

Abstract
Background & Aims
Epitranscriptomic modification of RNA has emerged as the most prevalent form of regulation of gene expression which affects development, differentiation, metabolism, viral infections, and most notably cancer. We have previously shown that Hepatitis B virus (HBV) transcripts are modified by N6 methyladenosine (m6A) addition. HBV also affects m6A modification of several host RNAs, including phosphatase and tensin homolog (PTEN), a well-known tumor suppressor. PTEN plays a critical role in antiviral innate immunity and the development of hepatocellular carcinoma (HCC). Reports have shown that PTEN controlled IRF-3 nuclear localization by negative phosphorylation of IRF-3 at Ser97, and PTEN reduced carcinogenesis by inhibiting the phosphatidylinositol-3-kinase (PI3K)/AKT pathway.
Approach & Results
Here, we show that HBV significantly increases the m6A modification of PTEN RNA, which contributes to its instability with a corresponding decrease in PTEN protein levels. This is reversed in cells, in which m6A methyltransferases (METTL3/14) expression, is silenced. PTEN expression directly increases activated IRF-3 nuclear import and subsequent interferon (IFN) synthesis. In the absence of PTEN, IRF-3 dephosphorylation at Ser97 site is decreased and IFN synthesis is crippled. In chronic-HBV patient biopsy samples, m6A modified PTEN mRNA levels were uniformly upregulated with a concomitant decrease of PTEN mRNA levels. HBV gene expression also activated PI3K/AKT pathway by regulating PTEN mRNA stability in HCC cell lines.
Conclusions
Thus, m6A epitranscriptomic regulation of PTEN by HBV affects innate immunity via inhibiting IRF-3 nuclear import and the development of HCC through activating PI3K/AKT pathway. Our studies collectively provide new insights into the mechanisms of HBV-directed immune evasion and HBV-associated hepatocarcinogenesis via m6A modification of the host PTEN mRNAs.
Keywords: RNA chemical modification, Epitranscriptomics, Immune evasion, Hepatocarcinogenesis
Introduction
HBV infection leads to chronic hepatitis and carries the risk for the development of HCC (1). HCC is the third leading cause of deaths worldwide. HBV is a member of Hepadnaviridae family. HBV genome codes for the following proteins; surface (HBs), precore or ‘e’ (HBe) and core (HBc) antigen proteins, polymerase (pol, a reverse transcriptase) and X (HBx) proteins. Although HBV is a DNA containing virus, it replicates via an RNA intermediate termed pregenome RNA (pgRNA) to produce a relaxed circular DNA which ultimately transforms into a covalently closed (cccDNA) in the nucleus (2). While there is an effective vaccine available for HBV, there are about 350 million people infected with HBV worldwide with 600,000 deaths reported annually (2). The antiviral treatment of HBV has limited efficacy. Once the drug is withdrawn, HBV infection can resume as cccDNA which is unaffected by antivirals, maintains its presence, can resume the infectious process (3). The inability to totally eliminate HBV infection poses a great challenge today. HBV can trigger changes in host gene expression profile to optimize conditions for viral replication and increase long-term survival of the virus (3). Chronic hepatitis B evolves into liver cirrhosis and hepatocellular carcinoma through the regulation of changes in the host gene expression. In 8–20% of chronic hepatitis B patients, the infection progresses to liver cirrhosis within 5 years and 2–8% of those with cirrhosis advance to HCC (4). The exact mechanism(s) or trigger that leads to HBV related HCC remains to be defined.
PTEN is widely known as a tumor suppressor with reduced expressions in several tumors (5). PTEN is a metabolic regulator as well as a negative regulator of cell growth signaling pathways. It is a dual phosphatase capable of both protein and lipid phosphatase activities (5). PTEN regulates PI3K/AKT signaling pathways by dephosphorylating phosphatidylinositol-3, 4, 5-triphosphate (PIP3). PTEN-knockout in transgenic mice model developed HCC (6). In addition to its role as a tumor suppressor, PTEN regulates innate immune response activated by viral infections. PTEN induces IRF-3 dephosphorylation at Ser97 site, promoting IRF-3 nuclear translocation. The silencing of PTEN activity using siRNAs decreased p-IRF-3 nuclear import, which leads to reduce IFN signaling pathway (7).
Functional relationships between host gene expression and viral infections have important roles in modulating the viral life cycle. Viral infections induce alterations of cellular regulatory networks of host proteins. Altered host gene expression induced by viruses paves the way for successful establishment of the infectious process and subsequent disease pathogenesis (8, 9). Most notably, viruses affect cellular transcription, translation and host gene expression. Several chemical modifications on cellular RNA are known to regulate RNA stability and turnover. Among the RNA chemical modifications, the N6-methyladenosine modification (m6A) is the most prevalent internal mRNA modification of eukaryotic cells (10). Recent development of detection methods with high sensitivity combined with high-throughput sequencing reveals informative profiles of m6A RNA modifications in cells. Over 7000 transcripts contain m6A modifications that are typically enriched in the 3′-untranslated region (UTR) and near the stop codons of cellular mRNA. These modifications have been linked to various biological processes, including sex determination, stem cell differentiation, circadian clock, meiosis, stress response and cancer (10). The m6A modification is a dynamic co-transcriptional process that is reversibly catalyzed by m6A ‘writers’, such as METTL3, METTL14, and WTAP and ‘erasers’, such as fat mass and obesity-associated protein (FTO) and ALKBH5 (10). The m6A modified mRNA directly interacts with YTH-domain family proteins (YTHDF1, YTHDF2 and YTHDF3). Functionally, YTHDF1 enhances m6A modified-mRNA translation, while YTHDF2 promotes target mRNA degradation. The YTHDF3 regulates degradation and translation of m6A modified-mRNA through cooperation with YTHDF1 and 2 (11). RNA genomes of several RNA viruses, as well as the RNA transcripts of DNA viruses, have been reported to be m6A modified (12–17). m6A modification of viral RNAs has been shown to affect various aspects of the viral life cycle and associated pathogenesis. m6A forming enzymes (methyltransferases), METTL3/14, and reader proteins (YTHDFs) play important roles in regulating the life cycle of both DNA and RNA viruses (12–17). While m6A modification of viral transcripts has been identified, their effects on viral replication and translation are being characterized.
We previously reported that HBV transcripts are modified by m6A and this modification played a dual role in regulation of the viral life cycle (14, 18). In this study, we demonstrate that HBV alters host gene expression by regulating m6A modification of cognate RNAs. We found that m6A modification of host RNA is either increased or decreased by HBV gene expression. Among those altered genes, we focused on PTEN mRNA, because PTEN has a critical role in innate immune response as well as liver carcinogenesis. We further demonstrate that HBV increases m6A modification of PTEN mRNA, which leads to the destabilization of PTEN mRNA, and blocks the interferon signaling pathway. Reduction in PTEN expression may contribute to the development of HCC. Altogether, our results highlight a novel mechanism of HBV immune evasion and possible role in HBV-associated HCC via m6A modification of the host PTEN mRNAs.
Experimental Procedures
Plasmids, antibodies, and reagents
HBV 1.3-mer was a kind gift from Dr. Wang-Shick Ryu (Yonsei University) and obtained from the Addgene (Cambridge, MA, USA). FLAG-YTHDF1, 2, and 3 plasmids were a kind gift from Dr. Stacy M. Horner (Duke University Medical Center) (15). Antibodies were obtained as follow: anti-HBc antibodies from Dr. Haitao Guo (University of Pittsburgh, PA, USA) (19), anti preS2, anti-YTHDF3, anti-GAPDH antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-METTL3 antibody from Proteintech Group (Rosemont, IL, USA), anti-METTL14 antibody from Sigma-Aldrich, San Jose, CA, USA), anti-FTO and anti-YTHDF2 antibodies from Abcam (Cambridge, MA, USA), anti-FLAG, anti-PTEN, anti-IRF-3, and anti-phospho-IRF-3 antibodies from Cell Signaling Technology (Danvers, MA, USA).
Cell culture and transfection
The Huh7 and HepG2 cells were obtained from ATCC (Manassas, VA, USA). The HepAD38 cells were provided by Dr. C. Seeger, Philadelphia (20). The cells were maintained as described previously (21). Primary human hepatocytes (PHH) were obtained from Gibco (Waltham, MA, USA). PHH were cultured according to the manufacturer’s protocol. HepG2 cells were transfected with plasmids and Poly (I:C) using Mirus TransIT-LT1 and Mirus TransIT-mRNA kit (Madison, WI, USA) according to the manufacturer’s protocol, respectively. siRNAs were transfected using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol.
