

SUMMARY
The ubiquitin system regulates the DNA damage response (DDR) by modifying histone H2A at Lys15 (H2AK15ub) and triggering downstream signaling events. Here, we find that phosphorylation of ubiquitin at Thr12 (pUbT12) controls the DDR by inhibiting the function of 53BP1, a key factor for DNA double-strand break repair by non-homologous end joining (NHEJ). Detectable as a chromatin modification on H2AK15ub, pUbT12 accumulates in nuclear foci and is increased upon DNA damage. Mutating Thr12 prevents the removal of ubiquitin from H2AK15ub by USP51 deubiquitinating enzyme, leading to a pronounced accumulation of ubiquitinated chromatin. Chromatin modified by pUbT12 is inaccessible to 53BP1 but permissive to the homologous recombination (HR) proteins RNF169, RAD51, and the BRCA1/BARD1 complex. Phosphorylation of ubiquitin at Thr12 in the chromatin context is a new histone mark, H2AK15pUbT12, that regulates the DDR by hampering the activity of 53BP1 at damaged chromosomes.
Graphical Abstract

In Brief
Modification of ubiquitin by phosphorylation is a new and largely unexplored concept in the field. Walser et al. found that Thr12 ubiquitin phosphorylation at histone H2AK15 regulates the DNA damage response by inhibiting 53BP1-mediated DNA repair. This study highlights new levels of complexity in the crosstalks among histone modifications.
INTRODUCTION
Ubiquitination regulates most cellular processes by controlling the stability, activity, and localization of many proteins through its degradative and signaling functions (Komander and Rape, 2012). Among all pathways regulated by ubiquitin, the DNA damage response (DDR) requires the highest levels of modulation to enable prompt activation/inactivation, signal amplification, and restrictions to defined subcellular compartments (Schwertman et al., 2016). The DDR has been best characterized in response to DNA double-strand breaks (DSBs), which elicit a signaling cascade triggered by phosphorylation of the histone variant H2A.X (referred to as γH2A.X), resulting in ubiquitination of his-tone H2A Lys13 and Lys15 (H2AK13ub and H2AK15ub) mediated by the ubiquitin ligase RNF168 (Gatti et al., 2012; Mattiroli et al., 2012). RNF168 is essential for activation of downstream DNA damage signaling and DNA repair. H2AK15ub is selectively recognized by 53BP1 (Fradet-Turcotte et al., 2013), which accumulates in nuclear foci and facilitates the recruitment of other proteins that restrict DNA end resection at DNA break sites, thereby favoring DNA repair by non-homologous end joining (NHEJ) over homologous recombination (HR) (Panier and Boulton, 2014). While HR is inhibited in the G1 phase of the cell cycle, when no sister chromatids are available, both pathways are active during S and G2 phases. However, it is still unclear how NHEJ is actively restricted in S and G2 phases to allow DNA end resection and the initiation of repair via HR.
In recent years, different proteomic studies have revealed that ubiquitin itself can be modified by small chemical groups, like phosphate (Bennetzen et al., 2010; Kettenbach et al., 2011; Lee et al., 2009; Zhou et al., 2013), further expanding the complex picture of its regulatory roles (Swatek and Komander, 2016), though it is not clear which of these modifications are functionally significant. Even though the discovery of ubiquitin phosphorylation has caused tremendous excitement, until now, only ubiquitin phosphorylation at Ser65 has been functionally characterized and shown to regulate the activity of Parkin E3 ubiquitin ligase and mitochondrial homeostasis (Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014; Wauer et al., 2015a).
In the present study, we investigated the role of ubiquitin phosphorylation in the context of chromatin ubiquitination and identified ubiquitin Thr12 as a novel player of the DDR signaling cascade. We discovered that phosphorylation of ubiquitin Thr12 in chromatin (i.e., H2AK15pUbT12) prevents the recruitment of 53BP1 to damaged chromatin by interfering directly with 53BP1 binding to the nucleosome. 53BP1 exclusion from chromatin caused by 53BP1 phosphorylation that blocks its recognition of H2AK15ub has previously been shown to attenuate the DDR and prevent chromosomal rearrangements during mitosis (Lee et al., 2014; Orthwein et al., 2014). Our data uncover an additional level of regulation of the DDR via formation of chromatin regions modified by pUbT12 and inaccessible to 53BP1 but permissive to RAD51 and BRCA1/BARD1, revealing phospho-ubiquitin as a novel signaling event in genome stability.
RESULTS
UbT12 Regulates the Formation of 53BP1 Foci
To unveil the possible role of ubiquitin phosphorylation in the DDR, we examined the known phosphorylation sites of ubiquitin, previously identified by mass spectrometry (Bennetzen et al., 2010; Kettenbach et al., 2011; Lee et al., 2009; Zhou et al., 2013), and tested the ability of phosphorylation-deficient forms to promote chromatin ubiquitination within the framework of the DNA damage signaling cascade. We found that while most of the ubiquitin mutants behave like wild-type (WT) ubiquitin, the mutation of Thr12 into Ala (UbT12A) consistently increased the level of chromatin ubiquitination (Figure 1A). This effect does not reflect an uneven expression of exogenous versus endogenous ubiquitin forms (Figure S1A). Next, we assessed whether UbT12A-induced chromatin ubiquitination is functional for DDR activation by evaluating the formation of 53BP1 foci. We observed that UbT12A expression in human osteosarcoma (U2OS) cells promotes accumulation of 53BP1 foci in most unperturbed cells, as measured by quantitative imaging (Figures 1B–1D). This effect cannot be explained by subtle differences in expression of WT versus T12A mutant or in the cell-cycle distribution (Figures S1B and S1C). We further confirmed this result using a ubiquitin replacement strategy in U2OS cells (Xu et al., 2009), wherein all endogenous copies of ubiquitin are depleted by doxycycline (dox)-inducible RNAi while simultaneously expressing short hairpin RNA (shRNA)-resistant hemagglutinin (HA)-tagged WT ubiquitin and UbT12A (Figures S1D–S1H). Importantly, 53BP1 accumulation in foci is not due to increased DNA damage, as demonstrated by similar levels of γH2A.X staining in cells expressing exogenous WT ubiquitin and UbT12A (Figure 1E), and is strictly dependent on the hydrophobic patch of ubiquitin, centered on Ile44, which is responsible for most ubiquitin functions in the cell (Figure 1F).
Figure 1. UbT12 Regulates 53BP1 Foci Formation, which Depends on RNF8 and RNF168 and Is Counteracted by RNF169.
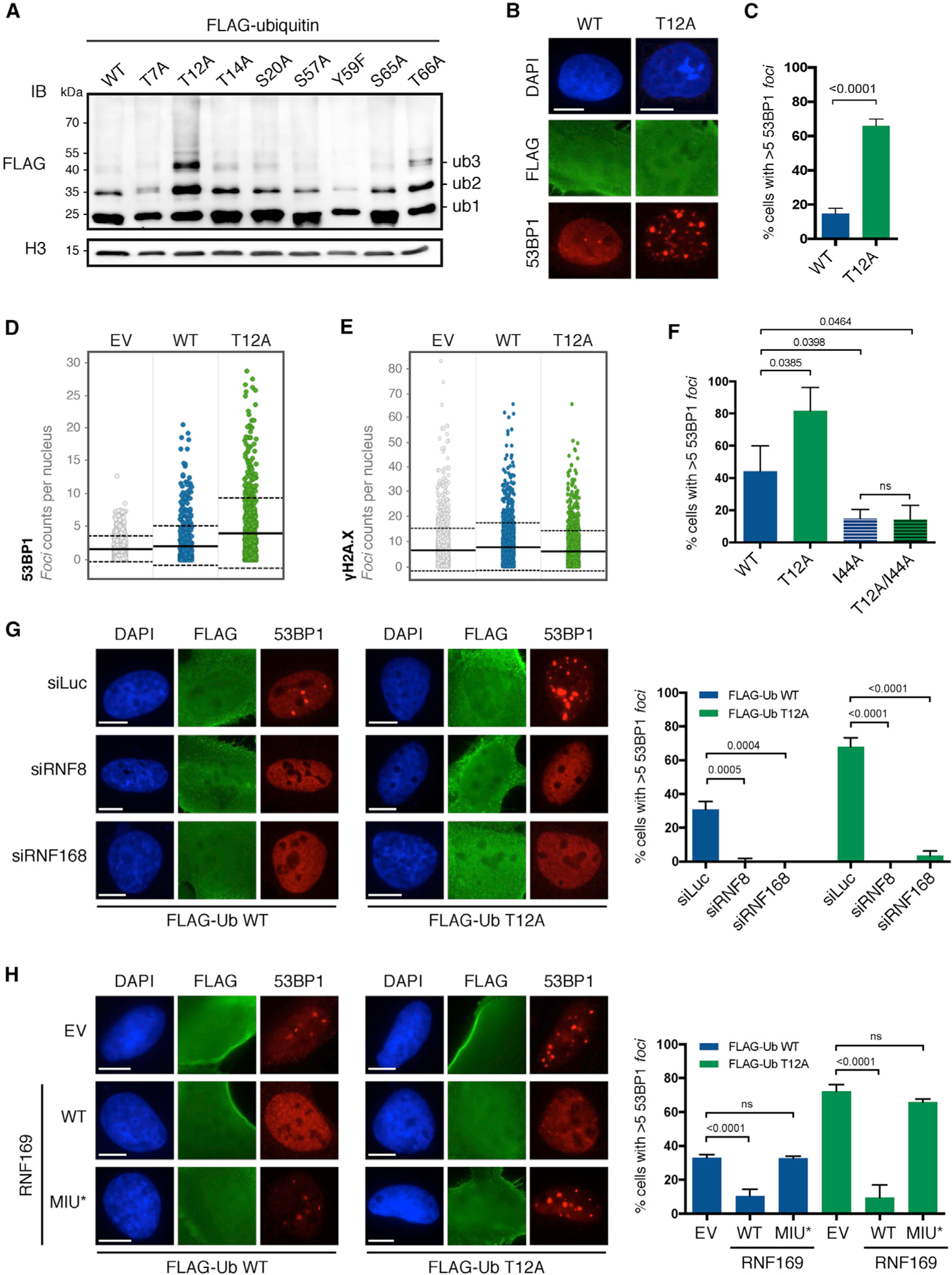
(A) Chromatin ubiquitination in HEK293T cells upon 24-h overexpression of the indicated FLAG-ubiquitin wild-type (WT) and point mutants. After acidic extraction, chromatin fractions were analyzed by FLAG and H3 immunoblot (IB). Ubiquitinated forms of histones are indicated (ub1, ub2, and ub3).
(B and C) 53BP1 foci formation in untreated U2OS cells after 24-h transfection of FLAG-ubiquitin WT or T12A and immunostained for FLAG, 53BP1, and DAPI. Representative images (B) and quantification of cells with more than five 53BP1 foci (C) in FLAG-positive cells are shown. Scale bars, 10 μm (B). At least 50 cells per condition were counted in each replicate, and data are represented as mean + SD (n = 3).
(D and E) Quantitative image-based cytometry (QIBC) shows the distribution of 53BP1 foci (D) and γH2A.X foci (E) in FLAG-positive cells transfected and stained as in (B). EV, empty vector. For each condition, images containing at least 1,000 cells per experiment were acquired (n = 3). Mean (solid line) and SD from the mean (dashed lines) are indicated.
(F–H) Quantification of cells with more than five 53BP1 foci in FLAG-positive U2OS cells transfected for 24 h with indicated FLAG-ubiquitin mutants (n = 3) (F) upon 72-h knockdown of RNF8 and RNF168 (siRNF8 and siRNF168; n = 3) (G) and upon 48-h overexpression of RNF169 WT or MIU-defective mutant (MIU*; n = 3) (H). Cells were immunostained for FLAG, 53BP1, and DAPI. At least 50 cells per condition were counted, and data are represented as mean + SD. Scale bars, 10 μm.
See also Figure S1.
UbT12A-Dependent Accumulation of 53BP1 Foci Depends on RNF8 and RNF168 and Is Counteracted by RNF169
53BP1 recruitment to chromatin is highly regulated, requiring the physical interaction of the TUDOR domain and ubiquitin-dependent recognition (UDR) motif of 53BP1 with di-methylated Lys20 of histone H4 (H4K20me2) (Botuyan et al., 2006; Pei et al., 2011) and H2AK15ub (Fradet-Turcotte et al., 2013; Hu et al., 2017; Wilson et al., 2016), respectively. To assess whether UbT12A is able to target H2AK15, as suggested by 53BP1 foci accumulation, we queried the ubiquitination status of histone H2A carrying the K118Q and K119Q amino acid substitutions. These substitutions prevent extensive C-terminal ubiquitination of histone H2A expected to also occur with UbT12A and allow detection of the markedly less abundant ubiquitination of Lys13 and Lys15 at the N terminus of H2A. Although essential for the activation of the DDR signaling cascade, ubiquitination of Lys13 and Lys15 is barely detectable even upon DNA damage (Gatti et al., 2012). Expression of UbT12A enhances the ubiquitination of histone H2A at Lys13 and Lys15 compared to expression of WT ubiquitin (as observed in the K118/119Q H2A mutant; Figure S1I). Expectedly, mutations of both C- and N-terminal sites (i.e., K13,15,118,119Q; 4K/Q) abolish H2A ubiquitination. UbT12A therefore accumulates detectably at H2AK13/K15. Since ubiquitination of H2AK13/K15 strictly depends on RNF168, we also tested the effect of depleting RNF168, or the upstream E3 ubiquitin ligase RNF8, epistatic to RNF168, on 53BP1 localization. Depletion of either ligase prevents 53BP1 chromatin recruitment induced by UbT12A, similarly to WT ubiquitin (Figures 1G and S1J), indicating that UbT12A conjugation to H2A is dependent on the canonical RNF8/RNF168 pathway. Moreover, overexpression of the DDR factor RNF169, a potent competitor of 53BP1 for binding to H2AK15ub (Hu et al., 2017; Poulsen et al., 2012), inhibits 53BP1 foci formation in cells expressing WT ubiquitin or UbT12A and is dependent on the integrity of RNF169 ubiquitin-binding domain (UBD) MIU (motif interacting with ubiquitin; Penengo et al., 2006; Poulsen et al., 2012) (Figure 1H), further confirming that ubiquitination with UbT12A occurs at H2AK15. Taken together these data indicate that ubiquitin Thr12 plays a regulatory role in chromatin ubiquitination and that its substitution to Ala leads to extensive chromatin ubiquitination and consequent accumulation of 53BP1 foci in the absence of exogenous DNA damage.
