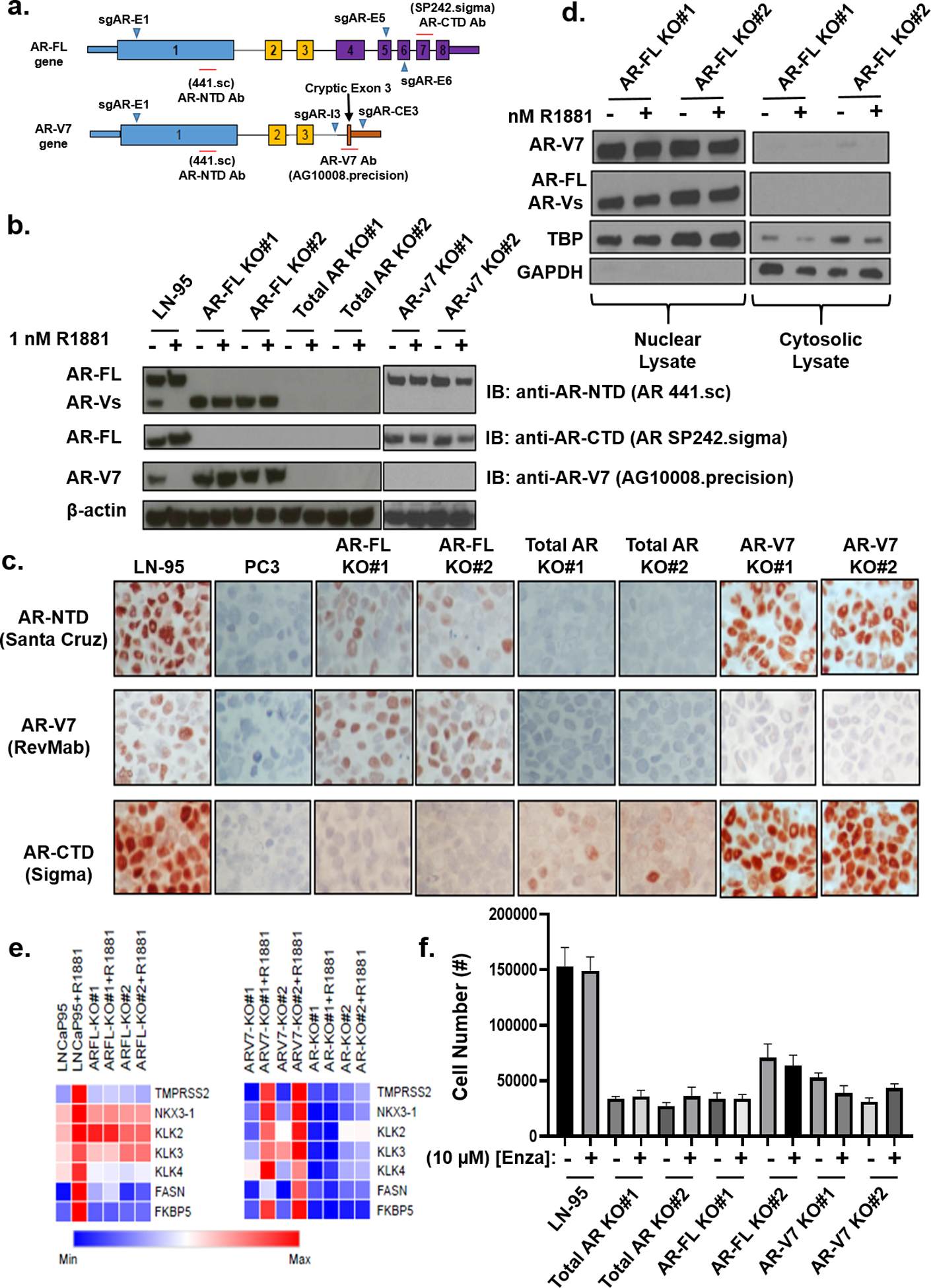
a) Overview of the CRISPR-Cas9 approach used to knockout AR-FL and/or AR-V7 in LN-95 cells. b) Western blot documenting knockout of AR-FL, AR-V7, or both in multiple LN-95 clones. c) IHC (200X) staining of parental LN-5 cells expressing both AR-FL and AR-V7 vs. AR-negative PC-3 cells and the relevant AR-knockout clones. d) Immunoblot documenting nuclear localization of LN-95 cell clones expressing only AR-V7 expressing (i.e. AR-FL KO) clones. e) RNAseq-based analysis of AR-target genes in parental, AR-FL, AR-V7, and total AR KO clones. f) In vitro growth after 6 days of the parental LN-95 cells vs. AR-FL, AR-V7, and total AR KO clones in 10% CS-FBS media.
