

SUMMARY
Vesicular- or vacuolar-type adenosine triphosphatases (V-ATPases) are ATP-driven proton pumps comprised of a cytoplasmic V1 complex for ATP hydrolysis and a membrane embedded Vo complex for proton transfer. They play important roles in the acidification of intracellular vesicles, organelles, and extracellular milieu in eukaryotes. Here we report cryo-electron microscopy structures of human V-ATPase in three rotational states at up to 2.9 Å resolution. Aided by mass spectrometry, we build all known protein subunits with associated N-linked glycans, and identify glycolipids and phospholipids in the Vo complex. We define ATP6AP1 as a structural hub for Vo complex assembly as it connects to multiple Vo subunits and phospholipids inside the c-ring. The glycolipids and the glycosylated Vo subunits form a luminal glycan coat critical for V-ATPase folding, localization and stability. Collectively, our studies provide mechanisms of V-ATPase assembly and biogenesis that rely on the integrated roles of ATP6AP1, glycans and lipids.
Graphical Abstract
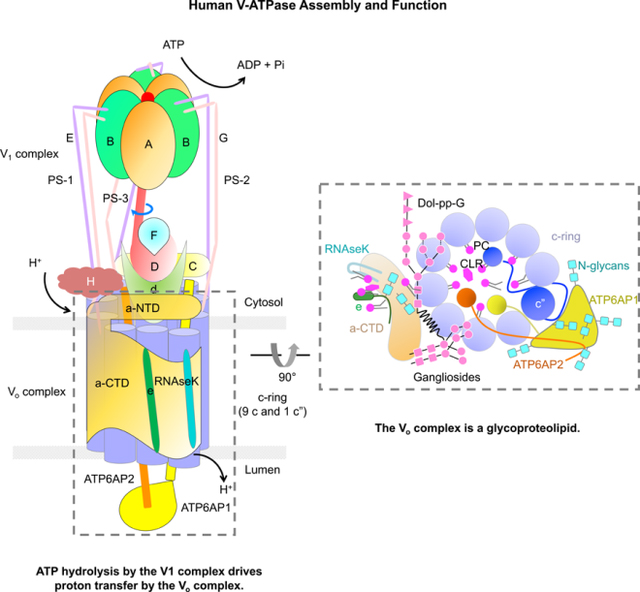
In Brief
Wang et al. report the mechanisms of human V-ATPase assembly. Cryo-EM structures of human V-ATPase combined with mass spectrometry analysis reveal that the protein subunits, glycans, glycolipids, and phospholipids are essential components of V-ATPase. Of note, subunit ATP6AP1, glycans, and lipids play important roles in V-ATPase assembly and biogenesis.
INTRODUCTION
As ATP hydrolysis-driven proton pumps that acidify intracellular vesicles, organelles, and extracellular milieu, V-ATPases play important roles in the regulation of fundamental physiological processes in all eukaryotes. Vesicular and organellar V-ATPases are essential in establishing and maintaining the pH homeostasis of endosomes and lysosomes and in supporting intracellular membrane trafficking and protein degradation, which in turn are important for immune responses, cell signaling, and neurotransmitter release (Casey et al., 2010; Cruciat et al., 2010; Forgac, 1992, 2007; Hinton et al., 2009; Pamarthy et al., 2018; Sun-Wada and Wada, 2015; Zoncu et al., 2011). V-ATPases have also been shown to directly associate with and regulate signaling complexes in the Notch, Wnt, and mTOR pathways (Cruciat et al., 2010; Pamarthy et al., 2018; Sun-Wada and Wada, 2015; Zoncu et al., 2011). Plasma membrane V-ATPases carry out extracellular acidification in specialized organs, which are critical for bone resorption, sperm maturation and acid excretion by the kidney (Forgac, 2007; McGuire et al., 2017). Increased V-ATPase expression and relocation to the plasma membrane have been suggested as a mechanism for maintaining an alkaline intracellular pH and an acidic extracellular pH to facilitate cancer cell proliferation and metastasis (Pamarthy et al., 2018). Thus, malfunction of V-ATPases can cause numerous human diseases including microbial infection, neurodegeneration, renal tubular acidosis, osteoporosis, and cancer (Hinton et al., 2009; Pamarthy et al., 2018).
As a well-studied model, the yeast V-ATPase was shown to contain 31 subunits that assemble into two structural modules: a cytoplasmic V1 ATPase and a membrane embedded Vo proton pump (Toei et al., 2010; Zhao et al., 2015). The V1 ATPase is composed of three copies of subunits A, B, E, and G, and one copy of subunit C, D, F, and H (Zhao et al., 2015), whereas the Vo proton pump contains subunit a, c8, c’, c’’, d, e, f, and Voa1 (Mazhab-Jafari et al., 2016). Structures of complete S. cerevisiae and M. sexta V-ATPases have been published at up to 6–7 Å resolution, and the S. cerevisiae V1 and Vo subcomplexes have been reconstructed to a high resolution of 3.5 Å (Mazhab-Jafari et al., 2016; Rawson et al., 2015; Roh et al., 2018; Zhao et al., 2015). During the preparation and revision of our current manuscript, cryo-electron microscopy (cryo-EM) structures of V-ATPases purified from rat or bovine brains were reported at overall resolutions of 3.9 Å and 3.4 Å respectively, revealing the structural architecture of mammalian V-ATPases (Abbas et al., 2020; Wang et al., 2020). Previous structural studies have provided insights into an elegant working mechanism of V-ATPases. In this mechanism, ATP hydrolysis by the cytoplasmic V1 ATPase drives the rotation of the membrane embedded, ring-shaped Vo proton pump to allow cycles of protonation and deprotonation of lipid-exposed glutamic acid residues for coupled proton transfer (Abbas et al., 2020; Mazhab-Jafari et al., 2016; Rawson et al., 2015; Roh et al., 2018; Toei et al., 2010; Zhao et al., 2015). However, little is known about the assembly mechanism of V-ATPases and no structures of human V-ATPases have been reported.
Here, we report cryo-EM structures of a human V-ATPase in three different states at up to 3.1 Å resolution overall, with 2.9 Å for focused-refined V1 and 3.0 Å for focused-refined Vo. The cryo-EM maps allowed us to fit and build complete atomic models for almost all subunits; in particular, they enabled the de novo tracing of the luminal domain of ATP6AP1. In addition, we observed and built many N-linked glycans, glycolipids and phospholipids. Based on our structural analysis, we propose a model for human V-ATPase assembly, regulation and function, providing mechanistic insights into the working principles of this fascinating molecular machine in higher organisms.
RESULTS
Structure Determination and Overall Architectures
We purified an endogenous human V-ATPase from the human embryonic kidney cell line HEK293F by affinity purification using a bait of N-terminally Flag-tagged SidK, which is a Legionella pneumophila effector protein shown to bind V-ATPases with high-affinity and partially inhibit them (Xu et al., 2010; Zhao et al., 2017) (Figure S1A and S1B). Because of the limited quantity of the human V-ATPase we could obtain, we used carbon-coated grids to concentrate the sample for cryo-EM data collection (Figure S1C). We collected two data sets on Titan Krios microscopes equipped with K3 direct electron detectors. 3D reconstruction of the second data set using the map obtained from the first data set as an initial model gave rise to three different rotational states of the V-ATPase with 38% of the total particles in state 1, 19% in state 2, and 11% in state 3 (Figure S1D and S1E; Table S1). The overall resolutions of the structures are 3.1 Å, 3.4 Å, and 3.6 Å respectively for the three states. Focused refinement of the 3.1 Å map allowed us to improve the resolution to 2.9 Å for the V1 complex and 3.0 Å for the Vo complex, which enabled us to fit and build an atomic model for almost all the subunits of the human V-ATPase (Figure 1A, 1B, S1F, S2A, S2B, and S2C; Table S1).
Figure 1. Cryo-EM Structure of Human V-ATPase.
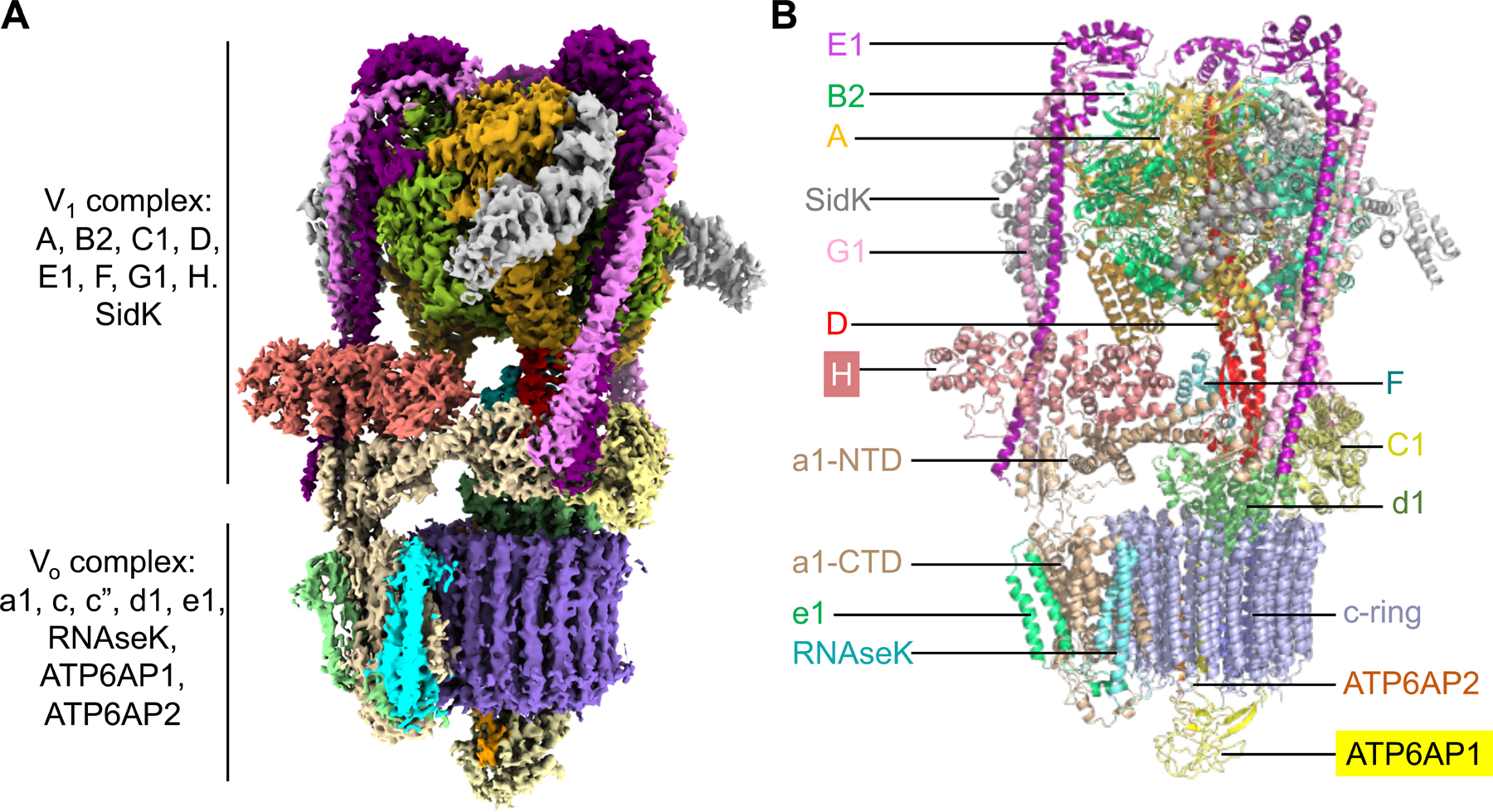
(A) Cryo-EM density of human V-ATPase (state 1) with subunits color coded.