UbT12 Is Phosphorylated in Chromatin Extracts and Accumulates in Nuclear Foci
Based on our results above and previous proteomic studies detecting phosphorylation of ubiquitin Thr12 (referred to as pUbT12) in cells (Bennetzen et al., 2010; Lee et al., 2009; Zhou et al., 2013), we addressed the potential function of pUbT12 in the DDR. We adopted a phage display strategy to develop pUbT12-specific human monoclonal antibodies (Silacci et al., 2005) (clone 2.B5; herein referred to as hu-pUbT12) (Figure S2A). In parallel, we obtained standard rabbit polyclonal antibodies (herein referred to as rb-pUbT12). The specificity and sensitivity of both antibodies were extensively validated by enzyme-linked immunosorbent assay (ELISA) and biochemistry (Figures 2A and S2B–S2D) using pUbT12 protein that we produced by linear solid-phase peptide synthesis (El Oualid et al., 2010) and recombinant ubiquitin mutants expressed in E. coli. With these tools, we could visualize ubiquitin phosphorylation at Thr12 in HEK293T and U2OS cell extracts, mainly in chromatin fractions (S3 fractions in Figures S2E–S2G). The pUbT12 antibody detection patterns had bands of different sizes, raising the issue of specificity of these antibodies in complex samples as opposed to purified proteins. For specificity assessment, we treated chromatin extracts with calf intestinal phosphatase (CIP) and lambda phosphatase and observed a dramatic reduction of pUbT12 signals. The patterns corresponding to ubiquitin conjugates did not change (Figures S2E–S2H), indicating that most of the signals detected with pUbT12 antibodies depend on phosphorylation. Moreover, we performed additional control assays with the recently characterized leader protease Lbpro of foot-and-mouth disease virus, which efficiently cleaves ubiquitin (Swatek et al., 2019). We first verified that Lbpro is also active on ubiquitin when phosphorylated at Thr12 by incubating Lbpro with the histone H2A-H2B dimer (Hu et al., 2017) modified at H2AK15 by pUbT12 using in vitro ubiquitination (Figure S2I; see STAR Methods for details). Then, we incubated Lbpro with chromatin extracts from cells and found that most of pUbT12 signals, as well as ubiquitin signals, were cleared in Lbpro-treated samples (Figure S2J). These results highlight the specificity of the pUbT12 signals detected in these samples and reveal that there are different species of pUbT12 conjugates on chromatin. Among the species recognized by pUbT12 antibodies, there is one sensitive to phosphatase and Lbpro treatment and detectable at a size corresponding to the mono-ubiquitinated form of H2A (Figure 2B). Importantly, we directly determined that pUbT12 is present at H2AK13/K15 by immunoprecipitation of FLAG-H2A from cells followed by pUbT12 immunoblot (Figure 2C). We detected a clear pUbT12 signal corresponding to the mono-ubiquitinated form of H2A. This signal is highly reduced when H2A is mutated at the N-terminal ubiquitination sites (K13,15Q) and completely absent when the N- and C-terminal H2A ubiquitination sites are mutated (K13,15,118,119Q). This result strongly indicates that phosphorylation of ubiquitin Thr12 occurs at the N-terminal ubiquitination site of histone H2A, defining a new histone mark, i.e., H2AK15pUbT12.
Figure 2. UbT12 Is Phosphorylated in Chromatin Extracts at H2AK15ub and Accumulates in Nuclear Foci.
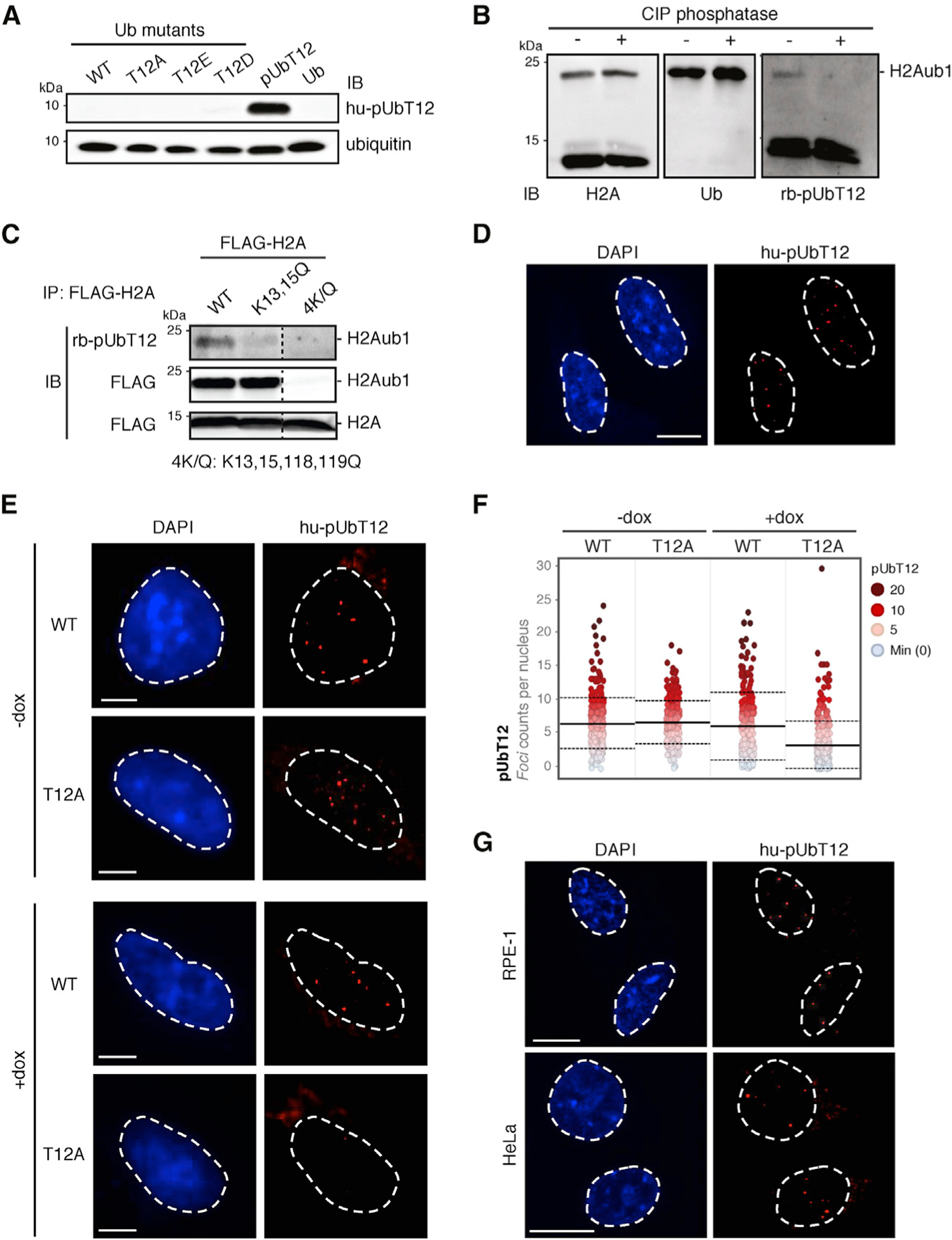
(A) Validation of pUbT12-specific antibody immunoglobulin G1 (IgG1) (clone 2.B5, referred as hupUbT12) by IB on chemically synthesized full-length pUbT12 and recombinant ubiquitin (Ub) mutants expressed in E. coli, as indicated.
(B) pUbT12 was detected in chromatin fraction of HEK293T cells by pUbT12 IB (H2Aub1; rb-pUbT12). H2A and ubiquitin IB are used as reference. Fractions were treated with calf intestinal phosphatase (CIP) to assess the specificity of the signal.
(C) Chromatin fractions of HEK293T cells transfected with FLAG-H2A WT and the indicated mutants were immunoprecipitated with FLAG antibody. pUbT12 and FLAG signals were detected by IB, and irrelevant lanes were digitally removed from the blot image (dashed line). Mono-ubiquitinated (H2Aub1) and unmodified H2A are indicated.
(D) Confocal analysis of nuclear pUbT12 foci in untreated U2OS cells stained with hu-pUbT12 and DAPI. Scale bar, 10 μm.
(E and F) pUbT12 foci in U2OS WT and T12A ubiquitin replacement cells, after 4 days of doxycycline (dox) induction, were analyzed by hu-pUbT12 and DAPI staining. Representative images (E) and quantification of nuclear pUbT12 foci (F) are shown. Scale bars, 5 μm (E). Each dot (F) represents a single cell color-coded according to pUbT12 foci number. For each condition, images containing at least 150 cells per experiment were acquired (n = 3). Mean (solid line) and SD from the mean (dashed lines) are indicated.
(G) Confocal analysis of nuclear pUbT12 foci in untreated RPE-1 and HeLa cells stained with hupUbT12 and DAPI. Scale bars, 10 μm.
See also Figure S2.
Next, we performed immunofluorescence analysis in unperturbed U2OS cells, which showed that pUbT12 localizes to discrete nuclear foci (Figure 2D), suggesting that ubiquitin phosphorylation occurs at defined chromatin sites. The specificity of nuclear pUbT12 immunofluorescence staining was assessed by quantitative microscopy in cells expressing either WT or T12A ubiquitin (Figures 2E and 2F) or upon competition with an excess of pUbT12 peptide (Figure S2K) and by co-staining with the rbpUbT12 antibody (Figure S2L). The presence of pUbT12-positive nuclear foci is also detectable in retinal epithelial cells (RPE-1) and cervical cancer cells (HeLa; Figure 2G), indicating that pUbT12 is a widespread modification occurring in different cell types.
pUbT12 Can Be Conjugated to Nucleosomes but Cannot be Cleaved by the Deubiquitinating Enzyme USP51
We addressed the molecular details of the conjugation and deconjugation of pUbT12. It was previously shown that phosphorylation at Ser65 can inhibit ubiquitin recognition by E3 ubiquitin ligases and deubiquitinating enzymes (Wauer et al., 2015b). To test whether alterations of ubiquitin Thr12, by phosphorylation or mutations, affect ubiquitin conjugation mediated by E1, E2, and E3 enzymes, we performed in vitro ubiquitination assays using the previously described H2A-H2B covalently fused dimer as a substrate (Hu et al., 2017). Interestingly, RNF168 efficiently conjugates pUbT12 (Figure 3A), as well as the phospho-inhibitory and phospho-mimetic mutants of ubiquitin Thr12 (i.e., T12A, T12D, and T12E), to H2A-H2B (Figure 3B). This result demonstrates that alteration of Thr12 does not affect chromatin ubiquitination catalyzed by RNF168. Next, we evaluated the effect of overexpressing the ubiquitin Thr12 mutants on chromatin ubiquitination in cells in the presence of endogenous RNF168 (Figure 3C). Overexpression of RNF168 was used as a reference for increased level of chromatin ubiquitination (RNF168 O/E +). We observed that not only UbT12A but also the phosphomimetic mutant UbT12E, and to a lesser extent UbT12D, increase in vivo chromatin ubiquitination compared to WT ubiquitin. These findings unveil a new regulatory mechanism occurring in cells that is associated with a dramatic accumulation of chromatin ubiquitination when ubiquitin Thr12 is mutated.
Figure 3. pUbT12 Can Be Conjugated to Nucleosomes but Cannot be Cleaved by the Deubiquitinating Enzyme USP51.
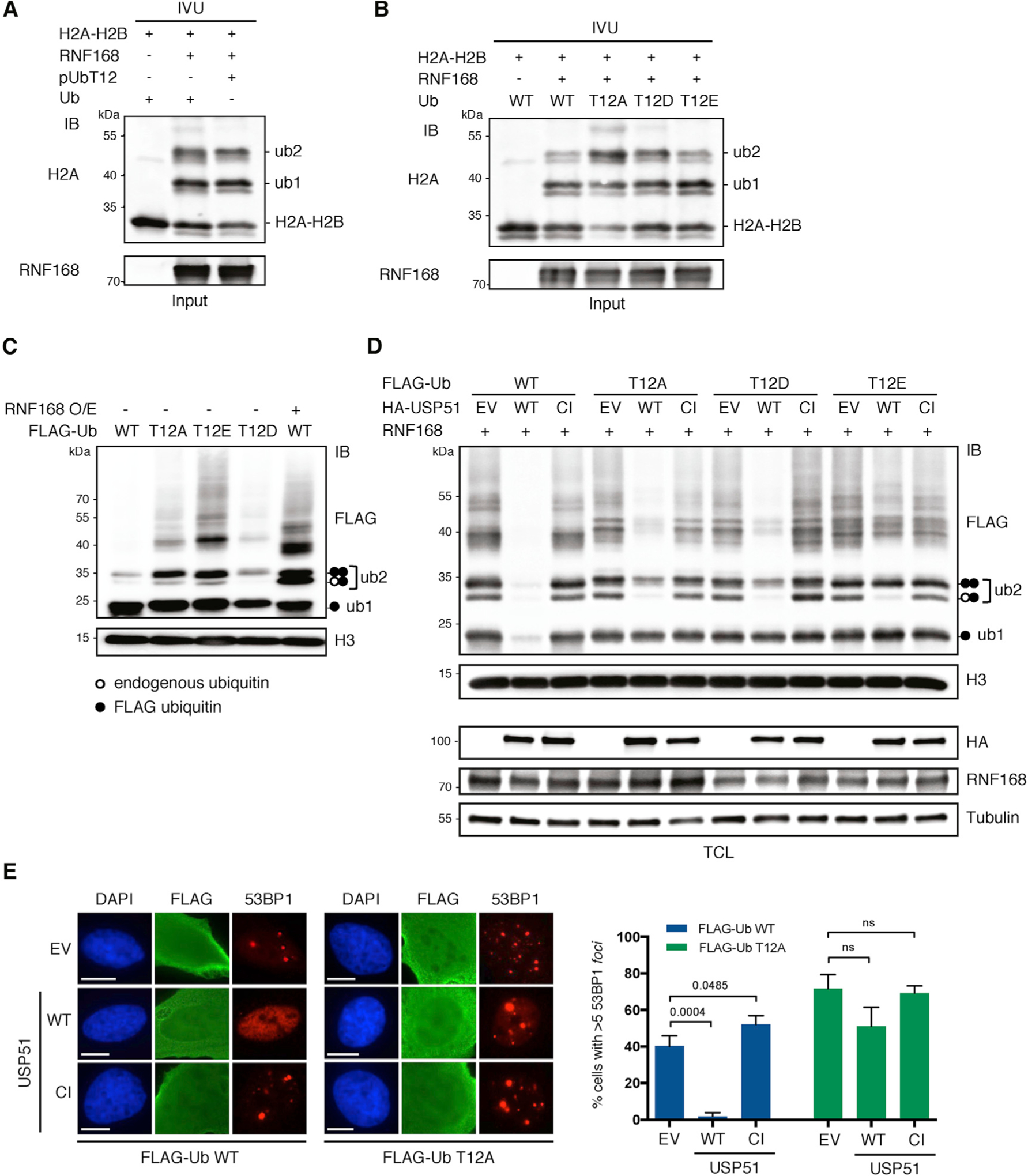
(A and B) In vitro ubiquitination assay (IVU) using purified E1, E2 (UbcH5c), E3 (GST-RNF168), and H2A-H2B dimer, together with ubiquitin (Ub), chemically synthetized pUbT12 (A) or bacterially expressed ubiquitin mutants (B), as indicated. Ubiquitinated H2A-H2B was detected by IB using anti-H2A antibody; unmodified, mono- and di-ubiquitinated forms of H2A-H2B dimer are indicated (H2A-H2B, ub1, and ub2).