The in vitro growth of LN-95 cells is not dependent on, but is augmented by AR-signaling as documented by the >75% reduction (p < 0.05) in growth of the total AR-KO vs. parental LN-95 cells in the androgen-depleted C/S media (Fig. 6f). As expected, Enza-treatment did not decrease further the depressed growth of the total AR-KO cells (Fig. 6f). Similar growth depression (p < 0.05) also occurs in both AR-FL and AR-V7 single KO cells and as expected Enza has no effect upon the depressed growth of AR-FL single KO cells expressing only AR-V7 (Fig. 6f). Significantly, Enza treatment had no effect upon the depressed growth of the LN-95 AR-V7 KO cells expressing only AR-FL (Fig. 6f). This is consistent with their growth already being maximally depressed by the loss of AR-V7. These results document that signaling from both AR-FL and AR-v7 is required for maximal growth of LN-95 cells in the androgen-depleted C/S media.
This conclusion is supported by RNAseq analysis. There are a series of 32 signature genes whose expression is significantly (i.e. >1.4-fold) AR stimulated vs. 19 genes AR repressed (Tbl. 2) in parental LN-95 cells growing optimally in androgen-depleted C/S media vs. AR-null (i.e. total AR KO) cells whose growth is maximally depressed. Thirteen out of the 32 (41%) AR-stimulated signature genes and 8 of 19 (42%) AR-repressed signature genes cannot be attributed specifically to either AR-FL or AR-V7 (i.e. they were not affected by KO of either AR-FL or AR-V7 alone). There are 10 of the 32 (31%) stimulated and 9 of 19 (47%) repressed genes, however, whose expression is regulated only by AR-FL (i.e. they were affected by KO of AF-FL but not AR-V7), consistent with the need for signaling by both receptors for maximal growth. Conversely, there is only 1 of the signature stimulated genes (i.e. PRKACB) whose expression is repressed only in AR-V7 expressing cells. There are several AR-stimulated and AR-repressed genes (e.g. IGFBP3, and PSD4) whose expression is repressed by AR-FL, but stimulated by AR-V7 (Tbl. 2). The data are consistent with overlapping and also distinct functional roles characterized previously and suggest the need for signaling by both receptors for maximal growth.
Table 2:
Genes whose transcription is stimulated or repressed in parental LN-95, AR-FL KO, or AR-V7 KO cells vs. AR-null (i.e. total AR KO) cells growing in androgen-depleted C/S media.
