

Abstract
Primary liver cancers, including hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA), are highly lethal and frequent tumors worldwide, with few effective treatment options. The mammalian target of Rapamycin (mTOR) complex is a central regulator of cell growth and metabolism by its ability to integrate inputs from amino acids, nutrients and extracellular signals. The mTOR protein is incorporated into two distinct complexes: mammalian target of Rapamycin complex 1 (mTORC1) and mammalian target of Rapamycin complex 2 (mTORC2). Specifically, mTORC1 regulates protein synthesis, glucose and lipid metabolism, and autophagy, whereas mTORC2 promotes liver tumorigenesis via modulating the AGC family of serine/threonine kinases, especially the AKT (protein kinase B) proteins. In human HCC and iCCA samples, genomics analyses have unraveled the frequent de-regulation of the mTOR complexes. Both in vitro and in vivo studies have demonstrated the key role of mTORC1 and mTORC2 in liver tumor development and progression. The first generation mTOR inhibitors have been evaluated for effectiveness in liver tumor treatment and provided unsatisfactory results. Current research efforts are devoted to generating more efficacious mTOR inhibitors and identify biomarkers for patient selection as well as for novel combination therapies. Here, we provide a comprehensive review of the mechanisms leading to deregulated mTOR signaling cascade in liver cancers, the mechanisms whereby the mTOR pathway contributes to HCC and iCCA molecular pathogenesis, the therapeutic strategies, and the challenges that we need to overcome to effectively inhibit mTOR in liver cancer treatment. In conclusion: Deregulated mTOR signaling significantly contributes to HCC and iCCA molecular pathogenesis. mTOR inhibitors, presumably administered in association with other drugs, might be effective against subsets of human liver tumors.
Keywords: Hepatocellular carcinoma, Cholangiocarcinoma, mTORC1, mTORC2, mTOR inhibitor
Introduction
Primary liver cancer, including HCC and iCCA, ranked as the seventh most common cancer in the world.(1) HCC refers to tumors characterized by hepatocellular features at the morphological and molecular level, whereas iCCA exhibit biliary epithelial cell characteristics. Most liver cancers are diagnosed in advance stage, when limited therapeutic options are available. For patients with advanced stage HCC, the multi-kinase inhibitors sorafenib and lenvatinib are the first-line treatment drugs, with an overall survival benefit of 3 to 5 months when compared to placebo treatment.(2) Therapeutic options for advanced stage iCCA are even more limited. The first-line treatment strategy, namely chemotherapy with gemcitabine plus cisplatin or other platin-based drugs, has demonstrated partial benefits for advanced-stage iCCA.(3) Recent TCGA studies have provided a comprehensive picture of the genetic landscape of HCC and iCCA.(4, 5) In HCC, mutations in TP53, CTNNB1, ARID1A/2, AXIN1 and TSC1/2 genes, and amplification of CCND1/FGF19, c-MET and c-MYC, are frequently observed.(4) In iCCA, mutations in TP53, IDH1/2, KRAS and SMAD4 genes have been often detected.(5) These genetic abnormalities provide the unique opportunity for precision medicine for liver cancer treatment. Nonetheless, additional mechanistic studies are required to fully elucidate how these genetic events lead to liver tumorigenesis in order to develop novel therapeutic approaches against HCC and iCCA.
The mTOR is a central regulator of cell growth and metabolism in response to growth factors, nutrients, and cellular stress. Both in vitro and in vivo studies have demonstrated the key role of the mTOR pathway in regulating HCC and iCCA development and progression.(6) Consequently, multiple mTOR inhibitors have been or are currently being tested for liver cancer treatment. Here, we provide a comprehensive review of the mTOR signaling cascade, how it contributes to liver cancer development, and how it can be targeted for liver cancer prevention and treatment.
mTOR complexes: mTORC1 and mTORC2
mTOR is an atypical serine/threonine protein kinase, which regulates cell growth and metabolism via phosphorylation of its downstream effectors. mTOR forms two distinct complexes, namely mTORC1 and mTORC2 (Figure 1). Specifically, mTORC1 is characterized by its sensitivity to the macrolide Rapamycin and consists of mTOR, Raptor (regulatory protein associated with mTOR), mLST8, and two inhibitory companions, namely PRAS40 and DEPTOR.(7) mTORC2 is instead insensitive to Rapamycin and consists of mTOR, mLST8, Rictor (Rapamycin insensitive companion of mTOR), DEPTOR, mSIN1 and Protor1/2 proteins.(7)
Figure 1.
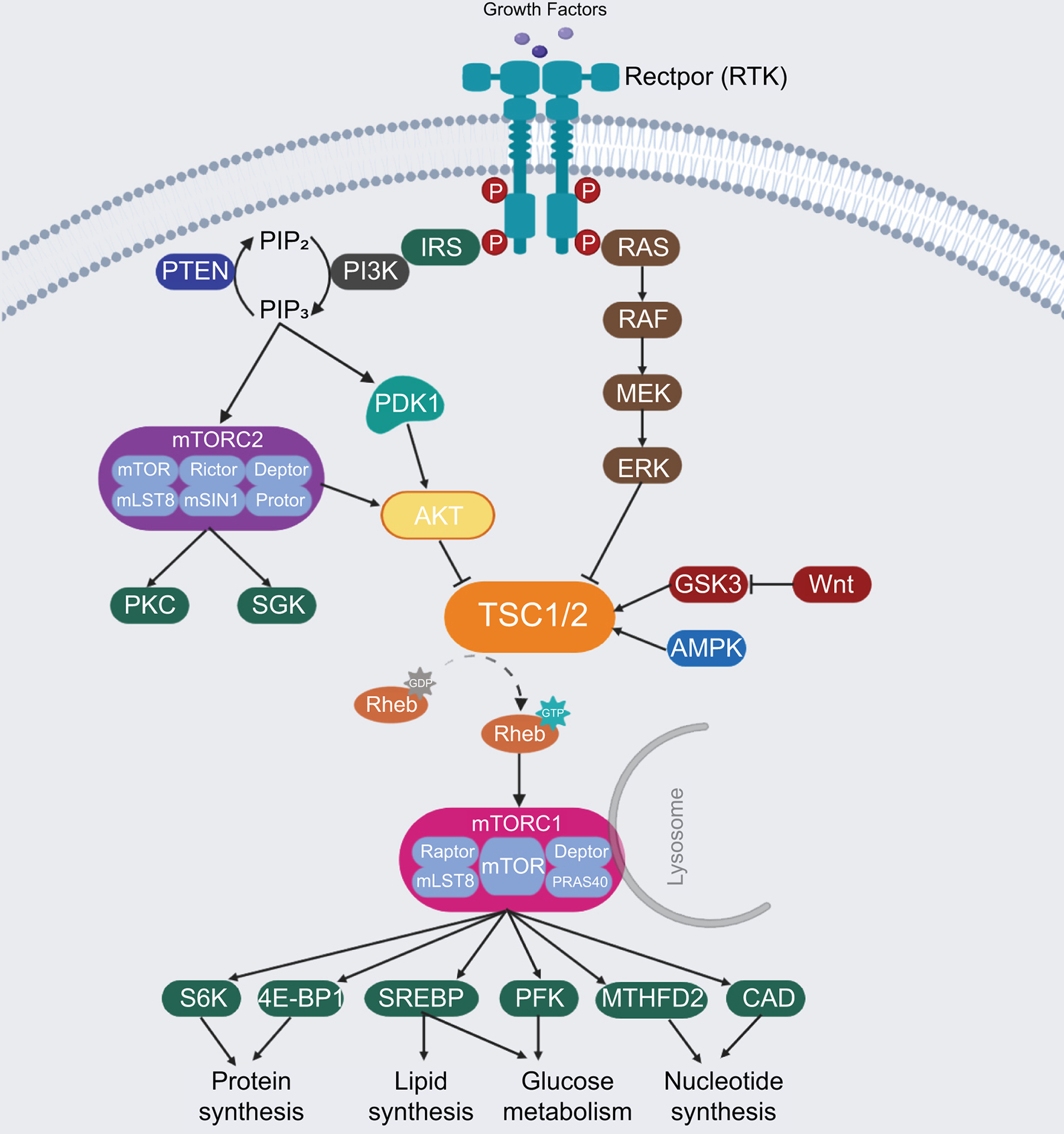
The mTOR signaling pathway network. RTK, receptor tyrosine kinase; IRS, Insulin receptor substrate; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PTEN, phosphatase and tensin homolog; PDK1, phosphoinositide-dependent kinase 1; AKT, protein kinase B; PKC, protein kinase C; SGK, serum- and glucocorticoid-regulated kinase; TSC, tuberous sclerosis complex; MEK, mitogen-activated protein kinase kinase; ERK, extracellular signal-regulated kinase; GSK3, glycogen synthase kinase 3; AMPK, AMP-activated kinase; Rheb, ras homolog enriched in brain; mTORC1, mammalian target of Rapamycin Complex 1; mTORC2, mammalian target of Rapamycin Complex 2; Raptor, regulatory protein associated with mTOR; Rictor, Rapamycin insensitive companion of mTOR; S6K, P70-S6 Kinase; 4E-BP1, eukaryotic translation initiation factor 4E binding protein 1; SREBP, sterol regulatory element binding protein; PFK, phospho-fructo kinase; MTHFD2, methylenetetrahydrofolate dehydrogenase 2; CAD, carbamoyl-phosphate synthetase.