(C) Chromatin ubiquitination in HEK293T cells upon 24-h overexpression of the indicated FLAG-ubiquitin forms together with RNF168 (RNF168 O/E; +) or the empty vector (−). After acidic extraction, chromatin fractions were analyzed by FLAG and H3 IB. Ubiquitinated forms of histones, modified by endogenous (white circle) or exogenous (FLAG tagged; black circle) are indicated (ub1 and ub2).
(D) IB of HEK293T chromatin extracts upon 48-h overexpression of RNF168 and the indicated FLAG-ubiquitin forms, together with USP51 WT or the catalytic inactive (CI) mutant. TCL, total cell lysate; EV, empty vector.
(E) 53BP1 foci formation in untreated U2OS cells upon 24-h overexpression of FLAG-ubiquitin WT or T12A and USP51 WT or CI mutant, stained for FLAG, 53BP1, and DAPI. Representative images (left) and quantification of cells with more than five 53BP1 foci (right) in FLAG-positive cells are shown. Scale bars, 10 μm. At least 50 cells per condition were counted in each replicate, and data are represented as mean + SD (n = 3).
See also Figure S3.
It was recently demonstrated that the deubiquitinating enzyme USP51 removes ubiquitin from the H2AK15 site, counteracting RNF168 activity (Ai et al., 2019; Wang et al., 2016). We therefore asked whether the accumulation of chromatin ubiquitination observed upon expression of the ubiquitin Thr12 mutants is due to impaired cleavage by USP51. First, we confirmed that WT USP51 efficiently clears chromatin ubiquitination, both in the context of endogenous RNF168 (Figure S3A) and upon its ectopic expression (Figure 3D). We also verified that ubiquitin cleavage depends on the integrity of USP51 catalytic domain (compare empty vector [EV], WT and catalytic inactive [CI] in FLAG-Ub WT). Strikingly, we found that the activity of USP51 on ubiquitinated chromatin is highly reduced in cells expressing any of the Thr12 ubiquitin mutants, as indicated by the high levels of mono-ubiquitinated (ub1) and di-ubiquitinated (ub2) histones in Thr12 mutants compared to WT ubiquitin (Figures 3D and S3A). Of note, the endogenous ubiquitin detected at the size of di-ubiquitinated histones (ub2, white and black circles)is efficiently cleaved byUSP51, while the FLAG-ubiquitin (ub2, black circles) is only removed when WT ubiquitin, but not the Thr12 mutants, is expressed. Also, impairment of USP51 activity was clearly observed on ubiquitination of H2AK13/K15, as revealed by the levels of mono- and diubiquitination of HA-tagged histone H2A mutated at C-terminal sites (HA-H2A K118,119Q; Figure S3B). In accordance with these findings, overexpression of USP51 inhibits the chromatin recruitment of 53BP1 in cells expressing WT ubiquitin, but not UbT12A (Figure 3E). Taken together, these data demonstrate that pUbT12 does not markedly affect the ubiquitination activity of RNF168. Rather, increased chromatin ubiquitination observed upon expression of the UbT12 mutants is likely due to defective cleavage by USP51.
pUbT12 Is Induced by DNA Damage in an RNF168-Dependent Manner
Being linked to the RNF8/RNF168/53BP1 pathway, we asked whether ubiquitin phosphorylation at Thr12 is fostered by DNA damage. After induction of DSBs by treatment with topoisomerase-2 inhibitor etoposide (eto) or ionizing radiation (IR), there was a significant increase in the number of nuclear pUbT12 foci, which quickly decreased to basal level after removal of DNA damage (Figures 4A, 4B, and S4A). Importantly, pUbT12 foci are quantitatively comparable to H2AK15ub foci in both untreated and etoposide-treated cells (Figure 4C). Quantitative imaging of cells labeled with the nucleotide analog 5-ethynyl-2-deoxyuridine (EdU) showed that the number of pUbT12-positive foci increases throughout the cell cycle (Figures S4B and S4C). This observation indicates that pUbT12, and likely the histone mark H2AK15pUbT12, are not diluted during DNA replication, as in the case of other, non-DNA-damage-induced histone marks required for 53BP1 recruitment (e.g., H4K20me2) (Pellegrino et al., 2017; Saredi et al., 2016), but are rather actively promoted during S and G2 phases of the cell cycle, similarly to the DDR markers γH2A.X and H2AK15ub (Pellegrino et al., 2017).
Figure 4. pUbT12 Is Induced by DNA Damage and Depends on RNF168.
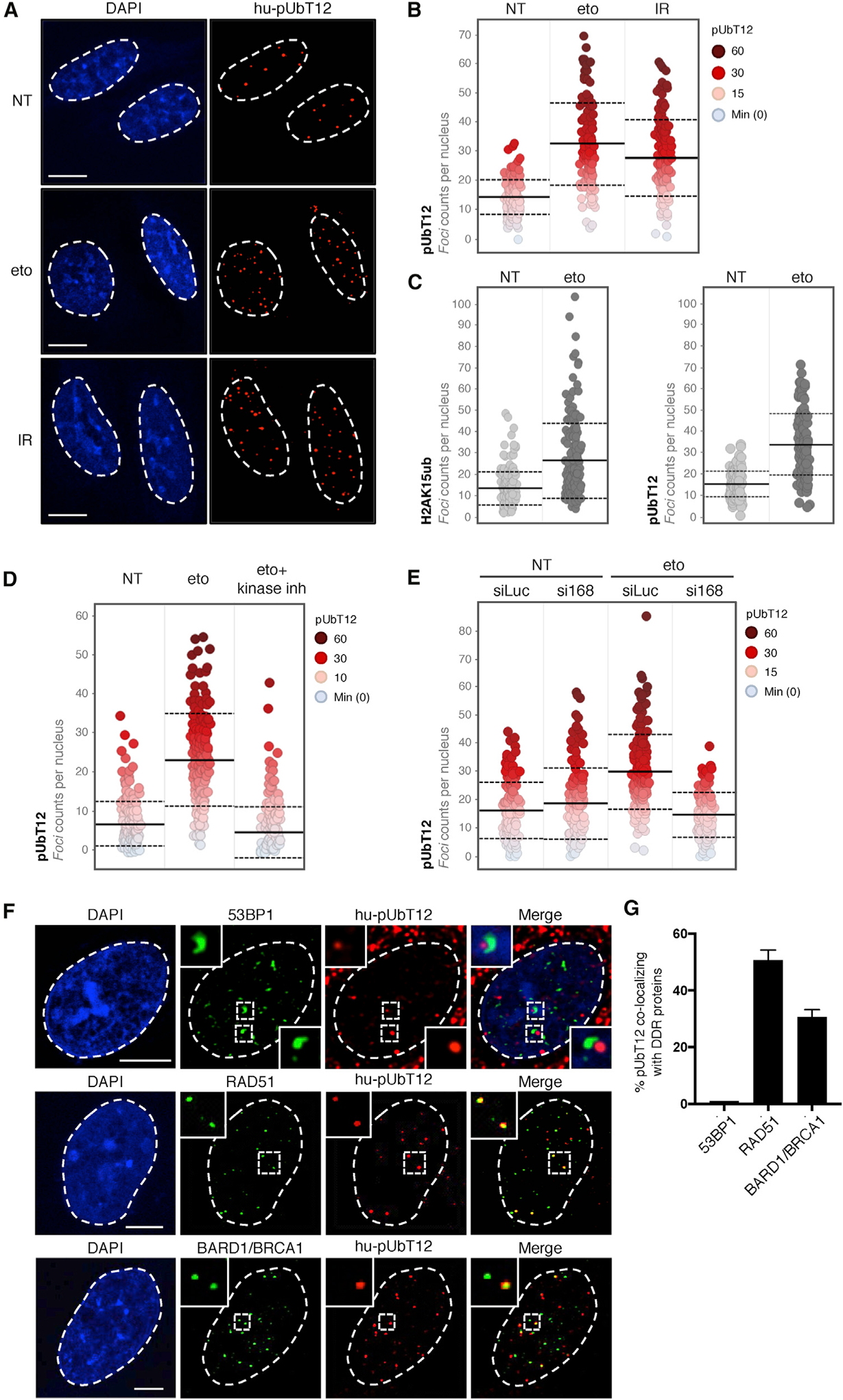
(A and B) Confocal analysis (A) and quantification (B) showing the distribution of nuclear pUbT12 foci in U2OS cells treated for 1 h with etoposide (eto; 5 μM), irradiated (IR; 1 Gy), or left untreated (NT). Each dot (B) represents a single cell color-coded according to pUbT12 foci number. Scale bars, 10 μm. For each condition, images containing at least 150 cells per experiment were acquired (n = 3). Mean (solid line) and SD from the mean (dashed lines) are indicated.
(C) Quantification of nuclear H2AK15ub and pUbT12 stained foci in U2OS cells treated for 1 h with 5 μM etoposide or left untreated. Each dot represents a single cell, and images containing at least 150 cells per condition were acquired. Mean (solid line) and SD from the mean (dashed lines) are indicated.
(D) Quantification of nuclear pUbT12 foci in U2OS cells treated for 1 h with 5 μM etoposide, left untreated, or pretreated with kinase inhibitors (ATM inhibitor KU-55933, 10 μM; ATR inhibitor VE-821, 10 μM; and DNA-PK inhibitor KU-57788, 2 μM) for 2 h before etoposide treatment. Each dot represents a single cell color-coded according to pUbT12 foci number. For each condition, images containing at least 150 cells per experiment were acquired (n = 3). Mean (solid line) and standard deviation from the mean (dashed lines) are indicated.
(E) Quantification showing the distribution of nuclear pUbT12 foci in U2OS cells treated for 1 h with 5 μM etoposide after 96-h depletion of RNF168 (si168). siLuc served as a control. Each dot represents a single cell color-coded according to pUbT12 foci number. For each condition, images containing at least 150 cells per experiment were acquired (n = 3). Mean (solid line) and SD from the mean (dashed lines) are indicated.
(F and G) Confocal analysis of co-localization between pUbT12-positive foci and 53BP1, RAD51, or BARD1 in U2OS cells treated for 1 h with etoposide (5 μM).
Representative images (F) and quantification of colocalizing signals (G) are shown. Scale bars, 5 μm. At least 250 pUbT12-positive foci per condition were counted in each replicate (n = 3).
See also Figure S4.
Next, we addressed whether pUbT12 can be modulated by interfering with the activity of key factors of DDR. Therefore, we treated U2OS cells with inhibitors of the DDR apical kinases, namely ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3-related protein (ATR), and DNA-dependent protein kinase (DNA-PK), prior to DNA damage induction. Interestingly, the etoposide-induced pUbT12 foci were abolished by inhibition of the DDR kinases (Figure 4D), as well as by depletion of RNF168, which is responsible for the generation of the H2AK15ub histone mark (Figures 4E and S4D–S4H), further corroborating the notion that ubiquitination of the H2AK15 site is required for the occurrence of UbT12 phosphorylation induced by DNA damage.
To gain additional insight into the role of ubiquitin phosphorylation in the DDR, we probed the co-localization of pUbT12 with different DDR markers by confocal microscopy. Due to the harsh procedure required for pUbT12 staining, several markers accumulating at DNA damage sites, such as γH2A.X and H2AK15ub, cannot be monitored. Nevertheless, we were able to perform co-localization studies with 53BP1 and key factors of the HR-mediated DNA repair, the recombinase RAD51 and the BRCA1/BARD1 complex (detected by BARD1 immunostaining; see Figures S4I and S4J). Strikingly, we found that in cells treated with etoposide, pUbT12 and 53BP1 signals are mutually exclusive, with almost no pUbT12-positive foci co-localizing with 53BP1. Conversely, pUbT12 is permissive for the localization of RAD51 and BRCA1/BARD1, showing 50% and 30%, respectively, of pUbT12 foci that are positive for these HR factors (Figures 4F and 4G). These data suggest the intriguing possibility that pUbT12 specifically counteracts 53BP1 recruitment to damaged chromatin, while other DNA repair factors are unaffected.
The pUbT12 Modification Limits Recruitment and Function of 53BP1 to Damaged Chromatin
The mutually exclusive nature of pUbT12 and 53BP1 foci may imply that chromatin modified by pUbT12 is a physical block to 53BP1 recruitment to DSBs. Consistent with this hypothesis, the cryoelectron microscopy structure of 53BP1 in complex with the nucleosome core particle (NCP) carrying H2AK15ub (NCP-H2AK15ub) suggests that phosphorylation of UbT12 would interfere with 53BP1 binding (Figure 5A). Indeed, Asp1620 in the UDR motif of 53BP1, an important residue for the interaction with ubiquitin attached to H2AK15ub (Wilson et al., 2016), points toward Thr12 (Figure 5A). Hence, phosphorylation of Thr12 is expected to prevent the 53BP1/NCPH2AK15ub interaction. To experimentally test this possibility, we generated NCP-H2AK15ub, which is also di-methylated on H4K20 to allow the interaction with the TUDOR domain, using either WT ubiquitin or pUbT12 (Figure S5A), and performed a glutathione S-transferase (GST) pull-down assay with a TUDOR-UDR 53BP1 construct (GST-53BP1 in Figure 5B). While GST-53BP1 interacted with NCP-H2AK15ub harboring WT ubiquitin, binding was completely abolished when pUbT12 was used to generate NCP-H2AK15ub (Figure 5B, Ub versus pUb). The inhibition of 53BP1/NCP-H2AK15ub interaction by pUbT12 parallels our in vivo observation that chromatin marked by pUbT12 hinders 53BP1 chromatin localization. Interestingly, and in line with our prediction based on the RNF169/NCP-H2AK15ub structural model (Hu et al., 2017), the interaction between RNF169 and NCP-H2AK15ub was retained even in the presence of pUbT12 (Figure 5C), suggesting a specific inhibitory function for pUbT12 on 53BP1 binding.
Figure 5. pUbT12 Limits Recruitment and Function of 53BP1 to Damaged Chromatin.
