

Abstract
Human NK cells are effectors of the innate immune system that originate from hematopoietic precursors in the bone marrow. While stromal cell lines that support NK cell development from hematopoietic precursors are often used to generate mature NK cells from lymphoid precursors in vitro, the nature of contributing factors of these stromal cells to the generation of functionally mature NK cells has been poorly described. Previous studies have shown that developing NK cells adhere to, and migrate on, developmentally supportive stroma. Here, we describe the generation of cell-derived matrices (CDMs) from a commonly used murine fetal liver stromal cell line. These CDMs are derived directly from the same EL08.1D2 stromal cell line known to support NK cell differentiation and contain ECM structural components fibronectin and collagen. We demonstrate that CDMs support NK cell adhesion and migration with similar properties as intact cells. Further, we show that CDMs support NK cell maturation from lymphoid precursors in vitro, albeit with reduced cell survival compared to intact cell-based differentiation. Together, these results describe a cell-free system that supports NK cell development and that can serve as a useful model for studying the nature of the biochemical interactions between NK cell developmental intermediates and developmentally supportive substrates.
Keywords: cell migration, extracellular matrix, NK cell development
1. INTRODUCTION
Human NK cells originate from common lymphoid progenitors in the bone marrow, but likely undergo further maturation in peripheral tissues, of which secondary lymphoid tissue is the best described.1–4 While the contact-dependent signals that drive NK cell differentiation from precursors are not fully defined, this process can be recapitulated in vitro as a method to study NK cell development and ultimately define minimal requirements for the generation of mature, functional NK cells.
NK cell development in vitro frequently includes the use of stromal cell lines for co-culture with precursors in the presence of cytokines. Such stromal cell line co-culture methods lead to significant expansion of relatively mature NK cells that are functional for production of cytokines and lysis of susceptible target cells.5–8 The best characterized cell lines that support NK cell development are the OP9 or OP9-DL1 lines, derived from newborn mouse bone marrow,8 and EL08.1D2 murine fetal liver stromal cell lines.9 In addition, co-culture with bone marrow stroma, human splenic fibroblasts, mesenchymal stromal cells, and other stromal cell lines such as M2–10B4 and AFT024 can support NK cell differentiation from CD34+ precursors.10–14 Cell-free methods have also been designed and include the use of heparin-based Glycostem media, which functions to bind cytokines and help prevent their degradation while avoiding the use of xenogenic co-culture.15–17 While heparin-based methods can produce mature NK cells at the same frequency as stromal cell based methods, the use of stromal cells increases the expansion of developing NK cells and remains the most efficient means of generating the greatest number of mature cells.15 Notably, the effect of co-culture with supportive stromal cell lines is dependent upon direct cell-cell contact and the use of transwell barriers prevents NK cell development in these systems.5,10
In our previous studies, we sought to characterize contact-dependent interactions between developing NK cells and these stromal cells, and we previously defined and characterized cell migration of NK cell developmental intermediates while undergoing differentiation and migration. We showed that NK cell migratory phenotype correlates with developmental stage and that NKDI derived in vitro from CD34+ stem cells exhibited progressively greater cell velocity and less frequent cell arrest as they matured.18,19 With this knowledge, we hypothesized that cell-cell adhesions between NKDI and EL08.1D2 stroma drive cell migration while concurrently provide developmentally supportive signaling to promote NK cell development. We additionally observed that this cell migration was not disrupted following the loss of stromal cells from the co-culture, leading us to hypothesize that the extracellular matrix components that remained supported the ongoing migration of NK cell developmental intermediates.18
Here, we sought to generate and characterize cell-derived extracellular matrices from the EL08.1D2 stromal cell line. The generation of cell-free matrices has been previously described as a means of studying the mechanical properties of the ECM, which is comprised of structural proteins such as collagen, fibronectin, and laminin, and serves as a scaffold for cells in vivo.20,21 These studies have justified the use of cell-derived matrix as a model of cell activity within tissue as a preferable alternative to synthetic hydrogels such as Matrigel or collagen gels as they replicate the complexity of the “matrisome” with greater fidelity.22 Here, we use ECM derived directly from EL08.1D2 as a minimal model system in which only NK cell-ECM interactions determine cell adhesion, migration, and development. Using this model system, we partially elucidate requirements for NK cell differentiation and expansion, and identify ECM production by EL08.1D2 stroma as a contributing component of their support of NK cell development.
2 |. METHODS
2.1 |. CD34+ precursor and primary NK cell isolation
T and B cell lineage depletion was performed using RosetteSep Human Hematopoietic Progenitor Cell Enrichment Cocktail (Stemcell Technologies) and Ficoll-Paque density gradient centrifugation from routine red cell exchange apheresis performed at Texas Children’s Hospital or Columbia University New York Presbyterian Hospital. Following pre-incubation with RosetteSep, apheresis product was layered on Ficoll-Paque for density centrifugation at 900×g (2000 rpm) for 20 min (no brake). Cells were harvested from the interface and washed with PBS by centrifugation at 1500 rpm for 5 min then resuspended in FCS for cell sorting. T- and B-cell depleted cultures were incubated with Abs for CD34 (clone 561, PE conjugate, Biolegend, 1:100) prior to sorting. FACS sorting was performed using a BD Aria II cytometer with an 85 μm nozzle at 45 psi. Purity after sorting was >80%. Primary NK cells for short-term imaging were similarly enriched with NK cell RosetteSep and enrichment was confirmed by flow cytometric analysis.
For FACS analysis of NK cell developmental intermediates, a 6-color flow cytometry panel was performed on a Bio-Rad ZE5 Cell Analyzer using Abs for CD56 (clone HCD56, Brilliant Violet 605, Biolegend, 1:200), CD3 (clone SK7, Brilliant Violet 711 or Brilliant Violet 786, Biolegend, 1:100), CD16 (clone 3G8, PE-CF594 conjugate, BD, 1:300), CD94 (clone DX22, APC conjugate, Biolegend, 1:100), CD117 (clone 104D2, PE/Cy7 conjugate, Biolegend, 1:10 or PE conjugate, Biolegend, 1:100), perforin (clone dG9, Brilliant Violet 711, Biolegend, 1:100), and a Zombie Aqua Viability Dye (1:100, Biolegend). For intracellular perforin staining, cells were fixed and permeabilized with CytoFix/CytoPerm (BD Biosciences). Flow cytometry data analysis was performed with FlowJo 10 (BD Biosciences).
2.2. Cell culture
EL08.1D2 cells stromal cells (a gift from Dr. J. Miller, University of Minnesota) were maintained on gelatinized culture flasks at 32°C in 40.5% α-MEM (Life Technologies), 50% Myelocult M5300 (STEMCELL Technologies), 7.5% heat-inactivated FCS (Atlanta Biologicals) with β-mercaptoethanol (10−5 M), Glutamax (Life Technologies, 2 mM), penicillin/streptomycin (Life Technologies, 100 U/ml), and hydrocortisone (Sigma, 10−6 M). Culture media was supplemented with 20% conditioned supernatant from previous EL08.1D2 cultures.
NK92 cells (ATCC) were maintained in 10% FBS Myelocult H5100 (Stemcell Technologies) with IL-2 (200 U/ml). Cell lines were authenticated by flow cytometry and confirmed monthly to be mycoplasma free.