mTORC1 plays a central role in protein, lipid, glucose as well as other biomass syntheses. The activation of mTORC1 predominantly relies on nutrients (amino acids, glucose, nucleotides, fatty acids and lipids) and growth factors. Nutrients facilitate the translocation of mTORC1 from the cytoplasm to the lysosomal surface, leading to its subsequent activation by the phosphatidylinositol 3-kinase (PI3K)/AKT signaling. Growth factors, like insulin, phosphorylate and activate AKT via phosphoinositide-dependent kinase 1 (PDK1) and PI3K proteins. In basal status, AKT inhibits the tuberous sclerosis complex (TSC), a hetero dimer composed of TSC1, TSC2 and TBC1D7, which act as a GTPase-activating protein on the RHEB GTPase.(8) Once activated, AKT inhibits the combination of TSC1 and TSC2 by phosphorylating the Ser939 and Thr1462 residues of TSC2. This molecular event allows the GTP-bound RHEB to directly bind and activate mTORC1 at the lysosome surface.(9) P70-S6 Kinase 1 (S6K1) and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1) are two key protein synthesis effectors of activated mTORC1.(10) Living cells also need enough lipids, glucose and nucleotides supply for anabolic processes and the inhibition of catabolic processes such as autophagy. In these processes, mTORC1 promotes the expression of metabolic genes such as sterol regulatory element binding protein 1 (SREBP1) and SREBP2, central transcriptional factors in fatty acid and cholesterol biosynthesis.(11) Also, mTORC1 promotes glucose metabolism by increasing the expression of glycolytic enzymes, such as phospho-fructo kinase (PFK).(12) In addition, mTORC1 enhances nucleotides synthesis that is required for DNA replication and ribosome assembly by increasing the expression of methylenetetrahydrofolate dehydrogenase 2 and carbamoyl-phosphate synthetase, two critical players in the purine and pyrimidine synthesis pathways, respectively (Figure 1).
mTORC2 controls cell proliferation and survival via phosphorylating several members of the AGC protein kinase family, including AKT, PKC (protein kinase C), and SGK (serum/glucocorticoid regulated kinase) kinases.(13) It has been shown that mTORC2 functions as a downstream effector of the insulin/PI3K cascade, directly or indirectly.(13) Once mTORC2 is induced, it phosphorylates and activates AKT, as well as SGK and PKC kinases, which in turn promote cell survival and proliferation. Activated AKT by mTORC2 may also subsequently activate mTORC1 via phosphorylating and inhibiting TSC1/2, as mentioned previously, thus inducing a positive feedback loop between mTORC1 and mTORC2 cascades in regulating cell growth, metabolism, and cell survival (Figure 1).
Deregulation of the mTOR pathway in HCC
In 2008, Villanueva A et al published the first study analyzing in detail the aberrant activation of the mTOR pathway in HCC.(6) Specifically, using p-RPS6 as the biomarker for mTORC1 activation, the authors found that the mTORC1 cascade was activated in about half of human HCC cases. Importantly, the TCGA study provided a comprehensive picture on how the mTOR pathway is deregulated along hepatocarcinogenesis.(4) Indeed, it is now clear that mutations in CTNNB1, TSC1/2, PIK3CA, loss of PTEN (phosphatase and tensin homolog), amplification of c-MYC, and activation of c-MET may lead to the activation of the mTOR pathway in this tumor type. In human HCCs, TSC1 and TSC2 mutations occur in 4% and 5% of HCC samples, respectively.(4) All these mutations lead to the activation of mTORC1 (Figure 2). The PI3K/PTEN/AKT cascade has been established as the major signaling event upstream of TSC1/2.(14) Among these genes, mutations in PIK3CA, RPS6KA3 and PTEN were detected in a small percentage of human HCCs.(4) Loss of PTEN protein expression has been shown to take place in about 50% of human HCCs.(15) c-MET is a key receptor tyrosine kinases (RTKs) that has been implicated in HCC pathogenesis. Using a c-MET activation gene expression signature, it was found that c-MET is activated in ~50% of human HCC samples.(16)
Figure 2.