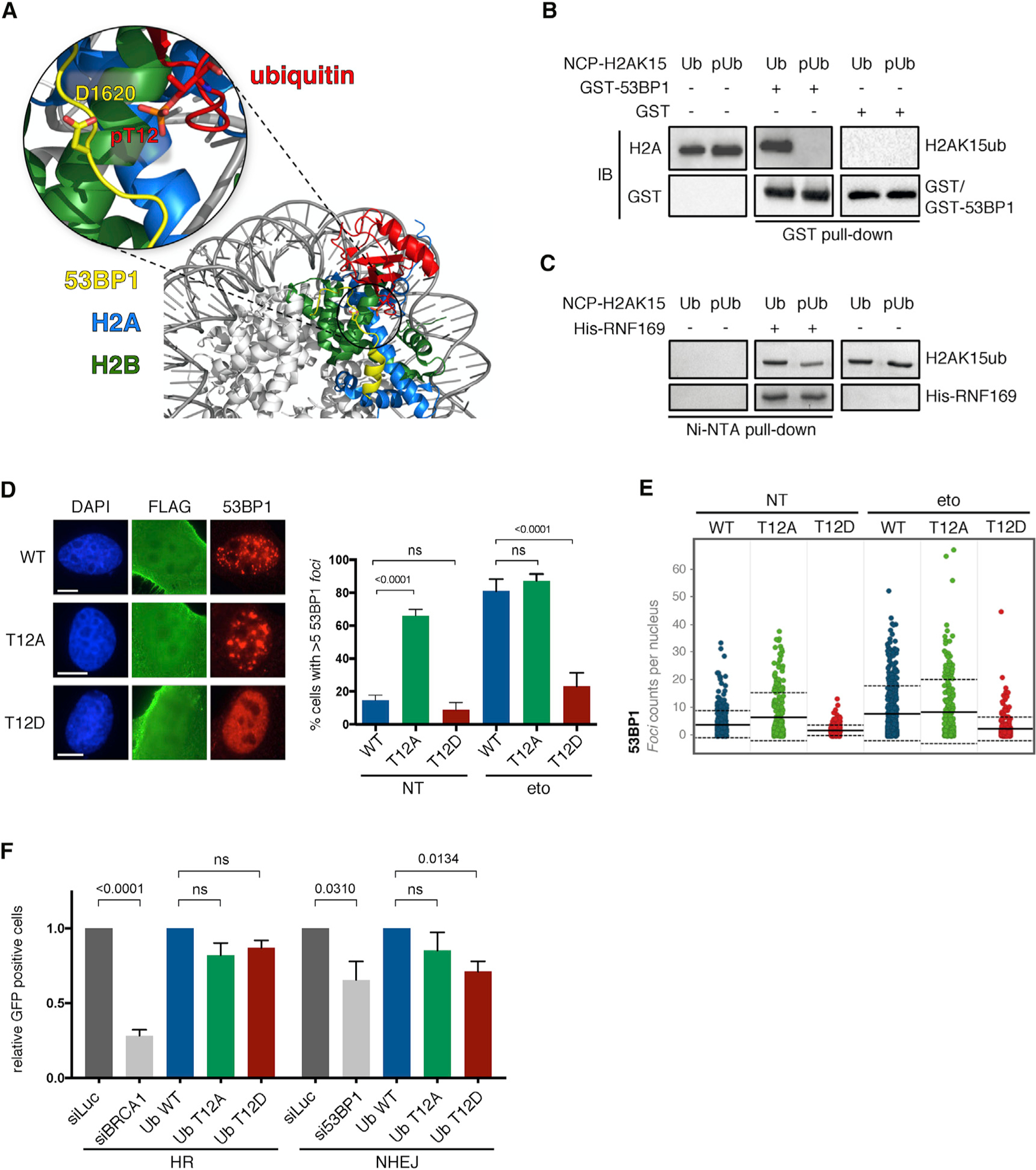
(A) Structural prediction of the interference of ubiquitin phosphorylation at Thr12 in the binding of 53BP1 with the nucleosome, using a cryoelectron microscopy model (NCP-H2AK15ub-53BP1).
(B) H2A and GST IB of GST pull-down assays using GST or GST-53BP1 with nucleosomes modified on H2AK15 by ubiquitin (Ub) or pUbT12 (pUb).
(C) Coomassie blue staining of Ni-NTA pull-down assays with NCP-H2AK15ub using Ub or pUbT12 and His-RNF169.
(D) 53BP1 foci formation in untreated (NT) and etoposide (eto) treated (1 h, 5 μM) U2OS cells upon 48-h FLAG-ubiquitin overexpression and 18-h ubiquitin knockdown immunostained for FLAG, 53BP1, and DAPI. Representative images (left) and quantification of cells with more than five 53BP1 foci (right) in FLAG-positive cells are shown (n = 3). At least 50 cells per condition were counted in each replicate, and data are represented as mean + SD (n = 3). Scale bars, 10 μm.
(E) Nuclear 53BP1 foci analyzed by high-content microscopy in cells as in (D). For each condition, images containing at least 1,000 cells per experiment were acquired (n = 3). Mean (solid line) and SD from the mean (dashed lines) are indicated.
(F) U2OS reporter cells, stably integrated with DR-GFP or EJ5-GFP to measure HR and NHEJ, respectively, were transfected with I-SceI and transduced with lentivirus (MOI = 1) for the indicated ubiquitin forms. For knockdown of BRCA1 (siBRCA1), 53BP1 (si53BP1), and control (siLuc), cells were transfected twice (72 h and 24 h before I-SceI transfection) with small interfering RNA (siRNA). After 72 h of I-SceI overexpression, GFP-positive cells were analyzed by FACS, and relative changes to ubiquitin WT or siLuc were calculated (n = 3). Data are represented as mean + SEM.
See also Figures S5 and S6.
Finally, we reckoned that if 53BP1 is unable to bind ubiquitinated NCPs when modified by pUbT12, mimicking pUbT12 in cells should prevent 53BP1 localization to damaged chromatin, ultimately affecting its activity. We therefore investigated the effect of the phospho-mimetic mutants T12D and T12E on 53BP1 localization upon induction of DNA damage in the presence or absence of endogenous ubiquitin. We found that expression of UbT12D largely prevents accumulation of 53BP1 to chromatin upon treatment with etoposide (Figures 5D, 5E, S5B, and S5C) in both the presence and absence of endogenous ubiquitin (Figure S5D, siLuc and siUb). Similar results were obtained by expressing the UbT12E phospho-mimetic mutant upon ubiquitin depletion, although it did not show a dominant effect when endogenous ubiquitin was present (Figure S5D). These results indicate that 53BP1 recruitment to damaged chromatin is strongly impaired by mimicking phosphorylation of ubiquitin Thr12.
We note that ubiquitin Thr12 is an essential residue for the vegetative growth of yeast cells (Sloper-Mould et al., 2001), making it likely that mutating Thr12 would interfere with mammalian cell physiology. It is therefore technically very challenging to properly address the function of Thr12 by mutagenesis in biological assays that typically last several days. Furthermore, Thr12 mutation leads to accumulation of USP51-resistant chromatin ubiquitination (Figures 3D, 3F, S3A, and S3B), which may have unpredictable effects on cell physiology. In an attempt to gain insights into the biological role of pUbT12, we manipulated the ubiquitin replacement system, as in Figure S1D, to express the different UbT12 mutants, and we probed cell viability over time. Expression of all ubiquitin mutants showed growth defects already after 4 days of dox induction. Longer treatment revealed high toxicity even upon complementation with WT ubiquitin (Figure S5E), likely due to the inability of the system to efficiently replace the pool of intracellular ubiquitin. Seeking alternative approaches to overcome this technical limitation, we developed a lentiviral-based system that expresses the different ubiquitin forms (Figure S6A) without depletion of endogenous ubiquitin. With this system, at early time points after viral transduction (3 days), cells expressing UbT12 mutants exhibited viability comparable to that of WT ubiquitin, although prolonged expression of the mutants (7 and 10 days) showed high toxicity, which is extreme with the T12E mutant (Figure S6B). We therefore used this system to examine how pUbT12 affects the activity of 53BP1. As a readout of 53BP1 functionality, we first measured the ability of cells to repair DSBs via HR and NHEJ by using an established I-SceI-based DNA repair reporter assay (Gunn and Stark, 2012). We found that in line with the impairment of 53BP1 chromatin recruitment observed upon expression of the phospho-mimetic mutants (Figures 5D and S5D), expression of UbT12D reduces the ability of cells to repair DSBs via NHEJ, while HR-mediated repair was not significantly affected (Figures 5F, S6C, and S6D). One of the hallmark of 53BP1 loss is the restoration of HR and acquisition of resistance to poly(ADP-ribose) polymerase (PARP) inhibition in BRCA1-deficient cells (Bunting et al., 2010). Thus, we next tested the effect of the UbT12 mutants in this context and found that expression of the phospho-mimetic UbT12D reduces the sensitivity of BRCA1-deficient cells to the PARP1 inhibitor olaparib (Figure S6E), suggestive of an impaired activity of 53BP1. Although relatively mild due to the high toxicity of the UbT12 mutants, this effect is consistent in different experiments. Overall, these results show that phosphorylation of ubiquitin Thr12 regulates the DDR by specifically limiting 53BP1 recruitment to damaged chromatin and reducing its NHEJ activity.
DISCUSSION
Large-scale proteomic studies have shown that a key protein modifier, ubiquitin, can itself be regulated by post-translational modifications (PTMs), such as phosphorylation and acetylation, at multiple sites. This intriguing new concept has fostered huge interest among scientists from different fields. As a paradigm of the importance of PTMs on ubiquitin, it was discovered that phosphorylation of ubiquitin Ser65 (pUbS65) by the kinase PINK1 plays a crucial regulatory role in mitochondrial homeostasis (Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014; Wauer et al., 2015a). To date, however, no other relevant ubiquitin phosphorylation event has been functionally characterized, likely due to the lack of specific tools to study such modifications under physiological conditions and their low abundance. By combining a variety of technical approaches and complementary expertise, we report a crucial and unexpected role of phosphorylation of ubiquitin at Thr12 in the regulation of chromatin ubiquitination and DDR. Several lines of evidence support the role of pUbT12 in DDR: (1) induction by DNA damage, (2) dependency on the activity of upstream DDR factors (e.g., the DDR apical kinases and RNF168), (3) similarity with the DDR histone mark H2AK15ub, and (4) co-localization with the HR factors RAD51 and the BRCA1/BARD1 complex.
Conjugation and Recognition of pUbT12
The dynamics of substrate conjugation are different for pUbT12 and pUbS65. It was shown that the ubiquitin ligase activity of Parkin is inhibited in vitro when the only ubiquitin species available is pUbS65, even if pUbS65 is required to release the inhibitory effect of the ubiquitin-like domain of Parkin and stabilize its open conformation via allosteric activation (Wauer et al., 2015a, 2015b). Conversely, RNF168 efficiently uses pUbT12 in vitro to promote auto-ubiquitination (data not shown) and ubiquitinate its specific target, histone H2A at Lys15, both in the H2A-H2B dimer and in the NCP (Figures 3A and S5A). However, whether or not RNF168 has a functional impact on the physiological turnover of pUbT12 on chromatin in cells is still unknown.
The conformation of pUbS65 does not impact ubiquitin recognition by small UBDs (Wauer et al., 2015b). Similarly, the DDR protein RNF169, recently reported to promote DSB-end resection and single-strand annealing (SSA) repair (An et al., 2018), can still bind NCP-H2AK15ub (by means of the MIU domain) when ubiquitin Thr12 is phosphorylated (Figure 5C). This is different from 53BP1, which cannot bind pUbT12-modified NCP-H2AK15ub. RNF169 and 53BP1 do not use the same ubiquitin-binding mechanism, as shown by 3D structures (Hu et al., 2017; Kitevski-LeBlanc et al., 2017; Penengo et al., 2006; Wilson et al., 2016). Ubiquitin recognition by 53BP1 is unusual, involving sandwiching of the 53BP1 UDR motif by the nucleosome and ubiquitin (Wilson et al., 2016). Phosphorylation of Thr12, located at the interface of UDR and ubiquitin in the 53BP1/NCP-H2AK15ub complex, dramatically destabilizes this interaction (Figure 5B). Moreover, the 3D structure of 53BP1/NCP-H2AK15ub suggests that phosphorylation of Thr66, which is close in space to Thr12, could also disrupt complex formation. Indeed, mutation of Thr66 (T66A) exerts similar but less pronounced effects on 53BP1 recruitment than modification of Thr12 (Figure 1A; data not shown). A compelling question is whether or not specificreaders recognize the unconventionalH2AK15pUbT12 phospho-site (the X/Y factors in the model; Figure 6). A systematic study is needed to address this important point.
Figure 6. Model Depicting the Regulation of DDR Events on Chromatin Mediated by pUbT12.
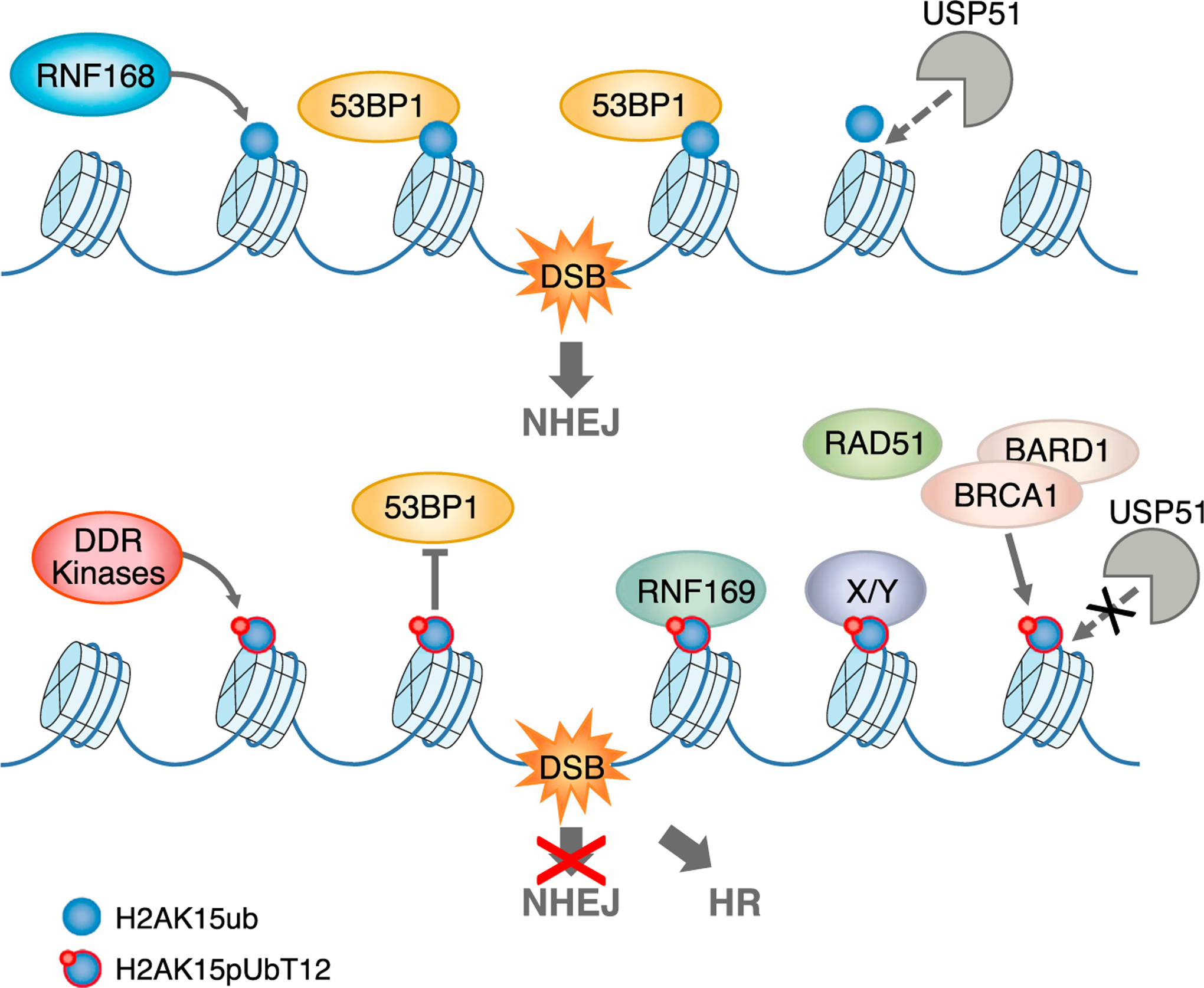
Top: DNA double-strand break (DSB) induces RNF168-dependent ubiquitination of histone H2A at Lys15 (H2AK15ub, blue sphere), which is directly bound by 53BP1, leading to DNA repair via non-homologous end joining (NHEJ). The deubiquitinating enzyme USP51 removes this mark, which attenuates the signaling, once the damage is repaired. Bottom: ubiquitin phosphorylation of H2AK15ub at Thr12, which depends on the upstream DDR kinases, generates the new epigenetic mark H2AK15pUbT12 (blue sphere with red circle), which impedes the recruitment of 53BP1 to damaged chromatin, blocking DNA repair via NHEJ. Importantly, H2AK15pUbT12 is permissive to the binding of RNF169 and the recruitment of RAD51 and the BRCA1/BARD1 complex, allowing DNA repair via homologous recombination (HR). Alteration of H2AK15ub by pUbT12 prevents the clearance of ubiquitin from chromatin by USP51, leading to sustained chromatin (phospho)ubiquitination. Proteins X/Y represent hypothetic factors that specifically recognize the H2AK15pUbT12 mark.
