

SUMMARY
We previously demonstrated that leukemia stem cells (LSCs) in de novo acute myeloid leukemia (AML) patients are selectively reliant on amino acid metabolism and that treatment with the combination of venetoclax and azacitidine (ven/aza) inhibits amino acid metabolism, leading to cell death. In contrast, ven/aza fails to eradicate LSCs in relapsed/refractory (R/R) patients, suggesting altered metabolic properties. Detailed metabolomic analysis revealed elevated nicotinamide metabolism in relapsed LSCs, which activates both amino acid metabolism and fatty acid oxidation to drive OXPHOS, thereby providing a means for LSCs to circumvent the cytotoxic effects of ven/aza therapy. Genetic and pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in nicotinamide metabolism, demonstrated selective eradication of R/R LSCs while sparing normal hematopoietic stem/progenitor cells. Altogether, these findings demonstrate that elevated nicotinamide metabolism is both the mechanistic basis for ven/aza resistance and a metabolic vulnerability of R/R LSCs.
In Brief
Disease recurrence is a major cause of death for cancer patients. Jordan et al. show that leukemia stem cells isolated from relapsed acute myeloid leukemia patients have unique metabolic properties, including elevated nicotinamide metabolism. Increased nicotinamide metabolism causes therapy resistance and can be targeted to eradicate relapsed leukemia stem cells.
Graphical Abstract
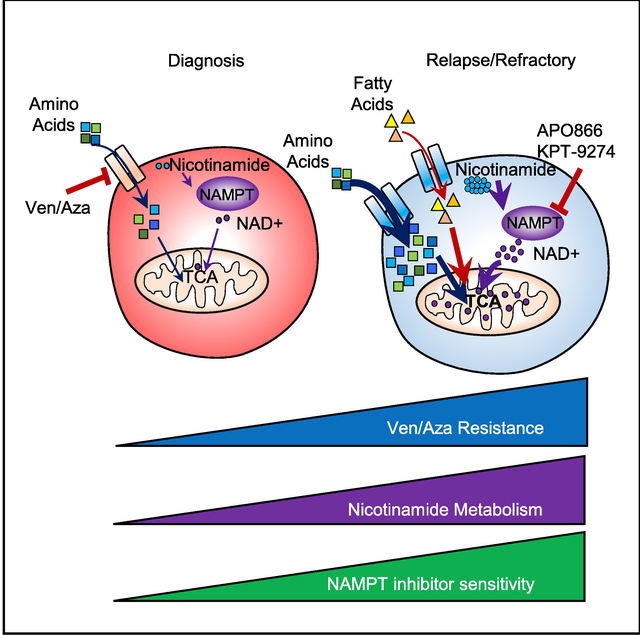
INTRODUCTION
Because of their central role in disease pathogenesis, leukemia stem cells (LSCs) have been extensively studied with the clinical goal of targeting this population in acute myeloid leukemia (AML) patients (Bonnet and Dick, 1997; Guan and Hogge, 2000; Hope et al., 2004; Ishikawa et al., 2007; Jordan et al., 2006; Lapidot et al., 1994; Pollyea and Jordan, 2017; Reya et al., 2001; Shlush et al., 2014, 2017; Terpstra et al., 1996; van Rhenen et al., 2005). Although many strategies have been described, a vulnerability of LSCs that appears central to AML biology is preferential reliance on oxidative phosphorylation (OXPHOS) (Lagadinou et al., 2013; Sriskanthadevan et al., 2015). Reports have described inhibition of OXPHOS as a strategy for AML LSCs, as well as tumor-initiating cells in multiple other forms of cancer (Janiszewska et al., 2012; Lee et al., 2017; Sancho et al., 2015; Viale et al., 2014). Thus, treatment regimens developed with a view toward targeting OXPHOS may have applicability for a range of cancers.
One strategy to inhibit OXPHOS in AML cells is through the use of BCL-2 inhibitors. We have previously demonstrated in primary human LSCs that OXPHOS at least partially depends on BCL-2 activity (Lagadinou et al., 2013). Similarly, other approaches that disrupt mitochondrial respiration target the LSC population (Chan et al., 2015; Cole et al., 2015; Jones et al., 2018, 2019; Liyanage et al., 2017; Skrtić et al., 2011; Sriskanthadevan et al., 2015), supporting a central role for OXPHOS in LSC survival. These preclinical findings have led to significant interest in the use of such strategies for AML patients. To this end, a series of recent clinical studies examined use of the BCL-2 inhibitor venetoclax in combination with conventional agents: azacitidine and decitabine (DiNardo et al., 2018a; Pollyea et al., 2018). Venetoclax with either of these agents showed promising activity in de novo AML patients deemed otherwise unfit for conventional aggressive chemotherapy (DiNardo et al., 2018a). Detailed analysis of patients undergoing treatment with venetoclax and azacitidine (ven/aza) showed in vivo inhibition of OXPHOS and efficient eradication of the LSC population (Jones et al., 2018; Pollyea et al., 2018). Altogether, these findings indicate that metabolic targeting of LSCs may provide an exciting approach to AML therapy.
From a mechanistic perspective, the sensitivity of LSCs to ven/aza treatment appears to reflect their distinct metabolic phenotype. Specifically, analysis of LSCs from de novo AML patients shows an aberrant reliance on the uptake and catabolism of amino acids as a means to drive the tricarboxylic acid (TCA) cycle and thereby promote OXPHOS (Jones et al., 2018). LSCs are unable to effectively use common metabolic fuels such as glucose or fatty acids (Jones et al., 2018). Treatment with ven/aza inhibits amino acid metabolism, thus decreasing OXPHOS and leading to LSC-specific death (Jones et al., 2018). Although many basic aspects of these findings remain to be elucidated, the empirical observations indicate that LSCs in de novo AML patients are relatively inflexible with regard to energy metabolism pathways (Jones et al., 2018), suggesting that metabolic targeting strategies may be fruitful for this patient population.
Despite promising results in previously untreated AML patients, reports on the use of venetoclax-based regimens for relapsed/refractory (R/R) patients indicate less efficacy, with objective response rates of only 21% (9 of 43 patients), and median overall survival of 3 months (DiNardo et al., 2018b). Although the reasons for reduced efficacy in the R/R setting may be complex, one component of the problem likely lies in the fundamental biology of LSCs. LSCs are the origin of relapsed AML (Shlush et al., 2017) and have been shown to undergo dramatic phenotypic changes during progression from de novo to relapsed disease (Ho et al., 2016). Furthermore, through investigation of LSC metabolism in R/R AML patients, we have described upregulation of fatty acid metabolism as a potential compensatory metabolic pathway by which the effects of ven/aza treatment may be negated (Jones et al., 2018). Consequently, we hypothesized that an altered metabolic phenotype in R/R AML patients may be responsible for the lack of ven/aza efficacy.
To explore our hypothesis, we performed in the present study a detailed comparative analysis of the LSC metabolome in de novo versus relapsed patients. We sought to determine the following: (1) Are LSCs isolated from R/R AML patients metabolically distinct from de novo patients? (2) Do metabolic differences mediate ven/aza resistance? (3) Can metabolic vulnerabilities be targeted to specifically eradicate R/R LSCs?
RESULTS
Relapse Predicts a Worse Response to ven/aza
We first sought to corroborate whether R/R AML patients had inferior responses to ven/aza compared with previously untreated or de novo AML patients, as previous single-center studies have suggested (DiNardo et al., 2018b). In our single-institution experience treating 49 de novo AML patients with ven/aza both in the context of the multi-institutional study NCT02203773 (N = 33) and with off-label use (N = 16), 40/49 (82%) achieved complete remission (CR) or CR with incomplete count recovery (CRi). In contrast, in our single-institution, off-label experience in the R/R setting (N = 12), only 3/12 (25%) R/R patients achieved a CR/CRi. R/R patients received multiple treatments before ven/aza; however, all received cytotoxic chemotherapy or a hypomethylating agent at some point before ven/aza (Table S1). Using Fisher’s exact test to compare response rates between de novo and R/R patients, the difference is highly significant (p = 0.0004). R/R and de novo patients had similar baseline characteristics, with the exception that R/R patients were more likely to be younger (p = 0.0075) (Table S2). Univariate logistic regression analysis showed cytogenetic risk and R/R disease to be the sole predictors of response to ven/ aza (Table 1); a multivariate logistic regression analysis constructed by including all covariates whose p values were 0.10 or below showed only R/R disease status to be predictive of response (p = 0.0024). Thus, our clinical experience with ven/ aza in R/R patients is consistent with reports from other single-institution studies (DiNardo et al., 2018b).
Table 1.