(B) Ribbon diagram of human V-ATPase structure (state 1) with subunits color coded and labeled. Subunits H and ATP6AP1 which are absent or not fully traced in the rat V-ATPase structure (15) are highlighted.
See also Table S1 and Figures S1 and S2.
Many subunits of human V-ATPases have multiple isoforms in different tissues and organelles. Mass spectrometry analysis, together with sequence identification from our maps, indicated that the V1 complex is composed of three copies of subunits A, B2, E1, and G1, and one copy of subunits C1, D, F, and H, and that the Vo complex comprises subunits a1, d1, e1 (equivalent to yeast vma9p), RNAseK (equivalent to yeast f), c (9 copies), c”, ATP6AP1 (also known as Ac45, and equivalent to yeast Voa1), and ATP6AP2 (also known as prorenin receptor, mammal-specific) (Figure 1B, S2D, S2E, and S2F).
Assembly of the V1 and Vo Complex
The V1 complex is composed of a top A3B3 head, a central stalk of D and F subunits (DF stalk), three peripheral stalks of E and G subunits, and a bottom collar that is formed by subunits H, C, and the N-terminal domain of a (a-NTD) from the Vo complex (Figure 2A). In the top head, three A subunits and three B subunits are alternatively arranged around a pseudo six-fold symmetry axis with three bacterial SidK molecules decorating the A subunits (Figure 2B, S3A, and S3B). The central stalk of subunit D and an associated subunit F connects the A3B3 head in the V1 complex to the Vo complex for torque transmission (Figure 2C). In the human V-ATPase structure, subunits E and G in each of the three peripheral stalks (PS-1, PS-2, and PS-3) (Guo and Rubinstein, 2018; Zhou and Sazanov, 2019) form long and curved coiled-coils at their N-terminal domains (NTDs) (Figure 2D), and their C-terminal domains (CTDs) adopt a similar compact configuration to engage the B subunits of the A3B3 head (Figure S3C). The ends of the NTDs of the three PSs associate like a collar with different partners in the complex, and thus adopt somewhat different conformations (Figure 2E and S3D). PS-1 links to the foot domain of a-NTD and subunit H (Figure S3D). PS-2 connects to both the head domain of a-NTD and the foot domain of subunit C (Figure S3D). PS-3 interacts with the head domain of subunit C (Figure S3D). The overall curvatures of the three PSs are different despite having the same sequences, which reflect their intrinsic structural plasticity and may serve to accommodate the tilting and twisting of the A3B3 head during ATP binding and hydrolysis.
Figure 2. Assembly of the V1 and Vo Complex.
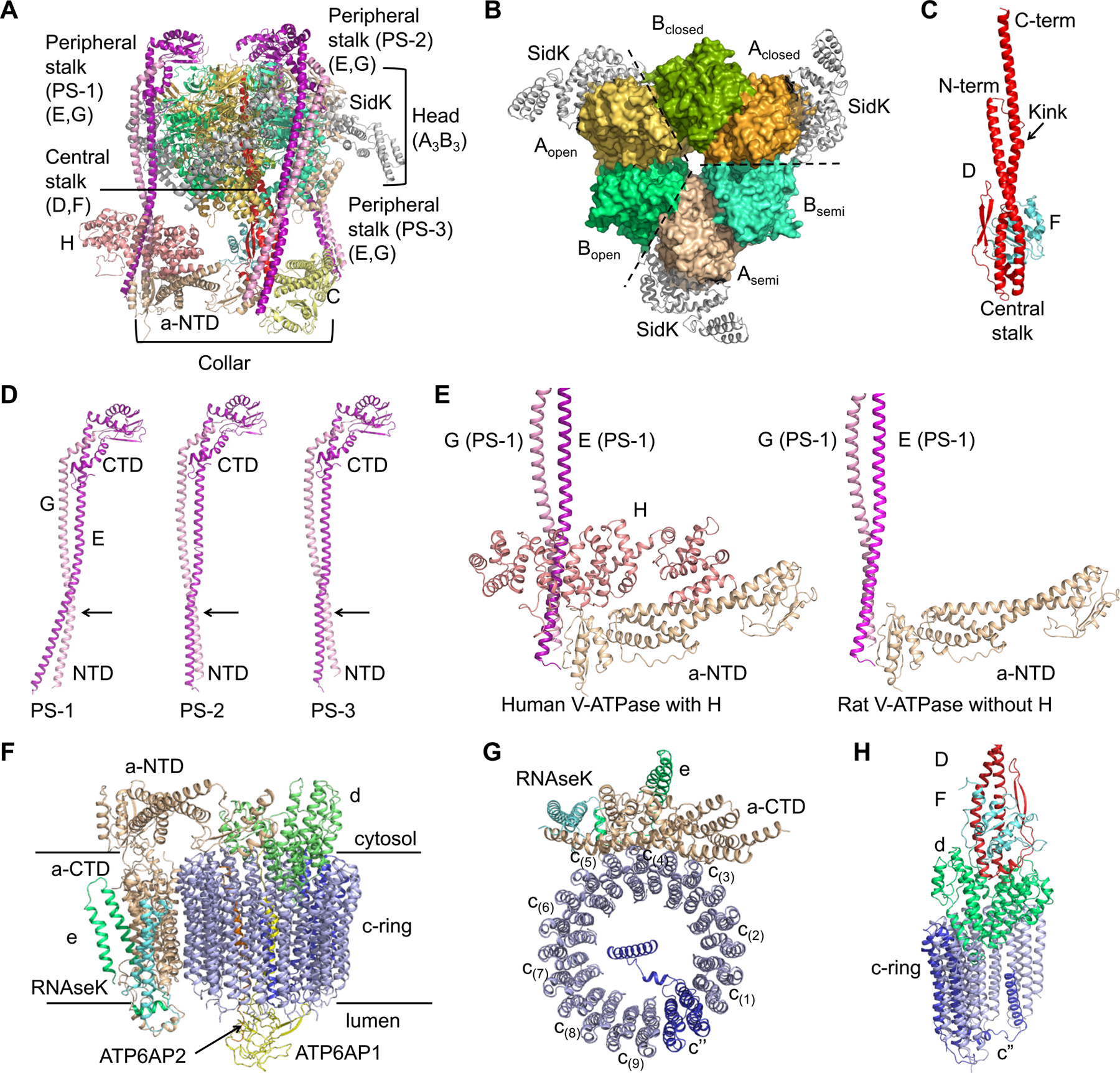
(A) Ribbon diagram of the V1 complex with each subunit colored as in Figure 1.
(B) The A3B3 hexamer surrounded by SidK (grey). The A3B3 hexamer is composed of three pairs of AB heterodimer in three conformations, closed, semi-open (semi) and open.
(C) Ribbon diagram of the central stalk that is composed of subunit D (red) and F (cyan). The proline containing kink in subunit D is indicated by an arrow.
(D) Conformational differences of the three peripheral stalks, with the CTDs of subunit E (purple) and subunit G (pink) aligned.
(E) Comparison between human and rat V-ATPase structures at the region where H is localized.
(F) Ribbon diagram of the Vo complex with each subunit colored as in Figure 1.
(G) Ribbon diagram of c-ring (blue) with CTD of subunit a (wheat), e (green) and RNAseK (cyan). The c” subunit is colored in marine.
(H) Interactions of subunit d (green) with the DF stalk (red and cyan) and the c-ring (blue).
See also Figure S3.
In comparison with the rat V-ATPase structure (Abbas et al., 2020), our human V-ATPase structure contains the essential subunit H, which interacts extensively with PS-1 and a-NTD (Figure 2E and S3E–F). These interactions across the V1 and Vo complexes may explain the ability of H to couple V1’s ATPase activity to Vo’s proton pumping activity in the intact V-ATPase (Diab et al., 2009; Liu et al., 2005; Sharma et al., 2018). The presence of subunit H appears to correlate with stabilization of state 1 relative to states 2 and 3 in our human V-ATPase sample (Figure S3G), as well as in the asymmetrical particle distribution observed in yeast V-ATPase with subunit H (~50%, 23% and 27% for states 1, 2 and 3 respectively) (Zhao et al., 2015). By contrast, the rat V-ATPase without subunit H displayed an evenly distributed rotational states (~37%, 31% and 32% for states 1, 2 and 3 respectively) (Abbas et al., 2020).