Molecular Understanding and Implications of pUbT12 Clearance from Chromatin
Alterations of Thr12 prevent the USP51-mediated hydrolysis of ubiquitin from chromatin, leading to the accumulation of ubiquitinated conjugates (Figures 3C, 3D, S3A, and S3B). This result is reminiscent of previous observations that several deubiquitinating enzymes belonging to the USP (ubiquitin-specific protease) family contact the first two β strands of ubiquitin, where Thr12 is located (Ye et al., 2011), thereby suggesting that mutations of this region would impair the cleavage by this class of deubiquitinating enzymes (DUBs). Intriguingly, it was shown that the closest relative of ubiquitin, NEDD8, which is not cleaved by USP21 (used as prototype of USPs), bears the replacement of Thr12 with Glu12 (T12E), hence preventing cleavage and allowing USP21 to discriminate between ubiquitin and NEDD8 (Ye et al., 2011). This finding has important implications for signaling, suggesting that phosphorylation at Thr12 may prolong DDR-induced chromatin ubiquitination, leading to sustained recruitment of the downstream factors that are activated by H2AK15ub, but not inhibited by pUbT12, such as RNF169 (as indicated in the model; Figure 6). An important consideration is that accumulation of chromatin ubiquitination at H2AK15 in cells expressing UbT12 mutants reveals that this histone mark is highly dynamic and far more frequent than expected based on experimental detection, even in absence of exogenous DNA damage. This observation further corroborates the key role of RNF168 and H2AK15ub in other basic processes, such as DNA replication of intrinsically unstable sequences (Schmid et al., 2018).
Dynamics of pUbT12 and Its Role in DDR
The induction of DNA damage leads to a cascade of protein phosphorylation events in cells, targeting many different proteins. One of the most prominent substrates is the histone variant H2A.X, the phosphorylation of which is triggered by many types of genotoxic stress and extends to over 20 Mb of damaged chromatin (Iacovoni et al., 2010). Conversely, pUbT12 depends not only on upstream phosphorylation events, but also on the ubiquitin ligase activity of RNF168, which is highly regulated in cells, as revealed by the limited number of H2AK15ub-positive foci detectable in cells upon formation of DSBs (Figure 4C). Notably, pUbT12 and H2AK15ub modifications are qualitatively and quantitatively very similar in terms of cell-cycle distribution, number of foci, and dependency on RNF168 and DDR kinases, and both are, expectedly, far less abundant than γH2A.X. These data strongly support a functional link between these two modifications in the context of DDR, with one (H2AK15ub) being required for the other (pUbT12). Interestingly, we observed that the significant co-localization of pUbT12 with RAD51 and the BRCA1/BARD1 complex (50% and 30%, respectively, is comparable to that obtained for H2AK15ub and 53BP1, with 50% of H2AK15ub foci being positive for 53BP1 (Figure S4K). On the other hand, the considerable fraction of 53BP1-negative H2AK15ub foci (~50%) may correspond to chromatin regions marked by pUbT12 and RAD51 and thereby committed to HR-mediated repair. Further molecular investigations will help unravel the specific function and crosstalk between these different histone modifications in the context of DDR and DNA repair.
Although the number of pUbT12-positive foci increases upon DNA damage, pUbT12 may not be restricted to H2AK15 ubiquitination. Besides the band detected in chromatin extracts at the size corresponding to mono-ubiquitinated H2A, we also observed phosphatase- and ubiquitin protease-sensitive pUbT12 signals at higher molecular weight (Figures S2E–S2I). In line with this finding, we clearly detect basal levels of Thr12 phosphorylation in unperturbed cells, which do not disappear upon treatment with DDR kinase inhibitors or depletion of RNF168 (Figure 4E and 4F). Altogether, this is suggestive of additional roles of pUbT12 in other nuclear processes that are not necessarily related to DDR-induced ubiquitination of chromatin at H2AK15.
Mechanistically, our finding that RNF168 is able to promote chromatin ubiquitination using pUbT12 does not necessarily imply that, in cells, free ubiquitin is first phosphorylated at Thr12 by a yet-elusive kinase and then conjugated to chromatin by RNF168. We envision two different scenarios, depending on whether the hypothetical kinase is targeting free ubiquitin or chromatin-conjugated ubiquitin (i.e., H2AK15ub). In the first case, activation of this ubiquitin kinase might increase the local concentration of free pUbT12, therefore favoring RNF168-dependent chromatin ubiquitination using pUbT12 over unmodified ubiquitin. Alternatively, since ubiquitin is a rather unusual substrate due to its small size, the unknown kinase might be able to phosphorylate ubiquitin only if attached to the target (i.e., histone H2A) in the chromatin context. This latter possibility would increase the spatial and temporal specificity of the signal. Regardless of the mechanism of ubiquitin phosphorylation, chromatin modification by H2AK15pUbT12 is highly regulated in cells, being strictly dependent on the local environment and requiring the activity of RNF168 on histone H2A. This high specificity might have evolved to avoid the spread of a phosphoubiquitin species that could be efficiently conjugated (Figure 3A), but not properly removed (Figures 3C and 3D).
Limitations of Study
Studying this DNA-damage-induced dual modification, consisting of ubiquitination and phosphorylation, in cells is extremely challenging for several reasons. First, pUbT12 is highly defined in space, occurring on specific chromatin regions marked by H2AK15ub, and predictably very dynamic, being potentially removed both by specific deubiquitinating enzymes and by phosphatases. Therefore, the H2AK15pUbT12 mark, as well as H2AK15ub, is expectedly limited in number compared to the abundant H2A.X phosphorylation. As a result, while the DSB-dependent induction of pUbT12 can be clearly observed by immunostaining, the dynamics of H2AK15pUbT12 proved hard to be detected by biochemistry. Additional optimization is required to develop more sensitive tools to monitor these important chromatin modifications. Moreover, the coexistence of two potential binding sites for interacting partners, consisting of ubiquitin- and phospho-binding domains, may render pUbT12 hardly accessible to antibodies for detection. This is supported by the harsh procedure required for pUbT12 immunostaining, which includes an antigen-retrieval step to unmask the epitope. Unfortunately, this treatment prevents monitoring of several DDR markers, such as γH2A.X and H2AK15ub, therefore limiting co-staining with important factors.
Additional difficulties are intrinsic to the ubiquitin system. Ubiquitin is encoded by different genes in mammalian cells (similarly to histone H2A); two genes, UBA52 and RPS27A, encode ubiquitin fused to ribosomal subunits, whereas UbB and UbC code for three and nine tandem-repeat, head-to-tail ubiquitin units. This complexity and the high conservation throughout evolution, with 96% sequence conservation from yeast to human, reflect the fundamental role of ubiquitin in cell viability and explain the toxicity of the ubiquitin mutants, making the genetic manipulation of ubiquitin very arduous. In case of UbT12 mutants, the toxicity is further exacerbated by the relevance of this residue for cell viability and the specific inability of the USP family of DUBs to cleave ubiquitin when altered at Thr12 of ubiquitin. As a consequence, while the results based on in vitro assay (53BP1 binding to NCP-H2AK15pUbT12) or short-term in vivo functional studies (accumulation of 53BP1 to damaged chromatin) clearly show the inhibitory effects of pUbT12 (and UbT12D/UbT12E mutants) on 53BP1, the outcome on long-term studies is less striking due to the intrinsic toxicity of UbT12 mutants.
Concluding Remarks
Since phosphorylation of ubiquitin at Thr12 impedes chromatin recruitment of 53BP1, this phosphorylation can be seen as a safe-lock mechanism that prevents 53BP1 binding to DNA lesions engaged in HR-mediated repair (Figure 6). It is noteworthy that recent evidence implicates a role for 53BP1 in limiting DSB resection, which fosters high-fidelity HR over mutagenic SSA repair (Ochs et al., 2016). Yet, a tight modulation of the levels of ubiquitin phosphorylation on chromatin is very important. While defective pUbT12 may promote inappropriate NHEJ when HR is favorable, excessive pUbT12 may stimulate uncontrolled RNF169 activation, leading to hyper-resection of DSBs and highly mutagenic SSA-mediated repair. Last, the finding that the phospho-inhibitory mutations (UbT12A and UbT12I) were found in patients with stomach adenocarcinoma and cutaneous melanoma (cBioPortal; https://www.cbioportal.org) provides an interesting link between alteration of ubiquitin Thr12 and cancer.
Our study highlights the intricate crosstalk between phosphorylation and ubiquitination of chromatin in the control of DDR and DNA repair, leading to the discovery of a novel and highly specific epigenetic mark at damaged chromatin, the phospho-ubiquitinated histone H2A, H2AK15pUbT12, which limits the access of 53BP1 to damaged chromatin. This finding opens new intriguing questions. What is the kinase targeting ubiquitin Thr12, and in what context (i.e., free ubiquitin or chromatin) is this phosphorylation occurring? Are there specific ubiquitin pro-teases able to cleave pUbThr12, or does ubiquitin first need to be dephosphorylated in order to be removed from chromatin? And if so, are there specific phosphatases acting on pUbT12 and/or specific for phospho-ubiquitinated histones? All of these represent important research avenues for future studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Lorenza Penengo (penengo@imcr.uzh.ch).
Materials Availability
All unique/stable reagents generated in this study will be made available upon request to the Lead Contact.
Data and Code Availability
Original data for figures in the paper are available at Mendeley Data: https://data.mendeley.com/datasets/k4fmj3nzfz/draft?a=c22150bc-d2e4-4095-a10a-08d376a16abf
METHOD DETAILS
Cell culture and drug treatments
U2OS, HEK293T, HeLa, RPE-1, SUM149PT and U2OS GFP DR and EJ5 reporter cells, kindly provided by J. Stark (Gunn and Stark, 2012), were grown in DMEM supplemented with 10% FBS. Stable U2OS ubiquitin replacement cells were grown in DMEM supplemented with 10% tetracycline-free FBS, puromycin (1 μg/ml) and G418 (400 μg/ml). All cell lines were grown in an atmosphere containing 6% CO2 at 37°C. U2OS cells were treated with etoposide (5 μM) and irradiated with 1 Gy (fixation after 45 min). For kinase inhibition, U2OS cells were pre-treated with ATM inhibitor KU-55933 (10 μM), ATR inhibitor VE-821 (10 μM) and DNA-PK inhibitor KU-57788 (2 μM) for 2 h before 1 h etoposide treatment (5 μM).
siRNA transfection
U2OS were transfected with siRNA at a final concentration of 40 nM for the indicated time using Oligofectamine™ Transfection Reagent according to manufacturer’s instructions. For siRNA targeting ubiquitin, both siRNA at a final concentration of 50 nM were transfected at the same time for 18 h. siRNA-resistant form of the different ubiquitin mutants were obtained by site-directed mutagenesis. The following siRNA were used:
siLuc 5′-CGUACGCGGAAUACUUCGAUU-3′
siRNF8 5′-CCATGTCCAGGATTCTGAGGCTCAA-3′
siRNF168 5′-CGUGGAACUGUGGACGAUAAUUCAA-3′
si53BP1 5′-GAACGAGGAGACGGUAAUA-3′
siBRCA1 5′-GGAACCUGUCUCCACAAAG-3′
siUb RPS27A 5′-AUGUAAAGGCCAAGAUCCAGGAUAA-3′
siUb UBA52 5′-CCAGUGACACCAUUGAGAAUGUCAA-3′
Generation of the ubiquitin replacement system
Stable U2OS ubiquitin replacement cell lines were generated as previously described (Xu et al., 2009) using U2OS-shUb cells (kindly provided by Z.J. Chen) transfected with RNAi-resistant expression vector (ubiquitin WT or T12A) using FuGENE® HD transfection according to manufacturer’s instruction. Forty-eight h after transfection, stable cells were selected with G418 (400 μg/ml) for 2 weeks and used for experiments. Replacement was induced by the addition of doxycycline (1 μg/ml) for the indicated time points. For cell viability, 400,000 cells were plated into a 6-well plate and viable cells (dead cells were stained with trypan blue) were counted after different time points.
Lentiviral expression system
For lentivirus production, HEK293T cells were transfected with the packaging plasmids pVSV, pMDL, pREV and the expression plasmid pR-EF1-FLAG-ubiquitin-IRES-DsRed (derived from pR-EF1 Cellecta Inc.) using jetPRIME® transfection according to the manufacturer’s instructions. The following day, medium was replaced with fresh DMEM medium, lentivirus was collected 72 h after transfection and stored at −80°C. Lentiviral titer was determined using FACS analysis (BD LSR II Fortessa) for DsRed positive cells 72 h after transduction. For immunofluorescence, U2OS cells grown on glass coverslips were transduced overnight with lentiviral particles (MOI = 1) in DMEM supplemented with 10 μg/ml polybrene. Cells were washed 18 h after transduction and fixed with 4% paraformaldehyde 72 h after transfection for immunofluorescence procedure (see below). For cell viability, U2OS cells were transduced as above and plated into a 96-well plate. After different time points, cells were fixed with 4% paraformaldehyde for 10 min and stained with 0.05% crystal violet for 30 min. Crystal violet was dissolved in 100% MeOH and absorbance was measured at 570 nm.
DNA repair assay
U2OS DR and EJ5 cells were transfected with pCBASceI using FuGENE® HD transfection according to manufacturer’s instruction. After 8 h, cells were transduced overnight with lentiviral particles (MOI = 1) in DMEM supplemented with 10 μg/ml polybrene. Cells were washed 18 h after transduction and fixed in 4% paraformaldehyde 72 h after transfection for subsequent FACS analysis (BD LSR II Fortessa) measuring DsRed and GFP positive cells. For control experiments, reporter cells were depleted for 53BP1 (U2OS EJ5) or BRCA1 (U2OS DR) by transfecting twice siRNA for a total of 6 days and I-SceI was expressed for 72 h before FACS analysis as described above.