Virus production and cell infection
HBV particles were harvested from the culture medium of HepAD38 cells. The supernatants were incubated with 5% polyethylene glycol (PEG) 8000 for overnight at 4 °C, and then centrifuged at 4,000 rpm for 30 min at 4 °C. Pellet was re-dissolved in a serum-free culture medium. PHH were incubated for 24 h with HBV particles, which are diluted in culture medium with 4% PEG 8000. After incubated with HBV particles, the cells were washed with culture medium. In vitro transcribed full-length HCV RNA was electroporated into Huh7 cells. At 72 h post-electroporation, the culture medium was passed through a 0.45 μm filter. For infection, Huh7 cells were incubated with filtered culture medium for 4 h. HIV particles were collected from pNL4.3 DNA transfected HEK293T cells. Infectious HIV-1 particles were used for infection in Jurkat cells.
qRT-PCR
Total RNA was isolated using Trizol reagent (Invitrogen, Waltham, MA, USA). The quantitative PCR was assessed with Ssoadvanced Universal SYBR Green supermix (Bio-Rad, Hercules, CA, USA). Each mRNA expression levels, normalized to GAPDH, were analyzed using the ΔΔCt method. The primers for qRT-PCR are shown in Table S1.
Methylated RNA immunoprecipitation (MeRIP) sequencing
MeRIP sequencing was performed as previously described (14). Libraries were sequenced to 1 x 50 base-pair reads on the Illumina HiSeq2500 at the Weill Cornell Medicine Epigenomics Core Facility. Reads were aligned to a combined human (hg38) using Spliced Transcripts Alignment to a Reference (STAR). Mean coverage was plotted for all three replicates using CovFuzze (https://github.com/al-mcintyre/covfuzze). The raw data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE114486. MeRIP qRT-PCR followed the same protocol, except that total RNA was not fragmented. Eluted RNA was reverse transcribed into cDNA and subjected to qRT-PCR.
Western blotting and immunoprecipitation
Cell pellets were lysed in NP-40 lysis buffer (1% NP-40, 50 mM Tris-HCl, pH 8.0, 150 mM NaCl) supplemented with a protease inhibitor and a phosphatase inhibitor (Thermo Scientific). Purified cell lysates were incubated with Anti-FLAG M2 Magnetic Beads (Sigma-Aldrich) for 2 h on a rotator at 4°C. Immunoprecipitates or lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane (Bio-Rad). Membranes were incubated by using various primary antibodies.
Subcellular fractionation
Cytoplasmic and nuclear protein fractions were prepared with NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific) according to the manufacturer’s protocol.
Ethics statement
Human liver biopsy specimens were collected from St. Mary’s Hospital, Catholic University, Seoul, South Korea from two healthy volunteers, three HCC anti-HBV-negative, and two HCC anti-HBV-positive patients. Liver biopsy samples were collected according to the institutional review board (IRB) protocols. The IRB has approved this study as a whole.
Statistical analysis
All results are representative of three independent experiments. For each result, error-bars represent the ± SD from at least three independent experiments. The p-value was calculated using a one-tailed unpaired Student’s t-test.
Results
HBV induces m6A modification of PTEN 3′-UTR
To determine whether HBV regulates m6A modification of host cellular RNA, we performed MeRIP assay with m6A-specific antibody in HepG2 cells expressing HBV 1.3-mer plasmid. Immunoprecipitated RNAs were analyzed by high-throughput sequencing. We found that HBV transfection either increased or decreased m6A modification of host cellular RNAs (S2 Table and S3 Table). The list shown represents a profile of 70 host cellular RNAs with either the higher or lower levels of m6A modification induced by HBV. Among the various host RNAs with m6A methylation levels altered by HBV, we focused on PTEN mRNA which displayed a dramatic 2-fold increase in m6A modification.
Most m6A modification is found within a consensus motif DRACH (where D= A, G, or U; R = G or A; A = denotes methylated adenosine; C = C; H = A, C, or U) and enriched in 3′-untranslated region (UTR) of cellular mRNAs (22). Since all DRACH motifs are not m6A methylated, we aligned m6A reads to PTEN 3′-UTR sequence to identify which DRACH motifs of the PTEN mRNA were m6A modified. We identified that PTEN 3′-UTR has eight different DRACH motifs and m6A peaks are enriched in 41–239-bp of 3′-UTR (Fig 1A). Interestingly, HBV significantly increased m6A peaks in 300–330-bp and 521–550-bp of PTEN 3′-UTR (Fig 1B) and as shown in Fig. 1C, m6A modification of PTEN mRNA was significantly increased in HBV expressing cells. We also checked the methylated PTEN mRNA levels in immunoprecipitated RNA by m6A-specific antibody using quantitative real-time RT-PCR (qRT-PCR) method. In the MeRIP-qRT-PCR assay (Fig 1D), m6A modified PTEN mRNA levels were increased by HBV. CREBBP (a cellular RNA known to contain m6A) and HPRT1 (a cellular RNA that does not contain m6A) were used as positive and negative controls, respectively (23).
Fig 1.
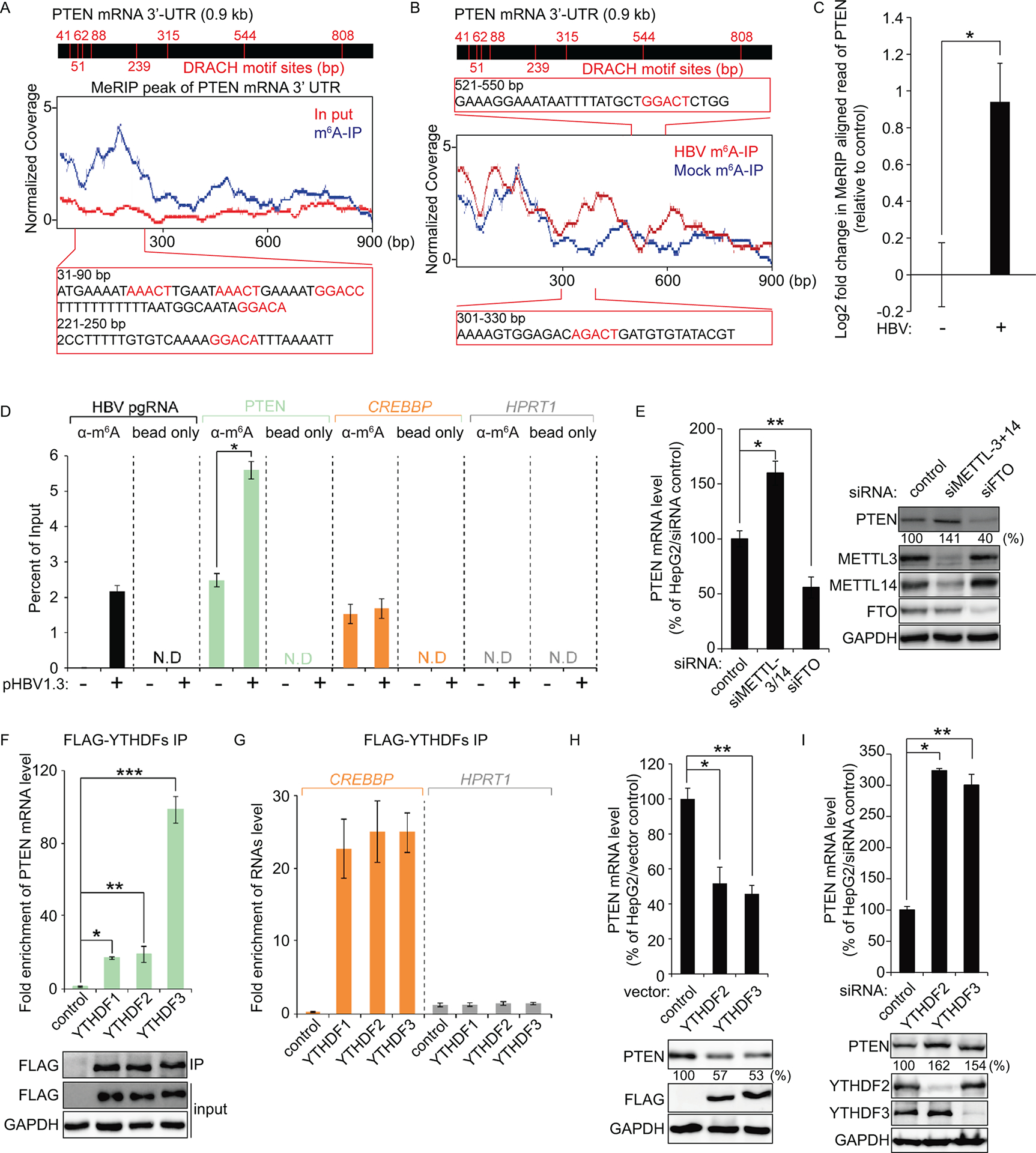