2.3 |. CDM preparation
To prepare plates for CDM production, 96-well cell culture plates were treated with 0.1% gelatin in ultrapure water (EmbryoMax, Millipore). Fifty microliters of 0.1% gelatin was deposited on 96-well plates and incubated at 37°C for 1 h. Following incubation, wells were washed twice with sterile PBS. Fifty microliters of 1% (v/v) glutaraldehyde (Sigma-Aldrich) was then added for a 30 min incubation at room temperature. Then, wells were washed twice with PBS and treated with 1 M glycine (Sigma) for 2 min at room temperature. Wells were washed with PBS and stored overnight at 4°C in EL08.1D2 cell culture media supplemented with 1% penicillin/streptomycin (Life Technologies).
EL08.1D2 cells were trypsinized and added to the gelatinized 96-well plates at a density of 104–105 cells/cm2 in a 150 μl volume. Cells were incubated at 32°C until the formation of a confluent monolayer. Cell culture media was then replaced with a solution of culture media supplemented with ascorbic acid (50 μg/ml; Sigma-Aldrich). Media was replaced every second day with freshly made ascorbic acid supplemented media for 7–10 days. After this treatment, cells were extracted by aspirating off the media and washing the cells three times with PBS. Stromal cells were denuded by adding 50 μL of extraction solution consisting of 2% (v/v) NH4OH (Sigma-Aldrich), 0.5% (v/v) Triton X-100 (Fisher Scientific) in PBS, which was first pre-warmed to 37°C. After a 3 min incubation at room temperature, extraction buffer was pipetted off and wells were washed twice with PBS. Next, 50 μL of DNase I (10 μg/ml; Sigma-Aldrich) was deposited and incubated at 37°C for 30 min. The DNase solution was then removed, followed by washing twice with PBS. The resulting CDMs were stored at 4°C in PBS supplemented with 1% (v/v) penicillin-streptavidin.
2.4 |. In vitro differentiation from CD34+ hematopoietic precursors
For in vitro CD34+ differentiation on EL08.1D2, 96-well plates were treated with 0.1% gelatin in ultrapure water to promote cell adherence. Fifty microliters of 0.1% gelatin was deposited on 96-well plates and incubated at room temperature for 30 min. Following incubation, wells were washed with PBS and left for an additional 60 min in a sterile culture hood to dry. Gelatinized 96-well plates were pre-coated with a confluent layer of EL08.1D2 cells at a density of 5–10 × 103 cells per well and then mitotically inactivated by irradiation at 300 rad. For in vitro CD34+ differentiation on CDM, 96-well plates containing cell-derived matrix were prepared as described above. For in vitro differentiation on low adhesion plates, low adhesion plates (Corning) were used without additional preparation.
Purified CD34+ hematopoietic precursors were cultured at a density of 2–20 × 103 cells per well on EL08.1D2 or CDM plates in Ham F12 media plus DMEM (1:2) with 20% human AB-serum, ethanolamine (50 μM), ascorbic acid (20 μg/ml), sodium selenite (5 μg/ml), β-mercaptoethanol (24 μM), and penicillin/streptomycin (100 U/ml) in the presence of IL-15 (5 ng/ml), IL-3 (5 ng/ml), IL-7 (20 ng/ml), Stem Cell Factor (20 ng/ml), and Flt3L (10 ng/ml) (all cytokines from Peprotech). Half-media changes were performed every 7 days, excluding IL-3 after the first week.
2.5 |. Western blotting
EL08.1D2 cells were lysed in RIPA buffer (Thermo Fisher) with 1% v/v HALT inhibitor (Thermo Fisher) at a concentration of 107 cells/ml. Samples were mixed with LDS buffer and DTT and separated on a 4–12% Bis-Tris gel. Proteins were transferred to a nitrocellulose membrane then blocked with skim milk. For CDM western blots, matrix was scraped directly into LDS/DTT solution and pre-heated at 95°C for 5 min. Fibronectin was detected with a polyclonal rabbit anti-fibronectin Ab (1:1000; Abcam) and actin with mouse anti-actin (1:5,000; Sigma) followed by either goat anti-mouse 680 or goat anti-rabbit 800 (1:10,000; LiCor). Proteins were detected on a LiCor Odyssey.
2.6 |. Acquisition of microscopy images
For tracking of NK developmental intermediates, cells were seeded at 2 × 103 cells per well on a 96-well ImageLock plate with confluent irradiated EL08.1D2 cells or cell-derived matrices, then imaged at 2 min intervals on the Incucyte ZOOM Live-Cell Analysis System (Sartorius) at 37°C in the phase-contrast mode (10× objective). Images were acquired for 120 min at an imaging frequency of 2 min and data were exported as TIFF image series for further analyses.
For microscopy, EL08.1D2 cells or cell-derived matrices were prepared on circular microscope coverslips in 24-well cell culture plates. Plates were blocked with heat denatured 2% BSA/PBS for 1 h at 37°C. Primary NK cells from lineage-depleted apheresis or NK92 cells were incubated on cells or cell-free matrix for 30 min in the presence of directly conjugated Abs to surface Ags CD56 (1:50, clone HCD56, Biolegend) and CD29 (1:50, clone TS2/16, Biolegend). Samples were fixed with 4% PFA/PBS for 20 min at room temperature, then washed 3 times with PBS and treated with 0.2% Triton X-100/PBS for 10 min. Samples were stained with polyclonal rabbit anti-collagen I Ab (1:100, Novus Biologicals), followed by phalloidin (Thermo Fisher). Samples were washed and coverslips were removed and mounted onto microscope slides in ProLong Gold (Life Technologies). Images were acquired on a Zeiss Axio Observer Z1 microscope with a Yokigawa W1 spinning disk and a 63× 1.40 NA objective. Illumination was by solid state laser and detection by Prime 95B sCMOS camera. Data were acquired in 3i Slidebook software and exported to Fiji23 for further analysis.
2.7 |. Image analysis and cell tracking
For analysis of collagen confluence, microscopy images showing collagen distribution were imported into Fiji, and a default auto threshold was applied to remove background which threshold was used. The confluence was then recorded based on the “Area Fraction” measurement. For actin footprint measurement, a freehand selection was drawn around cells and the area was then measured.
Cell tracking from Incucyte image series was performed using the TrackMate plugin in Fiji by manual tracking. Track statistics were exported from Fiji and the data were imported into MATLAB or Chemotaxis Tool, which was also used to generate rose plots. Mean speed, straightness, and arrest coefficient were calculated using MAT-LAB or Chemotaxis Tool. Mean speed was defined as the per-track average of all instantaneous speeds calculated at each frame. Accumulated distance was measured as the cumulative length of each track measured within a single imaging series. The straightness parameter was calculated by dividing the displacement by the total path length for each track. Arrest coefficient was defined as the percentage of time that the cell stays in arrest based on a threshold on instantaneous speed of 2 μm/min, or ≈1 cell body length per image interval.
2.8 |. Statistics
Statistical analysis was calculated using Prism 8.0 (GraphPad). Ordinary one-way ANOVA was used to compare track statistics. Mann-Whitney U test was used to compare actin footprint data. For all tests, P < 0.05 was considered significant.