Olaparib sensitivity
SUM149PT cells were transduced overnight with lentiviral particles (MOI = 1) in DMEM supplemented with 10 μg/ml polybrene. The following day, cells were seeded into a 6-well and treated with different concentrations of olaparib (NT, 100 nM, 250 nM, 500 nM and 1 μM). After 7–10 days, cells were fixed with 4% PFA, stained with 1% methylene blue for 10 min and colonies were counted manually.
Site-specific mutagenesis
Site-directed mutagenesis was performed using Pfu Turbo Polymerase, PCR product was digested with DpnI, transformed into E. Coli TOP10 and sequence verified. Oligonucleotides for mutagenesis are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FLAG M2 | Sigma-Aldrich | Cat# F3166 |
| H3 | Abcam | Cat# ab1791; RRID:AB_302613 |
| H2A | Millipore | Cat# 07–146; RRID:AB_11212920 |
| Ubiquitin P4D1 | Santa Cruz Biotechnology | Cat# sc-8017; RRID:AB_628423 |
| HA | Covance | Cat# MMS-101P; RRID:AB_2314672 |
| Tubulin | Sigma-Aldrich | Cat# T5201; RRID:AB_609915 |
| GST | Santa Cruz Biotechnology | Cat# sc-138; RRID:AB_627677 |
| RNF8 | R. Freire Instituto de Tecnologìas Biomédicas, Tenerife, Spain | N/A |
| RNF168 | R. Freire Instituto de Tecnologìas Biomédicas, Tenerife, Spain | N/A |
| hu-pUbT12 | This manuscript | N/A |
| rb-pUbT12 | This manuscript | N/A |
| 53BP1 | Abcam | Cat# ab36823; RRID:AB_722497 |
| γH2AX | Millipore | Cat# 05–636; RRID:AB_309864 |
| BRCA1 | Santa Cruz Biotechnology | Cat# sc-6954; RRID:AB_626761 |
| BARD1 | Richard Baer, Columbia, NY | N/A |
| Rad51 | BioAcademia | Cat# 70–002; RRID:AB_1056187 |
| H2AK15ub | Zhiguo Zhang, Mayo Clinic College of Medicine, Minnesota, USA | N/A |
| MYC 9E10 | Sigma-Aldrich | Cat# 05–419; RRID:AB_309725 |
| anti-mouse Alexa 488 | Thermo Fisher Scientific | Cat# A32723; RRID:AB_2633275 |
| anti-rabbit Alexa 555 | Thermo Fisher Scientific | Cat# A21428; RRID:AB_141784 |
| anti-human Alexa 546 | Thermo Fisher Scientific | Cat# A21089; RRID:AB_1500625 |
| anti-human Alexa 647 | Jackson ImmunoResearch | Cat# 709-605-149; RRID:AB_2340578 |
| anti-mouse HRP | Axon Lab | Cat# AC2115 |
| anti-rabbit HRP | Axon Lab | Cat# AC2114 |
| Anti-human HRP | Thermo Fisher Scientific | Cat# 31420 |
| Bacterial and Virus Strains | ||
| E. coli TOP10 | Thermo Fisher Scientific | Cat# C404010 |
| E. coli TG1 | Agilent | Cat# 200123 |
| E. coli Rosetta | Sigma-Aldrich | Cat# 709543 |
| E. coli BL21 pLysS | Sigma-Aldrich | Cat# 69451–3 |
| ETH-2-Gold phage display library | Dario Neri, ETHZ, Zurich | N/A |
| Lentivirus FLAG-Ub WT, T12A and T12D | This manuscript | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin | InvivoGen | Cat# ant-pr |
| G418 | Sigma-Aldrich | Cat# A1720 |
| etoposide | Sigma-Aldrich | Cat# E1383–25MG |
| Oligofectamine™ Transfection Reagent | Thermo Fisher Scientific | Cat# 12252011 |
| FuGENE® HD transfection | Promega | Cat# E2311 |
| doxycycline | Sigma-Aldrich | Cat# D9891 |
| jetPRIME® transfection | Polyplus | Cat# 114–07 |
| Polybrene transfection | Sigma-Aldrich | Cat# TR1003-G |
| Pfu Turbo Polymerase | Agilent Technologies | Cat# 600250 |
| DpnI | NEB | Cat# 7620011 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340–5ML |
| PhosSTOP | Roche | Cat# 7002220 |
| Calf Intestinal Phosphatase | NEB | Cat# 7620043 |
| Lambda phosphatase | Santa Cruz Biotechnology | Cat# sc-200312A |
| Lbpro | David Komander, WEHI, Australia | N/A |
| FLAG agarose beads | Sigma-Aldrich | Cat# A2220 |
| 3xFLAG-peptide | Sigma-Aldrich | Cat# F4799 |
| DAPI | Sigma-Aldrich | Cat# D9542–10MG |
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | Cat# 7001557 |
| Biotin-pUbT12 peptide | GenScript | N/A |
| Magnetic Streptavidin dynabeads | Invitrogen | Cat# M-280 |
| Human ubiquitin | R&D Systems | Cat# U-100H-10M |
| MaxiSorp plate | Thermo Fisher Scientific | Cat# 442404 |
| BM blue POD substrate | Sigma-Aldrich | Cat# 11484281001 |
| Protein A Sepharose | Sino Biological Inc. | Cat# 10600-P07E-RN |
| Biotin-ubiquitin | R&D Systems | Cat# UB-560–050 |
| (Biotin-) pUbT12 | This manuscript | N/A |
| Fmoc-Gly trityl resin | Rapp Polymere GmbH | Cat# RA1213 |
| PEG spacer | AK Scientific Inc. | Cat# V1167–5GR |
| HisPur™ Ni-NTA resin (E1, E2, H2A-H2B purification) | Thermo Fisher Scientific | Cat# 88221 |
| Ni-NTA resin (RNF169 purification) | QIAGEN | Cat# 30210 |
| Glutathione sepharose beads | Thermo Fisher Scientific | Cat# 11594935 |
| Prescission protease | Sigma-Aldrich | Cat# GE27-0843-01 |
| ATP | Sigma-Aldrich | Cat# A1852–1VL |
| Creatine kinase | Sigma-Aldrich | Cat# C9983–100UG |
| Creatine phosphate | Sigma-Aldrich | Cat# 27920–1G |
| Olaparib | LuBioScience | Cat# s1060–10MG |
| KU-55933 | Sigma-Aldrich | Cat# SML1109 |
| VE-821 | Sigma-Aldrich | Cat# SML1415–5MG |
| KU-57788 | Selleck Chemicals | Cat# S2638 |
| Methylene blue | Sigma-Aldrich | Cat# M9140–25G |
| Critical Commercial Assays | ||
| Click-iT® Plus EdU Imaging Kit | Thermo Fisher Scientific | Cat# C10640 |
| Gibson Assembly® Cloning kit | NEB | Cat# E5510S |
| Deposited Data | ||
| Raw imaging data | Mendeley Dataset | https://data.mendeley.com/datasets/k4fmj3nzfz/draft?a=c22150bc-d2e4-4095-a10a-08d376a16abf |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC® | Cat# HTB-96 |
| HeLa | ATCC® | Cat# CCL-2 |
| HEK293T | ATCC® | Cat# CRL-3216 |
| RPE-1 | ATCC® | Cat# CRL-4000 |
| SUM149PT | BIOIVT | RRID: CVCL_3422 |
| U2OS DR GFP reporter cells | Jeremy Stark, City of Hope Comprehensive Cancer Center, USA | N/A |
| U2OS EJ5 GFP reporter cells | Jeremy Stark, City of Hope Comprehensive Cancer Center, USA | N/A |
| U2OS shUb replacement cells | Zhijian Chen, University of Texas Southwestern Medical Center, USA | N/A |
| U2OS shUb ubiquitin WT replacement cells | This manuscript | N/A |
| U2OS shUb ubiquitin T12A replacement cells | This manuscript | N/A |
| Oligonucleotides | ||
| siLuc 5’-CGUACGCGGAAUACUUCGAUU-3’ | Microsynth AG | N/A |
| siRNF8 5’-CCATGTCCAGGATTCTGAGGCTCAA-3’ | Microsynth AG | N/A |
| siRNF168 5’-CGUGGAACUGUGGACGAUAAUUCAA-3’ | Microsynth AG | N/A |
| si53BP1 5’-GAACGAGGAGACGGUAAUA-3’ | Microsynth AG | N/A |
| siBRCA1 5’-GGAACCUGUCUCCACAAAG-3’ | Microsynth AG | N/A |
| siUb RPS27A 5’-AUGUAAAGGCCAAGAUCCAGGAUAA-3’ | Microsynth AG | N/A |
| siUb UBA52 5’-CCAGUGACACCAUUGAGAAUGUCAA-3’ | Microsynth AG | N/A |
| Recombinant DNA | ||
| pcDNA 3.1 | Thermo Fisher Scientific | Cat# V79020 |
| pcDNA 3.1 FLAG-ubiquitin WT and mutants | This manuscript | N/A |
| expression vector for ubiquitin replacement cells | Zhijian Chen, University of Texas Southwestern Medical Center, USA | N/A |
| pR-EF1, pVSV, pMDL, pREV | Beat Schäfer, University, Children’s Hospital Zurich, Switzerland | N/A |
| pCBASceI | Addgene | Cat# 26477 |
| USP51-FLAG | GenScript | Cat# OHu22284 |
| HA-USP51 | This manuscript | N/A |
| H2A mutants | This manuscript | N/A |
| RNF169 WT and MIU* | Michael Huen, University of Hong Kong, China | N/A |
| pVITRO1-dV-IgG1/λ | Addgene | Cat# 52214 |
| pcDNA 3.1 heavy chain pUbT12 antibody | This manuscript | N/A |
| pcDNA 3.1 light chain pUbT12 antibody | This manuscript | N/A |
| pGEX-6P2 | Amersham Pharmacia Biotech | Cat# 27-4598-01 |
| pET21d | Novagen | Cat# 69743–3 |
| Software and Algorithms | ||
| GraphPad Prism7 for MAC OS | GraphPad Software | https://www.graphpad.com/ |
| Fiji | ImageJ Software | https://imagej.net/Fiji |
| FlowJo 10.6.1 | FlowJo Software | https://www.flowjo.com/ |
| FusionCapt Advance Solo 7 17.02 | Vilber Lourmat | http://www.vilber.de/ |
| Spotfire data visualization software version 5.0.0 (QIBC data analysis) | TIBCO Software Inc. | https://www.tibco.com/products/tibco-spotfire |
| BD FACSDiva™ Software | Biosciences | https://www.bdbiosciences.com/en-us |
| Huygens Deconvolution Software | Scientific Volume Imaging | https://svi.nl/Huygens-Deconvolution |
| ScanR Image Analysis Software Version 3.0.0 | Olympus | https://www.olympus-lifescience.com/en |
| MassLynx Mass Spectrometry Software 4.1 | Waters | https://www.waters.com/waters/home.htm |
Cell fractionation
For cell fractionation, HEK293T and U2OS cells were collected in PBS containing protease inhibitor cocktail, PhosSTOP, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 10 μM N-ethylmaleimide (NEM). Cytosolic fraction (fraction S1) was removed by incubating cell pellet in hypotonic buffer (10 mM HEPES pH 7.5, 50 mM NaCl, 300 mM sucrose, 0.5% Triton-X, 1 mM PMSF, 10 μM NEM, protease inhibitor cocktail, PhosSTOP) for 15 min on ice followed by 5 min centrifugation at 1,500 g. Soluble nuclear fraction (fraction S2) was removed by incubating pellet with nuclear buffer (10 mM HEPES pH 7, 200 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM PMSF, 10 μM NEM, protease inhibitor cocktail, PhosSTOP) for 10 min on ice followed by 2 min centrifugation at 16,000 g. Chromatin fraction (fraction S3) was collected in lysis buffer (10 mM HEPES pH 7, 500 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF, 10 μM NEM, protease inhibitor cocktail, Phos-STOP) and sonicated for 15 min at low amplitude. The procedure was performed on ice unless stated otherwise.
Acidic chromatin extraction
Acidic extraction of HEK293T cells was performed 24 h or 48 h after transfection. Cells were collected in PBS containing protease inhibitor cocktail, 1 mM PMSF and 10 μM NEM. One-tenth of the samples were separately processed for total cell lysate (RIPA buffer supplemented with protease and phosphatase inhibitor cocktail), whereas the remaining was subjected to acidic extraction. Cell pellet was resuspended in PCA buffer (5% PCA, 1 mM PMSF and 10 μM NEM, protease inhibitor cocktail) and incubated for 10 min. After 10 min centrifugation at 13,000 rpm, supernatant was collected and cell pellet was processed two additional times as described above in PCA buffer (fraction S1). Thereafter, core histones were extracted by resuspending pellet three times in HCl buffer (0.4 N HCl, 10 μM NEM, protease inhibitor cocktail) for 15 min followed by 10 min centrifugation at 13’000 rpm. Core histones were precipitated from collected supernatants using TCA at a final concentration of 25%. After 30 min incubation and 20 min centrifugation at 13,000 rpm, protein pellet was washed with 100% Aceton/0.006% HCl and 100% Aceton. Pellet was dried at 37°C and resuspended in 50 mM Tris-HCl pH 8 (fraction S2). Samples were analyzed by SDS-PAGE and immunoblotting. The procedure was performed on ice unless stated otherwise.
Phosphatase and Lbpro treatment
For phosphatase treatments, samples after cell fractionation were incubated with Calf Intestinal Phosphatase (NEB) for 30 min at 37°C or lambda phosphatase (Santa Cruz) for 30 min at 30°C according to manufacturer’s instructions. For Lbpro treatment, in vitro ubiquitination reaction or chromatin fraction was incubated with 100 μM Lbpro kindly provided by D. Komander (Swatek et al., 2019) in cleavage buffer (50 mM NaCl, 50 mM Tris pH 8, 10 mM DTT) for 30 min or 1 h at 37°C and analyzed by immunoblotting.
Immunoprecipitation of FLAG-H2A
HEK293T cells were processed for cell fractionation as described above 48 h after transfection. FLAG-H2A was immunoprecipitated from chromatin fraction using FLAG agarose beads in immunoprecipitation buffer (20 mM HEPES pH 7.5, 10% glycerol, 150 mM NaCl, 0.1% Triton-X) for 1.5 h on a rotator at 4°C. Thereafter, beads were washed three times with immunoprecipitation buffer and proteins were eluted with 2 mg/ml 3xFLAG-peptide (in 50 mM Tris HCl (pH = 7.4) and 150 mM NaCl) for 30 min at room temperature. Eluted proteins were analyzed by immunoblotting.