The Vo complex comprises the c-ring of c(1)-c(9) and c”, the adjacent C-terminal domain of subunit a (a-CTD), e and RNAseK, the ATP6AP1 and ATP6AP2 subunits inside the c-ring, and subunit d at the cytosolic side of the c-ring to connect to the central stalk of the V1 complex (Figure 2F and 2G). Subunit d folds like a cone; the concave side of the cone accepts the lower part of the DF central stalk in a shape complementary manner (Figure 2H). There are extensive interactions between them, which may ensure a proper coupling of V1 and Vo, and facilitate torque transmission. The interface between subunit d and the c-ring (c(1), c” and c(9)) are also extensive, and composed of hydrophobic, hydrogen-bonding, and charged interactions (Figure 2H and S3H). Of note, subunit d utilizes two different surfaces to engage the same regions of two c subunits at residues Q123, R119, and K36 (Figure S3H).
V1-Vo Coupling and Torque Transmission
Comparison among the three observed states of human V-ATPase shows that the scaffolding components of the complex, including subunits C, E and G (PS), H, a, e, and RNAseK, undergo limited conformational changes during the ATP hydrolysis cycle (Figure 3A and 3B). Subtle changes are, however, visible and likely accommodated by the intrinsic structural plasticity of the components especially the long helical bundles of the PSs (Figure 3B). PSs are attached to the N-terminal domains (NTDs) of the A3B3 hexamer head. During the ATP hydrolysis cycle, NTDs stay mostly in place while the C-terminal domains (CTDs) of both A and B undergo conformational changes to form different AB pairs, closed (ADP-bound, or post-ATP hydrolysis), semi-open (nucleotide free) and open (nucleotide free in our structure, but should have high affinity for ATP) (Figure 3C and S3B). This ATP hydrolysis cycle triggers tilting and twisting of the A3B3 hexamer relative to the central axis of the V1 complex, leading to a conformational oscillation that was previously named as “precession” (Hahn et al., 2018; Zhao et al., 2017). Because the central stalk (subunits D and F) inserts into the middle hole of the A3B3 hexamer, the conformational precession drives rotation of the central stalk and the closely coupled d subunit and c-ring to achieve torque transmission (Figure 3C).
Figure 3. Molecular Coupling between V1 and Vo Complexes.
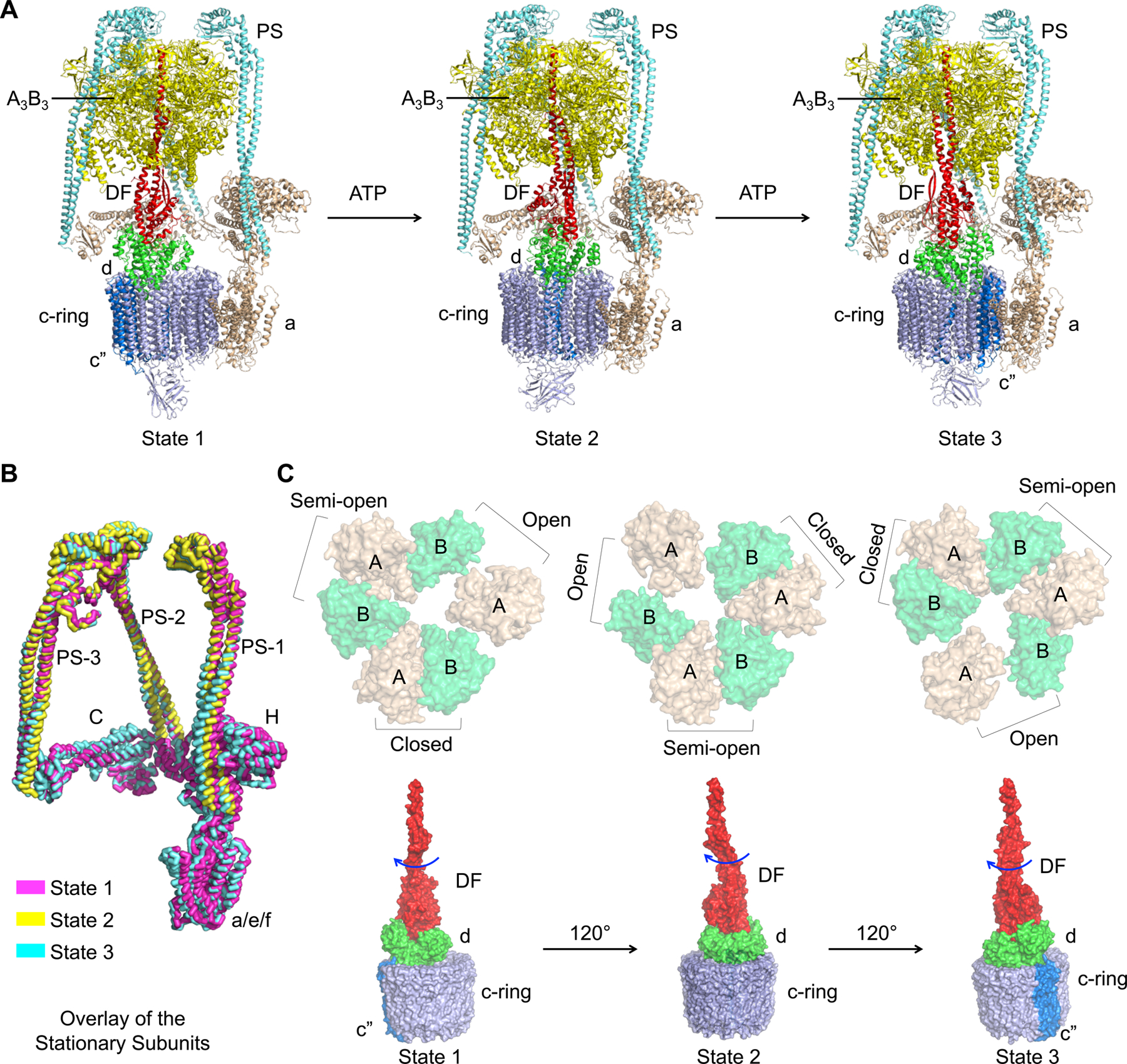
(A) Ribbon diagrams of the three rotational states of human V-ATPase with the central stalks in red, peripheral stalks in cyan, A3B3 head in tv-yellow, C, H, a, e and RNAseK in wheat, c-ring, ATP6AP1 and ATP6AP2 in blue, and d in green.
(B) Overlaid stationary subunits of human V-ATPase structures in three rotational states (state 1 in magenta, state 2 in yellow, and state 3 in cyan).
(C) Coordinated changes from the A3B3 head to the rotary DF-d-c-ring unit, shown in surface representations. The CTD hexamers of the A3B3 head (in wheat and green, top) are viewed from the Vo side, displaying the conformational precession between the states. The rotary units containing the central stalk (red), d (green) and the c-ring (blue) (bottom) are presented as side views, showing the ~120° rotation between states in response to the conformational precession at the A3B3 head.
The Unique ATP6AP1 Subunit: Vo Assembly and Stability
A striking difference between the human V-ATPase structure and all previous V-ATPase structures lies in the assembly subunit ATP6AP1 of the Vo complex, which comprises a transmembrane (TM) helix and a substantial luminal domain (LD) that folds as a globular β-prism structure (Figure 4A, 4B, and 4C). In yeast V-ATPase, the Voa1 subunit equivalent to human ATP6AP1 does not have a luminal domain (Roh et al., 2018), and in the rat V-ATPase structure, the ATP6AP1 luminal domain was not built due to limited resolution (Abbas et al., 2020) (Figure 4A). Nonetheless, the TM helices of human ATP6AP1, rat ATP6AP1 and yeast Voa1 exhibit conservation in structure and sequence (Figure S4A–B). The β-prism structure of the LD of ATP6AP1 is composed of 10 β-strands, in which β4, β5, β6, β7, and β8 form a planar β-sheet, and β1, β2, β3, β9, and β10 assemble into a bent β-sheet opposing the planar β-sheet (Figure 4C). Surprisingly, structural comparison using the DALI server (Holm and Sander, 1995) identified the lysosomal associated membrane protein (LAMP) domains of LAMP-1, −2 and −3 (Terasawa et al., 2016; Wilke et al., 2012) as significant structural homologs (Z-factors 11.5–13.3) despite complete lack of sequence homology (Figure 4D). Further, the LAMP-like LD of ATP6AP1 is conserved in different species (Figure S4C). Thus, the LD of ATP6AP1 is an evolutionarily conserved member of the LAMP family that comprises some of the most abundant protein components of lysosomal membranes (de Saint-Vis et al., 1998; Defays et al., 2011; Eskelinen, 2006; Rabinowitz and Gordon, 1991).
Figure 4. Structure of ATP6AP1 and Its Interaction Network.