| Upregulated Genes | |||||
|---|---|---|---|---|---|
| Gene | AR+ / AR- | AR-FL only / AR- | AR-V7 only / AR- | Function | |
| Both | KLK3 | 8.5 | 11.2 | 20.9 | Prostate-specific serine-type endopeptidase (Chymotrypsin) activity |
| NKX3–1 | 6.2 | 1.5 | 8.5 | Prostate-specific DNA-binding transcription factor | |
| PPP3CA | 4.8 | 2.6 | 4.6 | Calcineurin A protein phosphatase | |
| PPAP2A | 4.2 | 2.2 | 6.2 | Phosholipid phosphatase | |
| GPC6 | 3.8 | 1.5 | 3.9 | Glycosylphosphatidylinositol-anchored heparan sulfate proteoglycan | |
| BEX2 | 3.5 | 4.0 | 1.9 | Increases proliferation via the JNK/c-Jun pathway | |
| NEDD4L | 3.4 | 2.8 | 4.1 | E3 ubiquitin-ligase for TGFBR1 and Smad2 | |
| BTG1 | 2.7 | 2.0 | 3.4 | Enzyme binding and transcription coregulator activity | |
| KLK4 | 2.5 | 1.8 | 2.0 | Serine-type endopeptidase (Trypsin) activity and serine-type peptidase activity | |
| SLC25A36 | 2.4 | 2.2 | 1.6 | Mitochondrial function through transporting pyrimidine nucleotides for mtDNA/RNA synthesis | |
| TRPV6 | 1.9 | 1.6 | 1.9 | Calmodulin binding and calcium channel activity | |
| FKBP5 | 1.7 | 1.6 | 1.7 | Peptidyl-prolyl cis-trans isomerase activity and FK506 binding | |
| STEAP2 | 1.6 | 1.8 | 1.8 | Fe/Cu transporter activity and ferric-chelate reductase activity | |
| AR-FL only | UGT2B11 | 26.6 | 43.9 | 1.4 | Carbohydrate binding and glucuronosyltransferase activity |
| CD55 | 3.7 | 5.0 | 1.0 | Lipid binding and virus receptor activity | |
| UGT2B15 | 2.7 | 4.3 | 1.1 | Carbohydrate binding and glucuronosyltransferase activity | |
| CTAGE5 | 2.4 | 2.6 | 1.4 | Receptor in the endoplasmic reticulum required for collagen VII (COL7A1) secretion | |
| GTPBP2 | 1.8 | 2.8 | 1.1 | GTP binding and GTPase activity | |
| NEAT1 | 1.8 | 1.9 | 1.0 | Long non-coding RNA (lncRNA) | |
| SLC38A1 | 1.7 | 1.7 | 1.2 | Neutral amino acid transmembrane transporter activity and amino acid:sodium symporter | |
| BEST1 | 1.5 | 2.4 | 1.3 | Chloride channel activity. | |
| SLC43A1 | 1.5 | 1.8 | −1.3 | Sodium-independent, high affinity transport of large neutral amino acids | |
| NCOA1 | 1.4 | 2.9 | −1.1 | Transcriptional coactivator for steroid and nuclear hormone receptors | |
| AR-V7 only | KLK2 | 3.7 | 1.0 | 14.0 | Serine-type endopeptidase (Trypsin) activity |
| CALD1 | 2.5 | 1.4 | 2.6 | Actin- and myosin-binding protein | |