Univariate Logistic Regression Analysis to Determine Predictors of Response (CR/CRi) to Venetoclax with Azacitidine
| Predictor | Odds Ratio (95% CI) | p Value |
|---|---|---|
| Age | 1.04 (0.99, 1.10) | 0.1521 |
| Prior treatment (ref = no) | 0.075 (0.017, 0.334) | 0.0007 |
| Antecedent hematological disorder | 1.01 (0.30, 3.44) | 0.9918 |
| Treatment-related AML | 0.26 (0.05, 1.32) | 0.1048 |
| Cytogenetic risk group | 6.46 (1.75, 23.92) | 0.0120 |
| TP53 mutation | 0.58 (0.09, 3.80) | 0.9503 |
| FLT3 ITD mutation | 0.32 (0.08, 1.28) | 0.9752 |
| ASXL1 mutation | 0.57 (0.15, 2.07) | 0.9560 |
| European LeukemiaNet risk group | 0.11 (0.01, 0.91) | 0.9556 |
Relapsed LSCs Are Metabolically Distinct from De Novo LSCs
We recently demonstrated that ven/aza targets LSCs in de novo AML patients by decreasing energy metabolism (Jones et al., 2018; Pollyea et al., 2018). We therefore hypothesized that the inferior outcomes observed in R/R AML patients at least partially resulted from the differences in metabolism between de novo and R/R LSCs. To investigate this hypothesis, LSCs were enriched from primary human AML specimens by sorting cells for relatively low levels of reactive oxygen species (ROS) (Jones et al., 2018; Lagadinou et al., 2013; Pei et al., 2018; Pollyea et al., 2018). We have previously shown that relatively low ROS levels enrich LSCs in both de novo and R/R AML patient specimens (Lagadinou et al., 2013; Pei et al., 2018).
Global metabolomics analysis of LSCs isolated from 6 de novo and 6 R/R AML patients was performed by mass spectrometry. Because of cell number constraints, specimens were not paired except where indicated. This analysis revealed that de novo and R/R LSCs are metabolically distinct (Figure 1A). A total of 18 metabolites were found in higher abundance in R/R LSCs compared with de novo LSCs. Of the 18 metabolites, ten amino acids were increased in R/R LSCs compared with de novo LSCs. (Figure 1B). Because ven/aza targets LSCs at least partly by disrupting amino acid metabolism (Jones et al., 2018), it is possible that the increased level of amino acids observed in R/R LSCs contribute to ven/aza resistance in these cells. Indeed, we have previously reported that R/R LSCs are not sensitive to amino acid loss (Jones et al., 2018). We validated this finding in paired de novo and relapsed LSCs and AML blasts derived from the same patients. We found that amino acids were increased in relapsed LSCs compared with paired de novo LSCs (p = 1.75 × 10−11)(Figure 1C). Several other metabolites were determined to have increased abundance in R/R LSCs compared with de novo LSCs (Figure S1A), including the NAD+ precursor nicotinamide (Figure 1D). NAD+ levels were also increased in paired de novo/relapse LSCs (Figure 1E). NAD+ levels were not different in relapsed AML blasts compared with de novo AML blasts (Figure 1E), highlighting the importance of specifically analyzing the stem cell population within cancers like AML. NAD+ is a master regulator of cellular metabolism, including cancer cell metabolism (Chiarugi et al., 2012); therefore, we hypothesized that the increased levels of nicotinamide results in increased NAD+ production, which in turn may drive multiple components of metabolism in R/R LSCs.
Figure 1. R/R LSCs Have Increased Nicotinamide Metabolism.

(A) Principal-component analysis of de novo and R/R LSCs generated using MetaboAnalyst 4.0. AML specimens used in this analysis include AML1–AML9 and AML14–AML16.
(B) Heatmap of amino acids levels in de novo and R/R LSCs. AML specimens used in this analysis include AML1–AML9 and AML14–AML16.
(C) Amino acids levels in paired de novo and relapsed LSCs. AML specimens used in this analysis include AML17–AML20.
(D) Nicotinamide levels in de novo and R/R LSCs. AML specimens used in this analysis include AML1–AML9 and AML14–AML20. The left graph represents nicotinamide levels in unpaired specimens, and the right graph represents nicotinamide levels in paired specimens.
(E) NAD+ levels in paired de novo and relapsed LSCs and AML blasts (non-LSCs). AML specimens used in this analysis include AML17–AML21.
(F) Levels of SIL nicotinamide in de novo and R/R LSCs 12 h after [13C3,15N] nicotinamide incubation determined by mass spectrometry. AML2 and AML5 were used in this analysis.
(G) Levels of SIL NAD+ from nicotinamide in de novo and R/R LSCs 12 h after [13C3,15N] nicotinamide incubation determined by mass spectrometry. AML2 and AML5 were used in this analysis.
(H) Ratio of SIL NAD+/nicotinamide in de novo and R/R LSCs 12 h after [13C3,15N] nicotinamide incubation. AML2 and AML5 were used in this analysis. Statistical significance was determined using Student’s t test (B, D, F–H) or a paired Student’s t test (E). *p < 0.05, **p < 0.01, ***p < 0.005 ****p < 0.001.
To evaluate the role of nicotinamide, we first determined its contribution to the production of NAD+ in paired de novo and R/R LSCs. In eukaryotes, NAD+ is synthesized through the salvage pathway from nicotinamide (Kennedy et al., 2016) or the de novo synthesis pathway from the amino acid tryptophan (González Esquivel et al., 2017). To determine the relative contributions of the salvage and de novo synthesis pathways to NAD+ levels, LSCs isolated from de novo and R/R primary AML specimens were incubated with stable-isotope-labeled (SIL) 13C15N isotopes of nicotinamide or tryptophan. Labeled nicotinamide and tryptophan uptake and metabolism were subsequently measured by mass spectrometry. Nicotinamide was taken up (Figure 1F) and metabolized to NAD+ (Figure 1G) at significantly greater levels by relapsed LSCs compared with de novo LSCs. This resulted in an increased rate of NAD+ synthesis through the salvage pathway in relapsed LSCs (Figure 1H), suggesting an increase in the activity of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in the conversion of nicotinamide to NAD+ (Chiarugi et al., 2012). Despite increased NAD+ levels, no changes in NAMPT protein levels (Figure S1B) or NAMPT cofactor phosphoribosylpyrophosphate (PRPP)(Figure S1C) were observed in de novo and R/R LSCs. Furthermore, chemotherapy exposure did not alter cellular levels of NAD+ (Figure S1D). Overall, these data suggest that the most likely reason for increased NAD+ levels in R/R LSCs is increased nicotinamide uptake.
In contrast to the nicotinamide flux experiments, analysis of tryptophan metabolism reveal that tryptophan is metabolized to formyl-kynurenine, the first step in the NAD+ synthesis from tryptophan, in paired de novo and R/R LSCs (Figures S1E and S1F); however, tryptophan-derived 13C NAD+ was below detectable levels in these samples, indicating that tryptophan was not usedby de novo or R/R LSCs to produce NAD+ at detectable levels (Figure S1G). To confirm these findings, we incubated de novo and R/R LSCs with tryptophan and measured intercellular NAD+/H levels. We found no significant changes in total NAD+/H levels upon tryptophan addition (Figure S1H), suggesting that LSCs predominantly use the salvage pathway, not the de novo synthesis pathway, to synthesize NAD+.
Because NAD+ is an essential coenzyme used in various enzymatic oxidation/reduction reactions and plays indispensable roles in glycolysis, the TCA cycle, and fatty acid oxidation (FAO) (Chiarugi et al., 2012), we hypothesized that the increased NAD+ production observed in relapsed LSCs is likely to correlate with increased energy metabolism. Consistent with this hypothesis, ATP levels were increased in R/R LSCs compared with de novo LSCs (Figure 2A). To determine how R/R LSCs had increased ATP levels compared with de novo LSCs, SIL metabolic tracing was employed to investigate 13C15N-amino-acid catabolism, fatty acid metabolism (13C16 palmitate), and glycolysis (U-13C6 glucose) in paired de novo and relapsed LSCs (Figure 2B). R/R LSCs had increased amino acid uptake (Figure 2C) and catabolism into TCA cycle intermediates citrate, 2-oxogluta-rate, succinate, and malate compared with de novo LSCs (Figure 2D; Figure S2A). Fatty acid uptake was not different between de novo and R/R LSCs (Figure 2E); however, fatty acid metabolism to citrate and malate was elevated (Figure 2F; Figure S2B). Importantly, fatty acid metabolism to carnitines was not significantly different between de novo and R/R LSCs indicating that the increase in fatty acid metabolism was preferentially driving energy production through the TCA cycle (Figure S2B). Finally, glucose uptake (Figure 2G), glycolysis (Figure 2H), and 13C enrichment in citrate (Figure 2I) were increased in R/R LSCs compared with de novo LSCs. Overall, these data suggest that energy metabolism is increased in R/R LSCs compared with de novo LSCs.
Figure 2. R/R LSCs Have Increased Energy Metabolism Compared with De Novo LSCs.

(A) ATP levels from LSCs isolated from 3 de novo and 3 R/R AML patients determined by mass spectrometry. AML specimens used in this analysis include AML1– AML6. Statistical significance was determined using an unpaired Student’s t test.