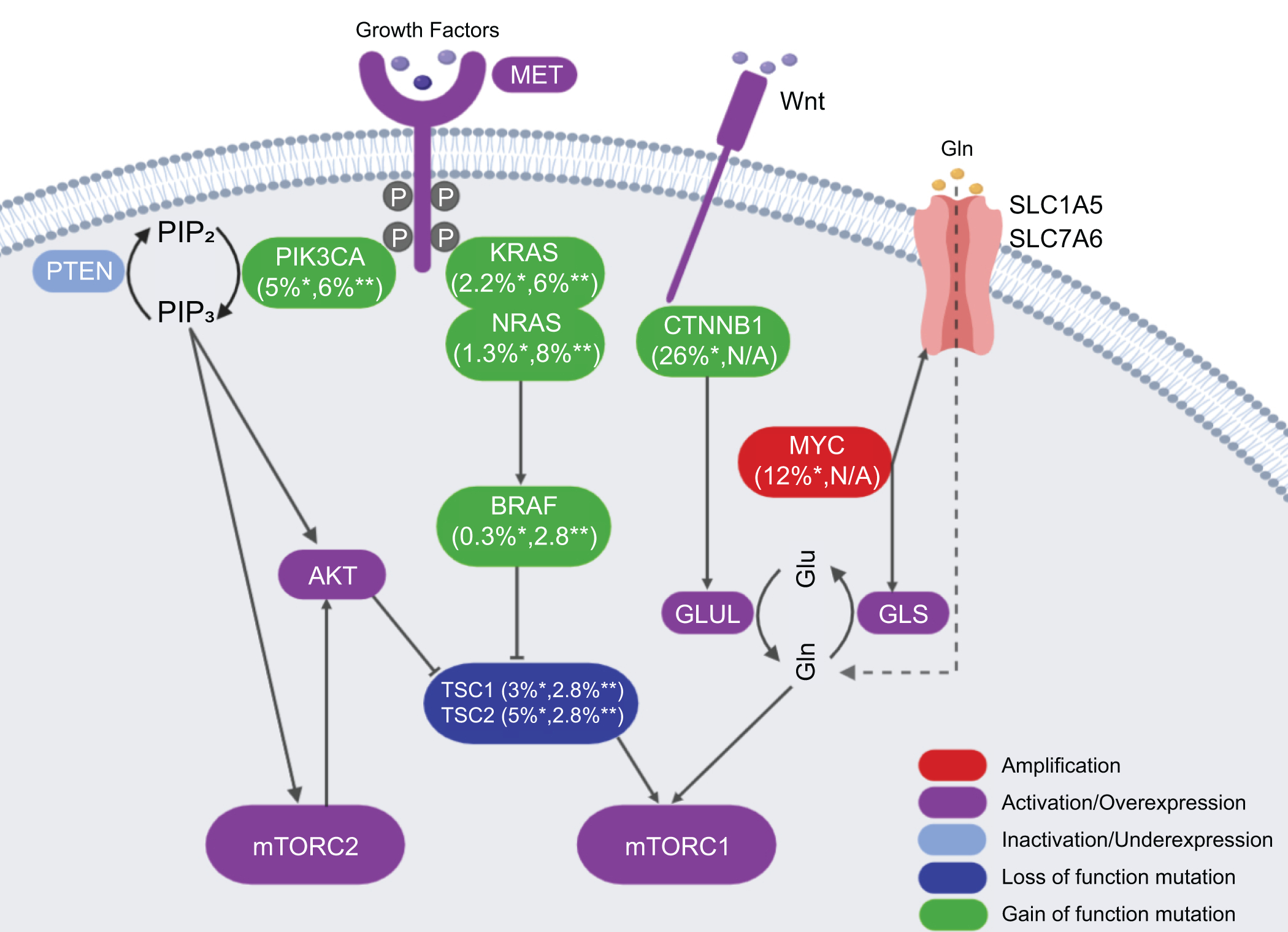
The common gene mutations and related rates in liver cancer. *indicates the mutation rate in HCC; **indicates the mutation rate in CCA. Mutation data are extracted from the published TCGA via cBioPortal (https://www.cbioportal.org/) of 372 HCC and 36 CCA samples. GLUL, glutamine synthetase; GLS, glutaminase; Glu, Glutamic acid; Gln, glutamine; SLC1A5, solute carrier family 1 member A5; SLC7A6, solute carrier family 7 member A6.
Besides the RTK/PI3K/PTEN/AKT cascade, amino acids, especially Leu, Gln and Arg, have been implicated in mTORC1 activation.(17) Therefore, genetic events leading to increased amino acids in the cell result in mTORC1 activation. For instance, c-MYC amplification is found in ~12% of human HCC samples.(4) c-MYC induces the expression of amino acid transporters, including solute carrier family 1 member A5 (SLC1A5) and solute carrier family 7 member A6 (SLC7A6), leading to increased amino acid uptake in HCC cells and mTORC1 activation.(18) In human HCC samples, a strong correlation between c-MYC protein levels and p-RPS6 as well as p-4E-BP1 has been described.(18) Mutations of the CTNNB1 gene, encoding β-catenin, occur in ~26% of human HCC samples (Figure 2).(4) Glutamine synthetase (GS) is a well-known target of activated Wnt/β-Catenin pathway. Recent studies have demonstrated that GS is able to promote glutamine-dependent mTORC1 activation in normal liver and HCC, thus underscoring the important interplay between the Wnt/β-Catenin and mTORC1 signaling cascades along liver carcinogenesis.(19)
It has been shown that p-AKT(S473), a biomarker for mTORC2 activation, is present in ~60% of human HCC samples.(20) The mechanisms leading to mTORC2 activation are poorly understood. No mutations in mTORC2 components have been identified. Chromosomal gain of RICTOR was detected in ~25% of human HCC samples, and high expression of RICTOR is associated with poor HCC prognosis.(21)
Deregulation of the mTOR pathway in iCCA
Much less is known about the mTORC1 and/or mTORC2 status and how they become activated in human iCCA. Studies have shown the frequent p-RPS6 expression in iCCAs with poor prognosis, and increased levels of p-AKT(S473) were detected in ~70% of human iCCA samples.(22, 23) Mutations that lead to mTOR activation, such as those affecting the TSC1/2 and CTNNB1 genes, occur very rarely in human iCCA samples. FGFR fusion mutations are uniquely found in iCCAs, but not in HCCs. As other RTKs, FGFR fusion mutations may lead to aberrant mTOR activation. Additional genetic events occurring in iCCA, such as KRAS or BRAF mutations, may also indirectly activate mTOR in this disease. Whether other mutations frequently identified in iCCA, such as mutations in TP53, IDH1/2 and SMAD4, can activate the mTOR signaling cascade remains to be addressed.
Functional role of mTORC1 in HCC and iCCA: evidence from mouse studies
Glucose metabolism is essential in cancer because malignant cells require glucose for their activities. Recent studies evidenced that in liver-specific TSC1 mutant mice, increased mTORC1 activation was accompanied by reduced glucose uptake, probably due to the downregulation of the PI3K/Akt pathway, which plays a role in glucose transport, via a mTORC1/S6K1-mediated negative feedback loop.(24) This finding is in line with the fact that TSC-deficient cells have been demonstrated to be hypersensitive to glucose withdrawal.(14)
A major transcriptional regulator of lipid metabolism-related genes is the SREBP family of transcription factors, as mentioned before. mTORC1 regulates SREBP via negative regulation of Lipin1, a phosphatidic acid phosphatase that represses SREBP activity.(25) mTORC1 signaling is necessary, but not sufficient, to activate SREBP1 and lipid synthesis in the liver, as also mTORC2 is required. In this regard, a recent study showed that liver-specific activation of mTORC1 promotes the expression of lipid synthesis genes and leads to the development of spontaneous HCC.(26) In this study, genetic mouse models developed spontaneous HCC along with an increased expression of SREBP1, ACC1 and FASN.