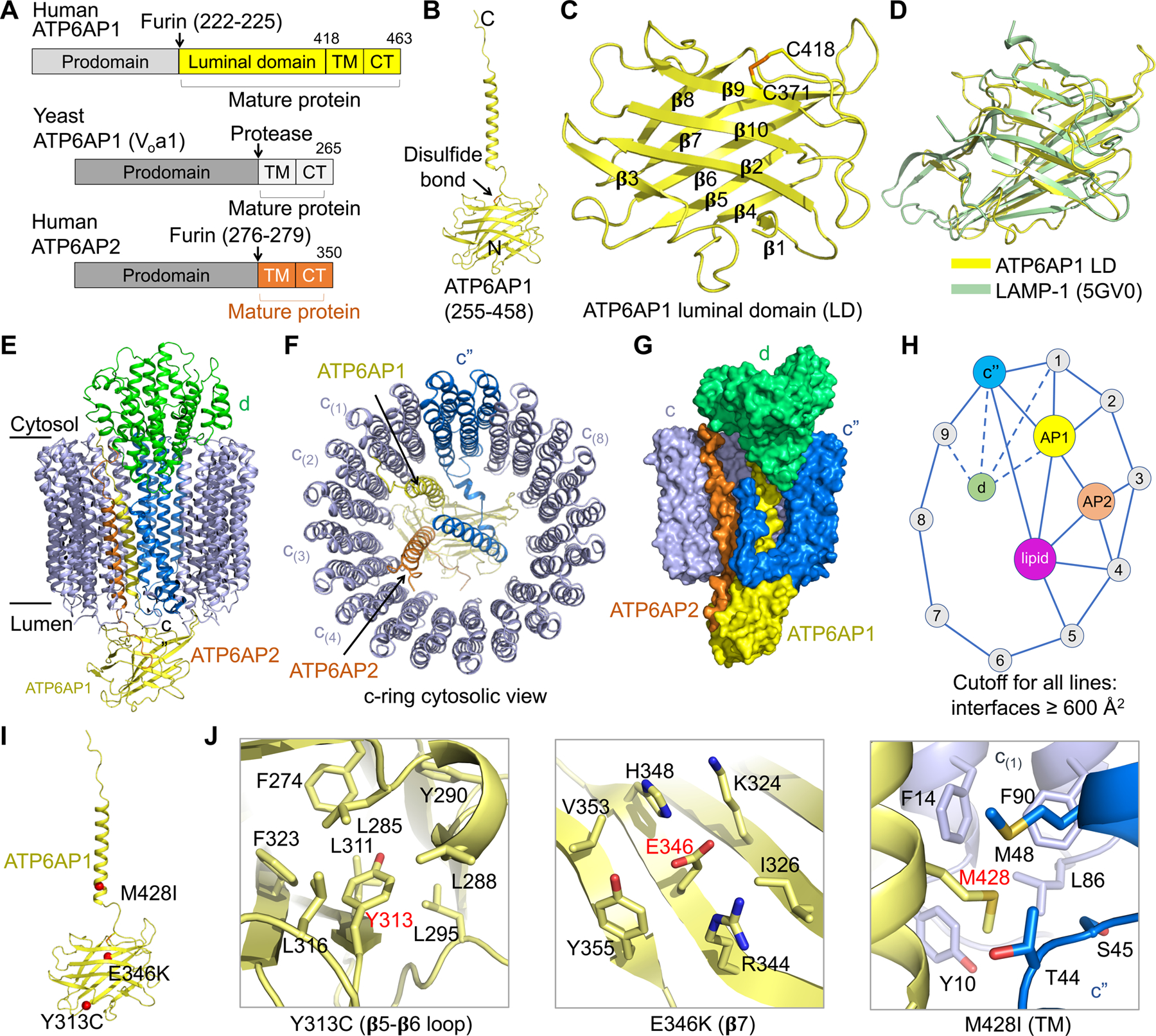
(A) Domain diagrams of human ATP6AP1 and ATP6AP2, and yeast ATP6AP1. The cleavage sites for furin or another protease in their luminal domains are shown.
(B) Ribbon diagram of mature ATP6AP1 in the human V-ATPase structure.
(C) Ribbon diagram of the luminal domain of ATP6AP1. Location of a conserved disulfide bond between C371 and C418 is marked. Secondary structures in the β-prism fold are labeled.
(D) Structural alignment of ATP6AP1 luminal domain (LD, yellow) with LAMP-1 (green) obtained from the DALI server (Holm and Sander, 1995).
(E, F) Side view (E) and cytosolic view (F) of the c-ring (c” in blue and c subunits in slate), ATP6AP1 (yellow), ATP6AP2 (orange), and subunit d (green), showing the overall interactions of ATP6AP1 with neighboring subunits.
(G) Surface representation of the c-ring, ATP6AP1, ATP6AP2, and subunit d, showing their interactions.
(H) A schematic diagram of the interaction network involving c-ring subunits, ATP6AP1, ATP6AP2, subunit d, and lipids. An interaction has to bury > 600 Å2 surface area to qualify. ATP6AP1 is the most connected subunit in the diagram.
(I) Missense disease mutations of ATP6AP1 (red) mapped onto its structure.
(J) Detailed interactions of the mutation sites within the ATP6AP1 subunit (Y313 and E346) and with neighboring subunits.
See also Figure S4.
Within the Vo complex, ATP6AP1 is centrally localized inside the c-ring, adjacent to ATP6AP2, c”, four c subunits, and d, with each paired interfacial surface area of up to 1,800 Å2 and a total buried surface area of over 7,000 Å2 between ATP6AP1 and all interacting partners (Figure 4E and 4F). In particular, the LAMP domain of ATP6AP1 engages with the N-terminal luminal tail of ATP6AP2, the linker in c” that connects its c-ring helix α2 to its single helix α1 inside the c-ring, and the luminal tails of subunits c(8) and c(9) (Figure S4D). The TM helix and the short cytosolic tail of ATP6AP1 pack against the TM helices of ATP6AP2, c”, c(1) and c(2), and interact with the part of subunit d inside the c-ring (Figure S4E and S4F). Collectively, ATP6AP1 interacts most extensively with ATP6AP2, c” and c(1) (>1,500 Å2), moderately with c(2) and d (> 800 Å2), and less extensively with c(8) and c(9). Using 600 Å2 burial surface area as a cutoff for the interactions, we display the interaction partners of ATP6AP1 (Figure 4G) as an interaction map for subunits of the c-ring and inside the c-ring (Figure 4H). ATP6AP1 is the single most connected subunit; in addition, it directly interacts with ordered phospholipid molecules inside the c-ring (Figure 4H) (also see below).
Missense mutations in the X-linked ATP6AP1 gene cause immunodeficiency in male patients with dominant clinical phenotypes including recurrent bacterial infections, hepatopathy, cognitive impairment, and abnormal protein glycosylation (Jansen et al., 2016). Furthermore, recurrent loss-of-function (nonsense, frameshift, splice-site, or in-frame deletion) somatic mutations in ATP6AP1 (63%) and ATP6AP2 (9%) account for 72% of granular cell tumors (GCTs) (Pareja et al., 2018). Three of the four missense ATP6AP1 mutations, Y313C, E346K and M428I, localize in the mature protein within either the luminal domain (Y313C, E346K) or the transmembrane domain (M428I). While Y313 is completely buried and packs against residues in the hydrophobic core, E346 of β7 forms salt bridges with R344 of β7 and K324 of β6 on the surface of the LAMP domain (Figure 4I and 4J). The change to a smaller and less hydrophobic residue in the Y313C mutant and the charge reversal in the E346K mutant may both destabilize the fold. The M428 residue localizes in the TM domain, whose side chain forms close hydrophobic contacts to both c(1) and c”, and the M428I mutation would likely compromise these interactions (Figure 4I and 4J). Consistent with their important structural roles, these three residues are highly conserved across different species of ATP6AP1 (Figure S4C). Thus, we conclude that missense disease mutations in ATP6AP1 cause reduced V-ATPase function by affecting its folding and assembly.
Our structural analysis elucidated that ATP6AP1 is crucially important for Vo assembly due to its ability to bring together multiple c-ring subunits, ATP6AP2, and lipids (see below for lipids). We further hypothesize that ATP6AP1, ATP6AP2 and the c-ring together provide a platform for the loading of subunit d during assembly of the Vo complex. Supporting these insights, genetic studies in yeast have previously indicated that the ATP6AP1-equivalent Voa1 subunit functions in the early step of Vo assembly in the ER as an assembly factor and is critical for the quality control of V-ATPase biogenesis (Jansen et al., 2016; Ryan et al., 2008). Structural studies of yeast V-ATPase also revealed that Voa1 remains as an integral component of the V-ATPase and that lack of Voa1 decreases subunit d recruitment as well as V-ATPase stability (Roh et al., 2018). Thus, for human and yeast V-ATPases alike, ATP6AP1 emerges as a central player in V-ATPase biogenesis, stability and function.
N-linked Glycosylation and Glycolipids: Folding, Trafficking and Stability
ATP6AP1 and subunits a and e in the Vo complex are all glycosylated at their luminal sides (Figure 5A). The LAMP domain structure of ATP6AP1 showed six N-linked glycans (N261, N273, N296, N303, N350, N357), consistent with previous studies indicating ATP6AP1 as a highly glycosylated protein (Chen et al., 2009), whereas subunits a and e each have one glycosylation site (N488 of a and N70 of e) (Figure 5B). These glycosylation sites are largely conserved across species (Figure S4C, S5A, and S5B); for subunit a isoforms (a1-4), glycosylation sites are universally present within the same region of the proteins but not exactly at the same sequence position (Figure S5A). We speculate that N-linked glycosylation plays multiple roles in V-ATPase function. First, previous studies revealed that mutations on the glycosylation sites of all a isoforms resulted in increased proteasomal degradation, ER retention and failed incorporation into V-ATPases, suggesting their critical roles in stability, trafficking and assembly (Esmail et al., 2018; Esmail et al., 2016). Second, N-linked glycosylation has been associated with inducing and/or stabilizing the formation of β-turns (Helenius and Aebi, 2001; O’Connor and Imperiali, 1996). In ATP6AP1, all six glycosylation sites occur at a β-turn (Figure 5B), suggesting that glycosylation may contribute to the proper folding of ATP6AP1 during V-ATPase biogenesis. Third, lysosomal proteins are usually heavily glycosylated because they have to withstand intra-lysosomal acidic pH < 5 and proteolytic activity of lysosomal hydrolases (Tokhtaeva et al., 2017). The glycans on subunits a, e, and ATP6AP1 may act as a shield to protect the V-ATPase from proteolysis in the lysosome (Figure 5A). Thus, N-linked glycosylation is an integral part of the V-ATPase function by promoting protein folding, proper subcellular localization and stability, in keeping with the classical roles of glycosylation (Helenius and Aebi, 2001; O’Connor and Imperiali, 1996).
Figure 5. N-linked Glycosylation and Glycolipids of the Vo Complex.