(B) Schematic of experimental design. LSCs were isolated from de novo and R/R AML patient specimens and then incubated with [13C6] glucose, 13C15N amino acids, or[13C16] palmitic acid for 1 or 8 h. Metabolites were then measured by mass spectrometry.
(C) Heatmap of 13C15N amino acids in de novo and R/R LSCs after a 1-h incubation. Statistical significance was determined using a paired Student’s t test. AML2 and AML5 were used in this analysis.
(D) TCA cycle intermediate citrate metabolized from 13C15N amino acids.
(E) Palmitic acid uptake from [13C16 palmitic acid.
(F) TCA cycle intermediate citrate metabolized from [13C16 palmitic acid. (G) Glucose uptake from [13C6] glucose.
(G) lucose uptake from [13C6] glucose.
(H) Pyruvae metabolized from [13C6] glucose.
(I) TCA cycle intermediate, citrae metabolized from [13C6] glucose.
(D–I) Statistical significance determined by two-way ANOVA, and AML2 and AML5 were used in the analysis. *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001.
Increased Nicotinamide Metabolism Mediates Energy Metabolism and ven/aza Resistance
As shown in Table 1 and Figures 1 and 2, R/R AML patients have an inferior response to ven/aza, increased nicotinamide, and increased energy metabolism compared with de novo LSCs. ven/aza targets de novo LSCs by decreasing energy metabolism (Jones et al., 2018; Pollyea et al., 2018). Therefore, we hypothesized that increased nicotinamide metabolism in R/R LSCs mediates ven/aza resistance. To explore this hypothesis, LSCs isolated from de novo AML patient specimens were incubated with nicotinamide. Subsequently, NAD+ levels, energy metabolism, and response to ven/aza were measured. Nicotinamide treatment increased levels of NAD+/H in de novo LSCs (Figure 3A). Next, the effect of nicotinamide pretreatment on energy metabolism was studied by measuring metabolic flux of SIL amino acids, [13C16] palmitic acid, and [13C6] glucose (sepa-rately) (Figure 3B). Nicotinamide pretreatment resulted in a significant increase in amino acid uptake (Figure 3C) and catabolism into malate but did not increase flow to 2-oxoglutarate (Figure 3D). Nicotinamide pretreatment did not increase fatty acid levels (Figure 3E) but did increase metabolism of fatty acids into TCA cycle intermediates 2-oxoglutarate and malate (Figure 3E). Furthermore, nicotinamide pretreatment increased glucose uptake (Figure 3F) but did not increase contribution of glucose metabolism to the TCA cycle (data not shown). Finally, nicotinamide pretreatment did not significantly alter other metabolic pathways (Figures S3A and S3B). Overall, these data show that by simply increasing nicotinamide levels in de novo LSCs, we are able to recapitulate many metabolic features of R/R LSCs.
Figure 3. Nicotinamide Metabolism Mediates Energy Metabolism and ven/aza Resistance.

(A) NAD+/H levels upon a 1-h incubation with 128 μM nicotinamide.
(B) Schematic of experimental design. LSCs were isolated from de novo AML patient specimens, incubated with 128 μM nicotinamide for 1 h, and then incubated with [13C6] glucose, 13C,15N amino acids, or [13C16] palmitic acid for 8 h. Metabolites were then measured by mass spectrometry.
(C) Heatmap of amino acid upon in vehicle control and nicotinamide pretreatment LSCs.
(D) TCA cycle intermediates metabolized from 13C15N amino acids.
(E) Palmitate and TCA cycle intermediates metabolized from [13C16] palmitic acid.
(F) Glucose and pyruvate metabolized from [13C6] glucose.
(G) Viability of de novo LSCs pretreated with 128 μM nicotinamide for 1 h and then ven (500 nM)/aza (2.5 μM) for 24 h where indicated. Statistical significance was determined by a two-way ANOVA. AML specimens 1–3, 11, and 12 were used in this analysis.
Statistical significance was determined using an unpaired Student’s t test (A and D–F) or a paired Student’s t test (C), and AML1–AML3 were used in the analysis. *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001.
We next asked whether increased nicotinamide induces ven/ aza resistance. LSCs isolated from de novo patient specimens were treated in vitro with ven/aza with or without nicotinamide pretreatment. As we have previously shown, LSCs isolated from de novo patients are sensitive to ven/aza (Jones et al., 2018; Pollyea et al., 2018); however, pretreatment with nicotinamide negates the cytotoxic effect of ven/aza, resulting in resistance to treatment (Figure 3G). Nicotinamide pretreatment also rescued the loss of viability observed upon venetoclax treatment alone (Figure S3C), but not azacitidine, cytarabine, or doxorubicin treatment (Figures S3D and S3E). However, the killing effect of azacitidine, cytarabine, or doxorubicin was less than that observed with venetoclax treatment, which may contribute to the lack of viability rescue. Altogether, these data suggest that R/R AML patients respond poorly to ven/aza therapy because of increased levels of nicotinamide and nicotinamide metabolism in R/R LSCs.
Inhibition of Nicotinamide Metabolism Decreases Survival of Relapsed LSCs
We next hypothesized that nicotinamide metabolism represents a potential metabolic vulnerability in R/R LSCs. An analysis of TCGA data shows that increased NAMPT expression correlates with poor survival outcomes in AML (Figure 4A). Several NAMPT inhibitors, such as APO866, have been developed and tested in preclinical and early-phase clinical trials (Sampath et al., 2015). Furthermore, APO866 treatment has been previously shown to target AML cells (Nahimana et al., 2009) but has not been evaluated in LSCs. Treatment of primary AML specimens with APO866 decreased NAD+/H levels in both de novo and R/R LSCs (Figure S4A), indicating successful inhibition of NAMPT activity. Despite decreased NAD+/H levels in both de novo and R/R LSCs, NAMPT inhibition only decreased viability and colony-forming potential of R/R LSCs (Figures 4B and 4C), indicating a selective reliance on NAMPT activity in relapsed AML. This finding was confirmed with a second NAMPT inhibitor, KPT-9274, which also decreased viability and colony-forming potential specifically in R/R LSCs (Figures 4D and 4E). Notably, KPT-9274 has been reported to target AML cells through inhibition of NAMPT (Mitchell et al., 2019). The selectively of APO866 and KPT-9274 for eradication of R/R LSCs was evident at relatively low doses (10 and 100 nM, respectively). In contrast, NAMPT inhibition did not decrease HSPC frequency or colony-forming ability at these doses (Figures 4F and 4G). Higher doses of APO866 and KPT-9274 decreased viability of both de novo and R/R LSCs (Figure S4B); however, higher doses also resulted in decreased normal HSPC colony-forming ability (Figure S4C). Furthermore, small interfering RNA (siRNA) knockdown of NAMPT in normal bone marrow cells decreased colony-forming ability (Figure S4D), suggesting that appropriate dosing of NAMPT inhibitor drugs will be important to achieve optimal clinical results. APO866 and KPT-9274 treatment also targeted ROS-high AML blasts (Figure S4E). To confirm that the cytotoxic effects observed upon NAMPT inhibition resulted from NAMPT targeting, we knocked down NAMPT using siRNA in three R/R patient specimens (Figure S4F) and measured viability 24 h after knockdown, as well as colony-forming potential. NAMPT knockdown decreased viability (Figure 4H) and colony-forming potential (Figure 4I) in each specimen, confirming the cytotoxic effects of the inhibitors resulted from NAMPT targeting.
Figure 4. Inhibition of Nicotinamide Metabolism Specifically Targets R/R LSCs.

(A) Percentage of survival of AML patients stratified by the 25% highest and lowest NAMPT expression. Data mined from TCGA.
(B) Viability of de novo and R/R LSCs after a 24-h treatment with 10 nM APO866. Each dot represents an individual patient specimen normalized to vehicle control. AML specimens used in this analysis are AML1–AML12.
(C) Colony-forming ability of de novo and R/R LSCs after treatment with 10 nM APO866. Each dot represents an individual patient specimen normalized to vehicle control. AML specimens used in this analysis are AML1, AML2, AML4–AML6, and AML8–AML11.
(D) Viability of de novo and R/R LSCs after a 24-h treatment with 100 nM KPT-9274. Each dot represents an individual patient specimen normalized to vehicle control. AML specimens used in this analysis include AML1–AML13.
(E) Colony-forming ability of de novo and R/R LSCs after treatment with 100 nM KPT-9274. Each dot represents an individual patient specimen normalized to vehicle control. AML specimens used in this analysis include AML1, AML2, and AML4–AML10.
(F) Frequency of normal HSPCs (CD34+/CD45+ cells) from normal mobilized peripheral blood after a 24-h treatment with 10 nM APO866 or 100 nm KPT-9274. Each dot represents an individual patient specimen.
(G) Colony number from normal mobilized peripheral blood after a 24-h treatment with 10 nM APO866 or 100 nM KPT-9274. Each dot represents an individual patient specimen.