Immunoblotting
Proteins were separated by SDS-PAGE and transferred onto PVDF or nitrocellulose membrane by wet transfer system. Membranes were blocked with TBS containing 5% milk or 5% BSA before overnight incubation with primary antibodies. After incubation with secondary horseradish peroxidase-coupled antibodies, signals were detected using ECL-based chemiluminescence. The following primary antibodies were used for western blot analysis: FLAG M2 (1:1,000), H3 (1:4,000), H2A (1:2,000), ubiquitin P4D1 (1:1,000), HA (1:1,000), Tubulin (1:8,000), GST (1:1,000), rb-pUbT12 (1:1,000), RNF8 (1:1,000) and RNF168 (1:1,000). The antibody recognizing pUbT12 was generated by rabbit immunization at Creative Biolabs.
Immunofluorescence
Cells were grown on sterile glass coverslips and transfected or treated as indicated. For 53BP1 (1:1,000), γH2AX (1:1,000), FLAG (1:10,000), HA (1:1,000), H2AK15ub (1:200; kindly gifted by Zhiquan Wang), BARD1 (1:500; kindly gifted by Richard Bauer) and BRCA1 (1:50) staining, cells were fixed with 4% PFA for 10 min and permeabilized with 0.2% Triton X-100/PBS for 5 min. After 2 h of blocking with 5% BSA/PBS, primary antibodies in 5% BSA/PBS were incubated for 1–2 h at room temperature followed by Alexa labeled secondary antibodies (1:100 5% BSA/PBS) for 30 min. After washing with PBS, DNA was stained by DAPI (0.5 μg/ml) for 10 min and cells were mounted with ProLong Gold Antifade. For cell cycle analysis, U2OS cells were labeled with 10 μM EdU for 30 min, fixed and permeabilized as described above. EdU detection was performed using the Click-iT® Plus EdU Imaging Kit according to manufacturer’s instructions. Cell cycle distribution was analyzed by QIBC.
For hu-pUbT12 staining, cells were fixed with 100% ice-cold methanol on ice for 10 min. Thereafter, antigen retrieval was performed using 0.1 M citric acid (pH 3) for 30 min at 80°C followed by 2 h blocking, incubation with hu-pUbT12 (2h), secondary antibody (30 min) and DAPI as described above. For co-localization experiments, cells were stained first with hu-pUbT12, fixed with 4% PFA for 5 min and then stained for 1 h with 53BP1, RAD51 (1:500), BARD1 (1:500) or rb-pUbT12 (1:200).
Images were acquired with a wide-field Leica DM6 B microscope (HCX PL APO 63x objective) or a Leica SP8 automated upright confocal laser scanning microscope (HCX PL APO CS2 63x immersion oil objective; NA 1.4). If necessary, Z series were deconvolved using Huygens Deconvolution software and a representative single Z slice is shown.
Quantitative image-based cytometry (QIBC) and ScanR analysis
Automated multichannel wide-field microscopy for QIBC was performed on an Olympus ScanR Screening System equipped with an inverted motorized Olympus IX83 microscope, a motorized stage, IR-laser hardware autofocus, a fast emission filter wheel with single band emission filters and a 12-bit digital monochrome Hamamatsu ORCA-FLASH 4.0 V2 sCMOS camera (2,048 × 2,048 pixels) using UPLSAPO 20 × objective (NA 0.75). For each condition, images containing at least 1,000 cells per experiment were acquired under non-saturating conditions at a single autofocus-directed z-position and identical settings were applied to all samples within one experiment. Images were analyzed with the inbuilt Olympus ScanR Image Analysis Software Version 3.0.0, a dynamic background correction was applied, nuclei segmentation was performed using an integrated intensity-based object detection module based on the DAPI signal and foci segmentation was performed using an integrated spot-detection module. All downstream analyses were focused on nuclei containing a 2C-4C DNA content as measured by total and mean DAPI intensities. Fluorescence intensities were quantified and are depicted as arbitrary units. Values were exported and analyzed with Spotfire data visualization software (TIBCO). Within one experiment, similar cell numbers were compared for the different conditions. Representative scatterplots and quantifications of independent experiments are shown.
For quantification of hu-pUbT12 foci, images were acquired with a wide-field Leica DM6 B microscope, a DMC2900 camera using a HCX PL APO 63x objective. For each experiment, images containing at least 150 cells per condition were acquired under non-saturating conditions at a single autofocus-directed z-position and identical settings were applied to all samples within one experiment. Images were converted and analyzed with the inbuilt Olympus ScanR Image Analysis Software Version 3.0.0 as described above.
Generation of human anti-pUbT12 antibody by phage display
Human pUbT12 antibody was selected using the ETH-2-Gold antibody phage display library as described previously (Silacci et al., 2005). One hundred and twenty pmol of biotinylated phospho-peptide (TLTGKpTITLEVE) were coupled to streptavidin dynabeads in PBS for 30 min at room temperature on a shaker. After washing with PBS, beads were blocked in 2% milk/ PBS for 1 h, washed with PBS and incubated for 1.5 h with phage library in 2% milk/ PBS containing 5 μM non-biotinylated ubiquitin as competitor. Thereafter, beads were washed with 0.1% Tween/PBS, PBS and bound phages were eluted with 100 mM TEA and neutralized with 1 M Tris-HCl pH 7.4. Exponentially growing TG1 E. coli were infected with eluted phages at 37°C and grown on 2xTY/ampicillin (100 μg/ml)/glucose (1%) agar plates overnight at 30°C. After growing to OD600 = 0.4 at 37°C, the culture was infected with VCSM13 helper phage for 30 min at 37°C and infected bacteria were grown in 2xTY/ampicillin (100 μg/ml)/kanamycin (33.3 μg/ml) overnight in a shaker at 30°C. Amplified phages from supernatant were precipitated using a final concentration of 4% PEG/0.5 M NaCl for 40 min on ice. After centrifugation, precipitation was repeated and pelleted phages were used for a second round of biopanning as described above. Thereafter, scFv expression of individual colonies from phage-infected TG1 was induced by 1 mM IPTG overnight at 30°C in a shaker and supernatant containing the soluble antibody was used for ELISA validation.
Therefore, a MaxiSorp plate was coated with 50 μg/ml avidin overnight at 4°C and 10−6 M biotin-pUbT12 and biotin-UbT12 peptides were immobilized by incubating 30 min at 37°C. After 2 h blocking with PBS/2% BSA, bacterial supernatant containing scFv was incubated together with 1 μg/ml MYC antibody (9E10) for 2 h followed by 1 h incubation with secondary anti-mouse HRP-coupled antibody. BM blue POD substrate was added for 5 min, reaction was stopped by adding sulphuric acid (final concentration 0.5 M) and OD was read at 650 nm and 450 nm (ODfinal = OD450-OD650).
Clones specifically recognizing pUbT12 peptide were sequenced, revalidated by ELISA and immunoblotting, and best clones were reformatted into human IgG1 by Gibson Assembly® Cloning according to the manufacturer’s instructions. Variable chains were amplified from phagemid vectors and constant chains were amplified from pVITRO1-dV-IgG1/λ. Heavy and light chains were cloned separately into pcDNA3.1 vectors, each containing a secretion sequence, according to (Zuberbühler et al., 2009). For antibody expression, HEK293T cells were transfected with both pcDNA3-HC and pcDNA3-LC in DMEM without FBS using jetPRIME® (poly-plus transfection) according to manufacturer’s instructions. IgG was purified from filtered supernantant collected after 24 h, 48 h by 4 h incubation with protein A Sepharose. Beads were washed with buffer A (100 mM NaCl, 0.1% Tween, 0.5 mM EDTA pH 8), buffer B (500 mM NaCl, 0.5 mM EDTA pH 8) and IgG was eluted with 0.1 M glycine (pH 2.2). After neutralization with 1 M ammonium bicarbonate to neutral pH, eluate was dialyzed against PBS overnight and stored at −80°C. Reformatted IgG was validated by immunoblot and ELISA (250 ng of biotin-pUbT12 or biotin-ubiquitin, as described above using secondary anti-human HRP-coupled antibody) and used for the experiments.
Chemical synthesis of pUbT12 and biotin-pUbT12
pUbT12 was synthesized on a Syro II MultiSyntech Automated Peptide synthesizer using standard 9-fluorenylmethoxycarbonyl (Fmoc) based solid phase peptide chemistry on a 25 μmol scale. Starting with the pre-loaded Fmoc-Gly trityl resin (0.18 mmol/g, Rapp Polymere GmbH), each successive amino acid (Novabiochem) was double-coupled in 4 molar excess for 25 min using PyBOP (4 equiv) and DiPEA (8 equiv) as coupling regents. The following six protected pseudoproline and DMB dipeptide building blocks were used during the synthesis: Fmoc-L-Ser(tBu)-L-Thr(ΨMe,Mepro)-OH, Fmoc-L-Leu-L-Ser(ΨMe,Mepro)-OH, Fmoc-L-Asp(OtBu)-(Dmb)Gly-OH, Fmoc-L-Ala-(Dmb)Gly-OH, Fmoc-L-Ile-L-Thr(ΨMe,Mepro)-OH, Fmoc-L-Leu-L-Thr(ΨMe,Mepro)-OH. The N-terminal methionine residue in the sequence was replaced by the known isoster norleucine. Deprotection of the Fmoc group was achieved with 20% piperidine in NMP (2 × 2 and 1 × 5 min). After completion of all coupling cycles, the peptide pUbT12 is cleaved from the resin or modified on the N terminus with a biotin-PEG moiety (biotin-pUbT12). In the latter case a PEG spacer (8-Fmoc-amino)-3,6-dioxaoctanoic acid (AK Scientific, Inc., Union city, CA, 4 equiv), is coupled to the N terminus using PyBOP (4 equiv) and DIPEA (4 equiv) in NMP for 25 min at ambient temperature. The Fmoc protection group is removed as described and biotin is coupled subsequently using HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, 4 equiv), HOBt (1-hydroxybenzotriazole, 4 equiv), DIPEA (8 equiv), and carboxy-functionalized biotin (Sigma-Aldrich, 4 equiv) in NMP and reacted for 2.5 h. The peptides were cleaved from the resin with TFA/H2O/phenol/iPr3SiH (90.5/5/2.5/2 v/v/v/v) for 3 h, precipitated in cold pentane/diethyl ether and washed 3 × with diethylether. The pellet was dissolved in H2O/CH3CN/formic acid (70/25/5; v/v/v) and lyophilized. The purity of the peptides was determined by LC-MS analysis. LC-MS measurements were performed on a system equipped with a Waters 2795 Separation Module (Alliance HT), Waters 2996 Photodiode Array Detector (190–750nm), Phenomenex Kinetex C18 (2.1×100, 2.6 μm) column and LCTTM Orthogonal Acceleration Time of Flight Mass Spectrometer. Samples were run using 2 mobile phases: A (0.1% formic acid in H2O/acetonitrile 99:1 v/v) and B (0.1% formic acid in H2O/acetonitrile 99:1 v/v) at a flow rate of 400 μL/min; gradient: 0–0.5 min, 5% B; 0.5–8 min, → 95% B; 8–10 min 95% B, 10–12 min, → 5% B. Data processing was performed using Waters MassLynx Mass Spectrometry Software 4.1 (deconvolution with Maxent1 function). The crude product was purified using RP-HPLC on a Waters Atlantis T3 C18 30×250 5 μm. Column Mobile phases: A = 0.05% aq. TFA and B = 0.05% TFA in CH3CN. Flow rate = 18 mL/min. Gradient: 20→45%B over 25 min. pUbT12 LC-MS: Rt 4.55 min; MS ES+ (amu) calculated: 8626 [M+H]+; found 8626 [M+H]+; biotin-pUbT12 LC-MS: Rt 4.72 min; MS ES+ (amu) calculated: 8997 [M+H]+; found 8998 [M+H]+.
Production of recombinant proteins in E. coli
E1 Uba1 was produced by transforming pET21d E1 into E. coli Rosetta, which were grown to OD600 = 0.6 and expression was induced overnight at 18°C with 1 mM IPTG. Cells were pelleted (15 min, 4,000 rpm), lysed with buffer A (50 mM NaH2PO4 pH 7.8, 300 mM NaCl, 10% glycerol, 10 mM imidazole, protease inhibitor cocktail) by sonication and purified with HisPur™ Ni-NTA resin for 2 h at 4°C on a rotator. Beads were washed with buffer A, buffer A + 1 M NaCl and buffer A + 20 mM imidazole, proteins were eluted with 300–500 mM imidazole and dialysed overnight using dialysis membranes against maintenance buffer (50 mM Tris HCl pH 7.4, 100 mM NaCl, 1 mM EDTA, 10% glycerol, 1 mM DTT). UbcH5c was produced in E. coli BL21 pLysS cells according to the protocol for the E1 Uba1 (see above) and purified onto a Superdex 75 size-exclusion chromatography column.
GST-ubiquitin and GST-RNF168 were produced by transforming pGEX-6P2 RNF168 or ubiquitin into E. coli BL21 pLysS, which were grown to OD600 = 0.6 (2xTY medium supplemented with 100 μg/ml ampicillin and 50 μg/ml chloramphenicol) and expression was induced over night at 18°C (GST-RNF168) or for 3 h at 37°C (GST-ubiquitin) with 0.5 mM IPTG. Cells were harvested by centrifugation (30 min 4,000 rpm), lysed in 50 mM HEPES pH 7.5, 200 mM NaCl, 1 mM EDTA, 0.1% NP-40, 5% glycerol, 1 mM PMSF, protease inhibitor cocktail. After sonication and clearing of the lysates, proteins were purified with glutathione Sepharose beads for 3 h at 4°C on a rotator. Thereafter, beads were washed three times with PBS/0.1% Triton X-100, twice with PBS/300 mM NaCl, twice with maintenance buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 10% glycerol, 1 mM EDTA, 1 mM DTT) and resuspended in maintenance buffer. Beads were equilibrated in cleavage buffer (50 mM Tris-HCl pH 7.4, 500 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.1% Triton X-100) and GST was removed by cleavage with prescission protease (1 U for 100 μg) for 3 h at room temperature.
Human RNF169 (residues 653–708) was cloned in a pET E. coli expression vector encoding an N-terminal His6-tag cleavable with TEV protease. Human 53BP1 (residues 1,484–1,635) encompassing the tandem TUDOR domains and UDR motif was expressed in E. coli as a fusion with an N-terminal GST tag as previously reported (Botuyan et al., 2018). RNF169 and 53BP1 were produced in BL21(DE3) E. coli cells grown at 37°C in LB media to an OD600 of 0.6. RNF169 expression was induced with 0.5 mM IPTG at 37°C for 3 h. For 53BP1, the expression was induced with 0.5 mM IPTG at 15°C for 16 h. Harvested cells were lyzed using an Avestin Emulsiflex C5 homogenizer. RNF169 was purified by Ni2+-nitrilotriacetic acid agarose beads (QIAGEN) in 50 mM sodium phosphate (NaPi), pH 7.5, 300 mM NaCl with 5, 20, and 250 mM imidazole as binding, washing and elution buffers, respectively. GST-tagged 53BP1 was loaded into a GSTPrep FF 16/10 column (GE Healthcare), washed with PBS (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 Mm KCl, 140 mM NaCl), pH 7.3, and eluted with 20 mM glutathione in PBS, pH 8.0. After elution, and cleavage of the His6-tag for RNF169, the proteins were purified by size exclusion chromatography using HiLoad 16/60 Superdex 75 and 200 columns (GE Healthcare) for RNF169 and 53BP1, respectively, with chromatography buffer consisting of 50 mM NaPi, pH 7.5, 300 mM NaCl. RNF169 was further purified by reversed-phase chromatography using a Jupiter 5 mm C18 300Å preparative column (Phenomenex).