3 |. RESULTS
3.1 |. Cell-derived matrices from EL08.1D2 contain common ECM components and support NK cell adhesion
Given our previous observation that cells migrated on previously EL08.1D2 confluent surfaces, we sought to define whether EL08.1D2 secrete extracellular components. Cell-derived matrices (CDMs) were generated from EL08.1D2 stroma using modified protocols from previously described methods.20,21,24 Briefly, cell culture plates were coated with gelatin to promote cell adhesion. As gelatin is soluble in aqueous solution, plates were then treated with glutaraldehyde to crosslink the gelatin and enhance its stability in solution.25 EL08.1D2 cells were added to the plates and allowed to adhere overnight. After a confluent monolayer was formed, ascorbic acid was cycled into the culture media for a period of 7 days in order to promote CDM formation. Cells were then denuded with extraction buffer containing NH4OH and Triton X-100 and the matrix was further treated with DNase to ensure that cellular and nuclear debris were removed (summarized in Fig. 1).
FIGURE 1. Workflow for generation of cell-free matrices from EL08.1D2 stroma.
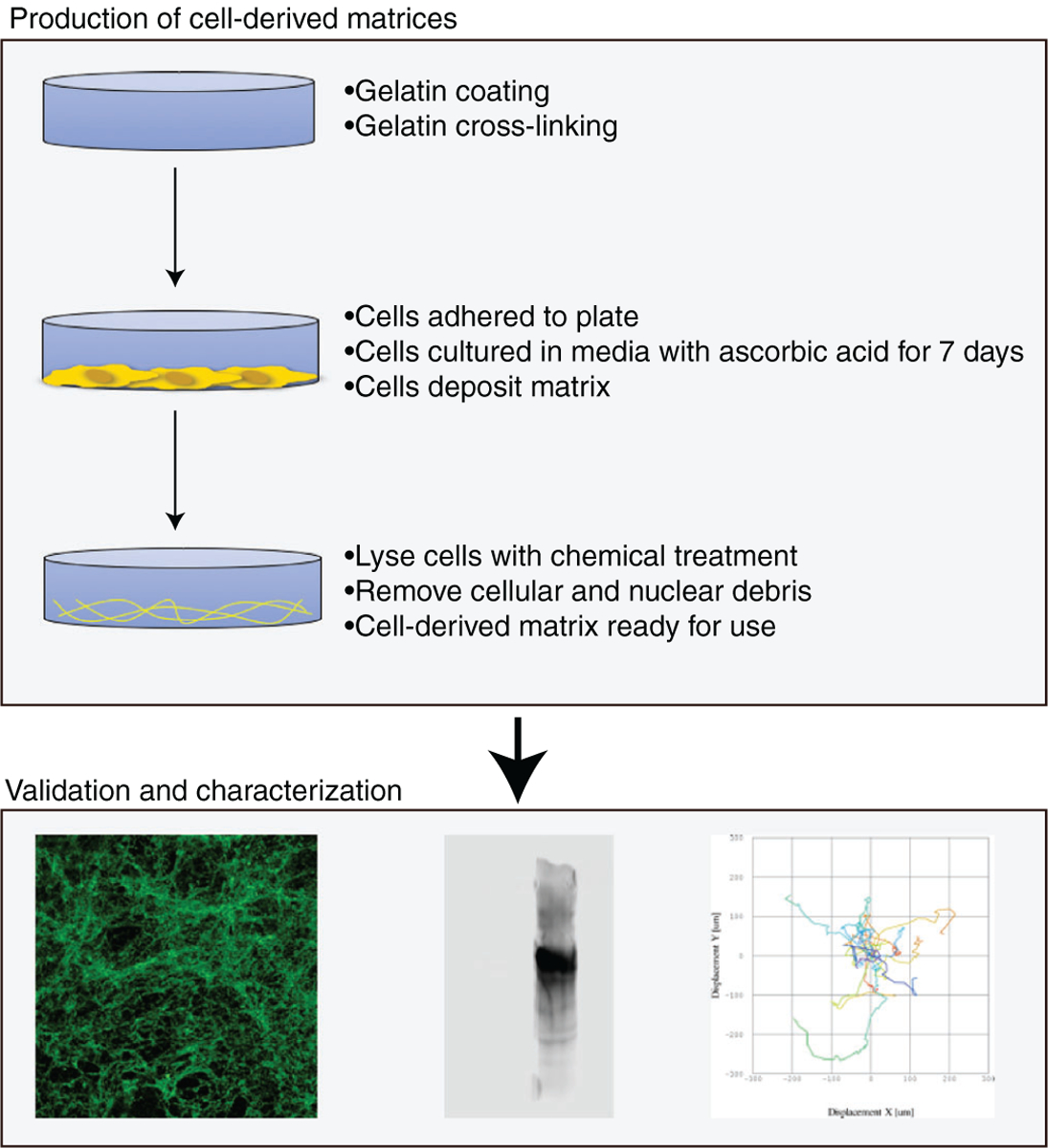
An overview of the protocol for the generation of CDM. Cell culture plates are coated with gelatin and seeded with EL08.1D2 stromal cells. Cells are allowed to adhere overnight and grown until confluent. Once confluent, ascorbic acid is cycled into the media for 7 days to promote matrix formation. During matrix extraction, cells are lysed with a chemical treatment and nuclear debris is removed with DNAse. CDMs can then be validated by microscopy and western blotting, or used for functional studies such as NK cell migration
To determine whether this culture method was inducing the secretion of ECM components from EL08.1D2 cells, CDMs were stained with collagen I Ab and imaged by confocal microscopy. Analysis of microscopy images revealed that cell-derived matrices contained a collagen network (Fig. 2A, top left). The network of collagen that we detected was significantly more confluent than that of intact EL08.1D2 stromal cells, on which we measured little or no detectable collagen I (Fig. 2A, bottom left). As a negative control for collagen I staining, we imaged microscopy slides with or without the treatment used for extracting cell-free matrices and found that collagen was not detected in either condition, confirming that treatment for matrix production and cell lysis did not induce nonspecific binding of Ab to the slides (Fig. 2A, right panels). Our observations were quantified by measuring the mean confluence of collagen I signal, which demonstrated that cell-free matrices were 13.76 ± 9.99% confluent versus 0.29 ± 0.50% confluence in intact cells (Fig. 2B). The negative control microscopy slides without EL08.1D2 stroma did not display a collagen network, even after treatment for CDM production, with mean confluences of 0.009 ± 0.016% and 0.467 ± 0.552% for cell-free controls with and without CDM treatment, respectively. To further confirm these results, we performed Western blots on solubilized CDM and EL08.1D2 cell lysates for fibronectin and actin content. Indeed, fibronectin content was detected from solubilized CDM but not EL08.1D2 lysates (Fig. 2C). These experiments, which additionally detected actin in lysates from intact EL08.1D2 cells but not CDM lysate, demonstrated that cell-derived matrices were being produced from EL08.1D2 stroma.
FIGURE 2. Cell-derived matrices contain ECM components and support NK cell adhesion.