(H) Viability of primary AML specimens 5, 8, and 9 after siRNA-mediated knockdown of NAMPT compared with scrambled control.
(I) Colony-forming ability of AML specimens 5, 8, and 9 after siRNA-mediated knockdown of NAMPT compared with scrambled control.
(J) Viability of LSCs isolated from paired de novo and relapsed specimens (AML specimens 17–20) after treatment with APO866, KPT-9274, ven/aza, or cytarabine compared with vehicle control.
Statistical significance was determined using Student’s t test (H and I), an unpaired Student’s t test (B–F), or ANOVA (J). *p < 0.05, **p < 0.01, ****p < 0.001.
Finally, we wanted to confirm that NAMPT inhibition preferentially targeted relapsed LSCs compared with diagnosis LSCs isolated from the same patient. To do this, we isolated LSCs from diagnosis and relapsed matched pairs and treated them with several drug regimens for 24 h. As shown in Figure 4J, relapsed LSCs were more sensitive to both APO866 and KPT-9274 treatment compared with diagnosis LSCs, as expected. In addition, we treated with same specimens with ven/aza and cytarabine. ven/aza preferentially targeted the diagnosis LSCs (Figure 4J), consistent with the clinical data. In general, cytarabine treatment had a minimal effect on LSCs (Figure 4J). Similar results were obtained in ROS-high AML blasts (Figure S4G). Notably, cytarabine killed diagnosis blasts better than what was observed in LSCs (Figure S4G).
To confirm that NAMPT inhibition was targeting functional R/R LSCs, specimens from the same patient obtained at diagnosis and relapse were cultured with APO866 or ven/aza for 24 h and engrafted into immunodeficient mice. As expected, de novo, but not relapsed, LSCs were targeted by ven/aza treatment, whereas R/R LSCs, but not de novo LSCs, were targeted by APO866 treatment (Figure 5A). These data indicate that functional R/R LSCs are insensitive to ven/aza but sensitive to NAMPT inhibition. Next, we determined whether NAMPT inhibition could target R/R LSCs in a primary R/R AML xenograft model (Figure 5B). Two weeks of APO866 or KPT-9274 treatment decreased R/R leukemia burden in vivo in immune-deficient mice (Figures 5C and 5D). NAMPT inhibitor treatment decreased secondary engraftment (Figure 5C), indicating that NAMPT inhibition targets functional LSCs in vivo. In vivo treatment of a de novo AML specimen did not decrease leukemic burden (Figure S5A). Chemotherapy treatment did decrease leukemic burden in the de novo AML specimen in vivo (Figure S5A), suggesting NAMPT inhibition is not an alternative to conventional chemotherapy for newly diagnosed AML patients. Furthermore, in vivo NAMPT inhibition using APO866 or KPT-9274 did not alter normal CD45+ hematopoietic cells (Figure 5E), HSPCs (Figure 5F), or monocyte and lymphocyte populations (Figures S5B–S5D), consistent with previous findings (Mitchell et al., 2019). Overall, these data demonstrate that NAMPT inhibition may be a viable approach to target R/R LSCs in AML patients while sparing normal HSPCs and hematopoietic cells. Finally, we determined whether NAMPT inhibition could potentiate the effect of venetoclax. The combination therapy was not better than single agents (Figure S5E), suggesting that there may be overlap in the mechanism by which ven/aza and NAMPT inhibitors target LSCs.
Figure 5. Inhibition of Nicotinamide Metabolism Specifically Targets R/R LSCs In Vivo.

(A) Paired specimens from the same patient, AML2 (de novo) and AML5 (relapse), were treated with ven/aza or APO866 for 24 h and then transplanted into immunodeficient mice. Engraftment was measured in the bone marrow.
(B) Experimental schema for the in vivo treatment and secondary engraftment assays.
(C) Leukemia burden in mouse bone marrow after 2 weeks of APO866 treatment and engraftment after in vivo APO866 treatment. Leukemia burden in mouse femur upon secondary engraftment.
(D) Leukemia burden in mouse femur after 2 weeks of KPT-9274 treatment.
(E) Levels of normal human cell engraftment (CD45+ cells) in mouse femur after 2 weeks of APO866 or KPT-9274 treatment.
(F) Normal HSPC (CD45+/CD34+) in mouse femur after 2 weeks of APO866 or KPT-9274 treatment.
(A and C–F) Statistical significance was determined using an unpaired Student’s t test. Each dot represents an individual mouse. **p < 0.01, ***p < 0.005, ****p < 0.001.
Inhibition of Nicotinamide Metabolism Decreases OXPHOS in Relapsed LSCs
To determine how NAMPT inhibition was specifically targeting R/ R LSCs, the effect of APO866 and KPT-9274 treatment on LSC energy metabolism was determined using a Seahorse assay. We observed that APO866 treatment minimally decreased basal OXPHOS (Figure 6A) but substantially decreased mitochondrial spare capacity (Figure 6B) in R/R LSCs as early as 4 h after treatment. No loss of basal or spare capacity was observed for LSCs from de novo patients. Consistent with these data, KPT-9274 treatment also decreased OXPHOS in R/R LSCs, but not de novo LSCs, 4 h after treatment (Figure 6C). Furthermore, a 24-h in vivo treatment of a primary R/R AML specimen with APO866 or KPT-9274 also decreased NAD+ levels (Figure 6D), basal OXPHOS (Figure 6E), and spare capacity (Figure 6F). Although NAMPT inhibition has previously been shown to decrease glycolysis (Watson et al., 2009; Xiao et al., 2013), we did not observe any effect of APO866 or KPT-9274 on glycolysis in vitro or in vivo (Figures S6A–S6C). These finding are consistent with our previous data that shows LSCs have low glycolytic capacity and do not depend on glucose for survival (Jones et al., 2018; Lagadinou et al., 2013), and they demonstrate that the mechanism by which NAMPT inhibition targets LSCs may be different from the mechanism by which it targets other cancer cell types or more mature AML blasts. NAMPT inhibition decreased glycolysis, but not OXPHOS, in ROS-high AML blasts, suggesting the NAD+ biology is critical for leukemic cell survival through different mechanisms in different cell populations (i.e., LSCs versus non-LSCs) (Figure S6D). NAMPT knockdown in total leukemia decreases OXPHOS in all AML specimens tested and glycolysis in 2 of the 3 AML specimens (Figure S6E).
Figure 6. Inhibition of Nicotinamide Metabolism Decreased OXPHOS in R/R LSCs.

(A and B) Oxygen consumption (A) and spare oxygen consumption capacity (B) determined by Seahorse assay in de novo and R/R LSCs after a 4-h treatment with 10 nM APO866. Each dot represents an individual patient specimen normalized to vehicle control. AML specimens used in this analysis are AML1–AML6, AML8, AML10, and AML11.
(C) Oxygen consumption determined by Seahorse assay in de novo and R/R LSCs after a 4-h treatment with 100 nM KPT-9274. Each dot represents an individual patient specimen normalized to vehicle control. AML specimens used in this analysis are AML1–AML6, AML8, and AML9.
(D) NAD+/H levels in leukemic cells isolated from mice treated with APO866 or KPT-9274 for 24 h.
(E) Oxygen consumption determined by Seahorse assay in leukemic cells isolated from mice treated with APO866 or KPT-9274 for 24 h.
(F) Spare oxygen consumption capacity determined by Seahorse assay in leukemic cells isolated from mice treated with APO866 or KPT-9274 for 24 h.
(G) Abundance of TCA cycle intermediates in de novo LSCs (red/pink) and R/R LSCs (blue/light blue) treated with vehicle control or APO866 for 4 h determined by mass spectrometry. Each dot represents an individual patient. AML specimens used in this analysis are AML1–AML6. Statistical significance was determined by a two-way ANOVA. Yellow stars indicate enzymes within the TCA cycle that are NAD+ dependent.
(H) Activity of 2-oxoglutarate dehydrogenase, isocitrate dehydrogenase, malate dehydrogenase, or hexokinase in R/R LSCs upon treatment with vehicle control or 10 nM APO866 for 4 h. Each dot represents an individual patient specimen. AML specimens used in this analysis are AML4–AML6.
(I) Enzyme activity was determined in de novo and R/R LSCs. AML1–AML3, AML5, AML6, and AML8 were used for this analysis. Each dot represents an individual AML specimen. Statistical significance was determined using a pair Student’s t test.
Statistical significance was determined using an unpaired Student’s t test (A–C and H) or ANOVA (D–F). *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001.