| PRKD1 | 2.1 | 1.4 | 3.0 | Serine/threonine-protein kinase involved in the regulation of MAPK8/JNK1 | |
| CD276 | 2.1 | 1.0 | 1.9 | Signaling receptor binding | |
| SSFA2 | 1.8 | 1.0 | 2.3 | Structural integrity and/or signal transduction | |
| GMNN | 1.8 | 1.0 | 2.2 | Geminin DNA replication inhibitor | |
| PRKCD | 1.5 | 1.3 | 2.8 | Calcium-independent, phospholipid- and diacylglycerol (DAG)-dependent serine/threonine-protein kinase | |
| IGFBP3 | 1.5 | −1.5 | 5.0 | Fibronectin binding and insulin-like growth factor I binding | |
| GULP1 | 2.0 | 1.1 | 1.9 | Modulates cellular glycosphingolipid and cholesterol transport | |
| Downregulated Genes | |||||
| Gene | AR+ / AR- | AR-FL only / AR- | AR-V7 only / AR- | Function | |
| Both | NR3C1 | −7.1 | −9.4 | 5.4 | Glucocorticoid Receptor |
| LDOC1 | −4.1 | −3.7 | −2.9 | Regulates the transcriptional response mediated by the nuclear factor kappa B | |
| NR4A2 | −3.7 | −4.5 | −2.2 | DNA-binding transcription factor activity and protein heterodimerization activity | |
| PLA2G2A | −2.8 | −1.5 | −1.4 | Calcium ion binding and phospholipase A2 activity | |
| GLI3 | −2.7 | −3.2 | −2.0 | Transcriptional activator and a repressor of the sonic hedgehog (Shh) pathway | |
| ZKSCAN3 | −2.6 | −1.8 | −2.7 | Transcriptional repressor of autophagy | |
| GRB10 | −2.5 | v2.8 | −2.5 | SH3/SH2 adaptor suppress signals from insulin and insulin-like growth factor receptors. | |
| GPC1 | −2.1 | −1.9 | −1.6 | Cell surface proteoglycan that inhibits FGF-mediated signaling | |
| AR-FL only | FAM198B | −2.3 | −4.7 | 1.4 | Golgi Associated Kinase 1B |
| SEMA6A | −2.0 | −2.4 | 1.1 | Cell surface receptor for PLXNA2 | |
| CAMK2N1 | −2.3 | −2.1 | −1.2 | Calcium/Calmodulin Dependent Protein Kinase II Inhibitor | |
| HOXB13 | −1.8 | −1.6 | −1.3 | Homeobox B13 which regulates AR activity | |
| QSOX1 | −1.8 | −2.2 | 1.2 | Protein disulfide isomerase activity and flavin-linked sulfhydryl oxidase activity | |
| CDK1 | −1.6 | −1.7 | −1.2 | Ser/Thr protein kinase | |
| JAG1 | −1.4 | −3.2 | 1.4 | Ligand for notch 1 receptor | |
| SESN1 | −1.3 | −2.3 | 1.2 | Intracellular leucine sensor that negatively regulates the TORC1 signaling pathway | |
| CAPNS1 | −1.2 | −1.6 | 1.4 | Calcium ion binding and calcium-dependent cysteine-type endopeptidase activity | |