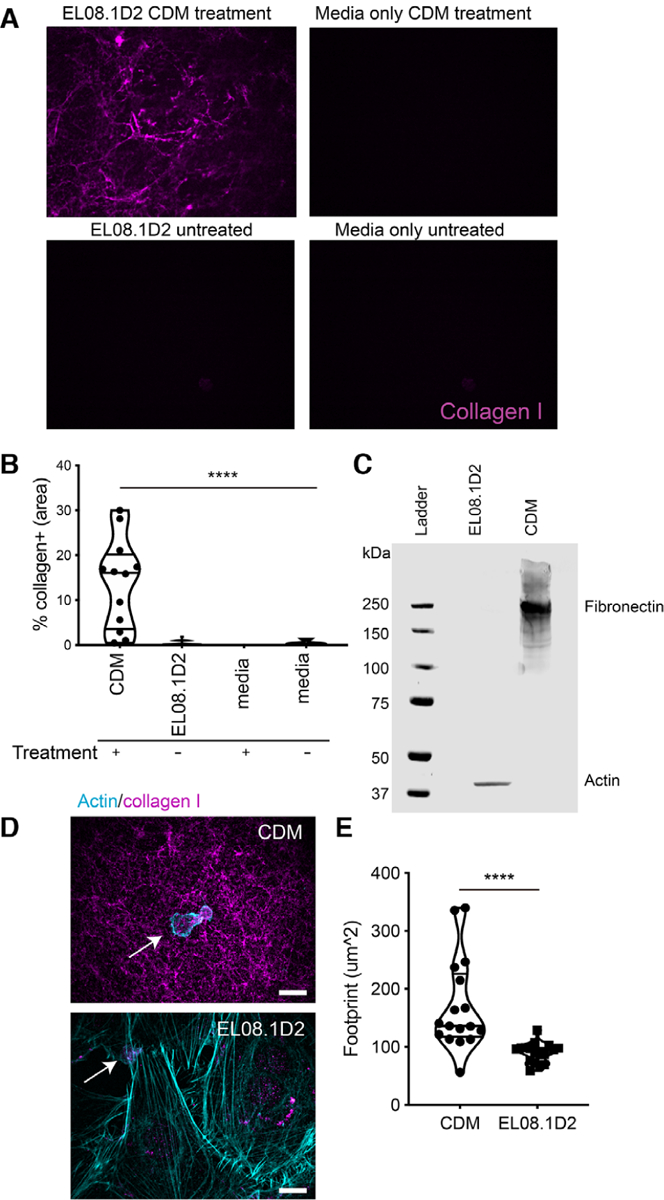
CDMs were generated as described in the text and compared with confluent intact EL08.1D2 cells or untreated control conditions. (A) Fluorescence microscopy images of conditions used to validate CDM production. Only slides containing EL08.1D2 that underwent treatment for CDM production (top left) produced a collagen network. Slides with EL08.1D2 that were untreated (bottom left), slides without EL08.1D2 that underwent treatment for CDM production (top right), and slides without EL08.1D2 that were untreated (bottom right) did not generate detectable collagen. (B) The area of the collagen network was measured for each condition and expressed as a frequency of the total field of view. Error bars represent sd. Means with significant differences were determined by one-way ANOVA with Tukey’s multiple comparison test (****P < 0.0001). n = 12–15 fields of view for CDM and EL08.1D2 conditions, 3–5 fields of view for negative controls. (C) CDM were solubilized in lysis buffer or cell lysates were prepared from intact EL08.1D2 stromal cells. Proteins were separated by SDS-PAGE and fibronectin or actin were detected by Western blotting. (D) Confocal microscopy images showing NK92 adhered to CDM or intact EL08.1D2 cells following immunostaining for collagen (magenta) or actin (cyan). Arrows denote NK92 cells, images shown as maximum projection of 29 Z slices. Scale bar = 10 μm. (E) Quantification of cell footprint in each condition. Error bars indicate sd. Significant differences between means were determined by Mann-Whitney U test (****P < 0.0001). n = 17 measurements per condition
To determine if CDMs supported NK cell adhesion, we imaged the human NK cell line NK92 cells incubated on CDM or EL08.1D2 stroma. NK92 were seeded on each substrate, allowed to adhere for 30 min, and then fixed and stained with phalloidin and Abs against collagen (Fig. 2D). Confocal microscopy revealed that NK92 adhered to both CDM and EL08.1D2 stroma, although NK92 on CDMs seemed to display greater spreading, including long, thin filopodia-like extensions. Quantitative image analysis confirmed that NK92 cells exhibited a significantly larger actin footprint on CDMs compared to EL08.1D2 stroma, with a mean footprint of 165.0 ± 60.4 μm2 on CDMs compared to 90.2 ± 16.9 μm2 on stroma (Fig. 2E). Together, these data demonstrate that EL08.1D2 secrete ECM proteins that form collagen and fibronectin-containing matrices that supports human NK cell adhesion and spreading.
3.2 |. CDMs support adhesion and migration of primary NK cells
Having confirmed that CDMs supported NK92 adhesion, we repeated these experiments with freshly isolated ex vivo NK cells to determine if primary cells also adhered to CDM. Ex vivo NK cells were isolated from human peripheral blood, incubated on CDM or EL08.1D2 stroma, and fixed and stained with phalloidin and Abs against, integrin β1 (CD29) and CD56. Confocal microscopy of cells again showed the presence of filopodia-like extensions characterized by integrin β1 and CD56 enrichment, demonstrating that integrin β1 is important for adhesion to both stroma and ECM (Fig. 3A). Z-stack information revealed that CDMs were somewhat 3-dimensional with a depth comparable to that of the EL08.1D2 monolayer as previously reported,20 and NK cells were capable of invading into the CDM. Once again, primary NK cells had greater spreading on CDMs, with a mean actin footprint of 40.0 ± 12.3 μm2 on CDM compared to 36.2 ± 11.8 μm2 on stroma (Fig. 3B).
FIGURE 3. Primary NK cells adhere to and migrate on cell-free matrices.