The role of mTORC1 in cancer protein metabolism is connected strictly with amino acid availability and uptake, protein production, and autophagy. Specifically, mTORC1 stimulates glutamine metabolism via regulation of transcription factors involved in the induction of glutaminolysis-related genes, like c-MYC, and liver-specific TSC1 knockout mice have reduced level of hepatic and plasma glutamine.(27) On the other hand, in liver tumors with mutations in CTNNB1, levels of GS (which is transcriptionally induced by β-catenin) are increased, and GS reduction prevents mTOR phosphorylation at Ser2448, leading to downregulation of mTOR.(19) Arginine is taken up into the normal cell mainly via the SLC7A1 transporter. Increased expression of this transporter has been reported in several cancers, including HCC lacking miR122 expression, leading to intracellular arginine accumulation and resistance to the multi-kinase inhibitor sorafenib.(28)
Nutrient deprivation, including amino acids and energy stress, are the main determinants of mTORC1-mediated autophagy in normal cells. In the liver, energy stress activates liver kinase B1 and AMP-activated kinase (AMPK) to suppress mTORC1 via inhibition of RAPTOR and activation of the TSC complex, but full activation of mTORC1 requires additional signaling induced by amino acids. It is not clear how amino acids trigger those mechanisms, and only a few molecular events have been elucidated. For instance, cellular uptake of L-glutamine by SLC1A5 and its subsequent rapid efflux by SLC7A5/SLC3A2, a bidirectional transporter that regulates the simultaneous efflux of L-glutamine and transport of leucine, are important for mTORC1 activation. In addition, loss of SLC1A5 function inhibits cell growth and activates autophagy presumably due to inhibition of leucine transport into cells.(29)
Several genetic alterations directly or indirectly involving PI3K/AKT/mTOR activation have been reported in advanced iCCA.(30) Moreover, gene expression profiling of invasive biliary cancer revealed the upregulation of downstream mediators of the mTOR pathway, such as S6K and EIF4E (Eukaryotic translation initiation factor 4E) as well as IGF1-R.(31) Previous reports showed that pan-mTOR kinase inhibitors were beneficial for the treatment of iCCA, even in tumors that are resistant to standard of care chemotherapeutics, especially in the subset exhibiting activated AKT/mTOR cascade.(22)
Functional role of mTORC2 in HCC and iCCA: evidence from mouse studies
As previously discussed, activation of mTORC2 occurs in ~60% human HCCs.(20) In vitro, it has been found that silencing of RICTOR inhibits p-AKT(S473) and HCC cell growth.(6) In vivo, overexpression of an activated form of AKT is sufficient to promote HCC development, supporting the oncogenic potential of the mTORC2/AKT cascade in HCC.(32) In addition, it has been shown that mTORC2 is activated in mouse HCCs induced by either simultaneous deletion of Pten and overexpression of c-Met (sgPten/c-Met)(15), overexpression of c-Myc and Mcl1 proto-oncogenes (c-Myc/Mcl1)(21) or loss of Tsc1 and Pten(33). Ablation of Rictor, the unique subunit of mTORC2, abolished mTORC2 activity and diminished p-AKT(S473) expression in the mouse liver.(13) Liver specific loss of mTORC2 led to multiple metabolic abnormalities, including constitutive gluconeogenesis, decreased lipogenesis and glycolysis, and defects in insulin signaling(13), suggesting that the mTORC2/AKT signaling is a major regulator of hepatic glucose and lipid metabolism during homeostasis. In sgPten/c-Met induced HCC, the deletion of Rictor completely prevented HCC formation in mice.(15) Similar results were obtained in the c-Myc/Mcl1 mouse HCC model.(21) In the liver, specific loss of Tsc1 and Pten induced HCC development; in this model, deletion of Rictor delayed, but did not completely prevent, HCC formation.(33)
The most important and profoundly characterized mTORC2 targets are the AKT kinases. In mammals, there are 3 AKT isoforms: AKT1, AKT2, and AKT3. AKT2 comprises of ~85% of total AKT proteins in the liver, while AKT1 comprises of the remaining 15%.(34) Studies have shown that AKT2 is the major AKT isoform transducing insulin signals in the liver.(35) Loss of Akt2 significantly inhibits loss of Pten induced HCC formation(36), whereas loss of Akt1, but not Akt2, completely abolishes c-Myc/Mcl1 driven hepatocarcinogenesis in vivo(21). In human HCC samples, p-AKT1 and p-AKT2 are expressed in different sample subsets, and p-AKT1 expression correlates with high c-MYC expression.(21) Overall, these studies suggest that mTORC2 may regulate different AKT isoforms to promote HCC development. Besides AKTs, other substrates of mTORC2 include PKC and SGK kinases. The functional roles of these kinases downstream of mTORC2 remain to be better characterized.
Few studies characterizing the functional role of mTORC2 in iCCA development and progression have been published. It has been shown that mTORC2 is activated in ~70% of human iCCAs, and that silencing of RICTOR inhibits human iCCA cell growth in culture.(22) The activated form of AKT is able to cooperate with activated Yap, Jag1 or loss of Fbxw7 to promote iCCA development in vivo(22, 37), thus supporting the oncogenic potential of mTORC2/AKT axis in iCCAs.
Targeting mTOR for HCC and iCCA treatment
As activation of mTOR has been identified in multiple tumor types, there has been great interest in targeting mTOR signaling pathway as a novel strategy for cancer treatment, including liver cancer.(38) The macrolide Rapamycin is the most famous mTOR inhibitor, initially widely used as an immunosuppressant drug in organ transplantation. Rapamycin binds to FRB-FK506 binding protein 12 (FKBP12), which subsequently binds to the FRB (FKBP12-Rapamycin binding) domain of the mTOR protein. This leads to the inhibition of T cell activation, as well as tumor cell growth and proliferation.(39) Subsequently, multiple Rapamycin analogs (Rapalogs) as well as other mTOR inhibitors have been developed (Figure 3).
Figure 3.
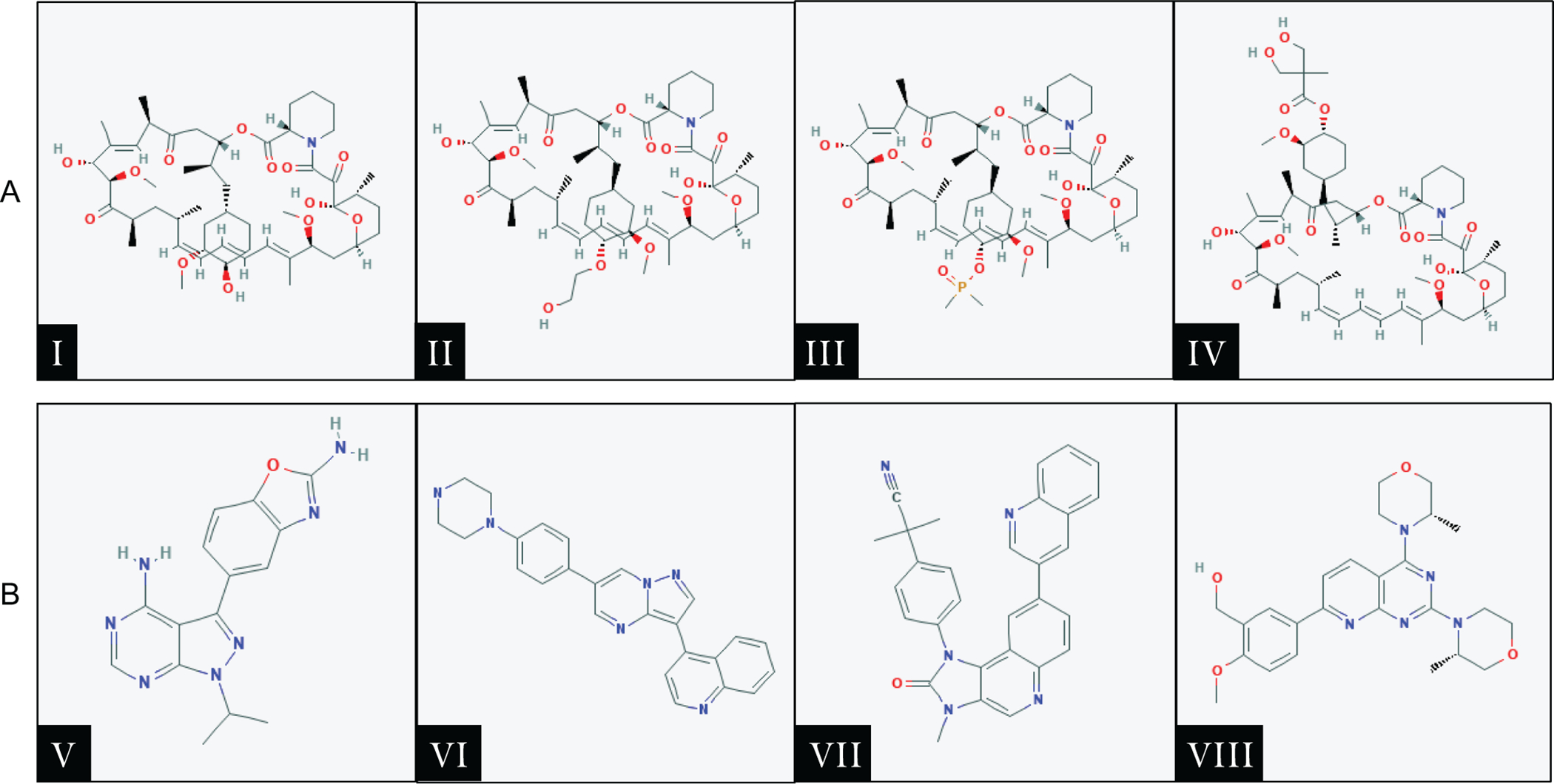
First and second generation mTOR inhibitors. A. First generation mTOR inhibitors (Rapamycin and Rapalogs): Ⅰ) Sirolimus, Ⅱ) Everolimus, Ⅲ) Deforolimus, Ⅳ) Temsirolimus. B. Second generation mTOR inhibitors: Ⅴ) MLN0128 (Sapanisertib), Ⅵ) CC-233, Ⅶ) NVP-BEZ235 (Dactolisib), Ⅷ) AZD-8055. Chemical structures of the drugs were obtained from the PubChem website (https://pubchem.ncbi.nlm.nih.gov).