PTEN 3′-UTR contains the m6A modification and its modification is regulated by HBV transfection. (A and B) Map of m6A-binding sites in the PTEN 3′-UTR region by MeRIP-seq (representative of three independent samples) from HBV expressing HepG2 cells. Blue coverage, normalized to the total number of reads mapping to the PTEN mRNA for each experiment, is in blue for MeRIP-seq and red for input MeRIP-seq. Eight DRACH motif sites were identified in PTEN 3′-UTR. Several m6A peaks were analyzed after normalizing for coverage. The Inset presents bp 31–90 and 221–250 of PTEN 3′-UTR region, with the m6A sites highlighted by red text (A). Read coverage, normalized to the total number of reads mapping to the PTEN mRNA for each experiment, is in red for HBV expressing cells RNA-seq and in blue for control cells RNA-seq. The Inset presents bp 301–330 and 521–550 of PTEN 3′-UTR region, with the m6A sites in HBV expressing cells highlighted by red text (B). (C) Fold change (log2) of the MeRIP reads of PTEN mRNA in the HBV expressing cells compared with the control HepG2 cells. Plotted in (C) are the relative reads number of PTEN (mean ± SD estimated from three independent samples). *P = 0.0058 (D) MeRIP-qRT-PCR of m6A modified HBV transcripts and PTEN mRNA from total RNA extracted from HepG2 cells transfected with HBV 1.3-mer plasmid. CREBBP and HPRT1 serve as positive and negative controls, respectively. *P = 0.00029 (E) Relative PTEN mRNA level in HepG2 cells transfected with siMETTL3 + 14 or siFTO at 2 days post-transfection. PTEN protein expression was assessed by immunoblotting. *P = 0.0015 ** P = 0.0035 (F) Enrichment of PTEN mRNA following immunoprecipitation of FLAG-tagged YTHDFs from extracts of HepG2 cells after 48 h transfection. Enriched PTEN mRNA was quantified by qRT-PCR as the percentage of input and graphed as fold enrichment relative to control. Immunoblot analysis of FLAG-YTHDFs in the input and IP is shown on bottom panels. *P = 0.00004 ** P = 0.0021 *** P = 0.00022 (G) RNA immunoprecipitation from FLAG-YTHDFs transfected HepG2 cells using anti-FLAG antibody, with qRT-PCR analysis of CREBBP and HPRT1 were quantified as relative enrichment RNA level. (H and I) HepG2 cells were transfected with FLAG-YTHDF2/3 or siRNAs of YTHDF2/3. After 48 h, cells were harvested to assess expression levels of PTEN mRNA and proteins levels (*P = 0.0015 ** P = 0.00013; E, *P = 0.00024 ** P = 0.0065; F). In (C – I), the error bars are the standard deviations of three independent experiments, each involving triplicate assays. Statistical significance of the difference between groups was determined via an unpaired Student’s t-test.
To determine whether m6A methylation of PTEN mRNA affects its RNA and protein levels, we depleted both METTL3 and 14 or FTO in HepG2 cells using siRNAs. The silencing of METTL3/14 elicited a 60 ± 10.90% increase in PTEN mRNA levels and a 41 ± 4.22% increase in PTEN protein levels. In contrast, the PTEN mRNA and protein levels were decreased by siFTO (45 ± 9.57% mRNA decrease; 53 ± 8.34% protein decreases; Fig 1E). These results suggest that m6A modification of PTEN mRNA negatively regulates its RNA and protein expression levels.
The stability and translation of m6A modified cellular RNAs are regulated by the “reader proteins” (YTHDFs) (10). Therefore, we next investigated whether the m6A modified PTEN mRNA is recognized by YTHDFs. We performed immunoprecipitation experiments using cell lysates from the HepG2 cells in which FLAG-YTHDF1, -YTHDF2 or -YTHDF3 was ectopically expressed. Fig 1F, demonstrates that PTEN mRNAs were enriched in FLAG-YTHDFs immunoprecipitates samples compared to control. CREBBP (positive) and HPRT1 (negative) RNAs were analyzed as control (Fig 1G). The m6A modified PTEN mRNA level recognized by YTHDF3 was a > 4-fold increase compared to YTHDF1 and 2. This result is supported by the finding that before YTHDF1 and 2 bind to m6A modified mRNAs, the target mRNAs are first recognized by YTHDF3 (11).
After determining that the m6A machinery regulates levels of PTEN mRNA and protein, we next analyzed whether the PTEN RNA and protein levels could be similarly affected by the m6A reader proteins. In HepG2 cells transiently expressing the YTHDF2 or YTHDF3, decreased PTEN mRNA (48 ± 9.26% and 55 ± 2.84% of control, respectively) and protein levels (46 ± 6.55% and 44 ± 14.64% of control, respectively) were observed (Fig 1H), whereas knockdown of either YTHDF2 or YTHDF3 dramatically increased levels of PTEN mRNA (a >3-fold and a >2-fold increases of control, respectively) and protein (162 ± 12.55% and 154 ± 14.64% of control, respectively; Fig 1I). These results together, reveal that PTEN mRNA is m6A modified, and this m6A modification is induced by HBV. In particular, m6A modification of PTEN mRNA reduces its RNA and protein levels.
HBV downregulates PTEN mRNA stability
Having found that HBV regulates m6A modification of PTEN mRNA, we analyzed whether HBV affects PTEN mRNA stability. The m6A modified PTEN mRNA levels were dramatically induced in chronic HBV patient samples (339 ± 50.53%, 468 ± 60.33%, and 1066 ± 164.25% increases in liver biopsies HBV-1, HBV-2, and HBV-3, respectively from patients infected with HBV) and PHH infected with HBV (195 ± 12.43%; Fig 2A and C), while total PTEN mRNA expression levels were reduced (47 ± 2.91%, 46 ± 9.59%, and 86 ± 9.59% decreases; Fig 2B, 48 ± 3.89%; Fig 2D). We confirmed these results using hepatoma cell lines transiently expressing HBV 1.3-mer and HepAD38 cells stably expressing HBV. Decreased PTEN mRNA and protein levels were observed in HBV expressing cells (Fig 2E, 2F, and S1 fig). HBV expressing-induced HepAD38 cells elicited a decrease of 55 ± 3.2 PTEN mRNA level and a 57 ± 6.0% percent decrease in PTEN protein level. HBV 1.3-mer transfection decreased 54 ± 7.2% of PTEN mRNA and 52 ± 5.2 PTEN protein levels in HepG2 cells and 58 ± 5.5% of PTEN mRNA and 57 ± 7.8 PTEN protein levels in Huh7 cells. We then determined the half-life of PTEN mRNA in HepG2 cells transfected with the HBV 1.3-mer following Actinomycin D treatment. Fig 1G shows that transiently HBV expressing reduced the half-life of PTEN mRNA, from about 9.2 ± 0.98 h to 7.2 ± 0.82 h and PTEN protein expression levels were also decreased in HBV expressing cells at each time points, compared with control cells. These results suggest that HBV negatively regulates PTEN mRNA stability in both chronic HBV patients and cultured HBV expressing cells.
Fig 2.