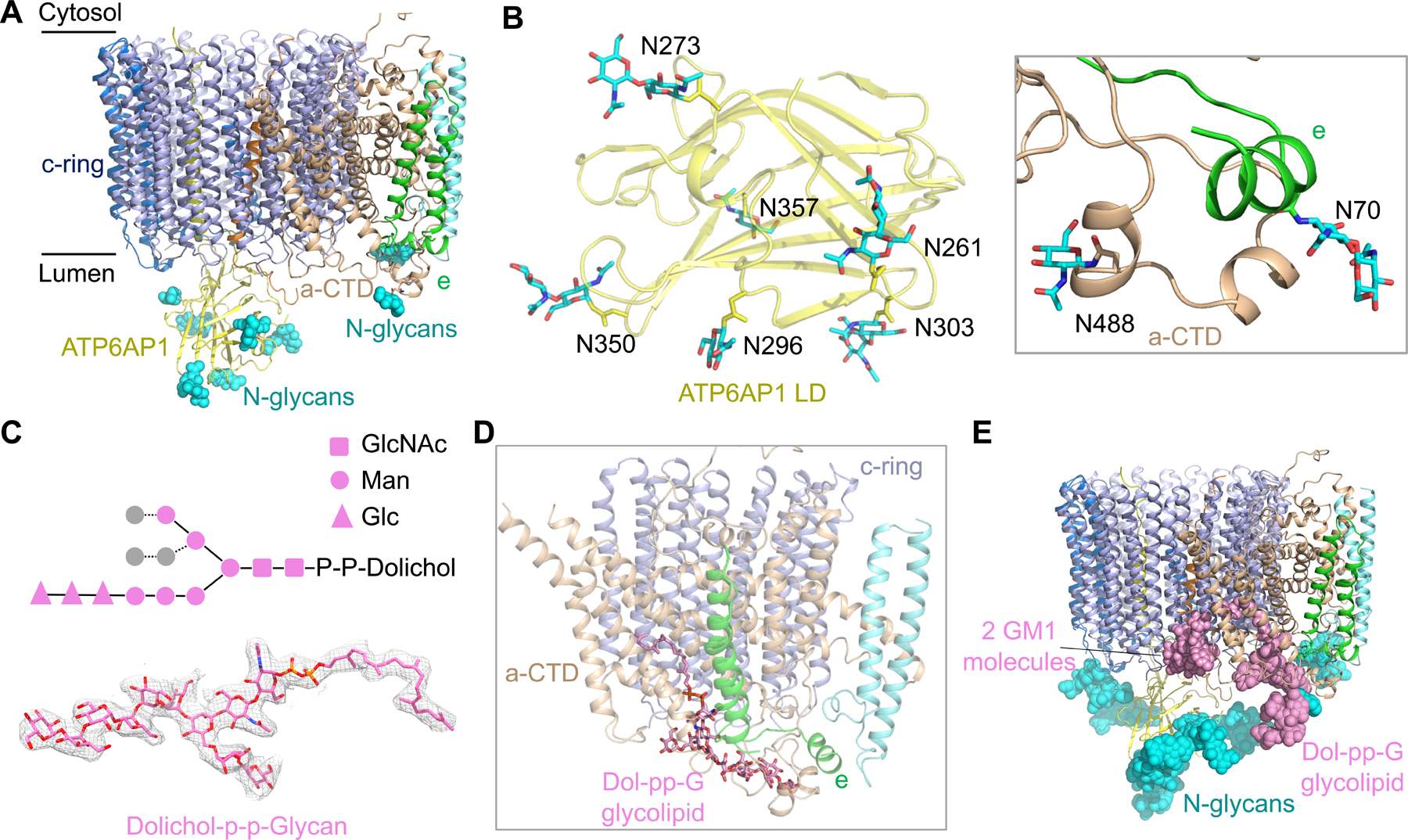
(A) N-linked glycans (cyan spheres) at the luminal side of the Vo complex from subunits ATP6AP1, a-CTD and e.
(B) Detailed structures of N-linked glycan units on subunits ATP6AP1, a-CTD and e.
(C) Diagram of the chemical structure, the model (in stick) and the cryo-EM density (2.0 σ) of the glycolipid dolichol-P-P-glycan (Dol-pp-G). Two copies of N-acetylglucosamine (GlcNAc) (square), six mannose (circle), and three glucose (triangle) units were resolved in the cryo-EM density map.
(D) Dol-pp-G bound to subunits c, a-CTD and e.
(E) The Vo complex shown with observed glycans at the luminal side. For each N-linked glycosylation site, a total of nine sugar units were modeled based on the complex-type N-glycan structure. GM1: monosialoganglioside.
See also Figure S5.
We identified an elongated density tightly associated with the Vo complex adjacent to a-CTD, e and the c-ring, and assigned it as a dolichol phosphate-linked glycan (Dol-pp-G) based on mass spectrometry evidence and the cryo-EM density (Figure 5C, 5D and S5C). Dolichol is a long-chain isoprenoid lipid that acts as glycosyl carrier lipids in the biosynthesis of N-linked glycoproteins, and dolichol-linked glycans are highly enriched in the ER. The Dol-pp-G molecule in our density has an acyl tail and a sugar head composed of at least 11 sugar units (Figure 5C and 5D). The first and second sugars are both N-acetylglucosamine (GlcNAc) and the sugar chain starts to branch at the third position, which is a mannose. The additional 6 sugar units on the main branch can all be traced, including three mannose (Man) and three glucose (Glc) units. For a minor branch, three additional mannose units was built. In comparison to the N-glycosylation precursor Dol-pp-GlcNAc2Man9Glc3, only three mannose units were invisible in the Dol-pp-G bound to the Vo complex. The hydrophilic sugar head makes many hydrogen bonds with a hydrophilic pocket formed by a-CTD and e, whereas the acyl tail inserts into a hydrophobic tunnel formed by a-CTD and the c-ring (Figure 5C and S5D). A similar density was also present in the rat V-ATPase and yeast V-ATPase structures, but was not assigned due to limited resolutions (Abbas et al., 2020; Vasanthakumar et al., 2019).
Sequence alignment showed that key residues coordinating Dol-pp-G are conserved across species from yeast to human (Figure S5E). This conservation suggests the functional significance of the bound Dol-pp-G glycolipid in Vo assembly and function, with possibilities including sealing the space between a-CTD and the c-ring to prevent proton leakage, controlling the subcellular localization of the Vo complex, and protecting the Vo complex from degradation by the hydrolases in the lysosome (Figure 5E). In line with this, mutation of K536 in yeast a, which is equivalent to K538 in human a1 that coordinates the phosphate group of Dol-pp-G (Figure S5F), led to ~60% loss of proton transfer activity (Toei et al., 2011). We also noticed densities between a-CTD and the c-ring, whose shape and mass spectrometry evidence suggested monosialoganglioside GM1 molecules, prototypical gangliosides in the glycolipid family (Figure 5E and S5G). Collectively, the glycolipids and the N-linked glycosylation may together regulate the biogenesis of V-ATPases and protect them from degradation by lysosomal hydrolyses (Figure 5E).
Ordered Lipid Molecules Are Integral Part of the Vo Complex
It has been increasingly appreciated that interactions of membrane proteins with lipids can be highly specific and play a critical role in maintaining the structural integrity and full function of membrane proteins (Bolla et al., 2019; Hunte and Richers, 2008; van Meer et al., 2008). However, limited understating on the role of lipids in the assembly and function of eukaryotic V-ATPases is currently available due to paucity of structural information. In human V-ATPase structures, we were surprised to find many ordered lipid molecules in the Vo complex: five associated with subunits a, e, and RNAseK (Figure 6A and 6B), and twenty four inside the c-ring (Figure 6C and 6D). Mass spectrometry analysis showed that phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS) are present in our human V-ATPase sample, with PC being the prevalent (Figure S6A and S6B), and that cholesterol (CLR) is also present (Figure S6C).
Figure 6. Lipid Molecules in the Vo Complex.
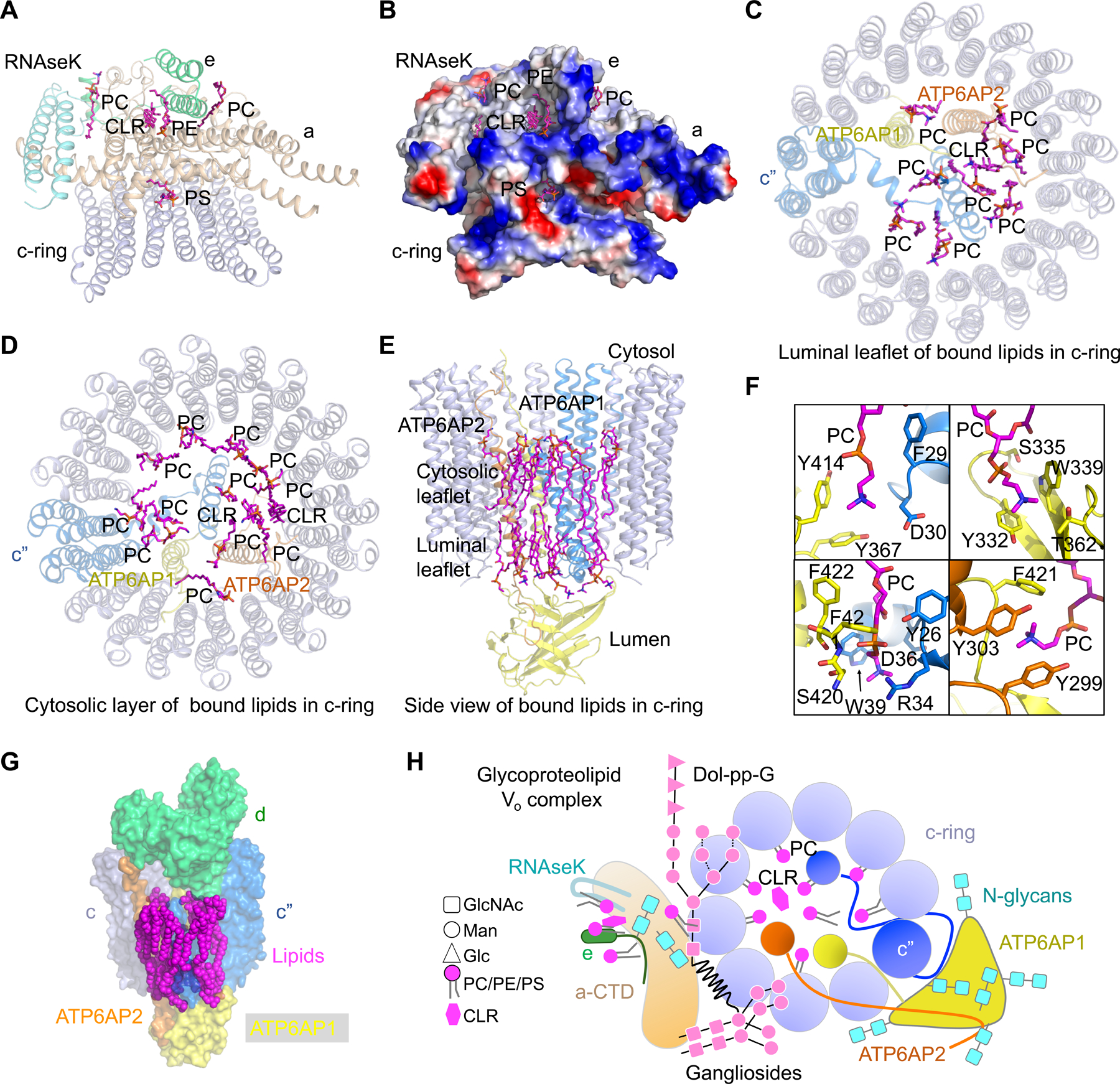
(A) Lipid molecules at the interfaces of subunits a, e, RNAseK, and the c-ring. These lipids establish extensive interactions with the protein subunits above. PC: phosphatidylcholine; PE: phosphatidylethanolamine; PS: phosphatidylserine; CLR: cholesterol.