First generation mTOR inhibitors: Rapalogs
The first generation of mTOR inhibitors are derived from Rapamycin, and they are referred to as Rapamycin analogs or Rapalogs. These drugs (Figure 3A) include Everolimus (RAD001), Deforolimus (AP23573), and Temsirolimus (CCI-779). Preclinical studies supported the effectiveness of these Rapalogs in inhibiting HCC cell growth. For instance, oral administration of RAD001 to mice bearing patient-derived HCC xenografts resulted in a dose-dependent inhibition of tumor growth.(40) Rapamycin and CCI-779 treatment of PLC/PRF/5 human HCC cells induced cell cycle arrest in G1 phase and reduced tumor growth in both colony formation assay and xenograft models.(41) In addition, long-term treatment with RAD001 significantly delayed DNA damage-induced liver tumor development.(42)
Due to these promising preclinical studies, Rapalogs were tested in clinical trials for advanced HCC treatment. A randomized phase 1/2 of Everolimus in advanced HCC clinical trial (NCT00390195) indicated that Everolimus (7.5mg/day) had acceptable tolerability and showed preliminary evidence of clinical activity.(43) In a phase 1/2 clinical study (NCT00321594) of Temsirolimus for patients with unresectable HCC, it failed to reach targeted progression free survival endpoint.(44) A global multicenter randomized Phase 3 clinical trial (NCT01035229) evaluating the effect of Everolimus on HCC patients who progressed or were intolerant to Sorafenib revealed that Everolimus did not improve overall survival in these patients.(45) These unsatisfactory results in different clinical trials strongly suggest that Rapalogs have very limited efficacy against advanced HCC as a single agent.
Liver transplantation (LTx) is considered a curative strategy for early stage liver cancer. Rapamycin and Rapalogs have been widely used as immunosuppressants to prevent transplant rejection. Studies have suggested that, in addition to their immunosuppressant activities, these mTOR inhibitors may also have anti-tumor activities. In a randomized, multicenter, open-label phase 3 clinical trial that evaluated Sirolimus use in liver transplant recipients with HCC, it was found that Sirolimus improved the recurrence-free survival and overall survival rates in the first 3 to 5 years, especially in a subset of low-risk patients, but thereafter it did not significantly modify the usual morbidity and mortality of the patients when compared to the conventional calcineurin inhibitor treatment.(46) In a meta-analysis study, it was discovered that compared with calcineurin inhibitors, mTOR inhibitors had decreased recurrence rates as well as improved overall survival rate.(47) However, this study mainly included retrospective series, which systematically exclude patients with drug withdrawal due to adverse events, combining the effects estimated from several studies (which is against the principles of meta-analysis). Further data on the efficacy of Rapalogs in LTx will be provided by two completed but yet to be published trials as well as two ongoing clinical trials (Table1).
Table 1.
Summary of completed and ongoing clinical trials with mTOR inhibitors in liver cancer*
| NCT No. | Status | Drug | Patient(s) | Gen. | Phase | HCC/ iCCA | Start | Completed | Result | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Single agent for advanced liver cancer | NCT00390195 | Completed | Everolimus | 39 | 1 | 1/2 | HCC | 2006 | 2011 | (43) | |
| NCT00321594 | Completed | Temsirolimus | 54 | 1 | 1/2 | HCC | 2006 | 2012 | (44) | ||
| NCT00516165 | Completed | Everolimus | 28 | 1 | 1/2 | HCC | 2007 | 2011 | |||
| NCT00973713 | Completed | Everolimus | 27 | 1 | 2 | iCCA | 2009 | 2012 | (48) | ||
| NCT01251458 | Completed | Temsirolimus | 46 | 1 | 1/2 | HCC | 2009 | 2016 | |||
| NCT01035229 | Completed | Everolimus | 546 | 1 | 3 | HCC | 2010 | 2013 | (45) | ||
| NCT01567930 | Completed | Temsirolimus | 26 | 1 | 2 | HCC | 2010 | 2013 | (49) | ||
| NCT00999882 | Completed | AZD8055 | 26 | 2 | 1 | HCC | 2009 | 2011 | |||
| NCT01177397 | Completed | CC223 | 173 | 2 | 1/2 | HCC | 2010 | 2016 | |||
| Adjuvant combination therapy for advanced liver cancer | NCT01010126 | Completed | Temsirolimus + Bevacizumab | 28 | 1 | 2 | HCC | 2009 | 2017 | (50) | |
| NCT02145559 | Completed | Sirolimus + Metformin | 24 | 1 | 1 | HCC/ iCCA | 2014 | 2018 | (51) | ||
| NCT00467194 | Completed | Sirolimus + Bevacizumab | 27 | 1 | 1 | HCC | 2006 | 2011 | (52) | ||
| NCT00775073 | Completed | Everolimus + Bevacizumab | 33 | 1 | 2 | HCC | 2008 | 2012 | |||
| NCT01687673 | Completed | Temsirolimus + Sorafenib | 28 | 1 | 2 | HCC | 2012 | 2019 | |||
| NCT01008917 | Completed | Temsirolimus + Sorafenib | 25 | 1 | 1 | HCC | 2009 | 2013 | (53) | ||
| NCT01005199 | Completed | Everolimus + Sorafenib | 106 | 1 | 2 | HCC | 2009 | 2016 | (54) | ||
| NCT01183663 | Completed | Temsirolimus + Lenalidomide | 2 | 1 | 1 | HCC | 2010 | 2016 | (55) | ||
| NCT01488487 | Completed | Everolimus + Pasireotide | 24 | 1 | 2 | HCC | 2011 | 2015 | |||
| NCT02724332 | Completed | Sirolimus + TACE | 300 | 1 | 1 | HCC | 2012 | 2015 | |||
| NCT01522820 | Completed | Sirolimus + Tumor Vaccine | 18 | 1 | 1 | HCC | 2012 | 2016 | |||
| NCT00949949 | Completed | Everolimus + Gemcitabine + Cisplatin | 38 | 1 | 1 | iCCA | 2009 | 2014 | |||
| NCT01888302 | Completed | Everolimus + Gemcitabine + Cisplatin | 1 | 1 | 1 | iCCA | 2013 | 2016 | |||
| Adjuvant combination therapy after LTx | NCT00328770 | Completed | Sirolimus + Immunosuppression reagents | 70 | 1 | 2/3 | HCC | 1996 | 2006 | ||