To determine how NAMPT inhibition effects OXPHOS, we used mass spectrometry to measure changes in metabolites following 4 h of APO866 treatment. Accumulation of citrate and fumarate, as well as shunting of 2-oxoglutarate to 2-hydroxy-glutarate specifically in R/R LSCs (Figure 6F), was observed upon APO866 treatment. Furthermore, APO866 treatment caused accumulation of malate in both de novo and R/R LSCs (Figure 6G). This TCA cycle intermediate pattern suggests decreased activity of NAD+-dependent TCA cycle enzymes, including isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase, and malate dehydrogenase (Figure 6G). The only other metabolic changes noted upon NAMPT inhibition were observed in both de novo and R/R LSCs (Figure S6F). Therefore, we hypothesized that APO866 treatment targets R/R LSCs by decreasing the activity of NAD+-dependent enzymes. To directly test this hypothesis, we measured the enzyme activities of isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase, and malate dehydrogenase in de novo and R/R LSCs. The activity of all three enzymes was significantly decreased in R/R LSCs (Figure 6H), but not in de novo LSCs (Figure S6G), upon APO866 treatment. Furthermore, the NAD+-independent enzyme hexokinase was not decreased upon APO866 treatment in R/R LSCs (Figure 6H) or de novo LSCs (Figure S6G), demonstrating that the effects of NAMPT inhibition are specific to NAD+-dependent enzymes. R/R LSCs have increased baseline activity of isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase, and malate dehydrogenase compared with de novo LSCs (Figure 6I), suggesting that NAD+-dependent TCA cycle enzymes require higher NAD+ levels for their activity. Altogether, these data show that NAMPT inhibition specifically targets R/R LSCs by decreasing NAD+-dependent TCA cycle enzymes, resulting in decreased OXPHOS levels.
NAMPT Inhibition Decreases OXPHOS by Inhibiting Amino Acid and Fatty Acid Metabolism
Next, we sought to determine which metabolic fuels were affected by NAMPT inhibition in R/R LSCs. To accomplish this, R/R LSCs were treated with APO866 for 4 h and incubated with SIL amino acids, palmitic acid, or glucose; subsequent incorporation in the TCA cycle was measured by mass spectrometry (Figure 7A). Amino acid levels were significantly decreased (Figure 7B), malate was significantly increased, and 2-oxoglutarate was significantly decreased upon APO866 treatment (Figure 7C). No changes in the 13C content of citrate from SIL amino acids were observed upon APO866 treatment (Figure S7A). Although palmitic acid uptake was not altered by APO866 treatment (Figure 7D), its metabolism into 2-oxogluta-rate was significantly decreased (Figure 6E). Citrate and malate levels were not significantly changed upon APO866 treatment from SIL palmitic acid (Figure S7B). Furthermore, no changes in glucose, pyruvate, or citrate from SIL glucose were observed upon APO866 treatment (Figures S7C and S7D), consistent with data from our Seahorse studies (Figure S6A). However, we did observe a minimal but significant decrease in SIL lactate levels (Figure S7D). Altogether, these data suggest that NAMPT inhibition targets R/R LSCs by decreasing OXPHOS specifically through decreased amino acid and fatty acid metabolism, which results from decreased TCA cycle enzyme activity.
Figure 7. Nicotinamide Metabolism Inhibition Decreases Amino Acid and Fatty Acid Metabolism.

(A) Schematic of experimental design. LSCs were isolated from R/R AML patient specimens, treated with vehicle control or 10 nM APO866 for 4 h, and then incubated with [13C6] glucose, 13C15N amino acids, or [13C16] palmitic acid for 8 h. Metabolites were then measured by mass spectrometry.
(B) Heatmap of 13C15N amino acids levels upon control or 10 nM APO866 treatment. AML5 was used in this analysis.
(C) Malate levels from 13C15N amino acids in control or 10 nM APO866-treated LSCs. AML5, AML8, and AML9 were used in this analysis.
(D) [13C16] palmitic acid levels in R/R LSCs treated with vehicle control or 10 nM APO866. AML5, AML8, and AML9 were used in this analysis.
(E) TCA cycle intermediates metabolized from 13C16 palmitic acid with vehicle or 10 nM APO866 treatment. AML5, AML8, and AML9 were used in this analysis.
(F) Viability of primary AML specimens 5, 8, and 9 after siRNA-mediated knockdown of HADH compared with scrambled control upon ven/aza or vehicle treatment.
(G) Colony-forming ability of primary AML specimens 5, 8, and 9 after siRNA-mediated knockdown of HADH compared with scrambled control upon ven/aza or vehicle treatment.
(H) OXPHOS levels of primary AML specimens 5, 8, and 9 after siRNA-mediated knockdown of HADH compared with scrambled control upon ven/aza or vehicle treatment.
(I) Diagram illustrating that LSCs isolated from R/R AML patients have increased nicotinamide metabolism and ven/aza resistance. Increased nicotinamide metabolism mediates overall energy metabolism in R/R LSCs, resulting in increased amino acid and fatty acid metabolism into the TCA cycle.
(J) Summary diagram showing that increased NAD+ levels in R/R LSCs are necessary for sufficient function of TCA cycle enzymes and that inhibition of nicotinamide metabolism decreases TCA cycle enzyme function, resulting in decreased levels of OXPHOS and LSC death.
Statistical significance was determined using Student’s t test (F–H), a paired Student’s t test (B), or an unpaired Student’s t test (C–E). *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001.
We have previously demonstrated that ven/aza can decrease amino acid levels in LSCs (Jones et al., 2018); therefore, we hypothesized that inhibition of NAD+-dependent fatty acid metabolism should resensitize resistant LSCs to ven/aza. Hydroxyacyl-coenzyme A dehydrogenase (HADH) is an NAD+-dependent enzyme involved in FAO. Knockdown of HADH alone (Figure S7E) did not affect R/R LSC viability (Figure 7F), colony-forming ability (Figure 7G), or OXPHOS (Figure 7H). However, knockdown of HADH targeted LSCs upon ven/aza treatment, resulting in decreased LSC viability (Figure 7F), colony-forming ability (Figure 7G), and OXPHOS (Figure 7H). These data suggest that the mechanism by which NAMPT inhibition targets R/R LSCs involves decreasing both amino acid and fatty acid metabolism.
Overall, our data demonstrate that nicotinamide metabolism mediates energy metabolism in R/R LSCs. We propose a model in which R/R LSCs have increased nicotinamide metabolism and resistance to ven/aza compared with de novo LSCs (Figure 7I). We have previously shown that de novo LSCs are metabolically inflexible and preferentially reliant on amino acid metabolism for survival, which can be targeted by ven/aza (Jones et al., 2018). In contrast, LSCs isolated from R/R AML patients have the ability to use both amino acids and fatty acids for energy production and therefore display increased metabolic flexibility. In this context, metabolic flexibility is the ability of cells to use different metabolic fuels (amino acids, sugars, fats, etc.) to produce energy, which appears to be a characteristic of R/R LSCs, unlike de novo LSCs. Furthermore, our data demonstrate that R/R LSCs have increased levels of amino acid and fatty acid contribution to TCA cycle function compared with de novo LSCs (Figure 7J). This appears to be driven at least partly by increased nicotinamide metabolism. Importantly, targeting nicotinamide metabolism effectively decreases both amino acid and fatty acid metabolism and in turn decreased TCA cycle function and OXPHOS (Figure 7J), providing a strategy to combat the increased metabolic flexibility observed in R/R LSCs in a clinically relevant manner.
DISCUSSION
Targeting disease-initiating LSCs has been a long-sought goal in AML research to provide better outcomes and perhaps even curative therapy for AML patients (Pollyea and Jordan, 2017). We reported that BCL-2-targeted BH3 mimetic venetoclax in combination with azacitidine is able to target LSCs in de novo AML patients who are ineligible for chemotherapy (Pollyea et al., 2018). LSC targeting was associated with superior outcomes for AML patients compared with other AML therapies (Pollyea et al., 2018). As a result of strong clinical data (DiNardo et al., 2018a; Pollyea et al., 2018), venetoclax-based regimens have been recently approved by the FDA for the chemotherapy-ineligible AML population.
Despite these promising clinical findings in de novo AML, it has been reported that R/R AML patients have inferior responses to ven/aza treatment (DiNardo et al., 2018b), a finding that was corroborated in the present study. We hypothesized that the differential response of de novo and R/R AML patients resulted from differences in fundamental aspects of LSC biology between de novo and R/R disease. Indeed, previous studies have demonstrated that LSC frequency and phenotypic diversity were dramatically increased in patients who relapse following chemotherapy, suggesting profound evolution of the LSC population as a consequence of initial treatment (Ho et al., 2016).
Because ven/aza targets LSCs isolated from de novo AML patients through a metabolic mechanism (Jones et al., 2018; Pollyea et al., 2018), we sought to understand the metabolic differences between de novo and R/R LSCs. We determined that R/R LSCs have increased nicotinamide levels and metabolism compared with de novo LSCs. We show that nicotinamide is metabolized to NAD+ through the salvage pathway in R/R LSCs. NAD+ levels were not increased in ROS-high AML blasts, demonstrating the importance of specifically interrogating the metabolic properties of malignant stem cell populations during disease pathogenesis. NAD+ is an essential cofactor in many enzymatic reactions and plays an important role in energy metabolism (Chiarugi et al., 2012); therefore, increased nicotinamide metabolism results in increased overall energy metabolism, which in turn leads to ven/aza resistance in R/R LSCs.