H2A-H2B fusion protein was produced by transforming pET21d H2A-H2B into E. coli BL21 (DE3), which were grown to OD600 = 0.6 and expression was induced for 3 h at 37°C with 1 mM IPTG. Cells were harvested by centrifugation (4,000 rpm 30 min), resuspended in buffer I (50 mM Tris-HCl pH 8, 500 mM NaCl, 1 mM PMSF, 5% glycerol, sonicated and centrifuged (27,000 g 20 min). Pellet was resuspended in buffer I supplemented with 6 M guanidine hydrochloride, centrifuged (27,000 g 20 min) and supernatant was incubated with HisPur™ Ni-NTA resin for 1 h at 4°C on a rotator. Beads were washed three times with buffer II (50 mM Tris-HCl pH 8, 500 mM NaCl, 6 M urea, 5 mM imidazole, 5% glycerol), once with buffer II supplemented with 20 mM imidazole and proteins were eluted with 250–500 mM imidazole. Eluted fractions were dialyzed overnight against buffer III (5 mM Tris-HCl pH 7.5, 5 mM β-mercaptoethanol). All steps were carried out at 4°C unless stated otherwise.
In vitro ubiquitination assay
For ubiquitination reactions, 3.2 μg of purified RNF168 was incubated with 0.35 μg purified E1 enzyme, 0.8 μg UbcH5c, 2.5 μg purified H2A-H2B and 2 μg purified ubiquitin in a buffer containing 25 mM Tris-HCl pH 7.4, 5 mM MgCl2, 100 mM NaCl, 1 μM DTT, 2 mM ATP, creatine kinase (1 U/ml) and creatine phosphate (10 mM) for 2 h at 30°C.
Production of nucleosomes
For in vitro binding assays, the nucleosomes, with and without H2AK15 ubiquitination and/or H4K20 dimethylation mimic (H4KC20me2), were assembled from recombinant DNA and recombinant human histones H3.1 and H4 and a fusion construct of his-tone H2A linked to the C terminus of histone H2B (H2A-H2B) as previously reported (Hu et al., 2017). Prior to nucleosome assembly, refolded H2A-H2B was ubiquitinated with recombinant WT ubiquitin and with chemically synthesized pUbT12, using E1 ubiquitin activating enzyme Uba1, E2 ubiquitin conjugating enzyme UbcH5c (Benirschke et al., 2010) and the RING domain of E3 ubiquitin ligase RNF168 (Figure S5) (Hu et al., 2017).
Pull-down of NCP with 53BP1
GST pull-down assays were carried out by first incubating 40 μL of 50% GST slurry in buffer 1 (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.05% NP-40, 0.1% BSA) with a bait (3 μg GST or equimolar amounts of GST-53BP1 or GST) on a nutator for 1 h at 4°C. Beads were then washed 3 times with buffer 1 (1 ml, 5 min), centrifuged (21,000 g, 2 min) between washes. Thirty-six μg of NCPH2AK15ubH4Kc20me2 (with ubiquitin WT or pUbT12) were added to the immobilized baits and mixed on a rotator for 2 h at 4°C. Beads were washed 5 × 5 min with buffer 1, the last wash omitting NP-40 and BSA, and excess liquid removed prior to adding 25 μL of 2 × Laemmli dye. Beads were boiled for 2 min and 10 μL of the supernatant was analyzed by immunoblotting.
Pull-down of NCP with RNF169
Fifty μl of Ni-NTA Agarose resin (50% slurry) were first equilibrated in high salt buffer (sodium phosphate 50 mM pH 7.5, NaCl 300 mM) and 7 μg of His-RNF169 were added to the resin and incubated at 4°C for 2 h on a nutator. The resin was then washed three times (1 ml, 5 min) with low salt buffer (sodium phosphate 50 mM pH 7.5, NaCl 150 mM), with centrifugation for 5 min at 21,000 g between washes. Twelve mg of NCP was added to the immobilized baits and mixed using a nutator at 4°C for 1 h. Beads were next washed 5 times for 5 min with 1 mL of low salt buffer containing 50 mM imidazole and 0.02% NP-40. The excess liquid was removed and 25 μL of Laemmli dye were added to the beads. Beads were boiled for 2 min and 10 μL were loaded on a 15% SDS-PAGE.
QUANTIFICATION AND STATISTICAL ANALYSIS
For quantification of cells with more than 5 53BP1 foci, at least 50 FLAG-positive cells were analyzed and % values were calculated. At least three independent experiments were pooled and represented as means +SD. QIBC and ScanR quantifications are depicted as boxplots or scatterplots (mild jittering). Cell cycle analysis was performed by gating cell cycle phases according to EdU (S) and DAPI (G1, G2) intensities. For pUbT12, 53BP1 and gH2A.X foci formation upon RNF168 depletion, relative changes in average pUbT12 foci counts compared to untreated siLuc condition from 3 independent experiments were pooled and represented as means +SEM. For co-localization quantification with 53BP1, RAD51 and BARD1, pUbT12 foci from 3 independent experiments with at least 30 cells and 250 foci each were analyzed for co-localization and shown as mean +SD. For co-localization quantification with rb-pUbT12, hu-pUbT12 foci from 36 cells were analyzed for co-localization and shown as scatterplot. For co-localization quantification of BRCA1 and BARD1 50 cells were analyzed, for H2AK15ub and 53BP1 co-localizations H2AK15ub foci from 3 independent experiments with at least 30 cells and 350 foci each were analyzed and shown as mean +SD. Representative immunofluorescence images were generated using ImageJ (1.50i). FACS data was analyzed using FlowJo 10.6.1 software (FlowJo LLC). For all statistical analyses, the unpaired t test was applied using GraphPad Prism 7, p < 0.05 was considered significant and p ≥ 0.05 was considered not significant.
Supplementary Material
Highlights.
Ubiquitin phosphorylation at Thr12 represents a novel histone mark, H2AK15pUbT12
Mutating UbT12 impairs removal of ubiquitin from core histones by the DUB USP51
pUbT12 increases upon DNA damage and depends on DDR kinases and ubiquitin ligase RNF168
pUbT12 blocks binding of 53BP1 to H2AK15ub, hampering 53BP1 activity at damaged DNA
ACKNOWLEDGMENTS
We thank Michael Walser, Beat Schäfer, Jeremy Stark, Richard Baer, Alessandro Sartori, Michael Huen, and David Komander for sharing useful reagents; Qi Hu for molecular modeling; Eliana Bianco and Stefano Ferrari for technical support; the Center for Microscopy and Image Analysis of the University of Zurich for technical assistance with imaging; and Richard Chahwan for discussion. This work was supported by the National Institutes of Health (NIH; grants CA132878 and GM116829) to G.M., the Netherlands Foundation for Scientific Research (VICI; grant 724.013.002) to H.O., the Swiss National Science Foundation (SNSF; grants 310030_184966 and 31003A_166370), the Novartis Foundation (grant 17A039) and the Krebsliga (grant KFS-4577-08-2018) to L.P.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.09.017.
DECLARATION OF INTERESTS
H.O. is a shareholder of UbiQ B.V.
REFERENCES
- Ai H, Guo Y, Sun D, Liu S, Qi Y, Guo J, Qu Q, Gong Q, Zhao S, Li J, and Liu L (2019). Examination of the deubiquitylation site selectivity of USP51 by using chemically synthesized ubiquitylated histones. ChemBioChem 20, 221–229. [DOI] [PubMed] [Google Scholar]
- An L, Dong C, Li J, Chen J, Yuan J, Huang J, Chan KM, Yu CH, and Huen MSY (2018). RNF169 limits 53BP1 deposition at DSBs to stimulate single-strand annealing repair. Proc. Natl. Acad. Sci. USA 115, E8286–E8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benirschke RC, Thompson JR, Nominé Y, Wasielewski E, Juranić N, Macura S, Hatakeyama S, Nakayama KI, Botuyan MV, and Mer G (2010). Molecular basis for the association of human E4B U box ubiquitin ligase with E2-conjugating enzymes UbcH5c and Ubc4. Structure 18, 955–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, and Andersen JS (2010). Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteomics 9, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, and Mer G (2006). Structural basis for the methylation state-specific recognition of histone H4–K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Cui G, Drané P, Oliveira C, Detappe A, Brault ME, Parnandi N, Chaubey S, Thompson JR, Bragantini B, et al. (2018). Mechanism of 53BP1 activity regulation by RNA-binding TIRR and a designer protein. Nat. Struct. Mol. Biol 25, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Oualid F, Merkx R, Ekkebus R, Hameed DS, Smit JJ, de Jong A, Hilkmann H, Sixma TK, and Ovaa H (2010). Chemical synthesis of ubiquitin, ubiquitin-based probes, and diubiquitin. Angew. Chem. Int. Ed. Engl 49, 10149–10153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, and Durocher D (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti M, Pinato S, Maspero E, Soffientini P, Polo S, and Penengo L (2012). A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle 11, 2538–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A, and Stark JM (2012). I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol 920, 379–391. [DOI] [PubMed] [Google Scholar]
- Hu Q, Botuyan MV, Cui G, Zhao D, and Mer G (2017). Mechanisms of ubiquitin-nucleosome recognition and regulation of 53BP1 chromatin recruitment by RNF168/169 and RAD18. Mol. Cell 66, 473–487.e479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, and Legube G (2010). High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, and Youle RJ (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol 205, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, and Muqit MM (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J 460, 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, and Gerber SA (2011). Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal 4, rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitevski-LeBlanc J, Fradet-Turcotte A, Kukic P, Wilson MD, Portella G, Yuwen T, Panier S, Duan S, Canny MD, van Ingen H, et al. (2017). The RNF168 paralog RNF169 defines a new class of ubiquitylated histone reader involved in the response to DNA damage. eLife 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, and Rape M (2012). The ubiquitin code. Annu. Rev. Biochem 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Na K, Kwon MS, Kim H, Kim KS, and Paik YK (2009). Quantitative analysis of phosphopeptides in search of the disease biomarker from the hepatocellular carcinoma specimen. Proteomics 9, 3395–3408. [DOI] [PubMed] [Google Scholar]
- Lee DH, Acharya SS, Kwon M, Drane P, Guan Y, Adelmant G, Kalev P, Shah J, Pellman D, Marto JA, and Chowdhury D (2014). Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol. Cell 54, 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, and Sixma TK (2012). RNF168 ubiquitinates K13–15 on H2A/H2AX to drive DNA damage signaling. Cell 150, 1182–1195. [DOI] [PubMed] [Google Scholar]
- Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, and Lukas C (2016). 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol 23, 714–721. [DOI] [PubMed] [Google Scholar]
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, and Durocher D (2014). Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193. [DOI] [PubMed] [Google Scholar]
- Panier S, and Boulton SJ (2014). Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol 15, 7–18. [DOI] [PubMed] [Google Scholar]
- Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, and Lou Z (2011). MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 470, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino S, Michelena J, Teloni F, Imhof R, and Altmeyer M (2017). Replication-coupled dilution of H4K20me2 guides 53BP1 to pre-replicative chromatin. Cell Rep 19, 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penengo L, Mapelli M, Murachelli AG, Confalonieri S, Magri L, Musacchio A, Di Fiore PP, Polo S, and Schneider TR (2006). Crystal structure of the ubiquitin binding domains of rabex-5 reveals two modes of interaction with ubiquitin. Cell 124, 1183–1195. [DOI] [PubMed] [Google Scholar]
- Poulsen M, Lukas C, Lukas J, Bekker-Jensen S, and Mailand N (2012). Human RNF169 is a negative regulator of the ubiquitin-dependent response to DNA double-strand breaks. J. Cell Biol 197, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saredi G, Huang H, Hammond CM, Alabert C, Bekker-Jensen S, Forne I, Reverón-Gómez N, Foster BM, Mlejnkova L, Bartke T, et al. (2016). H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature 534, 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid JA, Berti M, Walser F, Raso MC, Schmid F, Krietsch J, Stoy H, Zwicky K, Ursich S, Freire R, et al. (2018). Histone ubiquitination by the DNA damage response is required for efficient DNA replication in unperturbed S phase. Mol. Cell 71, 897–910.e8. [DOI] [PubMed] [Google Scholar]
- Schwertman P, Bekker-Jensen S, and Mailand N (2016). Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol 17, 379–394. [DOI] [PubMed] [Google Scholar]
- Silacci M, Brack S, Schirru G, Mårlind J, Ettorre A, Merlo A, Viti F, and Neri D (2005). Design, construction, and characterization of a large synthetic human antibody phage display library. Proteomics 5, 2340–2350. [DOI] [PubMed] [Google Scholar]
- Sloper-Mould KE, Jemc JC, Pickart CM, and Hicke L (2001). Distinct functional surface regions on ubiquitin. J. Biol. Chem 276, 30483–30489. [DOI] [PubMed] [Google Scholar]
- Swatek KN, and Komander D (2016). Ubiquitin modifications. Cell Res 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, Usher JL, Kueck AF, Gladkova C, Mevissen TET, Pruneda JN, Skern T, and Komander D (2019). Insights into ubiquitin chain architecture using Ub-clipping. Nature 572, 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang H, Liu J, Cheruiyot A, Lee JH, Ordog T, Lou Z, You Z, and Zhang Z (2016). USP51 deubiquitylates H2AK13,15ub and regulates DNA damage response. Genes Dev 30, 946–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Simicek M, Schubert A, and Komander D (2015a). Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, and Komander D (2015b). Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J 34, 307–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Benlekbir S, Fradet-Turcotte A, Sherker A, Julien JP, McEwan A, Noordermeer SM, Sicheri F, Rubinstein JL, and Durocher D (2016). The structural basis of modified nucleosome recognition by 53BP1. Nature 536, 100–103. [DOI] [PubMed] [Google Scholar]
- Xu M, Skaug B, Zeng W, and Chen ZJ (2009). A ubiquitin replacement strategy in human cells reveals distinct mechanisms of IKK activation by TNFalpha and IL-1beta. Mol. Cell 36, 302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Akutsu M, Reyes-Turcu F, Enchev RI, Wilkinson KD, and Komander D (2011). Polyubiquitin binding and cross-reactivity in the USP domain deubiquitinase USP21. EMBO Rep 12, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, and Mohammed S (2013). Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res 12, 260–271. [DOI] [PubMed] [Google Scholar]
- Zuberbühler K, Palumbo A, Bacci C, Giovannoni L, Sommavilla R, Kaspar M, Trachsel E, and Neri D (2009). A general method for the selection of high-level scFv and IgG antibody expression by stably transfected mammalian cells. Protein Eng. Des. Sel 22, 169–174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original data for figures in the paper are available at Mendeley Data: https://data.mendeley.com/datasets/k4fmj3nzfz/draft?a=c22150bc-d2e4-4095-a10a-08d376a16abf