| PSD4 | −1.3 | −1.4 | 1.5 | Phospholipid binding and ARF guanyl-nucleotide exchange factor activity | |
| AR-V7 only | PRKACB | 1.4 | 1.1 | −10.9 | Protein Kinase cAMP-Activated Catalytic Subunit Beta |
These results document that combined AR-FL plus AR-V7 dependent transcriptional regulation is needed for both growth stimulation under Abi-equivalent conditions and resistance to Enza. These results are not limited to the in vitro growth response. In xenograft studies, total AR-KO cells in Abi-equivalent castrated mice have no AR protein expression (Fig. 7a), and their growth is much slower than parental LN-95 cells expressing both AR-FL and AR-V7, but still faster than LNCaP (Fig. 7b). In contrast, growth of AR-FL KO cells retaining AR-V7 nuclear expression (Fig. 7a) is only minimally decreased in Abi-equivalent castrated mice (Fig. 7c). Growth of AR-V7 KO cells only expressing AR-FL is slower than in parental LN-95 and only slightly faster than total AR KO cells (Fig. 7d).
Figure 7: Characterization of AR-FL, AR-V7, vs. Total AR Knockout in LN-95 cells in vivo.
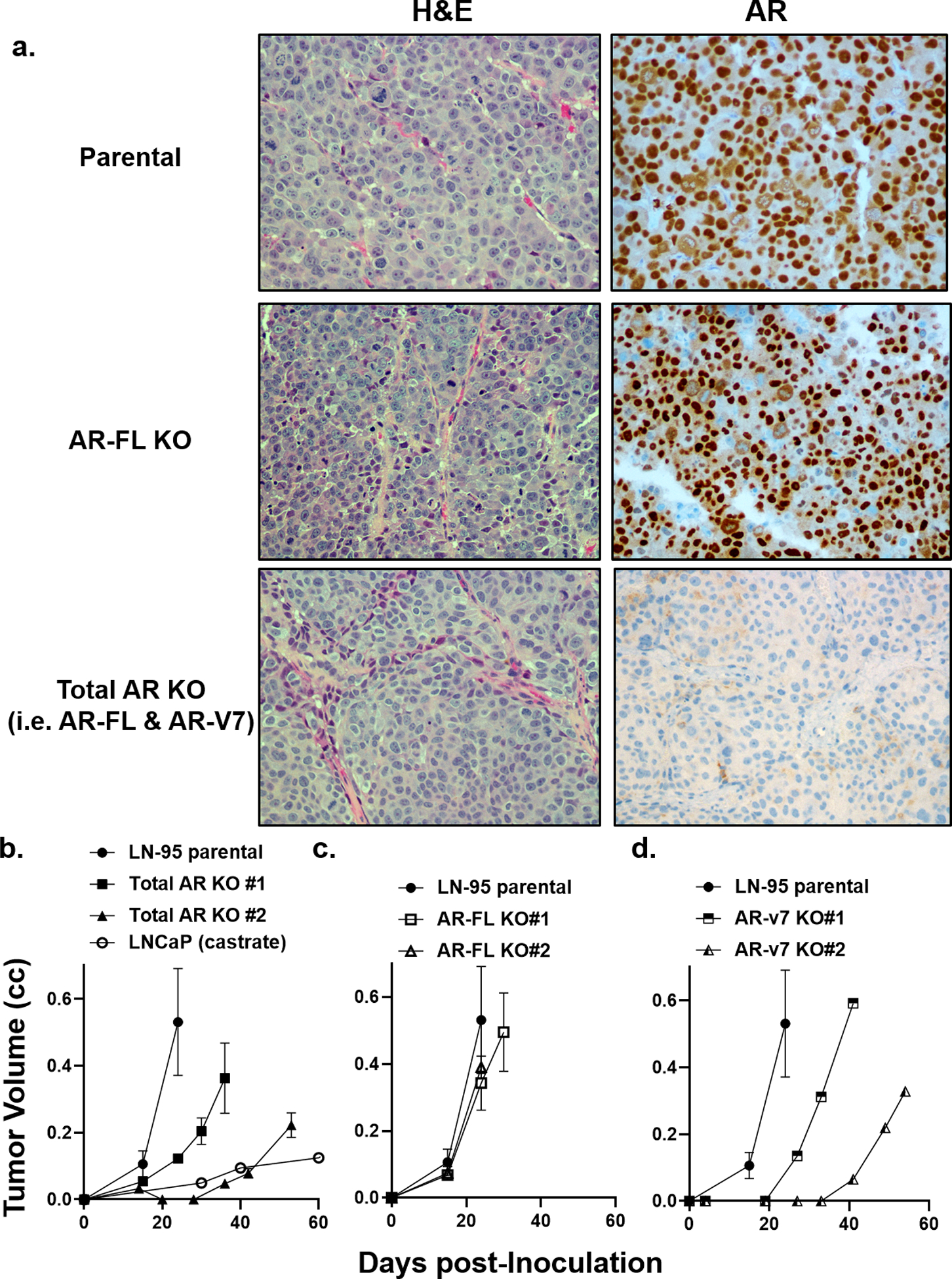
a) H & E histology and IHC for AR (200x) in parental LN-95 vs. AR-FL and total AR KO cells. b) Growth rate of parental LN-95 vs. total AR-KO clones in castrated hosts in vivo. c) Growth rate of parental LN-95 vs. AR-FL KO clones in castrated hosts in vivo. d) Growth rate of parental LN-95 s. AR-V7 KO clones in castrated hosts in vivo.
Discussion
The central question regarding the clinical significance of AR-V7 splice variant expression in CRPC is whether it is simply a marker of enhanced AR transcription characteristic of resistance to 2nd generation ARSi like Abi and Enza or whether it has a functional role in driving such resistance. To address this issue, the present study utilized independently derived PDXs in which the genetic and phenotypic changes could be followed before and after the development of ARSi resistance. While all of the PDXs lacked PTEN expression, there is not a consistent requirement for mutation in TP53, RB1, BRCA2, PIK3CA, or MSH2, or expression of SOX2 or ERG and ARSi-resistance. In contrast, the combined results document that elevated expression of AR-FL alone is sufficient for Abi- but not Enza-resistance. This is true even if AR-FL has a gain-of-function (GOF) mutation.