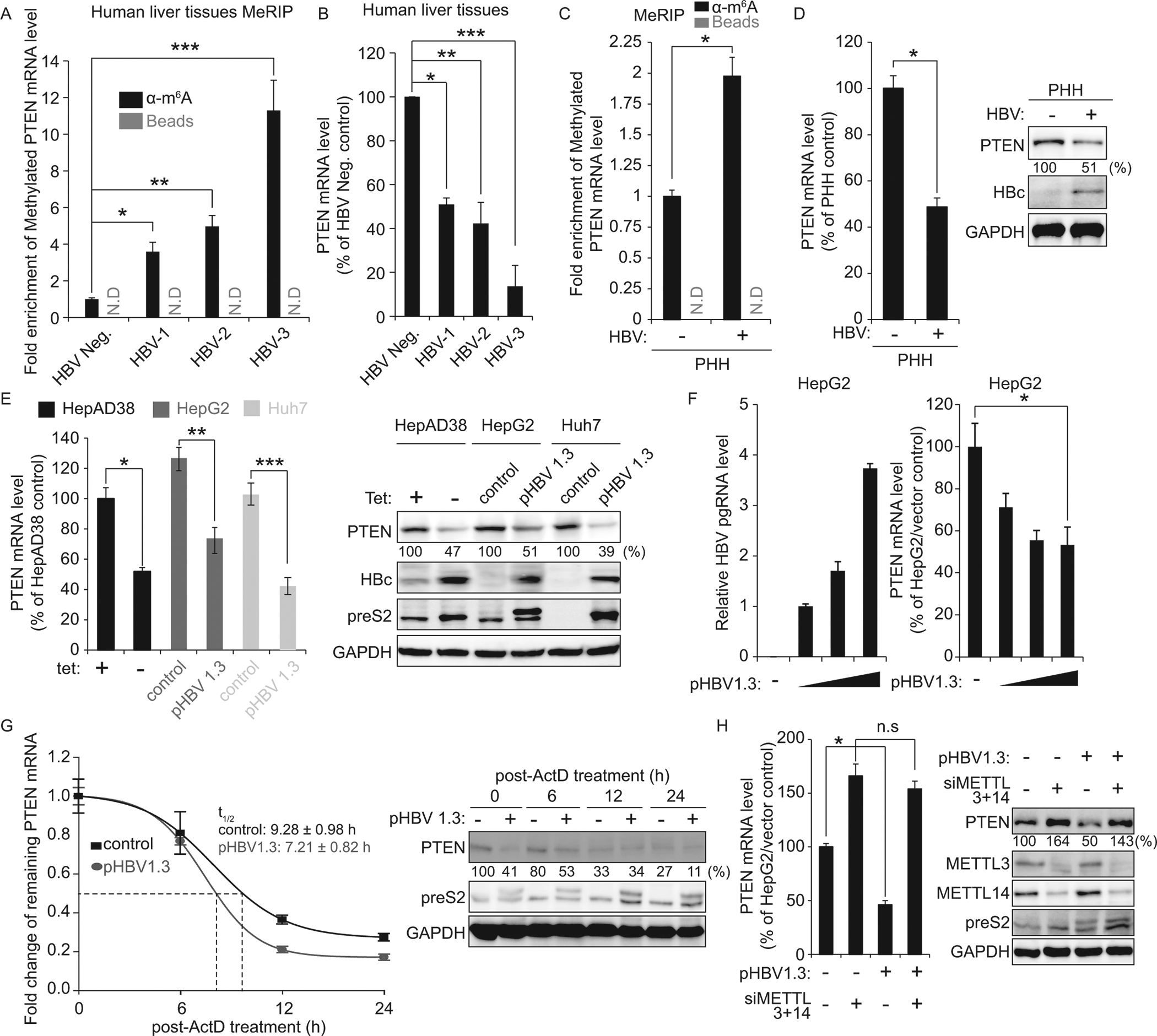
HBV reduces PTEN mRNA stability by regulating m6A modification. (A) MeRIP-qRT-PCR analysis of total RNA from chronic-HBV patients (HBV-1 to HBV-3) and healthy control (HBV-negative) using primers specific to PTEN mRNA. RNA was immunoprecipitated with anti-m6A antibody and eluted RNA was quantified by qRT-PCR. *P = 0.00051 ** P = 0.00043 *** P = 0.0011 (B) qRT-PCR quantification of PTEN mRNA levels normalized to GAPDH RNA in liver biopsies in (A). *P = 0.0036 ** P = 0.0061 *** P = 0.00027 (C and D) PHH were infected with HBV. After 72 h, PHH were harvested to assess expression levels of m6A modified PTEN mRNA, PTEN mRNA and protein (*P = 0.0047; C, *P = 0.0025; D). (E) Relative PTEN mRNA levels in HBV-induced HepAD38 and hepatoma cell lines (HepG2 and Huh7) transfected with HBV 1.3-mer plasmid at 2 days post-transfection. Shown in the right panels are the results of immunoblotting analysis for the indicated proteins in cell lysates. *P = 0.0044 **P = 0.0054 ***P = 0.0059 (F) HepG2 cells were transfected with increase amounts of HBV 1.3-mer (1, 5, and 10 μg). After 48 h, cells were harvested to assess expression levels of PTEN mRNA. *P = 0.00387 (G) qRT-PCR analysis of PTEN mRNA relative to GAPDH in the HBV 1.3-mer expressing HepG2 cells. The HBV 1.3-mer transfected HepG2 cells were treated with Actinomycin D at 24 h post-transfection. Cells were harvested at 0, 6, 12, and 24 h post Actinomycin D treatment and relative levels of remaining PTEN mRNA and proteins were analyzed. (H) The HBV 1.3-mer transfected HepG2 cells were depleted for METTL3 and 14 by siRNAs. After 48 h, total RNA and lysates were extracted to assess expression levels of PTEN mRNA and protein. *P = 0.0039 In (C – H), the error bars are the standard deviations of three independent experiments, each involving triplicate assays. P values were calculated using an unpaired t-test. n.s, not significant by unpaired Student’s t-test.
To verify whether HBV affects PTEN mRNA stability by increasing m6A modification of PTEN mRNA, we performed co-transfection with siMETTL3/14 and HBV 1.3-mer. Fig 2H demonstrates that HBV transfection destabilized PTEN mRNA, resulted in decreased PTEN protein level, but in the absence of METTL3/14, HBV did not affect PTEN mRNA and protein levels. Together, these results reveal that PTEN mRNA stability regulation is mediated through upregulating of m6A modification of PTEN mRNA by HBV.
HBV expression inhibits IRF-3 nuclear import, which specifically is regulated by PTEN protein expression
Recent studies have shown that IRF-3 has two phosphorylation sites. The S396 residue of IRF-3 is a positive phosphorylation site for IRF-3 activation, and the newly identified negative phosphorylation site S97 dictates IRF-3 nuclear import. PTEN can dephosphorylate IRF-3 S97 residue and facilitate its nuclear import for the IFN signaling pathway (7). Here, we sought to test the possibility that HBV affects PTEN protein expression to disrupt the IFN signaling pathway. First, we analyzed IFN-β mRNA levels in PTEN depleted cells. As expected, in the absence of PTEN, we observed that IFN-β mRNA level induced by poly (I:C) treatment was significantly reduced (a <4-fold decrease), and IRF-3 nuclear import was substantially inhibited (Fig 3 A and B). Next, we tested whether PTEN protein directly interacts with activated IRF-3. We performed co-immunoprecipitation experiments using cell lysates from the HepG2 cells in which FLAG-tagged PTEN protein was ectopically expressed. Fig 3C illustrates the ability of the PTEN protein to interact with activated IRF-3. Poly (I:C) treatment strongly induced the interaction between PTEN and phosphorylated IRF-3.
Fig 3.