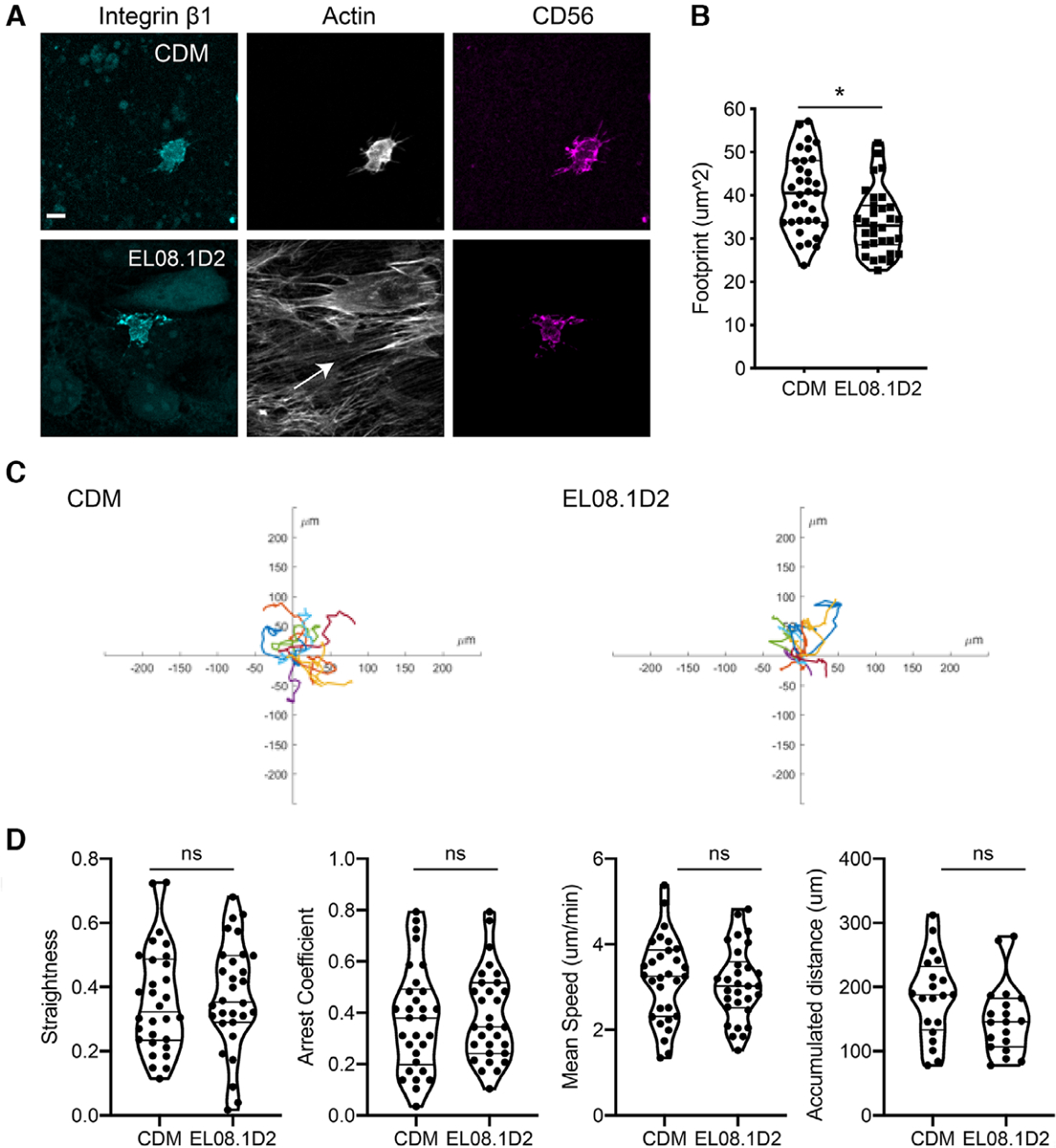
Freshly isolated primary NK cells were adhered to either EL08.1D2 or cell-derived matrix. (A) cells were co-incubated for 30 min and then fixed and immunostained as indicated followed by confocal microscopy. Images show maximum intensity projection of z-stacks. Scale bar = 10 μm. (B) Quantification of cell area in each condition. Error bars indicate sd. Means with significant differences were determined by Mann-Whitney U test (*P < 0.05). (C) Primary NK cells were isolated from peripheral blood and seeded on substrates and imaged every 2 min. Cell migration was tracked following image acquisition. Rose plots of primary NK cell tracks in each condition are shown. (D) Cells were tracked and migratory parameters were measured. n = 19, 20 cells from 2 individual healthy donors. Means were analyzed by Mann-Whitney test
As CDMs supported adhesion of both primary NK and NK92 cells, we hypothesized that cell migration of primary NK cells on CDM would be comparable to that of cells on stroma. Primary cells were seeded on either CDM, EL08.1D2, or low-adhesion plates coated with a hydrophilic, neutrally charged hydrogel to inhibit cell attachment. Immediately after cell addition, live-cell imaging was performed with image acquisition every 2 minutes. Cells were tracked from live cell imaging series (Fig. 3C). We found that freshly isolated primary NK cells had comparable migratory phenotypes on CDM and EL08.1D2 (Fig. 3D). In contrast, cells did not migrate or adhere on low adhesion plates and cell motions that were detected in imaging were predominantly drift of non-adherent cells (not shown). These results demonstrate that CDMs support integrin-dependent adhesion and migration of primary NK cells.
3.3 |. CDMs can partially support NK cell development from hematopoietic precursors
Having established that CDMs support NK cell adhesion and migration, and given that EL08.1D2 stroma support NK cell development, we sought to assess NK development on CDMs. CD34+ hematopoietic precursors were isolated from peripheral blood and seeded on either CDM, stroma, or low adhesion plates. Flow cytometry analysis for developmental markers was performed weekly. At 21 days, NKDI showed similar maturation on EL08.1D2 and CDMs but failed to survive on low adhesion plates (Fig. 4A). Expression of CD56 and CD94 was consistently observed for the first 2 conditions, denoting that NK cell developmental intermediates had successfully reached stage 4 of NK development. However, only ~63% of NKDIs in the CDM condition were positive for CD56 expression, whereas 80.3% of NKDIs were positive in the EL08.1D2 condition (Fig. 4B). Previous studies have identified the promotion of cell survival and proliferation by EL08.1D2 as a key contributing factor to their efficiency at generating mature NK cells15. In keeping with these data, cell counts showed that NK survival and proliferation was decreased in the CDM condition (Fig. 4C). While fewer cells were generated in the CDM condition, gating on CD56+ cells to define NK cell developmental stage 3 (CD117+CD94−CD16−), stage 4 (CD117+/−CD94+CD16−), and stage 5 (CD117−CD94+/−CD16+)4 showed no significant differences in their frequency from those generated on intact stroma (Fig. 4D). Furthermore, expression of perforin was similar between cells produced on CDMs when compared to those generated on EL08.1D2 cells, suggesting that cells that matured on CDMs had lytic potential (Fig. 4E).
FIGURE 4. CDM partially support NK cell development.