Enza-resistance requires both high AR-FL expression plus a critical level of AR-V7 expression. This conclusion is supported by several previous publications. For example, when Enza-sensitive LNCaP cells are engineered to express a 3-fold higher level of AR-FL protein, but without AR-V7 expression raising their total normalized AR protein to 99-fold greater than normal, the in vitro and in vivo growth of these cells remained Enza-sensitive40. In fact, this is the basis for the clinical development of Enza as a 2nd generation ARSi. An interesting corollary to these findings is that Abi-resistance of the CWR22-RH PDX involves a 25-fold elevated expression of GOF double mutated AR-FL compared to normal with no detectable expression of AR-V7 does not produce Enza-resistance. In contrast, in another CWR22 variant (i.e. CWR22Rv1), there is a genomic alteration (i.e. Exon 3 duplication) accompanying the gain of AR-V7 expression resulting in resistance to Enza23. In the current study, enzalutamide resistance requires both a >50-fold increase in AR-FL and AR-V7 protein expression at a level that is ~7–8-fold higher than AR-FL protein expression in normal prostate epithelium.
These results raise the question of the mechanism for enhanced AR-V7 expression in the lethal progression of CRPC. Along these lines, copy-number gains in the AR locus were reported more than 25-years ago as a major mechanism for resistance of metastatic prostate cancer to first-line ADT43. Recent studies determined that while rare in primary prostate cancer in hormonally-naïve patients, AR amplification occurs in the majority of mCRPCs, which is coupled with an amplified tandem duplication of a non-coding AR enhancer element located 624 kb upstream of AR33, 36, 44. This makes such co-amplification the most common molecular change in mCRPC and provides a mechanistic basis for the significant elevation in AR mRNA expression in most, but not all, mCRPCs. This elevation in overall AR transcription may be sufficient, even without additional changes in efficiency of AR mRNA splicing, to produce adequate expression of AR-V7 mRNA, and thus protein to drive ARSi-resistance. This is particularly possible since AR-V7 RNA is not a substrate of nonsense-mediated decay1.
There are however, additional genomic alterations and mRNA splicing changes that have been suggested to effect regulatory mechanisms for AR-V7 expression in mCRPC47. Resolving how these alterations affect AR-V7 expression is critical for identifying therapies for preventing and/or inhibiting enhanced AR-V7 expression from driving lethal progression of CRPC. Along these lines, the present LN-95 KO studies confirm earlier documentation13, 26 that there is autoregulatory negative feedback between the level of ligand dependent AR-FL transcription and AR-V7 expression. Such an autoregulatory negative feedback may explain paradoxical therapeutic response of prostate cancer patients resistant to 2nd generation ARSi to Bipolar Androgen Therapy (BAT) in which patients are rapidly cycled between a castrate to supraphysiologic level of T (SPT)14. In metastatic CRPC patients progressing on Enza, BAT results in resensitization when rechallenged with Enza38. This regaining of response to Enza is consistent with such SPT suppressing expression of AR-V7 thus preventing its transcriptional complementation with AR-FL transcriptional regulation needed for Enza-resistance. Presently, this is being tested.
Material and methods
Detailed procedures describing cell culture, proliferation assays, cytogenetic, genetic and epigenetic characterization, plasmid construction and transfection of CRISPR-Cas9 vectors, isolation of clonal cell lines by FACS, RNAseq, Western blot analysis, IHC, animal studies, and statistical analyses are included in the Supplemental Materials and Methods document, including Suppl. Fig. 1–3 and Suppl. Tbl. 1–2.
Supplementary Material
Acknowledgements
We are grateful to the patients and their families who participated in the Legacy Gift Rapid Autopsy program at Hopkins. We would like to acknowledge the Department of Defense Prostate Cancer Research Program W81XWH1810349 (JTI), W81XWH-17-1-0528 (WNB), W81XWH-18-2-0015 (AMD) and NIH Prostate SPOREs P50 CA058236 (JTI), Pathology Core from the Prostate SPORE P50 CA058236 (AMD) and P50 CA097186 (PSN), W81XWH-18-1-0347 (PSN), P01 CA163227 (PSN), Emerson Collective Cancer Research Fund [643396, (WNB)], Allegheny Health Network-Johns Hopkins University Cancer Research Fund (WNB), R01 CA185297 (JL and EA). Also, we wish to thank the Cell Imaging Facility, Animal Core Facility, Tissue Histology Core, Genetic Resource Core, Cytogenetics Core Facility, and the Autopsy Core from the CCSG grant supported by the SKCCC CCSG (P30 CA006973) for their services and assistance.