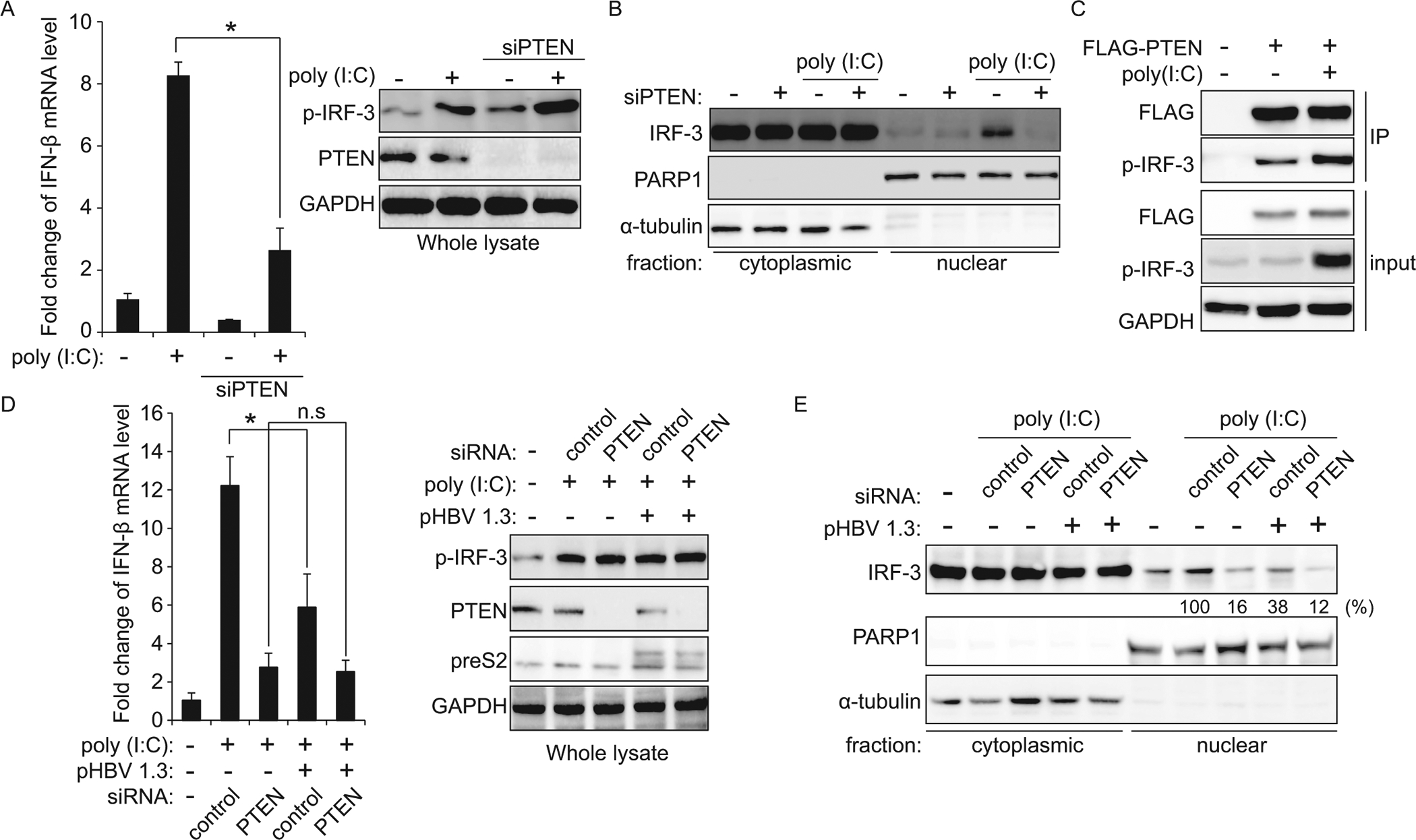
HBV inhibits IFN-β synthesis through regulating IRF-3 nuclear import. (A and B) HepG2 cells transfected with siPTEN or control siRNA were treated with poly (I:C). The IFN-β mRNA levels were determined by qRT-PCR at 12 h post poly (I:C) treatment, normalized with GAPDH. The p-IRF-3 protein levels were analyzed by immunoblotting in whole-cell lysates (*P = 0.00023; A). In (B), immunoblot analysis of isolated nuclear and cytoplasmic biochemical fractions from extracts of HepG2 cells co-transfected with siPTEN and poly (I:C). (C) HepG2 cells were transfected with FLAG-PTEN plasmid. After 36 h, cells were treated with poly (I:C) for 12 h. PTEN protein was immunoprecipitated using M2 FLAG antibody followed by immunoblotting with anti-p-IRF-3 and FLAG antibody. (D and E) HBV 1.3-mer expressing HepG2 cells were transfected with the control or PTEN specific siRNAs. After 36 h, the cells were stimulated with poly (I:C) for 12 h and cells were harvested to assess expression levels of IFN-β mRNA by qRT-PCR. Levels of p-IRF-3 and PTEN proteins were analyzed by immunoblotting. GAPDH was used as an internal control for loading (*P = 0.0088; D). In (E), the IRF-3 protein levels were determined by immunoblotting from isolated nuclear and cytoplasmic biochemical fractions (PARP1 – nuclear marker, α-tubulin – cytoplasmic marker). All experiments were performed in triplicate. Immunoblots shown are representative of three independent experiments. P values were calculated using an unpaired t-test. n.s, not significant by unpaired Student’s t-test.
After determining that PTEN protein regulates nuclear import of IRF-3, we next investigated whether HBV expression affects the IFN signaling pathway through inhibiting IRF-3 nuclear import by the downregulating PTEN mRNA stability. IFN-β mRNA level induced by poly (I:C) was decreased in HBV expressing cells (a <2-fold decrease; Fig 3D), but HBV failed to reduce IFN-β synthesis in PTEN depleted cells. The results presented in Fig 3D, indicate that IRF-3 nuclear import was inhibited by HBV expressing, while HBV did not affect IRF-3 nuclear translocation in PTEN depleted cells. The results demonstrate that HBV inhibits IRF-3 localization to the nucleus through destabilization of PTEN mRNA as a strategy of immune evasion.
HBV activates PI3K/AKT pathway through the upregulation of m6A modification of PTEN mRNA
It has been reported that PTEN inhibits cell survival and apoptosis signaling through blocking PI3K pathway leading to its inactivation enabling carcinogenesis (5). We sought to test the possibility that decreased PTEN protein expression by HBV affects liver tumorigenesis. Therefore, we analyzed PTEN mRNA and m6A modified PTEN mRNA levels in human liver biopsy specimens from a healthy individual (N-1), HBV-negative HCC patients (HCC-1 to HCC-3), and HBV-positive HCC patients (HCC-4 and HCC-5). PTEN mRNA expression levels were downregulated in both HBV-positive and negative HCC patients (34 ± 4.45%, 46 ± 2.55%, and 40 ± 4.92% decreases in liver biopsies HCC-1, HCC-2, and HCC-3, respectively from HCC patients; 64 ± 2.30% and 58 ± 7.82% decreases in liver biopsies HCC-4 and HCC-5, respectively from HBV-positive HCC patients; Fig 4A). In contrast, m6A methylated PTEN mRNA levels were significantly increased in all HCC patients samples (291 ± 4.51%, 376 ± 55.08%, and 289 ± 8.4% increases in liver biopsies HCC-1, HCC-2, and HCC-3; 441 ± 20.52% and 403 ± 26.48% increases in liver biopsies HCC-4 and HCC-5; Fig 4B). In particular, in HBV-positive HCC patients, the m6A modification of PTEN mRNA was on average about 28% higher than HBV-negative HCC patients. These results collectively indicate that decreased PTEN mRNA stability by m6A methylation is a critical factor in the development of HCC.
Fig 4.
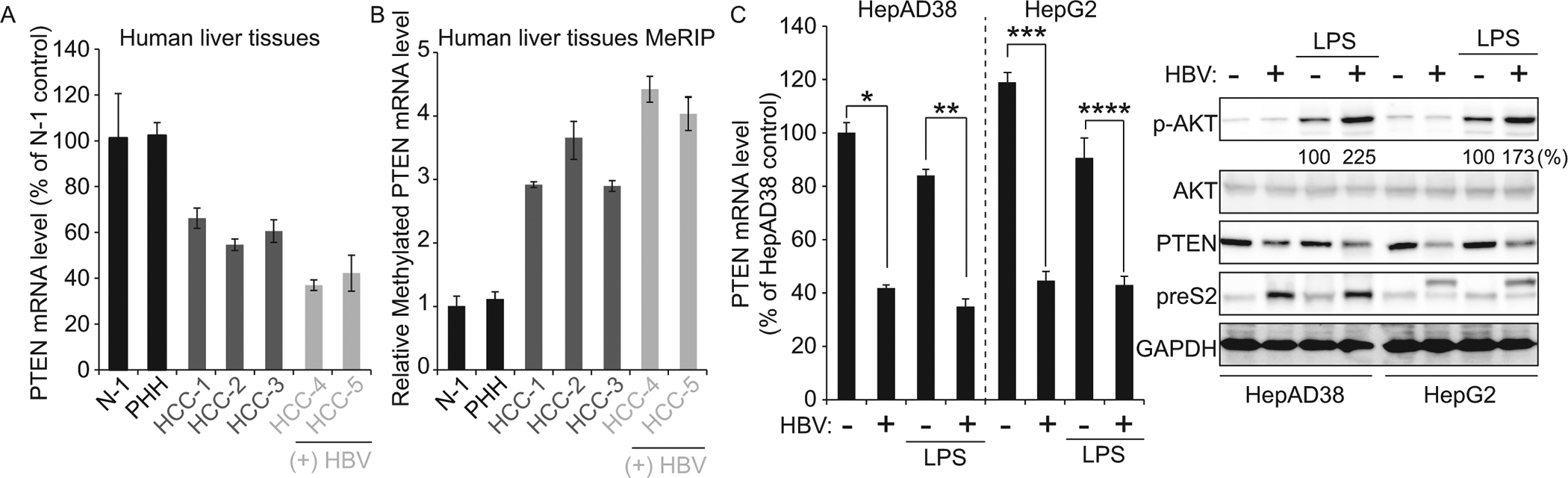
HBV activates PI3K/AKT pathway by regulating m6A modification on PTEN mRNA. (A) qRT-PCR quantification of PTEN mRNA levels normalized to GAPDH in liver biopsies from HBV-negative patients (HCC-1 to HCC-3) or HBV-positive (HCC-4 and HCC) HCC patients and healthy controls (N-1 and PHH). (B) MeRIP-qRT-PCR analysis of total RNA from liver biopsy samples in (A) using primers specific to PTEN mRNA. (C) HBV expressing HepG2 cells (HepAD38, HBV off and on, and transfected with HBV 1.3-mer) were treated with LPS. After 16 h post-treatment, the cells were harvested to assess expression levels of PTEN mRNA and indicated proteins by qRT-PCR and immunoblotting. *P = 0.00065 ** P = 0.00010 *** P = 0.0017 **** P = 0.00078
PI3K phosphorylates phosphatidylinositol-2, 4, 5,-triphosphate (PIP2), forming lipid-signaling second messenger PIP3 which in turn phosphorylates and activates AKT. PTEN disrupts the activity of PI3K by dephosphorylating PIP3 to generate PIP2 (6). To verify whether HBV affects PI3K activity by regulating PTEN expression, we checked phosphorylated AKT levels from HBV expressing cells (Fig 4C). Because p-AKT expression levels were low in HepAD38 cell lines and HepG2 cells, we treated HBV expressing cells with lipopolysaccharide (LPS), a PI3K activator. Interestingly, PI3K/AKT pathway was activated by HBV expression, suggesting that HBV activates PI3K pathway by decreasing PTEN expression levels, which acts as a negative regulator of PI3K pathway. These results imply that HBV infection can contribute to the development of HCC via the downregulation of PTEN mRNA stability.