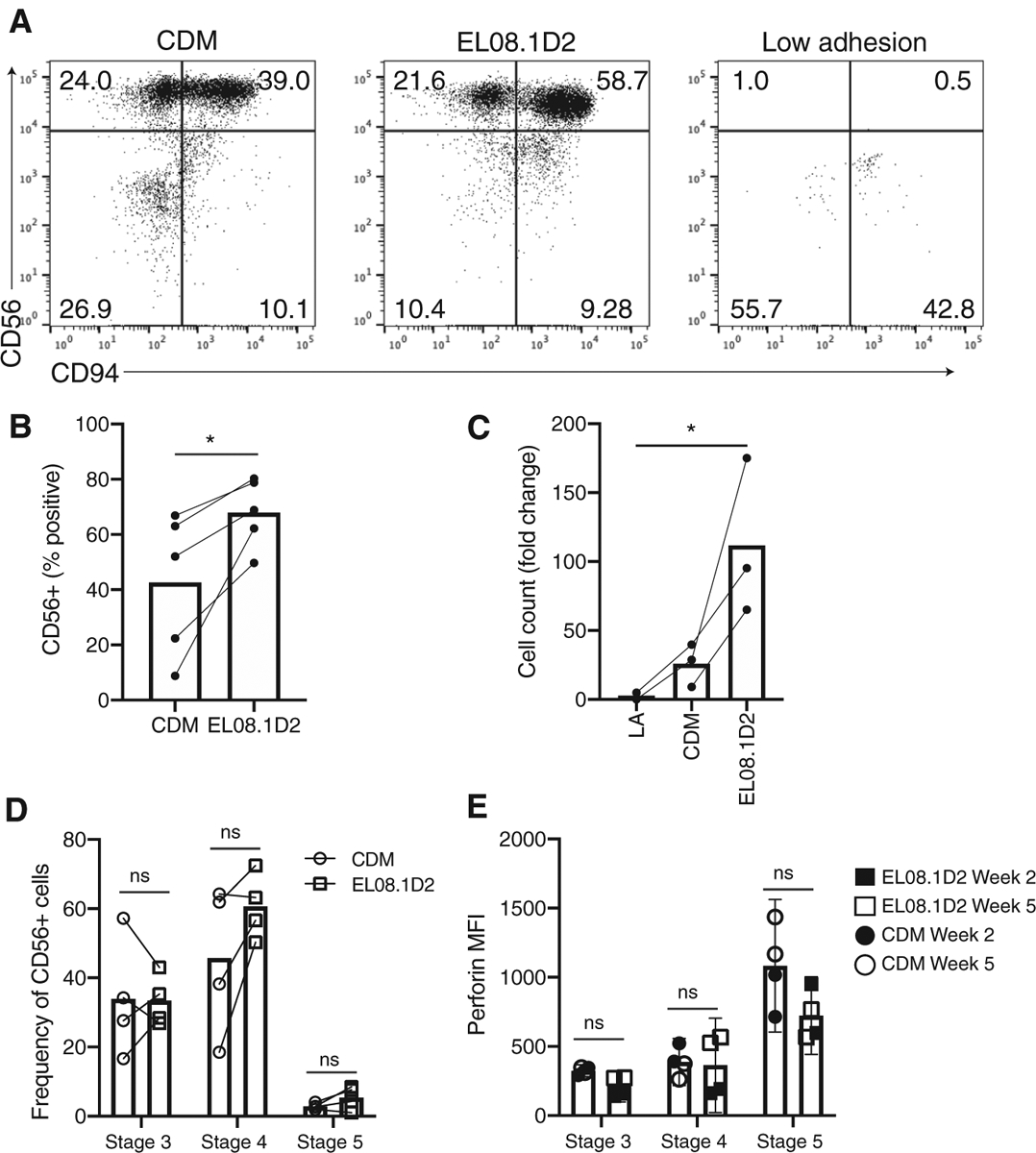
CD34+ HSCs were isolated from peripheral blood and seeded on CDM, EL08.1D2, or low adhesion plates. (A) After 21 days cells were harvested and analyzed by flow cytometry to determine cell maturation. Cells were gated on FSC/SSC and a live-dead marker for live lymphocytes. Representative flow cytometry plots are shown from 2 independent repeats with different donors. (B) Percentage of cells positive for CD94 or CD56 following culture of CD34+ precursors on paired CDM and EL08.1D2 conditions. n = 4 (CD94), 5 (CD56). *P < 0.05 by 2-tailed paired t-test. (C) Cell expansion shown as fold change from number of CD34+ precursors seeded at day 0. n = 2 (low adhesion), 3 (CDM and EL08.1D2) independent replicates from different healthy donors. *P < 0.05 by ordinary 1-way ANOVA with multiple comparisons. (D) Frequency of CD56+ cells in NK cell developmental stage 3 (CD56+CD117+CD94−), stage 4 (CD56+CD94+CD16−), or stage 5 (CD56+CD94+/−CD16+) at day 21 of differentiation. (E) Expression of perforin in mature NK cells differentiated on CDM or EL08.1D2. Cells were gated on stage 3 (CD56+CD117+CD94−) stage 4 (CD56+CD94+CD16−) or stage 5 (CD56+CD94+/−CD16+). n = 4 biological replicates analyzed at day 14 (closed symbols) or day 35 (open symbols) of culture. ns = not significant by two-way ANOVA with Sidak’s multiple comparisons test
Together, these results suggest a requirement for adhesion/migration for NK cell development; HSCs on low adhesion plates failed to develop into mature NK cells in contrast to those plated on CDM or EL08.1D2, which proliferated and matured. However, both cell maturation and survival of NKDI on CDM was inferior to that of the EL08.1D2 plates, suggesting that adhesion is necessary but not sufficient for NK development.
3.4 |. NK cell developmental intermediates undergo migration on CDM and EL08.1D2
Given that NKDI were capable of maturing on cell-derived matrices, we hypothesized that they would have similar migration behavior on CDM as has been previously described for cells undergoing maturation on EL08.1D2.18,19 CD34+ hematopoietic precursors were isolated from peripheral blood and seeded on either CDM or EL08.1D2. In conjunction with the weekly flow cytometry for developmental markers, cells were also imaged every 2 min and cell migration was tracked at weekly time points (Fig. 5A). We found that the properties of tracks did not significantly differ between the two conditions at each weekly time point measured (Fig. 5B–D). For example, the mean speed of tracks was 1.82 ± 1.18 μm/min on EL08.1D2 after 0 days, and 1.94 ± 1.20 μm/min on CDMs (Fig. 5C). After 21 days, this increased to 3.49 ± 0.89 μm/min on EL08.1D2 and 3.42 ± 1.18 μm/min on CDMs; this was consistent with previously reported migration speeds of mature NK cells in similar conditions.19,26 Similarly, after 0 days, mean straightness was 0.28 ± 0.19 on EL08.1D2 and 0.25 ± 0.14 on CDM, and mean arrest coefficient was 0.45 ± 0.26 on EL08.1D2 and 0.38 ± 0.13 on CDM (Fig. 5C and D). The only parameter in which we detected significant differences was arrest coefficient at 14 days, which was higher on CDMs. This difference was transient, however, as cells at 7 and 21 days had similar arrest coefficients. Taken together with our primary NK cell and cell line data, these data support the observation that CDM can support the migration of both mature and immature NK cells in a similar capacity as intact cells.
FIGURE 5. NKDIs have comparable migration on CDM and EL08.1D2.