| NCT00355862 | Completed | Sirolimus+CNI/MMF/Steroids | 525 | 1 | 3 | HCC | 2006 | 2014 | (46) | ||
| NCT01374750 | Completed | Sirolimus compare Tacrolimus or Cyclosporine | 45 | 1 | 2 | HCC | 2010 | 2016 | |||
| NCT02081755 | Recruiting | Everolimus and Tacrolimus VS. Tacrolimus and Myfortic or CellCept Imuran | 336 | 1 | 4 | HCC | 2014 | 2019 | |||
| NCT03500848 | Not yet recruiting | Tacrolimus +Sirolimus+MMF and/or steroids | 130 | 1 | 2/3 | HCC | 2018 | 2023 | |||
The completed and ongoing clinical trials (https://www.clinicaltrials.gov/) were included from their first start date until December 31, 2019. A search strategy was developed in combination with the Medical Subject Headings, Emtree and text terms, include ‘liver cancer’, ‘liver tumor’, ‘hepatocellular carcinoma’, ‘HCC’, ‘biliary cancer’, ‘cholangiocarcinoma’, ‘intrahepatic cholangiocarcinoma’, ‘ICC’, ‘iCCA’, ‘CCA’, ‘mammalian target of Rapamycin inhibitors’, ‘mTOR inhibitors’, ‘mTORi’, ‘Rapamycin’, ‘Rapamune’, ‘Sirolimus’, ‘Everolimus’, ‘RAD001’, ‘Deforolimus’, ‘AP23573’, ‘Temsirolimus’, ‘CCI-779’, ‘MLN0128’, ‘CC-233’, ‘NVP-BEZ235’, ‘AZD-8055’.
Due to the limited efficacy of Rapalogs as monotherapy against HCC, multiple clinical trials have been conducted or are ongoing to evaluate the therapeutic efficacy of Rapalogs in combination therapies (Table1). A phase 1 trial (NCT00467194) of Sirolimus in combination with Bevacizumab (Sirolimus 4mg/day, Bevacizumab 5 mg/kg q14) for advanced HCC patients posed promising clinical activity.(52) This study reported differences in dose-limiting toxicity among patients with cirrhosis, depending on degree of severity. A first line phase 2 trial (NCT01010126) of Bevacizumab plus Temsirolimus enrolled 28 advanced HCC patients reported a favorable Objective Response Rate (ORR) of 19 % and Overall Survival (OS) of 14 months.(50) To determine the maximum tolerated dose (MTD) of Everolimus in advanced HCC patients of Child-Pugh class A liver function is given with standard sorafenib treatment, a phase I study (NCT00828594) reported the MTD of Everolimus in combination with standard-dose sorafenib was 2.5mg, instead of 5mg per day (56). This dose failed to achieve an effective blood Everolimus concentration and did not move forward to phase II study. Of note, the 2.5mg and 5mg cohorts exhibited similar blood concentration of Everolimus. The researchers involved in the study attributed the MTD of a lower dose to patients’ mild liver cirrhosis determining the reduction of Everolimus clearance.
A randomized multicenter, multinational phase 2 trial (NCT01005199) on advanced HCC patients treated with Sorafenib either alone or in association with Everolimus did not demonstrate any survival benefit of combining the two drugs when compared with Sorafenib administration alone.(54)
An open-label randomized clinical trial (NCT02145559) in solid tumors, including HCC, for the treatment with Metformin, an AMPK inhibitor, and Sirolimus revealed that there were no significant changes in phospho-p70S6k and other PD biomarkers. This finding led to the termination of the clinical trial.(51) There are also several completed clinical trials of treating liver tumors with Rapalogs combined with Pasireotide, TACE, tumor vaccine and Gemcitabine plus Cisplatin, but the results have not been published to date (Table1, NCT01488487, NCT02724332, NCT01522820, NCT00949949, NCT01888302).
Data on the effectiveness of Rapalogs in iCCA are scanty. It has been shown that Everolimus inhibits the secretion of proinflammatory cytokines by cancer-associated myofibroblasts and suppresses the proliferation of HuCCT1 and TFK1 cholangiocarcinoma cell lines at low concentrations in vitro.(57) In addition, it has been reported that combining Gemcitabine and Everolimus resulted in a synergistic antigrowth effect in iCCA cells in vitro and in vivo.(58)
Second generation mTOR inhibitors
In order to overcome the shortcomings of Rapalogs and improve mTOR inhibitory efficacy, second-generation mTOR inhibitors have been developed. These include MLN0128 (Sapanisertib), CC-233, NVP-BEZ235 (Dactolisib), and AZD-8055 (Figure 3B). These inhibitors predominantly function to inhibit the mTOR kinase catalytic activity, therefore acting as ATP-competitive agents to mTOR. Unlike Rapalogs, these inhibitors exhibit high potential inhibitory efficacy of all phosphorylation events catalyzed by mTORC1, as well as mTORC2. The mTOR domain FATC, FAT, and kinase region structural display a great similarity to those of PI3K-related kinases. Several PI3K inhibitors can also efficiently inhibit the mTOR complexes and are called for this reason dual PI3K/mTOR inhibitors.
MLN0128 is one of the second-generation pan-mTOR inhibitors. Preclinical studies showed that MLN0128 effectively inhibits both mTORC1 and mTORC2, leading to the suppression of cell growth and induction of apoptosis in vitro and in vivo.(59) CC-233, another pan-mTOR inhibitor, exhibited significant cytotoxicity and growth suppression potency against HCC cell lines and primary human HCC cells.(60) Furthermore, CC-223 oral administration dramatically inhibited HepG2 xenografts growth.(60) In a rat model of insulin-induced hepatocarcinogenesis, the metabolic abnormalities and cell growth driven by the insulin signal pathway were effectively reverted by NVP-BEZ235, a dual PI3K/mTOR inhibitor.(61) Currently, there are two completed clinical trials of evaluating second-generation mTOR inhibitors, namely CC-223 and AZD-8055, in treating advanced HCC. However, no results of these clinical trials have been published to date (Table1).