Chronic HBV regulates m6A methylation of PTEN mRNA through inducing METTL3 expression
We investigated whether chronic HBV infection has any impact on host m6A machinery. We observed that METTL3 mRNA levels were increased in chronic HBV patient samples and PHH infected with HBV, whereas FTO mRNA levels were not changed (Fig 5A and B). METTL14 mRNA level was dramatically increased in HBV-3 sample, whereas increased METTL14 mRNA levels were not observed in samples from other HBV patients (HBV-1 and HBV-2) and HBV-infected PHH. These results correlated with Fig 2 A and B, which showed that m6A methylated PTEN mRNA was the highest in the HBV-3 patient. Because METTL14 mRNA expression level was regulated in only HBV-3 biopsy and this phenomenon was not observed in HBV-1 and -2 samples, suggesting that HBV infection regulates METTL3 expression, but METTL14 expression may be regulated by other factors.
Fig 5.
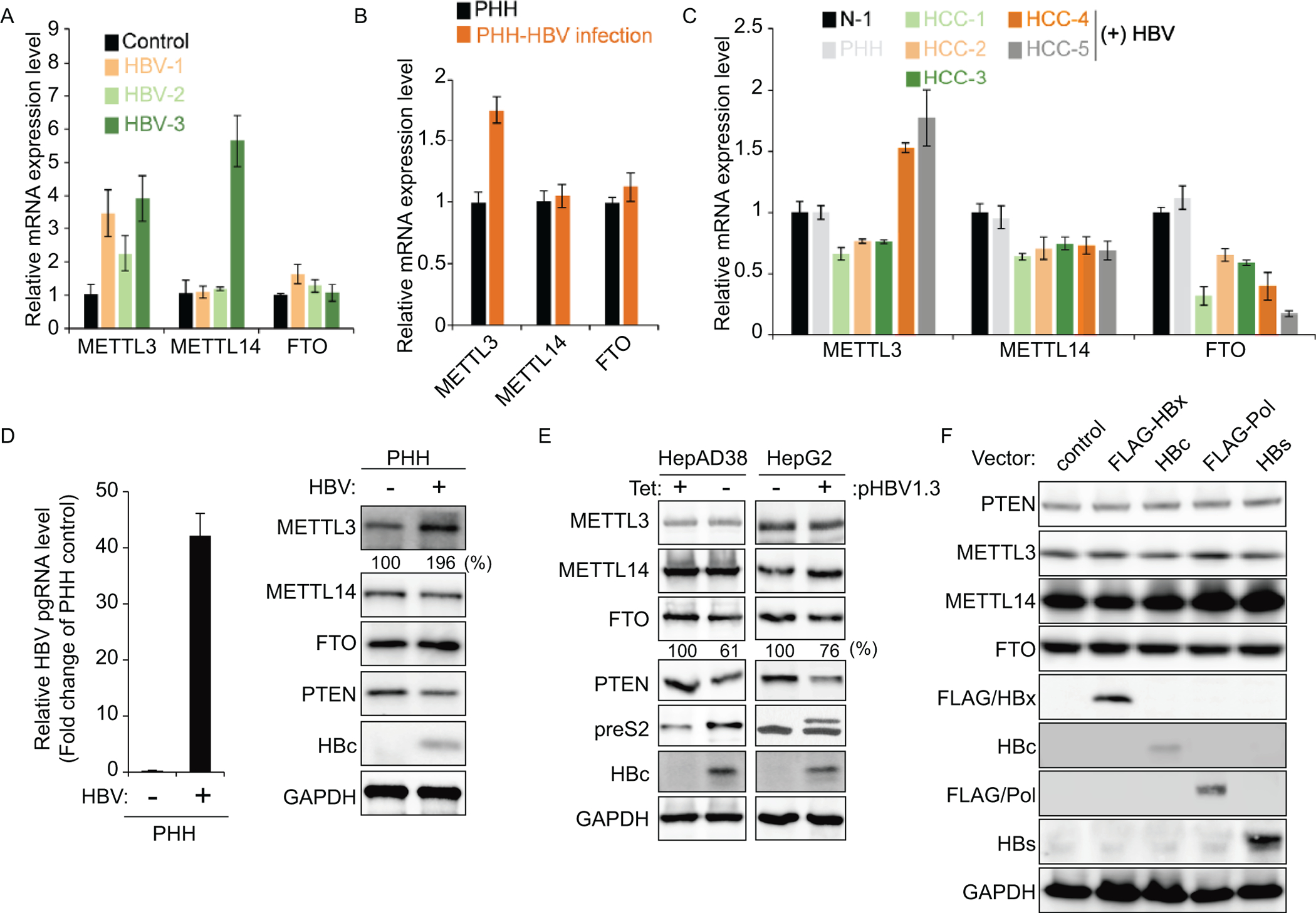
HBV infection induces METTL3 expression levels. (A – C) The m6A methyltransferase (METTL3/14) or demethylase (FTO) mRNA levels in liver biopsies from patients as described in Fig 2A, Fig 4A, and PHH infected with HBV. The METTL3, METTL14, and FTO mRNA levels were determined by qRT-PCR. (D) PHH were infected with HBV. After 72 h, PHH were harvested to assess expression levels of indicated proteins. (E) The indicated proteins were analyzed in lysates from HBV expressing HepG2 cells (HepAD38 and HBV 1.3-mer transfected cells). (F) The indicated proteins were analyzed in lysates from HepG2 cells that individually transfected with FLAG-HBx, HBc, FLAG-Pol and HBs.
In HBV-positive HCC samples, increased METTL3 mRNA expression levels were also observed, whereas in HBV-negative HCC patients, METTL3 was not induced (Fig 5C). The FTO expression levels were significantly reduced in both HBV-negative and positive HCC samples. Next, we confirmed m6A methyltransferases and demethylases protein expression levels in PHH infected with HBV and HBV expressing cell lines. In particular, METTL3 protein expression levels were induced only in HBV-infected PHH and not increased in HBV expressing hepatoma cell lines (Fig 5D and E). Decreased FTO protein expression levels were observed in HBV expressing cell lines, but not HBV-infected PHH. In HBV-infected PHH and chronic HBV patients (non-HCC), METTL3 expression was increased, while in liver cancer cell lines and HCC patients, FTO expression was decreased by HBV. These results suggest that HBV regulates host m6A machinery in different ways in HCC tissues/cell lines and non-HCC tissues/PHH, respectively.
To determine which HBV proteins affect host m6A machinery, we analyzed PTEN protein expression levels in cells expressing individual HBV genes (HBx, HBs, HBc, and HBV Pol; Fig 5F). PTEN protein expression levels were not altered in any of these HBV gene expressing cells. We also observed that FTO expression level was not affected by each HBV protein expression. These results suggest that m6A modification of PTEN might be regulated during HBV infection in a life cycle-dependent manner but not by any specific HBV gene product.
PTEN negatively regulates HBV replication
We investigated whether PTEN has any impact on HBV replication. HBV transcripts and protein levels were reduced in FLAG-PTEN plasmid-transfected cells (Fig 6A). Next, we assessed whether the silencing of PTEN can upregulate HBV replication, but we did not observe any changes in HBV pgRNA and protein levels in PTEN depleted cells (Fig 6B). In fact, HepG2 cell lines were established from hepatoblastoma cells. Thus, PTEN expression levels in HepG2 cell lines are expected to be lower compared to primary hepatocytes. As expected, we found that PTEN expression levels were dramatically decreased in HepG2 cells compared with PHH (S2 Fig). Therefore, we checked whether knockdown of PTEN in PHH affects HBV replication and found that HBV pgRNA and viral protein expression levels were significantly increased in PTEN depleted PHH (Fig 6C).