Conflicts of Interest
E. S. Antonarakis is a paid consultant/advisor to Janssen, Astellas, Sanofi, Dendreon, Pfizer, Amgen, AstraZeneca, Bristol-Myers Squibb, Bayer, Clovis, and Merck; has received research funding (to his institution) from Janssen, Johnson & Johnson, Sanofi, Dendreon, Genentech, Novartis, Bristol Myers-Squibb, AstraZeneca, Clovis, and Merck. E. S. Antonarakis and J. Luo are co-inventors of an AR-V7 biomarker technology that has been licensed to Qiagen.
References
- 1.Ajiboye AS, Esopi D, Yegnasubramanian S, Denmeade SR. Androgen Receptor Splice Variants Are Not Substrates of Nonsense-Mediated Decay. Prostate 2017; 77: 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014; 371: 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellur S, Van der Kwast T, Mete O. Evolving concepts in prostatic neuroendocrine manifestations: from focal divergent differentiation to amphicrine carcinoma. Hum Pathol 2018. [DOI] [PubMed] [Google Scholar]
- 4.Berchem GJ, Bosseler M, Sugars LY, Voeller HJ, Zeitlin S, Gelmann EP. Androgens induce resistance to bcl-2-mediated apoptosis in LNCaP prostate cancer cells. Cancer Res 1995; 55: 735–738. [PubMed] [Google Scholar]
- 5.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004; 10: 33–39. [DOI] [PubMed] [Google Scholar]
- 6.Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res 2015; 21: 1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 2008; 68: 5469–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esquenet M, Swinnen JV, Heyns W, Verhoeven G. LNCaP prostatic adenocarcinoma cells derived from low and high passage numbers display divergent responses not only to androgens but also to retinoids. J Steroid Biochem Mol Biol 1997; 62: 391–399. [DOI] [PubMed] [Google Scholar]
- 9.Guo Y, Kyprianou N. Restoration of transforming growth factor beta signaling pathway in human prostate cancer cells suppresses tumorigenicity via induction of caspase-1-mediated apoptosis. Cancer Res 1999; 59: 1366–1371. [PubMed] [Google Scholar]
- 10.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 2009; 69: 2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM et al. LNCaP model of human prostatic carcinoma. Cancer Res 1983; 43: 1809–1818. [PubMed] [Google Scholar]
- 12.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 2009; 69: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res 2012; 72: 3457–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isaacs JT, D’Antonio JM, Chen S, Antony L, Dalrymple SP, Ndikuyeze GH et al. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate 2012; 72: 1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Isaacs JT. Resolving the Coffey Paradox: what does the androgen receptor do in normal vs. malignant prostate epithelial cells? Am J Clin Exp Urol 2018; 6: 55–61. [PMC free article] [PubMed] [Google Scholar]
- 16.Jenster G, Trapman J, Brinkmann AO. Nuclear import of the human androgen receptor. Biochem J 1993; 293 (Pt 3): 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karan D, Schmied BM, Dave BJ, Wittel UA, Lin MF, Batra SK. Decreased androgen-responsive growth of human prostate cancer is associated with increased genetic alterations. Clin Cancer Res 2001; 7: 3472–3480. [PubMed] [Google Scholar]
- 18.Kurita T, Wang YZ, Donjacour AA, Zhao C, Lydon JP, O’Malley BW et al. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ 2001; 8: 192–200. [DOI] [PubMed] [Google Scholar]
- 19.Kyprianou N, English HF, Isaacs JT. Programmed cell death during regression of PC-82 human prostate cancer following androgen ablation. Cancer Res 1990; 50: 3748–3753. [PubMed] [Google Scholar]
- 20.Lam HM, McMullin R, Nguyen HM, Coleman I, Gormley M, Gulati R et al. Characterization of an Abiraterone Ultraresponsive Phenotype in Castration-Resistant Prostate Cancer Patient-Derived Xenografts. Clin Cancer Res 2017; 23: 2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leach FS, Velasco A, Hsieh JT, Sagalowsky AI, McConnell JD. The mismatch repair gene hMSH2 is mutated in the prostate cancer cell line LNCaP. J Urol 2000; 164: 1830–1833. [PubMed] [Google Scholar]
- 22.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275: 1943–1947. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 2013; 73: 483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin X, Tascilar M, Lee WH, Vles WJ, Lee BH, Veeraswamy R et al. GSTP1 CpG island hypermethylation is responsible for the absence of GSTP1 expression in human prostate cancer cells. Am J Pathol 2001; 159: 1815–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab 2003; 88: 2972–2982. [DOI] [PubMed] [Google Scholar]