(B) Electrostatic surface representation of subunits a, e, RNAseK, and c-ring, showing the interactions between the lipids and protein subunits.
(C, D) Lipid molecules inside the c-ring at the luminal leaflet (C) and the cytosolic leaflet (D). These lipids establish extensive interactions with ATP6AP1, ATP6AP2, and the c-ring.
(E) Side view of bound lipids inside the c-ring.
(F) Detailed interactions of lipid molecules with ATP6AP1 (yellow), c” (blue) and ATP6AP2 (orange).
(G) Surface representation of lipid molecules, c-ring, ATP6AP1, ATP6AP2, and subunit d, showing their interactions.
(H) A schematic diagram of the luminal view of the glycoproteolipid Vo complex, showing all protein subunits, N-linked glycans, glycolipids, and ordered PC and cholesterol lipids inside and outside the c-ring.
See also Figure S6.
Of the five lipid molecules associated with subunits a, e, and RNAseK, two were assigned as PC, one as PE, one as PS, and one as cholesterol (Figure 6A and S6D). These lipid molecules are nestled between a, e, and RNAseK, which may stabilize the assembly of the complex. The acyl chains of these lipids form hydrophobic interactions with a, e, and RNAseK, while their head groups establish hydrophilic interactions with them (Figure 6B). Of note, one PS molecule is localized at the interface between subunit a and c-ring, and may likely participate in the process of proton transfer. Lipids inside the c-ring reside both at the luminal membrane leaflet and the cytosolic membrane leaflet (Figure 6C, 6D, and 6E). Of the nine lipid molecules in the luminal leaflet, eight were assigned as PC and one as cholesterol based on the density (Figure 6C, S6E, and S6F). Of the fifteen lipid molecules in the cytosolic leaflet, thirteen were assigned as PC and two as cholesterol (Figure 6D, S6E, and S6F). These ordered lipids occupy the space between the inside surface of the c-ring and the transmembrane helices from ATP6AP1, ATP6AP2 and c”, and form extensive interactions with these protein subunits especially ATP6AP1 (Figure 6E and 4H). Detailed analysis showed that the acyl chains of these lipid molecules tightly pack against hydrophobic residues in c, c”, ATP6AP1, and ATP6AP2, and the charged headgroups form charge-charge interactions or hydrogen bonds with these same protein subunits (Figure 6F). We propose that the ordered lipid molecules, together with the central hub-subunit ATP6AP1, may stabilize and fine tune the interactions in the Vo complex of human V-ATPase (Figure 6G).
The lipids inside the c-ring are reminiscent of the ordered lipids observed in the crystal structure of the Enterococcus hirae V/A-ATPase for which no transmembrane helices exist inside the K-ring, the equivalent of c-ring in eukaryotic V-ATPases (Murata et al., 2005). A highly ordered layer of lipids lays inside the K-ring wall at both the cytosolic and the luminal halves, and two additional layers of less ordered lipids can be partially built inside the first layer (Murata et al., 2005). Initially the lipid molecules were assigned as phosphatidylglycerol molecules because they are the most abundant phospholipids in E. hirae. A more recent study by mass spectrometry showed that these lipid molecules are cardiolipins (Zhou et al., 2011). Like human V-ATPase, these lipids likely stabilize and modulate K-ring structure and function (Murata et al., 2005).
DISCUSSION
Our cryo-EM structures revealed important information about human V-ATPase in the following respects (Figure 6H). First, we de novo built the luminal domain of ATP6AP1, which despite lack of sequence identity, shares structural homology to the LAMP family that comprises some of the most abundant protein components of lysosomal membranes. Second, we discovered that ATP6AP1 interacts with multiple subunits, as well as c-ring lipids, in the Vo complex, making it a hub for bringing subunits together for assembly. Third, we identified important N-linked glycans on ATP6AP1 and other Vo subunits, as well as glycans from bound glycolipids (Dol-pp-G and GM1). These glycans are implicated in V-ATPase assembly, localization and stability, as well as in shielding V-ATPases from harmful lysosomal hydrolases. Fourth, we identified ordered phospholipid and cholesterol molecules inside the c-ring, many of which are stabilized by interactions with ATP6AP1 and may play vital roles in assembly and regulation of V-ATPases.
Thus, V-ATPases are emerging to be bona fide glycoproteolipid complexes, and our studies provided a platform for understanding the roles of glycans and lipids in physiological functions of V-ATPases. Our studies also raised important insights into the mechanism of V-ATPase biogenesis from the observation that ATP6AP1 is a central hub for V-ATPase assembly. How the different subunits of V-ATPases coordinate their assembly and function, and how V-ATPases can be targeted for therapeutic intervention require further structural, biochemical and cell biological investigations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Tian-Min Fu (fu.978@osu.edu).
Materials Availability
Plasmids generated in this study are available upon request from the Lead Contact with a completed Material Transfer Agreement.
Data and Code Availability
The cryo-EM maps included in this study have been deposited in the Electron Microscopy Data Bank with accession codes: EMD-21844, EMD-21845, EMD-21847, EMD-21848, and EMD-21849. The atomic coordinates have been deposited in the Protein Data Bank with accession codes: 6WM2, 6WM3, 6WM4, 6WLZ, and 6WLW.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The human V-ATPase sample was purified from HEK29F cells and its structures were determined by cryo-EM.
METHOD DETAILS
Expression and Purification of Human V-ATPase
The coding sequence of SidK (residue 14-273) was cloned into pET28a with N-terminal His and Flag tags. The recombinant plasmid was transformed into E. coli BL21(DE3) RIPL for expression. The protein expression was induced by 0.5 mM IPTG at 293K when the E. coli grew to a density of OD ~0.6. 16 hours post-induction, the cells were harvested, resuspended and sonicated in buffer A (20 mM Tris-HCl at pH 7.5, 300 mM NaCl, and 20 mM imidazole). After centrifugation to remove the cell debris, the supernatant was incubated with Ni-NTA resin at 4 °C for 30 minutes. The resin was then washed with 10 column volumes of buffer A and the protein was eluted using buffer A supplemented with 300 mM imidazole. SidK was further purified by gel filtration in 20 mM Tris-HCl at pH 7.5, 150 mM NaCl, and 1 mM tris(2-carboxyethyl)phosphine (TCEP).
For purification of the human V-ATPase, a 5 liter HEK293F cell pellet was incubated in 50 mM NaCl, protease inhibitor cocktail (Sigma-Aldrich), and 50 mM HEPES at pH 7.4 for 2 hours on ice. Cell debris was removed by centrifugation at 8,000 rpm for 30 minutes and the membrane fraction was pelleted by ultracentrifugation at 40,000 rpm for 1 hour. The membrane pellet containing the V-ATPase was resuspended using a Dounce homogenizer and was incubated with SidK in the buffer containing 150 mM NaCl, 1 mM TCEP, 1% lauryl maltose neopentyl glycol (LMNG), 0.1% cholesteryl hemisuccinate (CHS), protease inhibitor cocktail (Sigma-Aldrich), and 50 mM HEPES at pH 7.4 for 4 hours at 4 °C. After centrifugation at 17,000 rpm for 30 minutes, supernatant containing solubilized V-ATPase/SidK complex was then incubated with anti-Flag resin (Sigma-Aldrich) for 1 hour at 4 °C. The resin was collected and washed with 10 column volume of wash buffer containing 150 mM NaCl, 1 mM TCEP, 0.1% LMNG, 0.01% CHS, and 50 mM HEPES at pH 7.4. The V-ATPase/SidK complex was eluted using the wash buffer plus 100 µg/ml 3X FLAG peptide (Sigma-Aldrich). Further purification of V-ATPase/SidK was performed using glycerol gradient ultracentrifugation adapted from a previous method (Wang et al., 2018). The V-ATPase/SidK sample was flash-frozen and stored at −80 °C for further applications.
Cryo-EM Data Collection
A 3 µl drop of human V-ATPase sample at 0.05 mg/ml was applied to glow-discharged copper grids with lacey carbon support and a 3 nm continuous carbon film (Electron Microscopy Sciences), and incubated for 10 minutes. The grids were washed three times using 20 mM HEPES at pH 7.5 and 150 mM NaCl, blotted for 4.5 s in 100% humidity at 4 °C, and plunged into liquid ethane using an FEI Vitrobot Mark IV. All grids were screened using an FEI Talos Arctica microscope (Harvard Cryo-EM Center for Structural Biology). Two sets of data were collected on a 300 keV Titan Krios microscope (FEI) equipped with a K3 direct electron detector (Gatan). For dataset I, 4,608 movies were collected in counting mode, with 60 total frames per movie in 3 s, 63.4 electrons per Å2 accumulated dose, and 1.06 Å physical pixel size. For dataset II, 9,660 movies were collected in counting mode, with 40 total frames per movie in 3.2 s, a total dose of 50 electrons per Å2, and a physical pixel size of 1.08 Å.
Cryo-EM Structure Determination
For both datasets, movies were motion-corrected and dose-weighted using MotionCor2 (Zheng et al., 2017). Initial contrast transfer function (CTF) estimation was performed using CTFFIND4 (Rohou and Grigorieff, 2015). Initial particle picking was carried out using crYOLO (Wagner et al., 2019). Template-based particle picking was performed using Gautomatch (http://www.mrc-lmb.cam.ac.uk/kzhang/). Particles with a box size of 360 pixels were binned 6 times and subjected to 2D classification in Relion (Scheres, 2012). Particles from good classes were manually selected, unbinned and 2D classified a second time.