Fig 6.
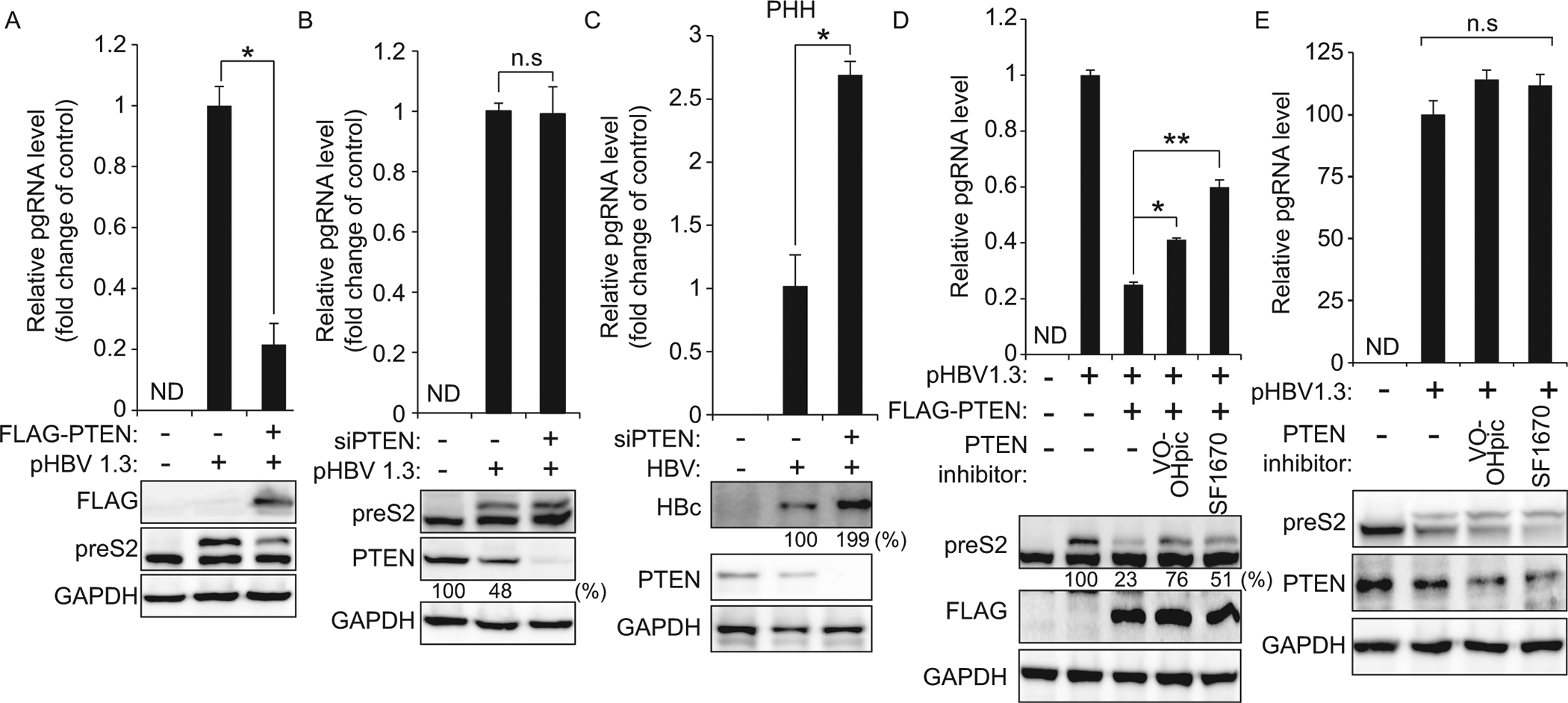
PTEN negatively regulates HBV replication and translation. (A) Relative HBV pgRNA level in HepG2 cells co-transfected with HBV 1.3-mer and FLAG-PTEN plasmid was determined 2 days after transfection. HBV preS2 protein level was analyzed by immunoblotting. *P = 0.00010 (B) Relative HBV pgRNA level in PTEN-silenced cells was analyzed by qRT-PCR at 48 h post-siRNA transfection. Levels of PTEN and HBV preS2 proteins were assessed by immunoblotting. (C) PTEN-silenced PHH were infected with HBV. After 72 h, total RNA and proteins were extracted to assess expression levels of HBV pgRNA and protein. *P = 0.0067 (D and E) HepG2 cells were co-transfected with the HBV 1.3-mer and FLAG-PTEN plasmid (D). HepG2 cells were transfected with HBV 1.3-mer (E). 12 h post-transfection, cells were treated with PTEN inhibitors (46 nM; VO-OHpic or 2 μM; SF1670) and 48 h later cells were harvested. HBV pgRNA level was analyzed by qRT-PCR and HBV preS2 protein level was analyzed by immunoblotting. *P = 0.00015 **P = 0.00026 n.s, not significant by unpaired Student’s t-test.
Next, we investigated whether the inhibitors of PTEN phosphatase activity affect HBV pgRNA and protein levels. We found that the treatment with PTEN inhibitors (VO-OHpic and SF1670) could rescue the inhibitory effects of ectopically PTEN protein expression on HBV replication (Fig 6D), whereas PTEN inhibitors did not affect the regulation of HBV replication and translation in un-transfected cells (Fig 6E). These results demonstrate that PTEN negatively regulates HBV replication, implying that HBV reduces PTEN expression to upregulate proviral activity.
Discussion
In this study, we examined the mechanism by which the cellular abundance of PTEN mRNA is downregulated by HBV infection. Diverse regulatory mechanisms, which include the transcriptional regulation and post-transcriptional turn over control of cellular mRNAs, might explain the differential expression of host mRNAs in HBV infected cells. The HBV affects m6A modification of multiple cellular mRNAs in addition to PTEN mRNA by increasing or decreasing the m6A modification (S2 Table and S3 Table). These cellular mRNAs did not have any conserved sequences, suggesting that the selection of the altered m6A modification of cellular mRNA by HBV lacks any specificity. In addition to the role of viral infection on changing host mRNA expression, the HBV is also known to be involved in the altering of miRNA expression (24). Because miR-21 is known to be induced by HBV (25) and miR-21 sequence has the DRACH motif, we analyzed whether HBV affects miR-21 expression through the regulation of m6A modification (S3 Fig). We found that miR-21 expression was increased in HBV transfected cells by reducing m6A modification. Thus, HBV-mediated change in miRNAs expression might be determined by m6A cellular machinery regulated by HBV. Interestingly, PTEN is the known target of miR-21 and it has been shown that miR-21 suppresses PTEN protein expression in several cancer cell lines (26). These results further suggest that HBV may regulate PTEN expression by altering the m6A modification of miR-21.
Because PTEN protein has a critical role in liver cancer by functioning as a tumor suppressor (5), we focused on PTEN among the m6A modified host mRNAs affected by HBV. We found that HBV reduced PTEN protein expression by increasing m6A methylation of PTEN mRNA (Fig 2 and 4). The tumor suppressor role of PTEN has been well characterized in cancer cells and PTEN knockout mice (6). Thus, our results suggest that HBV may contribute to the development of HCC by activating PI3K/AKT pathway, by decreasing PTEN mRNA stability. Decreased PTEN expression during the chronic HBV infection may in part explain the oncogenic phenotype previously observed in cell culture and human liver chimeric mice expressing HBV genes (27). Furthermore, the transgenic mice carrying the HBV gene and PTEN knockout mice are characterized by liver inflammation and fibrosis, followed by the development of HCC with age (5, 27), further supporting the role of the HBV infection in liver disease pathogenesis via its ability to causes PTEN mRNA destabilization by inducing increased m6A modification. In addition to liver cancer, PTEN expression levels are also reduced in various cancer tissues such as brain, breast, lung and prostate cancer, but it is not clear whether PTEN expression level reductions are mediated by m6A machinery in those cancer tissues (28). Thus, our results also imply that decreased demethylases (FTO) expression in various cancers may cause tumorigenesis by reducing PTEN expression (Fig 5C). It remains to be investigated whether the m6A modification of PTEN is regulated in several cancer tissues by decreasing FTO expression. If true, then induced m6A modification of PTEN by decreasing FTO expression can be a possible mechanism of reducing PTEN expression in cancer tissues. This hypothesis is supported by the finding that in several human cancer tissues, expression levels of m6A methyltransferases and demethylases are altered to regulate tumor cell proliferation, differentiation block, tumorigenesis, and metastasis (29).