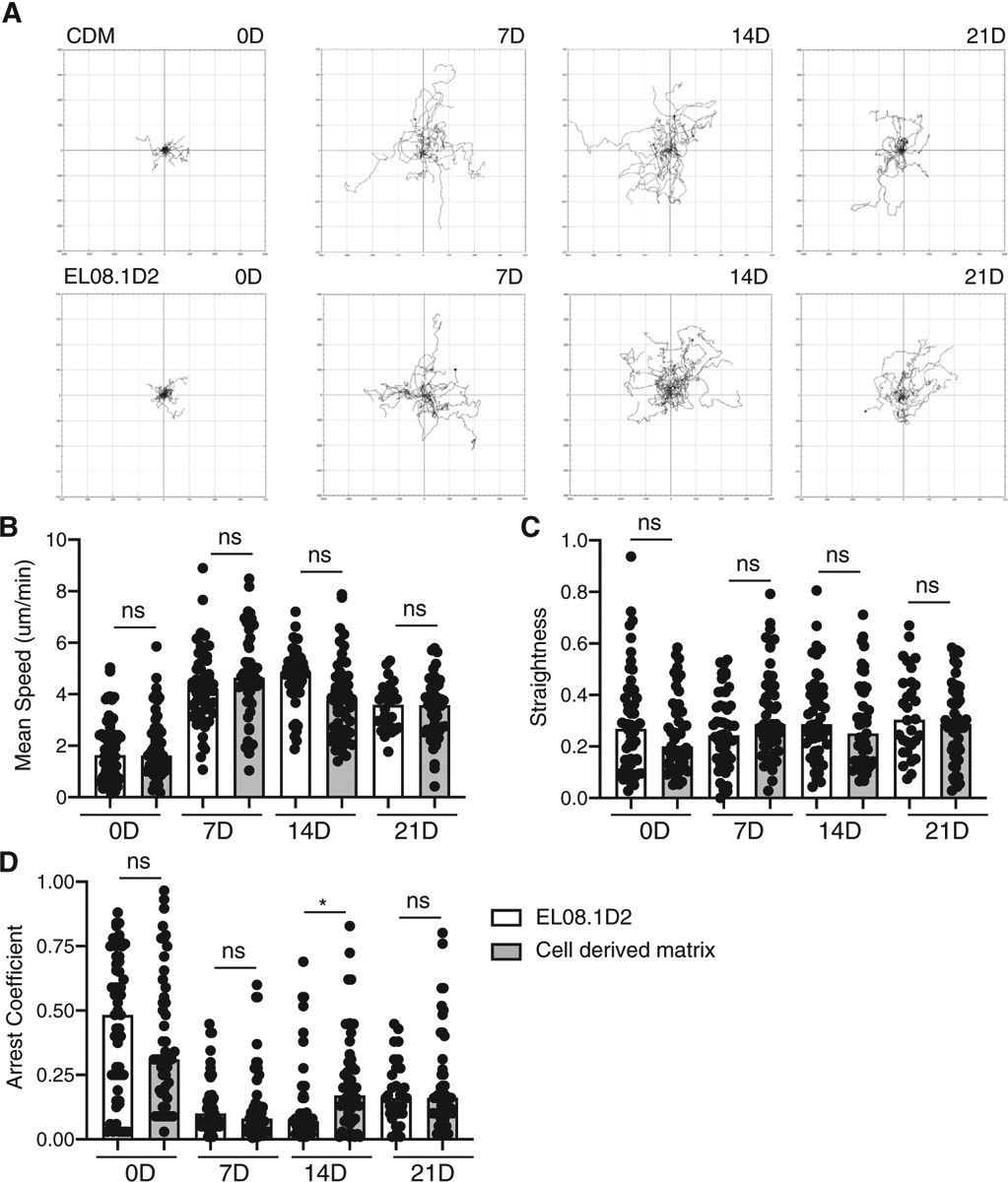
CD34+ HSCs were isolated from peripheral blood and seeded on CDM or EL08.1D2 stroma. Cell migration was imaged every 2 min by phase contrast imaging in an in-incubator imaging system. (A) Representative plots of cell tracks on CDM and EL08.1D2 at each week. The mean speed (B), straightness (C), and arrest coefficient (D) of cell tracks was determined. Error bars indicate sd. Means with significant differences were determined by ordinary 1-way ANOVA with Tukey’s multiple comparison test (*P < 0.05). n = 30–55 tracks pooled from 3 biological replicates
4. DISCUSSION
The use of stromal-based cell co-culture methods as a means of generating mature NK cells has been well described and has the potential to improve the immunotherapy approaches and provide insights into requirements for human NK cell development. However, the biochemical, physical and mechanical contributions of stromal cells to this process has not been fully described. Here, we describe the generation of fibronectin- and collagen-containing cell-derived matrices from EL08.1D2 stromal cells that can act as a substrate for NK cell adhesion and migration. We further show that these matrices can partially support in vitro NK cell development but with impaired expansion of NK cell developmental intermediates. These data extend our understanding of the nature of the contribution of stromal cells to lymphocyte development and support previous observations that stromal cells could be contributing ECM components to create a supportive microenvironment in co-cultures with NK cell developmental intermediates18.
Cell-free matrices have been well-described as a powerful tool for the studies of cell migration, tumor properties, and tissue regeneration.20,21,27 They provide a useful model that mimics both the complex composition and substrate stiffness28 of the in vivo cellular environment, while providing a substrate that is highly amenable to imaging by light microscopy and studies of cell motility and invasion. CDMs derived from various cell types have unique ECM components that reflect their unique matrisomes, however key ECM proteins including collagen, laminins, and fibronectin are conserved. While here we limited our characterization of CDMs to the presence of collagen and fibronectin, future studies will focus on higher-resolution analyses of both the matrix components and secreted factors from supportive and non-supportive cell lines.
Our detection of collagen and fibronectin in EL08.1D2-derived CDM, in conjunction with the adhesion and migration of primary NK cells and NK cell developmental intermediates, suggests that integrins are mediating interactions between cells and CDMs. Integrins are heterodimeric receptors that bind a number of ECM components, and the asparagine-glycine-aspartic acid (RGD) peptide is the basis of recognition of fibronectin by as many as 12 of the >20 known integrins. While the expression of integrins on hematopoietic cells is more restricted than that of adherent cells, human NK cells and developmental intermediates express integrin β1 and α4β1 (VLA-4) and α5β1 (VLA-5), which mediate adhesion of freshly isolated human NK cells to fibronectin.29 Their expression and our observations are consistent with the previously reported dependency of adherent cells on β1 integrin for adhesion and migration in 3D cell-free matrices.27 The increased cell spreading that we observed following short-term incubation of cells on CDMs relative to intact cells suggests that the properties that mediate this adhesion may differ from those of intact EL08.1D2 cells. This observation could reflect a relatively greater density of collagen and fibronectin on the CDMs when compared to intact cells, it could also be indicative of greater substrate stiffness induced by the glutaraldehyde cross-linking step.28 Regardless, it is notable that both primary and in vitro-derived NK cells migrated on CDM with no apparent differences in migratory properties from those cells migrating on intact EL08.1D2 cells. Specifically, we did not detect haptotaxis induced by matrix topology, as has been reported for cells migrating on fibroblast-derived ECM.20,30,31 This likely reflects differences in the composition or structure of the matrices between stromal cell-derived matrices and fibroblast-derived matrices, as well as different requirements for haptotaxis between immune cells and those that undergo non-amoeboid cell migration.32 Taken together with the adhesion data described above, however, the conserved patterns of migration seen on CDMs and intact cells suggests that the migration of NK cell developmental intermediates in an in vitro differentiation system can occur independently of cell-associated ligands, including ICAM and VCAM.
Whereas migration did not require the presence of intact cells, direct contact between NK cell developmental precursors and stroma greatly increased the expansion of precursors in our system as previously reported.15 The nature of the survival or proliferation signaling provided by stroma remains unknown, however previous studies have implicated conserved pathways such as Wnt and Notch, or secreted factors such as cytokines, in providing these signals.15 Additionally, bidirectional signaling between precursors and stroma may affect these interactions, as has been demonstrated for co-culture between EL08.1D2 and leukemic B cells.33 More explicit dissection of the effect of conditioned media or the use of previously co-cultured stroma would provide additional insight into the respective roles of soluble and contact-dependent signaling in these co-culture systems. Given the potential of NK cells as immunotherapeutic tools, the generation of mature NK cells in vitro without the use of xenogenic stroma remains an important challenge in the field34. As with other stroma-free methods, we sought to define whether the use of cell-derived matrix was sufficient for the production and proliferation of functional NK cells. We found here that, similarly to heparin-based media, while mature, perforin-expressing NK cells were produced on cell-derived matrices, the proliferation of these cells was significantly less than that of those on EL08.1D2 stromal cells. Whether the presence of CDMs offers a similar mechanism of support as heparin-based media, which is thought to support NK cell development at least in part by stabilizing cytokines, is unclear. However, with our system, we are now poised to leverage a reductionist approach whereby we test necessary and sufficient recombinant ligands that can support both differentiation and proliferation, potentially using cell-free matrix or ECM as a scaffold for additional ligands, as has been pursued for similar studies of T cell development.35 This approach, combined with further cell biological studies of the nature of receptor-ligand interactions between stromal cells and NK cell precursors, can ideally be used to better tailor systems that support the production of mature NK cells with desired functional phenotypes.