- 26.Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014; 33: 3140–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu C, Brown LC, Antonarakis ES, Armstrong AJ, Luo J. Androgen receptor variant-driven prostate cancer II: advances in laboratory investigations. Prostate Cancer Prostatic Dis 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moll JM, Kumagai J, van Royen ME, Teubel WJ, van Soest RJ, French PJ et al. A bypass mechanism of abiraterone-resistant prostate cancer: Accumulating CYP17A1 substrates activate androgen receptor signaling. Prostate 2019; 79: 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mostaghel EA, Cho E, Zhang A, Alyamani M, Kaipainen A, Green S et al. Association of Tissue Abiraterone Levels and SLCO Genotype with Intraprostatic Steroids and Pathologic Response in Men with High-Risk Localized Prostate Cancer. Clin Cancer Res 2017; 23: 4592–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mostaghel EA, Zhang A, Hernandez S, Marck BT, Zhang X, Tamae D et al. Contribution of Adrenal Glands to Intratumor Androgens and Growth of Castration-Resistant Prostate Cancer. Clin Cancer Res 2019; 25: 426–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naidu CK, Suneetha Y. Prediction and Analysis of Breast Cancer Related Deleterious Non-Synonymous Single Nucleotide Polymorphisms in the PTEN Gene. Asian Pac J Cancer Prev 2016; 17: 2199–2203. [DOI] [PubMed] [Google Scholar]
- 32.Pflug BR, Reiter RE, Nelson JB. Caveolin expression is decreased following androgen deprivation in human prostate cancer cell lines. Prostate 1999; 40: 269–273. [DOI] [PubMed] [Google Scholar]
- 33.Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ et al. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018; 175: 889. [DOI] [PubMed] [Google Scholar]
- 34.Sedelaar JP, Isaacs JT. Tissue culture media supplemented with 10% fetal calf serum contains a castrate level of testosterone. Prostate 2009; 69: 1724–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp A, Welti J, Blagg J, de Bono JS. Targeting Androgen Receptor Aberrations in Castration-Resistant Prostate Cancer. Clin Cancer Res 2016; 22: 4280–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeda DY, Spisak S, Seo JH, Bell C, O’Connor E, Korthauer K et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018; 174: 422–432 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol 1997; 11: 450–459. [DOI] [PubMed] [Google Scholar]
- 38.Teply BA, Wang H, Luber B, Sullivan R, Rifkind I, Bruns A et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol 2018; 19: 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tombal B, Denmeade SR, Gillis JM, Isaacs JT. A supramicromolar elevation of intracellular free calcium ([Ca(2+)](i)) is consistently required to induce the execution phase of apoptosis. Cell Death Differ 2002; 9: 561–573. [DOI] [PubMed] [Google Scholar]
- 40.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009; 324: 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vander Griend DJ, D’Antonio J, Gurel B, Antony L, Demarzo AM, Isaacs JT. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate 2010; 70: 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun 1990; 173: 534–540. [DOI] [PubMed] [Google Scholar]
- 43.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 1995; 9: 401–406. [DOI] [PubMed] [Google Scholar]
- 44.Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell 2018; 174: 433–447 e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang YC, Banuelos CA, Mawji NR, Wang J, Kato M, Haile S et al. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin Cancer Res 2016; 22: 4466–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Q, Rubenstein JN, Jang TL, Pins M, Javonovic B, Yang X et al. Insensitivity to transforming growth factor-beta results from promoter methylation of cognate receptors in human prostate cancer cells (LNCaP). Mol Endocrinol 2005; 19: 2390–2399. [DOI] [PubMed] [Google Scholar]
- 47.Zhu YL J Regulation of androgen receptor variants in prostate cancer. Asian J Urol 2020; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.