Hepatitis C virus (HCV) infection is also associated with the risk of the development of HCC (30). Decreased PTEN expression in HCV-infected cells was also observed through the upregulation of m6A modification, but HIV, a non-oncogenic virus, did not affect m6A modification on PTEN (S4 fig), suggesting that both hepatitis viruses (HBV, HCV) induce HCC by a shared mechanism(s) of destabilizing PTEN mRNA via altering m6A modification. PTEN inhibition can be one of the mechanisms accelerating tumor development by chronic virus infection.
Given that HBV transfection altered cellular m6A profiling, the important question remains as to how HBV alters m6A modification of cellular RNAs. Interestingly, we found that HBV infection upregulates METTL3 expression level in chronic HBV patients, HBV-positive HCC patients, and PHH infected with HBV (Fig 5), raising the possibility that HBV-mediated liver tumors might be caused by the upregulation of METTL3 expression. Supporting this hypothesis, we analyzed METTL3 expression levels in HBV negative HCC patient samples and HepG2 transfected with HBV. METTL3 expression levels were not changed in these samples (Fig 5). Notably, decreased FTO expression was observed in both HBV-positive and negative HCC patients, but not in chronic HBV patients (non-HCC) and PHH infected with HBV. These results clearly suggest that HBV induces METTL3 expression in normal hepatocytes and promotes m6A modification of PTEN mRNA to reduce its RNA stability and affect protein levels, which could aid in the development of liver neoplasia.
Despite the activation of RIG-I signaling by pgRNA, little is known about the molecular mechanism explaining how HBV infection does not induce IFN-α/β. Our laboratory has previously shown proposed that mitochondrial dynamics is altered by HBV in which Parkin translocation to mitochondria triggers massive ubiquitination of mitochondrial antiviral signaling (MAVS) protein thus clipping downstream IFN signaling (31, 32). In addition to its role as a tumor suppressor, the PTEN protein is also known to be involved in innate immune response activated by viral infections (7). The function of PTEN in innate immunity is well characterized in virus-infected cells. PTEN causes p-IRF-3 nuclear translocation by dephosphorylation at IRF-3 Ser97 residue, to activate the IFN signaling pathway. Thus, our observations explain that the reduced PTEN protein expression during HBV infection causes inhibition of p-IRF-3 nuclear import, which leads to the disruption of the IFN signaling pathway (Fig 3). HBV-induced elimination of PTEN can be one of the mechanisms of HBV immune evasion. Furthermore, over-expression of PTEN substantially decreased HBV replication and the negative effect of PTEN on HBV replication was rescued by inhibitors of PTEN phosphatase activity (Fig 6). These results together suggest that HBV reduces PTEN expression to evade innate immunity and positively regulates the viral life cycle.
In summary, our results unravel novel mechanisms by which HBV controls host gene expression to maintain persistent infection via regulating host m6A modification machinery. HBV infection suppresses innate immunity by the effects of PTEN in the IFN signaling pathway. Finally, the HBV-induced m6A modification of PTEN leading to its reduced levels of expression may contribute to the virus-associated hepatocarcinogenesis. Further, this work expands the role of m6A modification in HCC.
Supplementary Material
Financial Supports
Research is supported by an NIH grants AI125350 and AI139234 to A.S.
List of Abbreviations
- m6A
N6 methyladenosine
- PTEN
phosphatase and tensin homolog
- HCC
hepatocellular carcinoma
- PI3K
phosphatidylinositol-3-kinase
- HBV
hepatitis B virus
- pgRNA
pregenome RNA
- PIP2
phosphatidylinositol-2, 4, 5,-triphosphate
- PIP3
phosphatidylinositol -3, 4, 5-triphosphate
- YTHDF
YTH-domain family protein
- MeRIP
Methylated RNA immunoprecipitation
- UTR
untranslated region
- FTO
fat mass and obesity-associated protein
- IFN
interferon
- LPS
lipopolysaccharide
- HCV
hepatitis C virus
- MAVS
mitochondrial antiviral signaling protein
- PHH
primary human hepatocytes
References
- 1.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet 2018;391:1301–1314. [DOI] [PubMed] [Google Scholar]
- 2.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000;64:51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu J, Protzer U, Siddiqui A. Revisiting Hepatitis B Virus: Challenges of Curative Therapies. J Virol 2019;93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.An P, Xu J, Yu Y, Winkler CA. Host and Viral Genetic Variation in HBV-Related Hepatocellular Carcinoma. Front Genet 2018;9:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen CY, Chen J, He L, Stiles BL. PTEN: Tumor Suppressor and Metabolic Regulator. Front Endocrinol (Lausanne) 2018;9:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carnero A, Paramio JM. The PTEN/PI3K/AKT Pathway in vivo, Cancer Mouse Models. Front Oncol 2014;4:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li S, Zhu MZ, Pan RG, Fang T, Cao YY, Chen SL, Zhao XL, et al. The tumor suppressor PTEN has a critical role in antiviral innate immunity. Nature Immunology 2016;17:241–249. [DOI] [PubMed] [Google Scholar]
- 8.Lamontagne J, Mell JC, Bouchard MJ. Transcriptome-Wide Analysis of Hepatitis B Virus-Mediated Changes to Normal Hepatocyte Gene Expression. PLoS Pathog 2016;12:e1005438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim GW, Lee SH, Cho H, Kim M, Shin EC, Oh JW. Hepatitis C Virus Core Protein Promotes miR-122 Destabilization by Inhibiting GLD-2. PLoS Pathog 2016;12:e1005714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev 2015;29:1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res 2017;27:315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lichinchi G, Gao S, Saletore Y, Gonzalez GM, Bansal V, Wang Y, Mason CE, et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat Microbiol 2016;1:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzales-van Horn SR, Sarnow P. Making the Mark: The Role of Adenosine Modifications in the Life Cycle of RNA Viruses. Cell Host Microbe 2017;21:661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imam H, Khan M, Gokhale NS, McIntyre ABR, Kim GW, Jang JY, Kim SJ, et al. N6-methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc Natl Acad Sci U S A 2018;115:8829–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gokhale NS, McIntyre ABR, McFadden MJ, Roder AE, Kennedy EM, Gandara JA, Hopcraft SE, et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016;20:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kennedy EM, Bogerd HP, Kornepati AVR, Kang D, Ghoshal D, Marshall JB, Poling BC, et al. Posttranscriptional m(6)A Editing of HIV-1 mRNAs Enhances Viral Gene Expression. Cell Host & Microbe 2016;19:675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hesser CR, Karijolich J, Dominissini D, He C, Glaunsinger BA. N6-methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog 2018;14:e1006995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imam H, Kim GW, Mir SA, Khan M, Siddiqui A. Interferon-stimulated gene 20 (ISG20) selectively degrades N6-methyladenosine modified Hepatitis B Virus transcripts. PLoS Pathog 2020;16:e1008338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu YJ, Nie H, Mao RC, Mitra B, Cai DW, Yan R, Guo JT, et al. Interferon-inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem-loop structure of viral RNA. Plos Pathogens 2017;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, et al. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrobial Agents and Chemotherapy 1997;41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog 2013;9:e1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012;485:201–206. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014;505:117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamontagne J, Steel LF, Bouchard MJ. Hepatitis B virus and microRNAs: Complex interactions affecting hepatitis B virus replication and hepatitis B virus-associated diseases. World J Gastroenterol 2015;21:7375–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou ZH, Quan J. Hepatitis B virus X protein increases microRNA-21 expression and accelerates the development of hepatoma via the phosphatase and tensin homolog/phosphoinositide 3-kinase/protein kinase B signaling pathway. Molecular Medicine Reports 2017;15:3285–3291. [DOI] [PubMed] [Google Scholar]
- 26.Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007;133:647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Y, Chen WL, Louie SG, Yen TS, Ou JH. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology 2007;45:16–21. [DOI] [PubMed] [Google Scholar]
- 28.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer 2011;11:289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, Liu T. The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res 2019;79:1285–1292. [DOI] [PubMed] [Google Scholar]
- 30.Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 1998;4:1065–1067. [DOI] [PubMed] [Google Scholar]
- 31.Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, Watanabe T, et al. The RNA Sensor RIG-I Dually Functions as an Innate Sensor and Direct Antiviral Factor for Hepatitis B Virus. Immunity 2015;42:123–132. [DOI] [PubMed] [Google Scholar]
- 32.Khan M, Syed GH, Kim SJ, Siddiqui A. Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. Plos Pathogens 2016;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.