

Summary
Background and objectives:
The International Collaboration on Cancer Reporting (ICCR) is a not-for-profit to develop evidence-based, internationally agreed-upon standardized data sets for each anatomic site, to be used throughout the world. Providing global standardization of pathology tumor classification, staging, and other reporting elements will lead to improved patient management and enhanced epidemiological research.
Methods:
Pheochromocytoma and paraganglioma are uncommon and are frequently overlooked in registry data sets. Malignant criteria have previously been defined only when there was metastatic disease.
Results:
With recent recognition of a significant inheritance association and the development of risk stratification tools, this data set was created in order to obtain more meaningful outcomes and management data, using similar criteria across the global pathology community. Issues related to key core and non-core elements, especially clinical hormonal status, familial history, tumor focality, proliferative fraction, adverse or risk stratification features, and ancillary techniques, are discussed in the context of daily application to these types of specimens.
Conclusions:
The ICCR data set, developed by an international panel of endocrine organ specialists, establishes a pathology-standardized reporting guide for pheochromocytoma and paraganglioma.
Keywords: Checklist, Data set, Synoptic reporting, Structured report, Paraganglioma, Pheochromocytoma, ICCR
1. Introduction
An accurate pathology report sets in motion patient management decisions and therapeutic options by providing all of the key diagnostic criteria and as much predictive information as possible to inform patient care [1,2]. Standardized, checklist-type reporting provides meaningful pathology information that can be interpreted uniformly across all patient settings, no matter where treatment may be implemented. Standardized cancer reporting data sets have been developed for national use in the United Kingdom, the United States of America, and Australia, but they are not internationally standardized or directly comparable. Variations in data elements, terminology, the data set structure, or recommended methodology may compromise interoperability of core data for research or benchmarking in cancer management. The classification of tumors has been internationally standardized for decades with the publication of the World Health Organization (WHO) Tumour Classification series (https://tumourclassification.iarc.who.int/), but the international harmonization of cancer pathology reporting has not been previously well developed. The International Collaboration on Cancer Reporting (ICCR) is a not-for-profit organization founded in 2010 and sponsored by an ever-expanding number of pathology organizations who see the value of this type of data set development. The organizations include the Royal Colleges of Pathologists of Australasia and the United Kingdom, the College of American Pathologists, the Canadian Association of Pathologists in association with the Canadian Partnership Against Cancer, the European Society of Pathology, the American Society of Clinical Pathology, and the Faculty of Pathology, Royal College of Physicians of Ireland.
The goal of the ICCR is to reduce the global burden of cancer data set development and duplication of effort by different organizations, by producing standardized, internationally agreed-upon, evidence-based data sets for cancer pathology reporting throughout the world, providing international benchmarking in cancer management.
2. Methods
Under the governance of the ICCR Board and Dataset Steering Committee, a worldwide network of dedicated expert pathologists and clinicians works toward developing standardized, evidence-based data sets to support structured pathology reporting of cancer worldwide. The ICCR has stated guidelines for the development of the data sets (http://www.iccr-cancer.org/datasets/dataset-development). An elected series champion for a suite of related anatomic sites (i.e., endocrine organs) oversees the selection of a chair and domain experts for an organ or anatomic site who serve as the Dataset Authoring Committee (DAC). Each DAC is composed of an expert panel with international experience, particularly important in endocrine organ tumors wherein there are worldwide geographical differences in inheritance and syndrome presentation and prevalence of different tumor types. The pheochromocytoma and paraganglioma DAC was composed of 11 pathologists from 6 countries, with several members having previous experience in national data set development. In order to accurately incorporate the complex clinical and laboratory findings in endocrine organ tumors, two endocrinologists (from the Netherlands and Australia) were included on the panel, along with a member of the ICCR governance team, to help provide terminology harmonization across the suite of data sets. A series of teleconferences between all of the members engendered lively discussion and comment about criteria selection, with the final document reached by consensus of the DAC members. To ensure a timely and quality-assured approach with minimum disruption to participating expert clinicians, each data set was developed with the services of a dedicated ICCR Project Manager following established processes of evidentiary review, international expert participation, and, finally, open international consultation, after which comments were reviewed and, when necessary, incorporated into the final data set before publication. The ICCR pheochromocytoma and paraganglioma data set is specific to resection specimens and some biopsies of tumors correctly considered on a risk spectrum [3]. When developing the data set, the expert panel distinguished between reporting of core elements and non-core elements. Core elements are considered essential for clinical management, staging, or prognosis and thus are mandatory reporting items. Reporting of core elements is supported by the National Health and Medical Research Council evidence level III-2 (based on prognostic factors among patients in a single arm of a randomized control trial) and above [4]; given the rarity of cases, this level of evidence may not always be available, and in that circumstance, it must meet with unanimous agreement by members of the expert committee. Though not considered mandatory, non-core elements are agreed-upon reporting elements that may be clinically important and recommended as good clinical practice or are not yet fully validated. This review will summarize the ICCR histopathology reporting guidelines for pheochromocytoma and paraganglioma, focusing on a discussion of the core elements for inclusion, while also giving an overview of non-core, but still recommended, elements that show promise in future management, but as yet do not have widespread adoption.