From a functional perspective, we demonstrated that perturbation of nicotinamide metabolism through NAMPT inhibition selectively targeted R/R LSCs. Our data suggest that LSC reliance on NAD+ metabolism to sustain OXPHOS specifically at relapse resulted from increased activity of metabolic pathways, including increased activity of the NAD+-dependent TCA cycle enzymes, as well as increased amino acid, fatty acid, and glucose metabolism. Increasing the dose of NAMPT inhibitor used does allow targeting of de novo AML, as previously shown (Mitchell et al., 2019; Nahimana et al., 2009); however, this may be accompanied by unwanted toxicity on normal hematopoietic cells, which we showed in Figure S4 and has been reported previously in clinical trials (Goldinger et al., 2016). At the lower doses that target R/R LSCs in vivo, no toxicity was observed to normal HSPCs or other hematopoietic cells in our study. Further clinical evaluation of NAMPT inhibitors at doses that selectively target leukemic cells is needed. Overall, these data suggest that inhibition of NAMPT may be a useful therapeutic strategy for R/R AML. This is particularly significant because the R/R AML patient population has limited therapeutic options and, to the best of our knowledge, no therapeutic strategy has been developed specifically to target the LSCs in this patient population.
NAMPT inhibition resulted in a decrease in OXPHOS in R/R LSCs, but not a decrease in glycolysis. These data are inconsistent with previous reports in other cancer types that demonstrate that NAMPT inhibition kills cancer cells by decreasing glycolysis (Tan et al., 2013; Tolstikov et al., 2014) and thus suggest that different types of cancer may have distinct metabolic vulnerabilities that can each be targeted by NAMPT inhibition. Furthermore, we observed that NAMPT inhibition targets both LSCs and mature AML blasts but through different metabolic mechanisms, decreasing OXPHOS and glycolysis, respectively. These data highlight the importance of understanding metabolic heterogeneity within a tumor and the need to specifically examine cancer stem cell populations. NAMPT inhibition may also be a useful strategy to target other cancer stem cell types that have been shown to depend on OXPHOS for survival, including breast, glioblastoma, and pancreatic malignancies (Janiszewska et al., 2012; Lee et al., 2017; Sancho et al., 2015). Future clinical studies should be designed to evaluate the safety and efficacy of NAMPT inhibition for R/R AML patients and other OXPHOS-dependent cancers.
Finally, we demonstrated that NAMPT disrupts OXPHOS by decreasing both amino acid and fatty acid metabolism into the TCA cycle. This is consistent with our previous data (Jones et al., 2018) showing that R/R LSCs depend on both amino acids and fatty acid metabolism for OXPHOS and therefore LSC survival and that de novo LSCs, in contrast, rely on amino acid metabolism for OXPHOS and survival (Jones et al., 2018). Addition of ven/aza did not potentiate the effect of NAMPT inhibition in R/R LSCs. Based on the role of NAD+ as an upstream driver of both amino acid and fatty acid metabolism, we suggest that NAMPT inhibition supersedes the effects of ven/aza, thereby explaining the lack of increased cell death upon combination of these two modalities. This finding also underscores the importance of increased metabolic flexibility that occurs in R/R LSCs as a consequence of primary treatment regimens (Jones et al., 2018). We suggest that in the design of de novo cancer therapies, it may be important to avoid selective pressures that induce increased metabolic flexibility. The findings in AML also raise the question as to whether malignant stem cells in other forms of cancer may display varying degrees of metabolic flexibility, a consideration that may inform the development of improved regimens.
Limitations of Study
A limitation of our current study is the lack of validation of protein expression of NAMPT and HADH upon siRNA knockdown. Although RNA expression is provided, acquiring the needed cell numbers from primary AML specimens did not allow interrogation of NAMPT and HADH protein levels.
STAR*METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Craig T. Jordan (craig.jordan@ucdenver.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate new datasets.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Specimens
AML specimens were obtained from apheresis product, peripheral blood from AML patients, or bone marrow, or mobilized peripheral blood from healthy donors who gave informed consent for sample procurement on the University of Colorado tissue procurement protocol. See Table S3 for additional details on the human AML specimens. For prior treatment information on R/R AML specimens used in this study see Table S4.
METHOD DETAILS
Human Specimen Culturing
When culturing was required, all samples were cultured in a base media of MEM without amino acids and 5.5mM glucose (My BioSource, MBS752807) supplemented with physiologic levels of amino acids (Carolina, 84–3700) and 10nM human cytokines SCF (PEPROTech, 300–07), IL3 (PEPROTech, 200–03), and FLT3 (PEPROTech, 300–19) as previously described (Jones et al., 2018). In addition, the media was supplemented with low density lipoprotein (Millipore, 437744), BIT (Stem cell technologies, 09500) β-ME (GIBCO, 21985–023), penicillin/streptomycin.
Cell Sorting
Primary AML specimens were thawed, stained with CD45 (BD, 571875) to identify the blast population, CD19 (BD, 555413) and CD3 (BD, 557749) to exclude the lymphocyte populations, DAPI (EMD Millipore, 278298), and CellROX deep red (Thermo Fisher, C10422), and sorted using a BD FACSARIA. ROS-low LSCs were identified as the cells with the 20% lowest ROS levels and the ROS-high blasts were identified as the cells with the highest 20% ROS levels, as previously described(Jones et al., 2018; Lagadinou et al., 2013; Pei et al., 2018; Pollyea et al., 2018).
Global UHPLC-MS Metabolomics
Approximately 100,000–500,000 ROS-Low LSCs were sorted and metabolomics analyses were performed via ultra-high pressure-liquid chromatography-mass spectrometry (UHPLC-MS – Vanquish and Q Exactive, Thermo Fisher) as previously reported (Nemkov et al., 2015). Briefly, cells were extracted in ice cold methanol:acetonitrile:water (5:3:2 v/v/v) at a concentration of 2 million cells/mL of buffer. After vortexing for 30 min at 4°C, samples were centrifuged at 12,000 g for 10 min at 4°C and supernatants processed for metabolomics analyses. Ten microliters of sample extracts were loaded onto a Kinetex XB-C18 column (150 × 2.1 mm i.d., 1.7 μm – Phenomenex). A 5 min gradient (5%–95% B, phase A: water + 0.1% formic acid and phase B: acetonitrile with + 0.1% formic acid for positive ion mode; 0%–100% B, phase A: 5% acetonitrile + 5mM ammonium acetate and phase B: 95% acetonitrile + 5mM ammonium acetate for negative ion mode) were used to elute metabolites. The mass spectrometer scanned in Full MS mode at 70,000 resolution in the 65–975 m/z range, 4 kV spray voltage, 45 sheath gas and 15 auxiliary gas, operated in negative and then positive ion mode (separate runs). Metabolite assignment was performed against an in-house standard library, as reported (Nemkov et al., 2015, 2019).
Metabolic Flux
500,000 ROS-Low LSCs were sorted and incubated with stable isotope substrates including uniformly 13C,15N-labeled amino acids (Cambridge Isotope Laboratories, MSK-A2-US-1.2), [13C6] glucose (Sigma-Aldrich, 389374), or [13C16]palmitic acid (Sigma-Aldrich, 705573) where indicated. Metabolomics analyses were performed via UHPLC-MS using the 5 min method as described above and previously (Gehrke et al., 2019).
Viability Assays
Patient samples were sorted and cultured without amino acids or drugs for 24 hours. Viability was assessed by trypan blue (GIBCO, 15250–071) staining and manual cell counting or annexin V and 7AAD staining followed by flow cytometry.
Normal HSC Analysis
Mobilized peripheral blood samples from three individuals was thawed, cultured in indicated conditions for 24 hours and CD34+ (BD, 572577) and CD45+ (BD, 571875), double positive percentages were quantified by flow cytometry (FACsCaliber, BD).
CFU Assays
Primary AML samples or normal mobilized peripheral blood samples were cultured with indicated drugs for 24 hours before being plated in human methylcellulose (R&D systems HSC003). Colonies were counts 10–17 days after the initial plating.
Patient Derived Xenograft Models
To determine the effect of APO866, KPT-9274 in vivo NSGS mice were transplanted with human AML leukemia cells via tail vein injection (2 million/mouse). Six-eight weeks after transplantation, mice were treated five days on two days off with 20mg/kg twice daily of APO866, 20mg/kg twice daily KPT-9274, or saline for two weeks, and then the composition of the residual leukemia cells within the bone marrow was examined. For secondary engraftment assays, equal numbers of human leukemic cells from control and drug treated mice were transplanted into immune deficient mice, approximately 0.5 million per mouse depending on cell numbers. Six to ten weeks post transplantation mice were safrafice and the composition of the leukemic cells within the bone marrow was examined by flow cytometry.