For dataset I, 345,711 particles were selected after 2D classification and used for ab-initio model reconstruction. 3D refinement was performed in Relion and yield a 4.4 Å map (Scheres, 2012). Per-particle CTF corrections were calculated using Gctf (Zhang, 2016), which improved the resolution of the 3D reconstruction to 3.9 Å. Particles were then 3D classified into four classes with local search using small angler steps, resulting in 3 out of the 4 classes corresponding to 3 different states of the V-ATPase. Particles form a selected 3D class were “polished” through the Bayesian polishing process (Zivanov et al., 2019). “Polished” particles were imported into cryoSPARC to perform non-uniform 3D refinement (Punjani et al., 2017). 3D reconstruction of state 1 V-ATPase gives the best resolution, at 3.5 Å.
For dataset II, 782,583 particles were processed with the same procedures except that the 3.9 Å map with all particles from dataset I were used as a reference map, and the particles were 3D-classified into 6 classes. 3 classes out the 6 total corresponding to the 3 states of the V-ATPase were selected for non-uniform 3D refinement in cryoSPARC (Punjani et al., 2017). The map resolutions and particle numbers for the 3D reconstructed states are 3.1 Å (301,510 particles in state 1), 3.4 Å (150,243 particles in state 2), and 3.6 Å (90,369 particles in state 3), respectively.
Focused refinements of Vo and V1 regions were performed using the local refinement protocol in cryoSPARC (Punjani et al., 2017), which generated a 3.0 Å map of the Vo region in state 1 and a 2.9 Å map of the V1 region in state 1 with much more detailed features. Focused refinements of other states were also performed. The focused refinement of the H subunit and a-NTD were performed by density subtraction and 3D classification without alignment in Relion (Scheres, 2012). Classes were selected, reverted to original particles, and used for local 3D refinement, yielded a map at a resolution of 3.2 Å. All maps were sharpened within cryoSPARC (Punjani et al., 2017).
Cryo-EM Model Building and Refinement
Initial structures of human V-ATPase in state 1 were modeled using SWISS-MODEL (Biasini et al., 2014) based on yeast and rat V-ATPases (PDB ID:3J9T, 6O7U, 6VQ6) (Abbas et al., 2020; Roh et al., 2018; Zhao et al., 2015), and fitted as rigid bodies into cryo-EM maps using Chimera (Pettersen et al., 2004). ATP6AP1 was built de-novo based on the 3.0 Å map of the Vo region from focused refinement. The H subunit was modeled based on the crystal structure of the yeast H subunit (PDB ID: 1HO8) (Sagermann et al., 2001) using the 3.2 Å map from focused refinement. Lipids and sugars with well-defined densities were also modeled. The structures of human V-ATPase in state 2 and state 3 were modeled based on the model of human V-ATPase in state 1. Inspection, model building, and manual adjustment were carried out in coot (Emsley and Cowtan, 2004). Real-space refinement was performed using PHENIX (Adams et al., 2010). All representations of densities and structural models were generated using Chimera, ChimeraX (Goddard et al., 2018) and Pymol (Delano, 2002).
Lipidomics Analysis
The protein complexes with co-purified lipids were digested with trypsin at 37°C overnight and dried by a SpeedVac vacuum concentrator (Thermo Fisher Scientific). The peptides/lipids mixture was dissolved in sample buffer (45% acetonitrile, 30% isopropanol and 15% H2O) and sonicated for 10 min. For LC-MS/MS analysis, lipids were loaded on a C18 column (Acclaim PepMap 100, C18, 75 µm × 15 cm, Thermo Scientific) by a Dionex UltiMate 3000 RSLC nano System connected to an Eclipse Tribrid Orbitrap mass spectrometer (Thermo Scientific). A binary buffer system was used with buffer A of acetonitrile: H2O (60:40), 10 mM ammonium formate, 0.1% formic acid and buffer B of isopropanol: acetonitrile (90:10), 10 mM ammonium formate, 0.1% formic acid. Lipids were separated at 40°C with a gradient of 32% to 99% buffer B at a flow rate of 300 nl/min over 30 min. Spray voltage was set to 2.2 kV with funnel RF level at 40 and heated capillary temperature at 320°C. For data-dependent acquisition, full MS mass range was set to 300 to 2000 with a resolution of 120000 and AGC target of 100%. Fragment spectra were acquired in Orbitrap with a resolution of 15000 using higher-energy collisional dissociation (HCD) with stepped collision energy of 25, 30 and 35%. Cholesterol was detected in positive ion mode, the other lipids were detected in negative ion mode. The raw data was processed by LipiDex for phospholipid identification (Hutchins et al., 2018), cholesterol and glycolipids were manually identified by examining the fragment spectra. For dolichol-PP-oligosaccharides identification, a C8 column (Acclaim PepMap 100, C8, Thermo Scientific) was used instead of C18 column. The MS1 spectra at the end of the gradient (99% Buffer B) were summed and averaged, and the exact masses were used for dolichol-PP-oligosaccharide identification.
Proteomics Analysis
The protein complexes were fractionated by SDS-PAGE (NuPAGE 4-12% Bis-Tris gel, Thermo Fisher Scientific). The gel was cut into fourteen slides, reduced with dithiothreitol, alkylated with iodoacetamide and further digested with trypsin at 37 °C overnight. Peptides were dissolved in 0.1% TFA and separated on a C18 column (Acclaim PepMap 100, C18, 75 µm × 15 cm) by Dionex UltiMate 3000 RSLC nano System connected to an Eclipse Tribrid Orbitrap mass spectrometer. Spray voltage was set to 2.2 kV with funnel RF level at 60 and heated capillary temperature at 320°C. For data-dependent acquisition, full MS mass range was set to 350 to 2000 with a resolution of 60000 and AGC target of 100%. Fragment spectra were acquired in Orbitrap with a resolution of 15000 using HCD with normalized collision energy of 30%. Raw files were processed with Maxquant software (version 1.6.3.4) with human Uniprot Reference Proteome (Tyanova et al., 2016). Trypsin was set to specific enzyme with fixed modification of carbamidomethylation (cysteine) and variable modifications of oxidation (methionine) and acetylation (protein N-term). Intensity Based Absolute Quantification (iBAQ) value of each protein isoform was used for estimating their relative abundances.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis
For cryo-EM structure determination, the numbers of particles used for each of the 3D reconstructions are listed in Figure S1, and Fourier shell correlation analyses for resolution determination were performed in cryoSPARC (Punjani et al., 2017). For mass spectrometry (MS) analysis, the abundance of each subunit isoform was inferred using a linear model generated from external standards after each protein MS intensity was normalized by the corresponding number of theoretically observable peptides.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pen Strep | GIBCO | Cat# 15140-122 |
| Freestyle 293 medium | GIBCO | Cat# 12338-018 |
| Fetal bovine serum | GIBCO | Cat# 16000-044 |
| Lauryl Maltose Neopentyl Glycol | Anatrace | Cat# NG310 |
| Cholesteryl hemisuccinate | Anatrace | Cat# CH210 |
| Critical Commercial Assays | ||
| Superdex 200, 10/300 GL | GE Healthcare | Cat# 17-5175-01 |
| Deposited Data | ||
| V-ATPase (state 1) density map | This study | EMD-21847 |
| V-ATPase (state 1) atomic model | This study | PDB 6WM2 |
| V1 density map | This study | EMD-21845 |
| V1 atomic model | This study | PDB 6WLZ |
| Vo density map | This study | EMD-21844 |
| Vo atomic model | This study | PDB 6WLW |
| V-ATPase (state 2) density map | This study | EMD-21848 |
| V-ATPase (state 2) atomic model | This study | PDB 6WM3 |
| V-ATPase (state 3) density map | This study | EMD-21849 |
| V-ATPase (state 3) atomic model | This study | PDB 6WM4 |
| Experimental Models: Cell Lines | ||
| HEK293F GnTI- | ATCC | Cat# CRL-3022 |
| Recombinant DNA | ||
| SidK (14-273) | pET28a | N/A |
| Software and Algorithms | ||
| SerialEM | (Mastronarde, 2005) | http://bio3d.colorado.edu/SerialEM |
| RELION | (Scheres, 2012) | https://www2.mrc-lmb.cam.ac.uk/relion |
| UCSF Chimera | (Pettersen et al., 2004) | http://www.cgl.ucsf.edu/chimera |
| PHENIX | (Adams et al., 2010) | https://www.phenix-online.org |
| COOT | (Emsley and Cowtan, 2004) | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL | Molecular Graphics System, Version 1.8 Schrodinger | http://www.pymol.org |
| Maxquant | Maxquant 1.6.3.4 | https://www.maxquant.org |
| Other | ||
| 400 mesh Lacey carbon grids | Electron Microscopy Sciences | LC400-Cu-100 |
Highlights.
Cryo-EM structures of the human V-ATPase reveal the mechanism of its assembly
The luminal domain of ATP6AP1 adopts a β-prism fold and belongs to the LAMP family
Dol-pp-glycan and lipids are bona fide components of the human V-ATPase
Biogenesis of V-ATPases rely on the synergy among proteins, glycans, and lipids
ACKNOWLEDGMENTS
We thank Dr. Anant Menon and Alexander B. Tong for discussions. Cryo-EM grid screening and collection of dataset I were performed with the assistance of Richard Walsh, Sarah Sterling, and Shaun Dawson at the Harvard Cryo-Electron Microscopy Center for Structural Biology supported by grants from the National Institute of General Medical Sciences (GM103310). The cryo-EM dataset II was collected by Ulric Baxa, Adam D. Wier, and Thomas J. Edwards at the National Cancer Institute’s National Cryo-EM Facility, Frederick National Laboratory for Cancer Research under contract HSSN261200800001E. Tandem mass spectrometry was performed by Ross Tomaino and Christopher Jahns at Taplin Mass Spectrometry Facility. L.W. was supported by funding from an NIH T32 grant (5T32AI007512-34). T.-M.F. was supported by funding from an NIH T32 grant (5T32HL066987-18 to L.E.S.) and by start-up funds from the Ohio State University Comprehensive Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abbas YM, Wu D, Bueler SA, Robinson CV, and Rubinstein JL (2020). Structure of V-ATPase from the mammalian brain. Science 367, 1240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic acids research 42, W252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolla JR, Agasid MT, Mehmood S, and Robinson CV (2019). Membrane Protein-Lipid Interactions Probed Using Mass Spectrometry. Annu Rev Biochem 88, 85–111. [DOI] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, and Orlowski J (2010). Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 11, 50–61. [DOI] [PubMed] [Google Scholar]
- Chen R, Jiang X, Sun D, Han G, Wang F, Ye M, Wang L, and Zou H (2009). Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J Proteome Res 8, 651–661. [DOI] [PubMed] [Google Scholar]
- Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, Boutros M, and Niehrs C (2010). Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 327, 459–463. [DOI] [PubMed] [Google Scholar]
- de Saint-Vis B, Vincent J, Vandenabeele S, Vanbervliet B, Pin JJ, Ait-Yahia S, Patel S, Mattei MG, Banchereau J, Zurawski S, et al. (1998). A novel lysosome-associated membrane glycoprotein, DC-LAMP, induced upon DC maturation, is transiently expressed in MHC class II compartment. Immunity 9, 325–336. [DOI] [PubMed] [Google Scholar]
- Defays A, David A, de Gassart A, De Angelis Rigotti F, Wenger T, Camossetto V, Brousset P, Petrella T, Dalod M, Gatti E, et al. (2011). BAD-LAMP is a novel biomarker of nonactivated human plasmacytoid dendritic cells. Blood 118, 609–617. [DOI] [PubMed] [Google Scholar]
- Delano WL (2002). The PyMol Molecular Graphics System.