In iCCA, preclinical studies have shown that MLN0128 is able to inhibit iCCA growth in culture as well as to possess anti-tumor activities in an iCCA model driven by overexpression of activated AKT and YAP protooncogenes.(22) Mechanistically, MLN0128 effectively suppressed mTORC1 and mTORC2 signaling in iCCA cell lines, leading to profound cell apoptosis. Intriguingly, MLN0128 did not affect iCCA cell proliferation. Furthermore, MLN0128 synergized with Palbociclib, a CDK4/6 inhibitor, in determining a dramatic growth constraint of iCCA cells. Remarkable tumor regression was achieved upon combination therapy of these two drugs in the AKT/YAP mouse iCCA model.(62) Currently, there is no clinical trials testing the therapeutic efficacy of second-generation mTOR inhibitors for iCCA treatment.
Summary and Discussion
Mounting evidence points to a pivotal role of the mTOR signaling cascade in primary liver cancer. In particular, studies revealed that the mTOR cascade possesses several regulatory functions in liver tumorigenesis, affecting multiple features of the cancer cells, including proliferation, survival, invasion, and metabolism. In the same investigations, it has also been shown that blockade of the mTOR cascade is detrimental for the growth of HCC and iCCA cells. The current finding from human and experimental studies strongly support the development of innovative, safer and more potent anti-mTOR therapies for the treatment of liver cancer. It is worth to note that mild or severe liver cirrhosis often coexists with liver cancer, leading to either reduced clearance or insufficient intra-tumor concentration of mTOR inhibitors. In this scenario, the full anti-oncogenic activity of mTOR inhibitors might be hampered and compensatory therapeutic strategies should be developed.
As we have illustrated previously, despite the essential roles of mTOR in hepatocarcinogenesis, at the clinical level Rapalogs demonstrate limited therapeutic efficacy, either alone or in combination. It is important to underline that Rapamycin and Rapalogs are only partial inhibitors of mTORC1/2 and the new pan-mTOR inhibitors are more efficacious than Rapalogs, at least in in vitro and in vivo preclinical models. Since the success of anti-mTOR therapies seems to rely on multiple factors and, especially, on the functional crosstalk with other pathways, comprehensive studies should be conducted to address these important issues. In particular, a better understanding of the consequences of these molecular interactions is required to design tailored therapies, including combination therapies, where several pathways are concomitantly suppressed.
Another key issue is that even though the results from clinical trials have been negative so far, it is likely that a subset of patients whose tumors display activation of the mTOR pathway will still benefit from mTOR inhibition. Indeed, most of the clinical trials were conducted on an unselected patient population to date, whereas the use of the mTOR inhibitors should be limited to tumors exhibiting activated mTOR signaling. Therefore it is critical to identify novel reliable biomarkers of mTOR pathway activation to allow the selection of liver cancer patients who might benefit from mTOR suppression. Based on the current literature, mutation status of TSC1/2 may be the most promising biomarkers for selecting patients for anti-mTOR based therapies(63, 64). Nevertheless, clinical trials are required to test the hypothesis.
Financial Support:
This work is supported by NIH grants R01CA190606 and R01CA239251 to XC; P30DK026743 for UCSF Liver Center; China Scholarship Council PhD fellowship to Xinjun Lu (201906380153)
Abbreviations:
- HCC
hepatocellular carcinoma
- iCCA
intrahepatic cholangiocarcinoma
- mTOR
mammalian target of Rapamycin
- mTORC1
mammalian target of Rapamycin complex 1
- mTORC2
mammalian target of Rapamycin complex 2
- AKT
protein kinase B
- Rictor
Rapamycin insensitive companion of mTOR
- PI3K
phosphatidylinositol 3-kinase
- TSC
tuberous sclerosis complex
- SREBP
sterol regulatory element binding protein
- PTEN
phosphatase and tensin homolog
- GS
glutamine synthetase
- Rapalogs
Rapamycin analogs
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1).Global Burden of Disease Cancer C, Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F, et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 2018;391:1163–1173. [DOI] [PubMed] [Google Scholar]
- 3).Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273–1281. [DOI] [PubMed] [Google Scholar]
- 4).Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep 2017;18:2780–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008;135:1972–1983, 1983 e1971–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature 2013;497:217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 2012;47:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol 2013;203:563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol 2004;24:200–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 2008;8:224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017;168:960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 2012;15:725–738. [DOI] [PubMed] [Google Scholar]
- 14).Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 2002;10:151–162. [DOI] [PubMed] [Google Scholar]
- 15).Xu Z, Hu J, Cao H, Pilo MG, Cigliano A, Shao Z, et al. Loss of Pten synergizes with c-Met to promote hepatocellular carcinoma development via mTORC2 pathway. Exp Mol Med 2018;50:e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Kaposi-Novak P, Lee JS, Gomez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest 2006;116:1582–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol 2013;14:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Liu P, Ge M, Hu J, Li X, Che L, Sun K, et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017;66:167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Adebayo Michael AO, Ko S, Tao J, Moghe A, Yang H, Xu M, et al. Inhibiting Glutamine-Dependent mTORC1 Activation Ameliorates Liver Cancers Driven by beta-Catenin Mutations. Cell Metab 2019;29:1135–1150 e1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Hu J, Che L, Li L, Pilo MG, Cigliano A, Ribback S, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep 2016;6:20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Xu Z, Xu M, Liu P, Zhang S, Shang R, Qiao Y, et al. The mTORC2-Akt1 Cascade Is Crucial for c-Myc to Promote Hepatocarcinogenesis in Mice and Humans. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Zhang S, Song X, Cao D, Xu Z, Fan B, Che L, et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J Hepatol 2017;67:1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Andersen JB, Spee B, Blechacz BR, Avital I, Komuta M, Barbour A, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012;142:1021–1031 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Jiang X, Kenerson H, Aicher L, Miyaoka R, Eary J, Bissler J, et al. The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am J Pathol 2008;172:1748–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011;146:408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Li T, Weng J, Zhang Y, Liang K, Fu G, Li Y, et al. mTOR direct crosstalk with STAT5 promotes de novo lipid synthesis and induces hepatocellular carcinoma. Cell Death Dis 2019;10:619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Cornu M, Oppliger W, Albert V, Robitaille AM, Trapani F, Quagliata L, et al. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc Natl Acad Sci U S A 2014;111:11592–11599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Kishikawa T, Otsuka M, Tan PS, Ohno M, Sun X, Yoshikawa T, et al. Decreased miR122 in hepatocellular carcinoma leads to chemoresistance with increased arginine. Oncotarget 2015;6:8339–8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009;136:521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Hezel AF, Deshpande V, Zhu AX. Genetics of biliary tract cancers and emerging targeted therapies. J Clin Oncol 2010;28:3531–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Hansel DE, Rahman A, Hidalgo M, Thuluvath PJ, Lillemoe KD, Schulick R, et al. Identification of novel cellular targets in biliary tract cancers using global gene expression technology. Am J Pathol 2003;163:217–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011;140:1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017;32:807–823 e812. [DOI] [PubMed] [Google Scholar]