Seahorse Assays
XF96 (Agilent Technologies, 102417–100) extracellular flux assay kits were used to measure oxygen consumption (OCR) and glycolytic flux (ECAR). ROS-low LSCs were sorted, drug treated for four hours and plated into XF96 well plates. OCR and ECAR was measured according to the manufacture protocol and as previously described (Jones et al., 2018).
Enzyme Assays
Activities of α-ketoglutarate dehydrogenase, malate dehydrogenase, isocitrate dehydrogenase, and hexokinase were determined in LSCs as described in the manufacture protocol.
NAD+/H Assays
Levels of NAD+/H was determined in LSCs as described in the manufacturer’s protocol.
Quantitative RT-PCR
RNA was isolated with the RNeasy plus mini kit (QIAGEN) following manufactures protocol. cDNA was synthesized using the iScript One-Step RT-PCR kit (Bio-Rad). Quantitative real-time PCR was performed with LightCycler96 real-time PCR using SYBR Green I Master Mix reagent (Roche Applied Science).
Immunoblotting
Protein lysates were loaded on a polyacrylamide gel. Proteins were transferred to a polyvinylidene difluoride membrane using the mini trans-blot transfer system (Bio-Rad). To detect specific antigens, blots were probed with primary antibodies NAMPT (Invitrogen), GAPDH (Santa Cruz), and HADH (Abcam) on a shaker at 4°C, overnight, followed by 1 h of room temperature incubation with HRP-conjugated secondary antibodies (Santa Cruz). Chemoluminescence was recorded using the automated Gel Doc XR system (Bio-Rad).
Transfection of siRNA in primary AML
Primary AML specimens were transfected with siRNA constructs targeting NAMPT, HADH, or a non-targeting scrambled siRNA (Dharmacon) following established protocols (Brunetti et al., 2018). Specifically, 2×105 cells were electroporated using the Neon electroporator (Invitrogen) in Buffer T: R 1600, V 10 ms, 3 pulses.
QUANTIFICATION AND STATISTICAL ANALYSIS
Frequency data used to compare response rates between R/R and untreated patients were analyzed using a Chi-square Fisher’s Exact test. To test the association between response and the various independent variables of interest (age, prior treatment, prior MDS/MPN, t-aml, swog cyto category, tp53 mutation status, flt3 itd mutation status, ptpn mutation status, ras mutation status, asxl mutation status, and eln risk group), a univariate logistic regression model was run with response status as the outcome and each of the 11 independent variables assessed for significance in their own model. All independent variables meeting a p value threshold of 0.10 were retained for inclusion in a multivariate logistic regression model with response as the outcome to assess the effect of each of these variables on the outcome of interest while controlling for the presence of other significant or suggestively significant variables.
Error bars represent a standard deviation (SD). Biological factors were investigated for their significance using two-way Anova, two-tailed Student’s t test with paired or unpaired analysis. Specific analysis used is indicated in the figure legend of each figure. P value less than 0.05 was considered significant. Data with statistical significance (* p < 0.05, ** p < 0.01, *** p < 0.005, **** p < 0.001) are shown in Figures.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD45-FITC | BD Biosciences | Cat# 564585; RRID:AB_2732068 |
| CD3-PeCy7 | BD Biosciences | Cat# 557749; RRID:AB_396855 |
| CD19-Pe | BD Biosciences | Cat# 555413; RRID:AB_395813 |
| Cell ROX deep red | Thermo Fisher | Cat# C10422; RRID: N/A |
| CD34-PeCy7 | BD Biosciences | Cat# 560710; RRID: AB_1727470 |
| NAMPT | Invitrogen | Cat# PA5-23198; RRID:AB_2540724 |
| GAPDH | Santa Cruz | Cat#sc-32233; RRID:AB_627679 |
| Biological Samples | ||
| Human acute myeloid leukemia specimens | University of Colorado Hematologic Malignancies Tissue Bank | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Amino Acids | Carolina | Cat# 84-3700; RRID: N/A |
| Human SCF | PEPROtect | Cat# 300-07; RRID: N/A |
| Human IL3 | PEPROtect | Cat# 200-03; RRID: N/A |
| Human FLT3 | PEPROtect | Cat# 300-19; RRID: N/A |
| Low Density lipoprotein | Millipore | Cat# 437744; RRID: N/A |
| BIT9500 Serum Substitute | Stem Cell Technology | Cat# 09500; RRID: N/A |
| 13C,15N-labeled amino acids | Cambridge Isotope Laboratories | Cat# MSK-A2-US-1.2; RRID: N/A |
| 13C6 glucose | Sigma-Aldrich | Cat# 389374; RRID: N/A |
| 13C16 palmitic acid | Sigma-Aldrich | Cat# 705573; RRID: N/A |
| nicotinamide (2,6,CARBONYL-13C3; RING-1-15N) | Cambridge Isotopes | Cat#CNLM-9757; RRID: N/A |
| 13C11,15N2tryptophan | Sigma-Aldrich | Cat#574597; RRID: N/A |
| Cell-Tak | Corning | Cat# 324240; RRID: N/A |
| Human methylcellulose | R&D systems | Cat# HSC003; RRID: N/A |
| APO866 | SelleckChem | Cat# S2799; RRID N/A |
| KPT-9274 | Karyopharm | N/A |
| Oligomycin | Sigma-Aldrich | Cat# 871744; RRID: N/A |
| FCCP | Sigma-Aldrich | Cat# C2920; RRID: N/A |
| Antimycin A | Sigma-Aldrich | Cat# A8774; RRID: N/A |
| Rotenone | Sigma-Aldrich | Cat# R8875; RRID: N/A |
| NAMPT siRNA | Dharmacon | Cat# L-004581-00-0005; RRID: N/A |
| HADH siRNA | Dharmacon | Cat# L-008298-00-0005; RRID: N/A |
| Scrambled siRNA | Dharmacon | Cat# D-001810-01-05; RRID: N/A |
| Critical Commercial Assays | ||
| XF96 extracellular flux assay kits | Agilent Technologies | Cat# 102417-100; RRID: N/A |
| α-ketoglutarate dehydrogenase assay | Abcam | Cat# ab83431; RRID: N/A |
| malate dehydrogenase assay | Abcam | Cat# ab119693; RRID: N/A |
| Isocitrate dehydrogenase assay | Abcam | Cat# ab102528; ab102528 |
| Hexokinase activity assay | Abcam | Cat# ab136957; ab102528 |
| NAD/NADH Glo | Promega | Cat# 9071; RRID: N/A |
| Neon Transfection System | Thermo Scientific | N/A |
| Neon™ Transfection System Starter Pack | Thermo Scientific | Cat#MPK5000S; RRID: N/A |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74136; RRID: N/A |
| iSCRIPT cDNA Synthesis | Biorad | Cat# 1708890; RRID: N/A |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(CMV-IL3,CSF2,KITLG)1Eav/MloySzJ | The Jackson Laboratory | Cat# JAX:013062, RRID:IMSR_JAX:013062 |
| Oligonucleotides | ||
| NAMPT Forward: gttcctgagggctttgtcat | This paper | N/A |
| NAMPT Reverse: ggccactgtgattggatacc | This paper | N/A |
| GAPDH Forward: cagcaagagcacaagaggaa | This paper | N/A |
| GAPDH Reverse: gtgggggactgagtgtgg | This paper | N/A |
| Software and Algorithms | ||
| Flowjo | Flowjo | N/A |
| Metaboanalyst | http://www.metaboanalyst.ca/ | N/A |
| Morpheus | https://software.broadinstitute.org/morpheus/ | N/A |
| Maven | http://genomics-pubs.princeton.edu/mzroll/index.php | N/A |
Highlights.
Relapsed leukemia stem cells are metabolically unique
Nicotinamide metabolism is increased upon relapse in leukemia stem cells
Increased nicotinamide metabolism results in venetoclax resistance
Targeting nicotinamide metabolism eradicates relapsed leukemia stem cells
ACKNOWLEDGMENTS
The authors thank Karyopharm Therapeutics for providing KPT-9274 and guidance for its use in PDX models and the University of Colorado Hematology Clinical Trials Unit (HCTU) for help in acquisition of patient samples. We also acknowledge the Molecular and Cellular Analytical Core within the Colorado Nutrition and Obesity Research Center for the use of the Seahorse Analyzer. This work was supported by an award from the Leukemia and Lymphoma Society, American Cancer Society, and Cancer League of Colorado (to C.L.J.); the Evans MDS Foundation young investigator award (to B.M.S.); the University of Colorado Department of Medicine Outstanding Early Career Scholar Program and the Leukemia and Lymphoma Society clinical scholars award (to D.A.P.); T32CA190216 from the National Institutes of Health (NIH) (to M.A.); the Webb-Waring Early Career Award from the Boettcher Foundation, RM1GM131968 from the National Institute of General and Medical Sciences, and R01HL146442 and R01HL148151 from the National Heart, Lung, and Blood Institute (to A.D.); the St. Baldrick’s fellow award (to A.W.); V Foundation award T2017-012 and the Courtenay C. and Lucy Patten Davis Endowed Chair in Lung Cancer Research (to J.D.); the Ruth and Ralph Seligman Chair in Hematology (to C.S.); NIH National Cancer Institute (NCI) grants R01 CA200707 and R01 CA243452 (to C.T.J.); and a Leukemia and Lymphoma Society Specialized Center of Research grant (principal investigator, C.T.J.). C.T.J. is supported by the Nancy Carroll Allen Endowed Chair.