In summary, here we describe the generation of ECM components from the EL08.1D2 murine fetal liver cell line. Primary NK cells and NK cell developmental intermediates adhere to these cell-free matrices and undergo non-directed migration that is similar to migration on intact cells. Taken together, these data provide a greater understanding of the nature of developmental support provided by commonly used stromal feeder cells.
ACKNOWLEDGMENT
This work was supported by NIH-NIAID R01AI137073 to E.M.M. The authors wish to thank Dr. Michael Diehl for helpful discussions.
Abbreviations:
- CDM
cell-derived matrix
- NKDI
NK cell developmental intermediate
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Eissens DN, Spanholtz J, van der Meer A, et al. Defining early human NK cell developmental stages in primary and secondary lymphoid tissues. PLoS One. 2012;7:e30930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freud AG, Yokohama A, Becknell B, et al. Evidence for discrete stages of human natural killer cell differentiation in vivo. J Exp Med. 2006;203:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freud AG, Becknell B, Roychowdhury S, et al. A human CD34(+) subset resides in lymph nodes and differentiates into CD56brightNatural killer cells. Immunity. 2005;22:295–304. [DOI] [PubMed] [Google Scholar]
- 4.Freud AG, Keller KA, Scoville SD, et al. NKp80 defines a critical step during human natural killer cell development. Cell Rep. 2016;16: 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grzywacz B, Kataria N, Sikora M, et al. Coordinated acquisition of inhibitory and activating receptors and functional properties by developing human natural killer cells. Blood. 2006;108:3824–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woll PS, Martin CH, Miller JS, et al. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol. 2005;175:5095–5103. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann D, Spanholtz J, Osl M, et al. Ex vivo generated natural killer cells acquire typical natural killer receptors and display a cytotoxic gene expression profile similar to peripheral blood natural killer cells. Stem Cells Dev. 2012;21:2926–2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck RC, Padival M, Yeh D, et al. The notch ligands Jagged2, Delta1, and Delta4 induce differentiation and expansion of functional human NK cells from CD34 cord blood hematopoietic progenitor cells. Biol Blood Marrow Transplant. 2009;15:1026–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCullar V, Oostendorp R, Panoskaltsis-Mortari A, et al. Mouse fetal and embryonic liver cells differentiate human umbilical cord blood progenitors into CD56-negative natural killer cell precursors in the absence of interleukin-15. Exp Hematol. 2008;36:598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Briard D, Brouty-Boyé D, Azzarone B, et al. Fibroblasts from human spleen regulate NK cell differentiation from blood CD34+ progenitors via cell surface IL-15. J Immunol. 2002;168:4326–4332. [DOI] [PubMed] [Google Scholar]
- 11.Herrera L, Salcedo JM, Santos S, et al. OP9 feeder cells are superior to M2–10B4 cells for the generation of mature and functional natural killer cells from umbilical cord hematopoietic progenitors. Front Immunol. 2017;8:755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller JS, Alley KA, McGlave P, Differentiation of natural killer (NK) cells from human primitive marrow progenitors in a stroma-based long-term culture system: Identification of a CD34 7 NK progenitor. Blood. 1994;83:2594–2601. [PubMed] [Google Scholar]
- 13.Zhao X, Weinhold S, Brands J, et al. NK cell development in a human stem cell niche: kIR expression occurs independently of the presence of HLA class I ligands. Blood Adv. 2018;2:2452–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller JS, McCullar V, Punzel M, et al. Single adult human CD34/Lin−/CD38− progenitors give rise to natural killer cells, B-Lineage cells, dendritic cells, and myeloid cells. Blood. 1999;93:96–106. [PubMed] [Google Scholar]
- 15.Dezell SA, Ahn Y-O, Spanholtz J, et al. Natural killer cell differentiation from hematopoietic stem cells: a comparative analysis of Heparin- and Stromal cell-supported methods. Biol Blood Marrow Transplant. 2012;18:536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spanholtz J, Preijers F, Tordoir M, et al. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One. 2011;6:e20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spanholtz J, Tordoir M, Eissens D, et al. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS One. 2010;5:e9221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee BJ, Mace EM, Acquisition of cell migration defines NK cell differentiation from hematopoietic stem cell precursors. Mol Biol Cell. 2017;28:3573–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mace EM, Gunesch JT, Dixon A, et al. Human NK cell development requires CD56-mediated motility and formation of the developmental synapse. Nat Commun. 2016;7:12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaukonen R, Jacquemet G, Hamidi H, et al. Cell-derived matrices for studying cell proliferation and directional migration in a complex 3D microenvironment. Nat Protoc. 2017;12:2376–2390. [DOI] [PubMed] [Google Scholar]
- 21.Franco-Barraza J, Beacham DA, Amatangelo MD, et al. Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr Protoc Cell Biol. 2016;71:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naba A, Clauser KR, Ding H, et al. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biology. 2016;49:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hellewell AL, Rosini S, Adams JC, A rapid, scalable method for the isolation, functional study, and analysis of cell-derived extracellular matrix. J Vis Exp. 2017:55051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L-J, Ou Y-C, The micro patterning of glutaraldehyde (GA)-crosslinked gelatin and its application to cell-culture. Lab on a Chip. 2005;5:979. [DOI] [PubMed] [Google Scholar]
- 26.Khorshidi MA, Vanherberghen B, Kowalewski JM, et al. Analysis of transient migration behavior of natural killer cells imaged in situ and in vitro. Integr Biol. 2011;3:770–778. [DOI] [PubMed] [Google Scholar]
- 27.Cukierman E, Pankov R, Stevens DR, et al. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. [DOI] [PubMed] [Google Scholar]
- 28.Soucy PA, Werbin J, Heinz W, et al. Microelastic properties of lung cell-derived extracellular matrix. Acta Biomater. 2011;7:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gismondi A, Morrone S, Humphries MJ, et al. Human natural killer cells express VLA-4 and VLA-5, which mediate their adhesion to fibronectin. J Immunol. 1991;146:384–392. [PubMed] [Google Scholar]
- 30.Dozynkiewicz MA, Jamieson NB, Macpherson I, et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev Cell. 2012;22:131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hakkinen KM, Harunaga JS, Doyle AD, et al. Direct comparisons of the morphology, migration, cell adhesions, and actin cytoskeleton of fibroblasts in four different three-dimensional extracellular matrices. Tissue Eng Part A. 2011;17:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo X, Aoun L, Biarnes-Pelicot M, et al. Different integrins mediate haptotaxis of T lymphocytes towards either lower or higher adhesion zones. bioRxiv. 2019;509240. [Google Scholar]
- 33.Lutzny G, Kocher T, Schmidt-Supprian M, et al. Protein kinase C-β-Dependent activation of NF-κB in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. Cancer Cell. 2013;23:77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rezvani K, Rouce RH, The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. 2015;6:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shukla S, Langley MA, Singh J, et al. Progenitor T-cell differentiation from hematopoietic stem cells using Delta-like-4 and VCAM-1. Nat Methods. 2017;14:531–538. [DOI] [PubMed] [Google Scholar]