- 34).Pauta M, Rotllan N, Fernandez-Hernando A, Langhi C, Ribera J, Lu M, et al. Akt-mediated foxo1 inhibition is required for liver regeneration. Hepatology 2016;63:1660–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001;292:1728–1731. [DOI] [PubMed] [Google Scholar]
- 36).Galicia VA, He L, Dang H, Kanel G, Vendryes C, French BA, et al. Expansion of hepatic tumor progenitor cells in Pten-null mice requires liver injury and is reversed by loss of AKT2. Gastroenterology 2010;139:2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Wang J, Wang H, Peters M, Ding N, Ribback S, Utpatel K, et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J Hepatol 2019;71:742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol 2014;60:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Lang SA, Moser C, Fichnter-Feigl S, Schachtschneider P, Hellerbrand C, Schmitz V, et al. Targeting heat-shock protein 90 improves efficacy of rapamycin in a model of hepatocellular carcinoma in mice. Hepatology 2009;49:523–532. [DOI] [PubMed] [Google Scholar]
- 40).Huynh H, Chow KH, Soo KC, Toh HC, Choo SP, Foo KF, et al. RAD001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J Cell Mol Med 2009;13:1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Hui IC, Tung EK, Sze KM, Ching YP, Ng IO. Rapamycin and CCI-779 inhibit the mammalian target of rapamycin signalling in hepatocellular carcinoma. Liver Int 2010;30:65–75. [DOI] [PubMed] [Google Scholar]
- 42).Buitrago-Molina LE, Pothiraju D, Lamle J, Marhenke S, Kossatz U, Breuhahn K, et al. Rapamycin delays tumor development in murine livers by inhibiting proliferation of hepatocytes with DNA damage. Hepatology 2009;50:500–509. [DOI] [PubMed] [Google Scholar]
- 43).Shiah HS, Chen CY, Dai CY, Hsiao CF, Lin YJ, Su WC, et al. Randomised clinical trial: comparison of two everolimus dosing schedules in patients with advanced hepatocellular carcinoma. Aliment Pharmacol Ther 2013;37:62–73. [DOI] [PubMed] [Google Scholar]
- 44).Yeo W, Chan SL, Mo FK, Chu CM, Hui JW, Tong JH, et al. Phase I/II study of temsirolimus for patients with unresectable Hepatocellular Carcinoma (HCC)- a correlative study to explore potential biomarkers for response. BMC Cancer 2015;15:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Zhu AX, Kudo M, Assenat E, Cattan S, Kang YK, Lim HY, et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA 2014;312:57–67. [DOI] [PubMed] [Google Scholar]
- 46).Geissler EK, Schnitzbauer AA, Zulke C, Lamby PE, Proneth A, Duvoux C, et al. Sirolimus Use in Liver Transplant Recipients With Hepatocellular Carcinoma: A Randomized, Multicenter, Open-Label Phase 3 Trial. Transplantation 2016;100:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Grigg SE, Sarri GL, Gow PJ, Yeomans ND. Systematic review with meta-analysis: sirolimus- or everolimus-based immunosuppression following liver transplantation for hepatocellular carcinoma. Aliment Pharmacol Ther 2019;49:1260–1273. [DOI] [PubMed] [Google Scholar]
- 48).Yeung YH, Chionh FJM, Price TJ, Scott AM, Tran H, Fang G, et al. Phase II study of everolimus monotherapy as first-line treatment in advanced biliary tract cancer: RADichol. J Clin Oncol 2014;32:15_suppl:4101–4101. [Google Scholar]
- 49).Sachdev JC, Javed AY, Weir AB, Korn RI, Gulla SM, Newbold RG, et al. A phase II study of temsirolimus in previously treated advanced hepatocellular carcinoma (HCC). J Clin Oncol 2014;32:15_suppl:4098–4098. [Google Scholar]
- 50).Knox JJ, Qin R, Strosberg JR, Tan B, Kaubisch A, El-Khoueiry AB, et al. A phase II trial of bevacizumab plus temsirolimus in patients with advanced hepatocellular carcinoma. Invest New Drugs 2015;33:241–246. [DOI] [PubMed] [Google Scholar]
- 51).Sehdev A, Zha Y, Karrison TG, Janisch LA, Cohen EEW, Maitland ML, et al. A pharmacodynamic study of sirolimus and metformin in patients with advanced solid tumors. J Clin Oncol 2017;35:TPS11628–TPS11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52).Choo SP, Chowbay B, Ng QS, Thng CH, Lim C, Hartono S, et al. A Phase 1 dose-finding and pharmacodynamic study of rapamycin in combination with bevacizumab in patients with unresectable hepatocellular carcinoma. Eur J Cancer 2013;49:999–1008. [DOI] [PubMed] [Google Scholar]
- 53).Kelley RK, Lee MR, Hwang J, Gordan JD, Nimeiri HS, Bocobo AG, et al. Detection of circulating tumor cells (CTC) using a non-EpCAM-based, high-definition, single-cell assay in advanced hepatocellular carcinoma (HCC) for patients enrolled on phase I and II trials of sorafenib plus temsirolimus. J Clin Oncol 2017;35:4_suppl:311–311. [Google Scholar]
- 54).Koeberle D, Dufour JF, Demeter G, Li Q, Ribi K, Samaras P, et al. Sorafenib with or without everolimus in patients with advanced hepatocellular carcinoma (HCC): a randomized multicenter, multinational phase II trial (SAKK 77/08 and SASL 29). Ann Oncol 2016;27:856–861. [DOI] [PubMed] [Google Scholar]
- 55).Ganesan P, Piha-Paul S, Naing A, Falchook G, Wheler J, Fu S, et al. Phase I clinical trial of lenalidomide in combination with sorafenib in patients with advanced cancer. Invest New Drugs 2014;32:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56).Finn RS, Poon RT, Yau T, Klumpen HJ, Chen LT, Kang YK, et al. Phase I study investigating everolimus combined with sorafenib in patients with advanced hepatocellular carcinoma. J Hepatol 2013;59:1271–1277. [DOI] [PubMed] [Google Scholar]
- 57).Rattanasinganchan P, Leelawat K, Treepongkaruna SA, Tocharoentanaphol C, Subwongcharoen S, Suthiphongchai T, et al. Establishment and characterization of a cholangiocarcinoma cell line (RMCCA-1) from a Thai patient. World J Gastroenterol 2006;12:6500–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58).Lin G, Lin KJ, Wang F, Chen TC, Yen TC, Yeh TS. Synergistic antiproliferative effects of an mTOR inhibitor (rad001) plus gemcitabine on cholangiocarcinoma by decreasing choline kinase activity. Dis Model Mech 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59).Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012;485:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60).Xie Z, Wang J, Liu M, Chen D, Qiu C, Sun K. CC-223 blocks mTORC1/C2 activation and inhibits human hepatocellular carcinoma cells in vitro and in vivo. PLoS One 2017;12:e0173252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61).Evert M, Calvisi DF, Evert K, De Murtas V, Gasparetti G, Mattu S, et al. V-AKT murine thymoma viral oncogene homolog/mammalian target of rapamycin activation induces a module of metabolic changes contributing to growth in insulin-induced hepatocarcinogenesis. Hepatology 2012;55:1473–1484. [DOI] [PubMed] [Google Scholar]
- 62).Song X, Liu X, Wang H, Wang J, Qiao Y, Cigliano A, et al. Combined CDK4/6 and Pan-mTOR Inhibition Is Synergistic Against Intrahepatic Cholangiocarcinoma. Clin Cancer Res 2019;25:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63).Huynh H, Hao HX, Chan SL, Chen D, Ong R, Soo KC, et al. Loss of Tuberous Sclerosis Complex 2 (TSC2) Is Frequent in Hepatocellular Carcinoma and Predicts Response to mTORC1 Inhibitor Everolimus. Mol Cancer Ther 2015;14:1224–1235. [DOI] [PubMed] [Google Scholar]
- 64).Ho DWH, Chan LK, Chiu YT, Xu IMJ, Poon RTP, Cheung TT, et al. TSC1/2 mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut 2017;66:1496–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]