- Diab H, Ohira M, Liu M, Cobb E, and Kane PM (2009). Subunit interactions and requirements for inhibition of the yeast V1-ATPase. J Biol Chem 284, 13316–13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL (2006). Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med 27, 495–502. [DOI] [PubMed] [Google Scholar]
- Esmail S, Kartner N, Yao Y, Kim JW, Reithmeier RAF, and Manolson MF (2018). N-linked glycosylation of a subunit isoforms is critical for vertebrate vacuolar H(+) -ATPase (V-ATPase) biosynthesis. J Cell Biochem 119, 861–875. [DOI] [PubMed] [Google Scholar]
- Esmail S, Yao Y, Kartner N, Li J, Reithmeier RA, and Manolson MF (2016). N-Linked Glycosylation Is Required for Vacuolar H(+) -ATPase (V-ATPase) a4 Subunit Stability, Assembly, and Cell Surface Expression. J Cell Biochem 117, 2757–2768. [DOI] [PubMed] [Google Scholar]
- Forgac M (1992). Structure, function and regulation of the coated vesicle V-ATPase. J Exp Biol 172, 155–169. [DOI] [PubMed] [Google Scholar]
- Forgac M (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8, 917–929. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, and Rubinstein JL (2018). Cryo-EM of ATP synthases. Curr Opin Struct Biol 52, 71–79. [DOI] [PubMed] [Google Scholar]
- Hahn A, Vonck J, Mills DJ, Meier T, and Kuhlbrandt W (2018). Structure, mechanism, and regulation of the chloroplast ATP synthase. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, and Aebi M (2001). Intracellular functions of N-linked glycans. Science 291, 2364–2369. [DOI] [PubMed] [Google Scholar]
- Hinton A, Bond S, and Forgac M (2009). V-ATPase functions in normal and disease processes. Pflugers Arch 457, 589–598. [DOI] [PubMed] [Google Scholar]
- Holm L, and Sander C (1995). Dali: a network tool for protein structure comparison. Trends Biochem Sci 20, 478–480. [DOI] [PubMed] [Google Scholar]
- Hunte C, and Richers S (2008). Lipids and membrane protein structures. Curr Opin Struct Biol 18, 406–411. [DOI] [PubMed] [Google Scholar]
- Hutchins PD, Rusell JD, and Coon JJ (2018). LipiDex: an integtated software package for high-confidence lipid identification. Cell Syst 6, 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen EJ, Timal S, Ryan M, Ashikov A, van Scherpenzeel M, Graham LA, Mandel H, Hoischen A, Iancu TC, Raymond K, et al. (2016). ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun 7, 11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Tarsio M, Charsky CM, and Kane PM (2005). Structural and functional separation of the N- and C-terminal domains of the yeast V-ATPase subunit H. J Biol Chem 280, 36978–36985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Mazhab-Jafari MT, Rohou A, Schmidt C, Bueler SA, Benlekbir S, Robinson CV, and Rubinstein JL (2016). Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nature 539, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire C, Stransky L, Cotter K, and Forgac M (2017). Regulation of V-ATPase activity. Front Biosci (Landmark Ed) 22, 609–622. [DOI] [PubMed] [Google Scholar]
- Murata T, Yamato I, Kakinuma Y, Leslie AG, and Walker JE (2005). Structure of the rotor of the V-Type Na+-ATPase from Enterococcus hirae. Science 308, 654–659. [DOI] [PubMed] [Google Scholar]
- O’Connor SE, and Imperiali B (1996). Modulation of protein structure and function by asparagine-linked glycosylation. Chem Biol 3, 803–812. [DOI] [PubMed] [Google Scholar]
- Pamarthy S, Kulshrestha A, Katara GK, and Beaman KD (2018). The curious case of vacuolar ATPase: regulation of signaling pathways. Mol Cancer 17, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareja F, Brandes AH, Basili T, Selenica P, Geyer FC, Fan D, Da Cruz Paula A, Kumar R, Brown DN, Gularte-Merida R, et al. (2018). Loss-of-function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat Commun 9, 3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rabinowitz SS, and Gordon S (1991). Macrosialin, a macrophage-restricted membrane sialoprotein differentially glycosylated in response to inflammatory stimuli. J Exp Med 174, 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson S, Phillips C, Huss M, Tiburcy F, Wieczorek H, Trinick J, Harrison MA, and Muench SP (2015). Structure of the vacuolar H+-ATPase rotary motor reveals new mechanistic insights. Structure 23, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh SH, Stam NJ, Hryc CF, Couoh-Cardel S, Pintilie G, Chiu W, and Wilkens S (2018). The 3.5-A CryoEM Structure of Nanodisc-Reconstituted Yeast Vacuolar ATPase Vo Proton Channel. Mol Cell 69, 993–1004 e1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M, Graham LA, and Stevens TH (2008). Voa1p functions in V-ATPase assembly in the yeast endoplasmic reticulum. Mol Biol Cell 19, 5131–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagermann M, Stevens TH, and Matthews BW (2001). Crystal structure of the regulatory subunit H of the V-type ATPase of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 98, 7134–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Oot RA, and Wilkens S (2018). MgATP hydrolysis destabilizes the interaction between subunit H and yeast V1-ATPase, highlighting H’s role in V-ATPase regulation by reversible disassembly. J Biol Chem 293, 10718–10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun-Wada GH, and Wada Y (2015). Role of vacuolar-type proton ATPase in signal transduction. Biochim Biophys Acta 1847, 1166–1172. [DOI] [PubMed] [Google Scholar]
- Terasawa K, Tomabechi Y, Ikeda M, Ehara H, Kukimoto-Niino M, Wakiyama M, Podyma-Inoue KA, Rajapakshe AR, Watabe T, Shirouzu M, et al. (2016). Lysosome-associated membrane proteins-1 and -2 (LAMP-1 and LAMP-2) assemble via distinct modes. Biochem Biophys Res Commun 479, 489–495. [DOI] [PubMed] [Google Scholar]
- Toei M, Saum R, and Forgac M (2010). Regulation and isoform function of the V-ATPases. Biochemistry 49, 4715–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toei M, Toei S, and Forgac M (2011). Definition of membrane topology and identification of residues important for transport in subunit a of the vacuolar ATPase. J Biol Chem 286, 35176–35186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokhtaeva E, Mareninova OA, Gukovskaya AS, and Vagin O (2017). Analysis of N- and O-Glycosylation of Lysosomal Glycoproteins. Methods Mol Biol 1594, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, and Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, and Feigenson GW (2008). Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasanthakumar T, Bueler SA, Wu D, Beilsten-Edmands V, Robinson CV, and Rubinstein JL (2019). Structural comparison of the vacuolar and Golgi V-ATPases from Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 116, 7272–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T, Merino F, Stabrin M, Moriya T, Antoni C, Apelbaum A, Hagel P, Sitsel O, Raisch T, Prumbaum D, et al. (2019). SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun Biol 2, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Fu TM, Zhou Y, Xia S, Greka A, and Wu H (2018). Structures and gating mechanism of human TRPM2. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Long T, Hassan A, Wang J, Sun Y, Xie XS, and Li X (2020). Cryo-EM structures of intact V-ATPase from bovine brain. Nat Commun 11, 3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke S, Krausze J, and Bussow K (2012). Crystal structure of the conserved domain of the DC lysosomal associated membrane protein: implications for the lysosomal glycocalyx. BMC Biol 10, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Shen X, Bryan A, Banga S, Swanson MS, and Luo ZQ (2010). Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog 6, e1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Benlekbir S, and Rubinstein JL (2015). Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 521, 241–245. [DOI] [PubMed] [Google Scholar]
- Zhao J, Beyrakhova K, Liu Y, Alvarez CP, Bueler SA, Xu L, Xu C, Boniecki MT, Kanelis V, Luo ZQ, et al. (2017). Molecular basis for the binding and modulation of V-ATPase by a bacterial effector protein. PLoS Pathog 13, e1006394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. [DOI] [PMC free article] [PubMed]
- Zhou L, and Sazanov LA (2019). Structure and conformational plasticity of the intact Thermus thermophilus V/A-type ATPase. Science 365. [DOI] [PubMed] [Google Scholar]
- Zhou M, Morgner N, Barrera NP, Politis A, Isaacson SC, Matak-Vinkovic D, Murata T, Bernal RA, Stock D, and Robinson CV (2011). Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science 334, 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, and Scheres SHW (2019). A Bayesian approach to beam-induced motion correction in cryo-EM single-particle analysis. IUCrJ 6, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, and Sabatini DM (2011). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM maps included in this study have been deposited in the Electron Microscopy Data Bank with accession codes: EMD-21844, EMD-21845, EMD-21847, EMD-21848, and EMD-21849. The atomic coordinates have been deposited in the Protein Data Bank with accession codes: 6WM2, 6WM3, 6WM4, 6WLZ, and 6WLW.