Footnotes
DECLARATION OF INTERESTS
D.A.P. receives research funding from Abbvie and has served as a consultant for Abbvie. C.L.J. is currently employed at the Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada.
SUPPLEMENTAL INFORMATION Supplemental Information can be found online at https://doi.org/10.1016/j.stem.2020.07.021.
REFERENCES
- Bonnet D, and Dick JE (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737. [DOI] [PubMed] [Google Scholar]
- Brunetti L, Gundry MC, Kitano A, Nakada D, and Goodell MA (2018). Highly Efficient Gene Disruption of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. J. Vis. Exp 134, 57278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong W-J, Zhao F, Medeiros BC, Tyvoll DA, and Majeti R (2015). Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 21, 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A, Dölle C, Felici R, and Ziegler M (2012). The NAD metabolome—a key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752. [DOI] [PubMed] [Google Scholar]
- Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, et al. (2015). Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 27, 864–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, Arellano M, Frattini MG, Kantarjian H, Popovic R, et al. (2018a). Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-rando-mised, open-label, phase 1b study. Lancet Oncol. 19, 216–228. [DOI] [PubMed] [Google Scholar]
- DiNardo CD, Rausch CR, Benton C, Kadia T, Jain N, Pemmaraju N, Daver N, Covert W, Marx KR, Mace M, et al. (2018b). Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am. J. Hematol. 93, 401–407. [DOI] [PubMed] [Google Scholar]
- Gehrke S, Rice S, Stefanoni D, Wilkerson RB, Nemkov T, Reisz JA, Hansen KC, Lucas A, Cabrales P, Drew K, and D’Alessandro A (2019). Red Blood Cell Metabolic Responses to Torpor and Arousal in the Hibernator Arctic Ground Squirrel. J. Proteome Res. 18, 1827–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldinger SM, Gobbi Bischof S, Fink-Puches R, Klemke C-D, Dréno B, Bagot M, and Dummer R (2016). Efficacy and Safety of APO866 in Patients With Refractory or Relapsed Cutaneous T-Cell Lymphoma: A Phase 2 Clinical Trial. JAMA Dermatol 152, 837–839. [DOI] [PubMed] [Google Scholar]
- González Esquivel D, Ramírez-Ortega D, Pineda B, Castro N, Ríos C, and Pérez de la Cruz V (2017). Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 112 (Pt B), 331–345. [DOI] [PubMed] [Google Scholar]
- Guan Y, and Hogge DE (2000). Proliferative status of primitive hematopoietic progenitors from patients with acute myelogenous leukemia (AML). Leukemia 14, 2135–2141. [DOI] [PubMed] [Google Scholar]
- Ho T-C, LaMere M, Stevens BM, Ashton JM, Myers JR, O’Dwyer KM, Liesveld JL, Mendler JH, Guzman M, Morrissette JD, et al. (2016). Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 128, 1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope KJ, Jin L, and Dick JE (2004). Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 5, 738–743. [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, et al. (2007). Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat. Biotechnol. 25, 1315–1321. [DOI] [PubMed] [Google Scholar]
- Janiszewska M, Suvá ML, Riggi N, Houtkooper RH, Auwerx J, Clément-Schatlo V, Radovanovic I, Rheinbay E, Provero P, and Stamenkovic I (2012). Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 26, 1926–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, et al. (2018). Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 34, 724–740.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CL, Stevens BM, D’Alessandro A, Culp-Hill R, Reisz JA, Pei S, Gustafson A, Khan N, DeGregori J, Pollyea DA, and Jordan CT (2019). Cysteine depletion targets leukemia stem cells through inhibition of electron transport complex II. Blood 134, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan CT, Guzman ML, and Noble M (2006). Cancer stem cells. N. Engl. J. Med. 355, 1253–1261. [DOI] [PubMed] [Google Scholar]
- Kennedy BE, Sharif T, Martell E, Dai C, Kim Y, Lee PW, and Gujar SA (2016). NAD+ salvage pathway in cancer metabolism and therapy. Pharmacol. Res. 114, 274–283. [DOI] [PubMed] [Google Scholar]
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, et al. (2013). BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, and Dick JE (1994). A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648. [DOI] [PubMed] [Google Scholar]
- Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sá nchez V, Sanders ME, et al. (2017). MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab 26, 633–647.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage SU, Hurren R, Voisin V, Bridon G, Wang X, Xu C, MacLean N, Siriwardena TP, Gronda M, Yehudai D, et al. (2017). Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood 129, 2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SR, Larkin K, Grieselhuber NR, Lai T-H, Cannon M, Orwick S, Sharma P, Asemelash Y, Zhang P, Goettl VM, et al. (2019). Selective targeting of NAMPT by KPT-9274 in acute myeloid leukemia. Blood Adv. 3, 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahimana A, Attinger A, Aubry D, Greaney P, Ireson C, Thougaard AV, Tjørnelund J, Dawson KM, Dupuis M, and Duchosal MA (2009). The NAD biosynthesis inhibitor APO866 has potent antitumor activity against hematologic malignancies. Blood 113, 3276–3286. [DOI] [PubMed] [Google Scholar]
- Nemkov T, D’Alessandro A, and Hansen KC (2015). Three-minute method for amino acid analysis by UHPLC and high-resolution quadrupole orbitrap mass spectrometry. Amino Acids 47, 2345–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemkov T, Reisz JA, Gehrke S, Hansen KC, and D’Alessandro A (2019). High-Throughput Metabolomics: Isocratic and Gradient Mass Spectrometry-Based Methods. Methods Mol. Biol 1978, 13–26. [DOI] [PubMed] [Google Scholar]
- Pei S, Minhajuddin M, Adane B, Khan N, Stevens BM, Mack SC, Lai S, Rich JN, Inguva A, Shannon KM, et al. (2018). AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 23, 86–100.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollyea DA, and Jordan CT (2017). Therapeutic targeting of acute myeloid leukemia stem cells. Blood 129, 1627–1635. [DOI] [PubMed] [Google Scholar]
- Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, D’Alessandro A, Culp-Hill R, Riemondy KA, Gillen AE, et al. (2018). Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 24, 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF, and Weissman IL (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111. [DOI] [PubMed] [Google Scholar]
- Sampath D, Zabka TS, Misner DL, O’Brien T, and Dragovich PS (2015). Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 151, 16–31. [DOI] [PubMed] [Google Scholar]
- Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Graña O, et al. (2015). MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab 22, 590–605. [DOI] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al. ; HALT Pan-Leukemia Gene Panel Consortium (2014). Identification of pre-leukaemic hae-matopoietic stem cells in acute leukaemia. Nature 506, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, Medeiros JJF, Rao-Bhatia A, Jaciw-Zurakowsky I, Marke R, et al. (2017). Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 547, 104–108. [DOI] [PubMed] [Google Scholar]
- Skrtic M,Sriskanthadevan S, Jhas B,Gebbia M,Wang X,Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, et al. (2011). Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20, 674–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, Jhas B, Hurren R, Gronda M, Wang X, et al. (2015). AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 125, 2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Young DA, Lu ZH, Wang T, Meier TI, Shepard RL, Roth K, Zhai Y, Huss K, Kuo MS, et al. (2013). Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. J. Biol. Chem. 288, 3500–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpstra W, Ploemacher RE, Prins A, van Lom K, Pouwels K, Wognum AW, Wagemaker G, Lö wenberg B, and Wielenga JJ (1996). Fluorouracil selectively spares acute myeloid leukemia cells with long-term growth abilities in immunodeficient mice and in culture. Blood 88, 1944–1950. [PubMed] [Google Scholar]
- Tolstikov V, Nikolayev A, Dong S, Zhao G, and Kuo MS (2014). Metabolomics analysis of metabolic effects of nicotinamide phosphoribosyltransferase (NAMPT) inhibition on human cancer cells. PLoS ONE 9, e114019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, van der Pol MA, Waisfisz Q, Ossenkoppele GJ, and Schuurhuis GJ (2005). High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin. Cancer Res. 11, 6520–6527. [DOI] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. (2014). Oncogene ablationresistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M, Roulston A, Bélec L, Billot X, Marcellus R, Bédard D, Bernier C, Branchaud S, Chan H, Dairi K, et al. (2009). The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 29, 5872–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Elkins K, Durieux JK, Lee L, Oeh J, Yang LX, Liang X, DelNagro C, Tremayne J, Kwong M, et al. (2013). Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia 15, 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new datasets.