3. Scope
The data set was developed for the pathology reporting of adrenalectomy/partial adrenalectomy specimens for pheochromocytoma, other excisions for paragangliomas, and biopsies of related specimens (see Anatomic sites of paraganglia) [3]. Neuroblastoma, ganglioneuroblastoma, sarcoma, lymphoma, and metastasis to the adrenal medulla are not included. Adrenal cortical tumors are included in a separate data set [5]. The ICCR data sets include core and non-core elements, as highlighted previously, with the core elements considered to be the minimum reporting requirements for pheochromocytoma and paraganglioma, but including non-core elements to provide the flexibility to include additional elements that may be needed at the local level. There is significant variation in the strength of the evidence available for these tumors, with most data derived from retrospective case series due to the rare nature of the neoplasms. This review will summarize the ICCR Phaeochromocytoma and Paraganglioma Histopathology Reporting Guide in two sections, core and non-core elements, with a discussion of the requirements within each element, salient evidence, and practical issues around inclusion.
4. Anatomic sites of paraganglia
Paraganglia are neural crest–derived neuroendocrine organs that produce catecholamines as their usual hormonal product. They are divided into two groups, associated with sympathetic or parasympathetic nerves. Sympathetic paraganglia (sympathoadrenal) are further separated into two anatomic subgroups: the adrenal medulla and extra-adrenal sympathetic paraganglia. Tumors arising from the adrenal medulla are termed pheochromocytomas (Fig. 1), while tumors arising from extra-adrenal locations are called paragangliomas irrespective of their sympathetic or parasympathetic origins. While parasympathetic paragangliomas have traditionally been referred to as head and neck paragangliomas (carotid body [Fig. 2], jugulotympanic, vagal, and laryngeal), some sympathetic paragangliomas may arise from the cervical sympathetic chain.
Fig. 1.
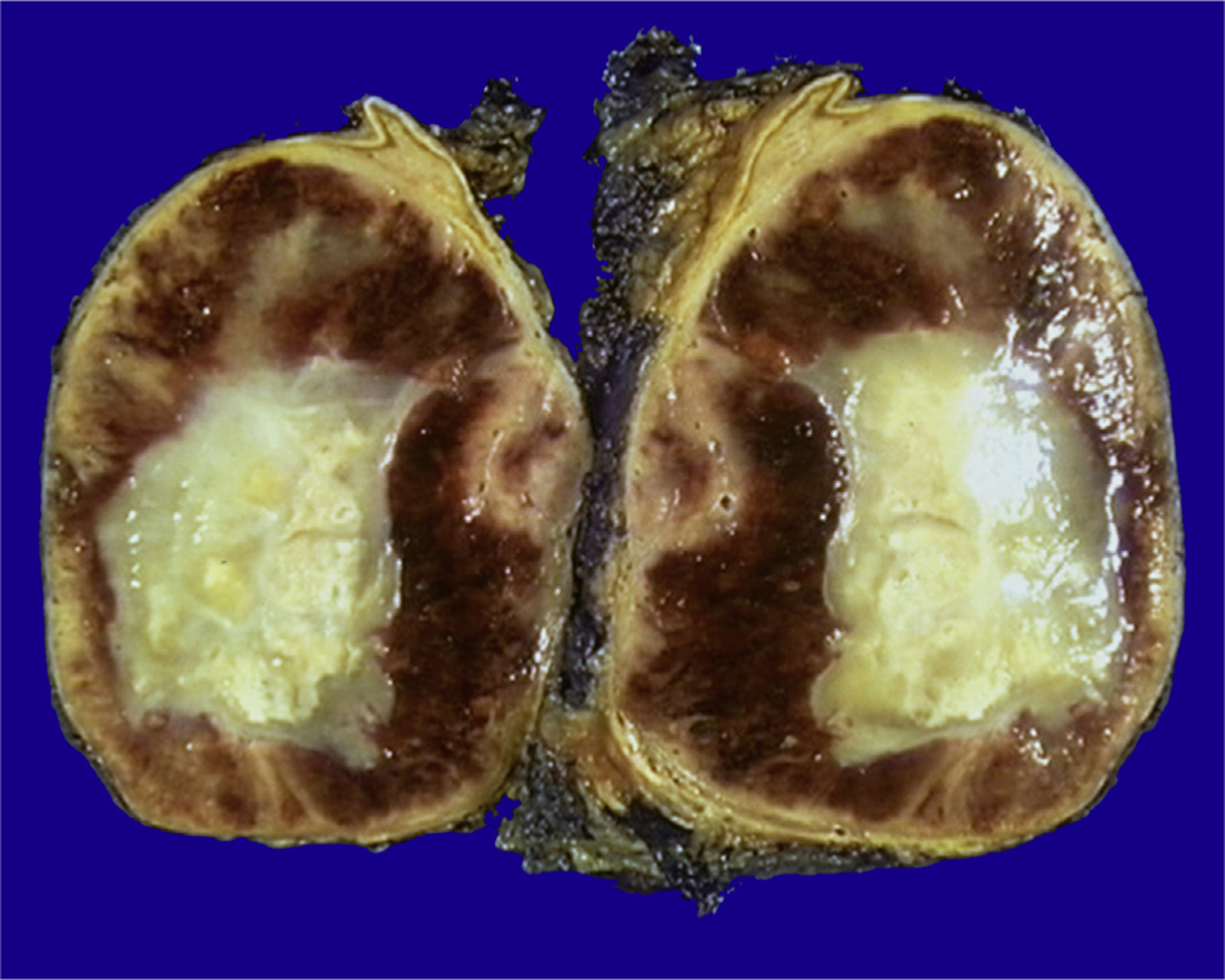
A gross specimen of adrenal gland pheochromocytoma with a yellow, necrotic center. The rim of the residual adrenal cortex can be seen at the edge of the tumor.
Fig. 2.
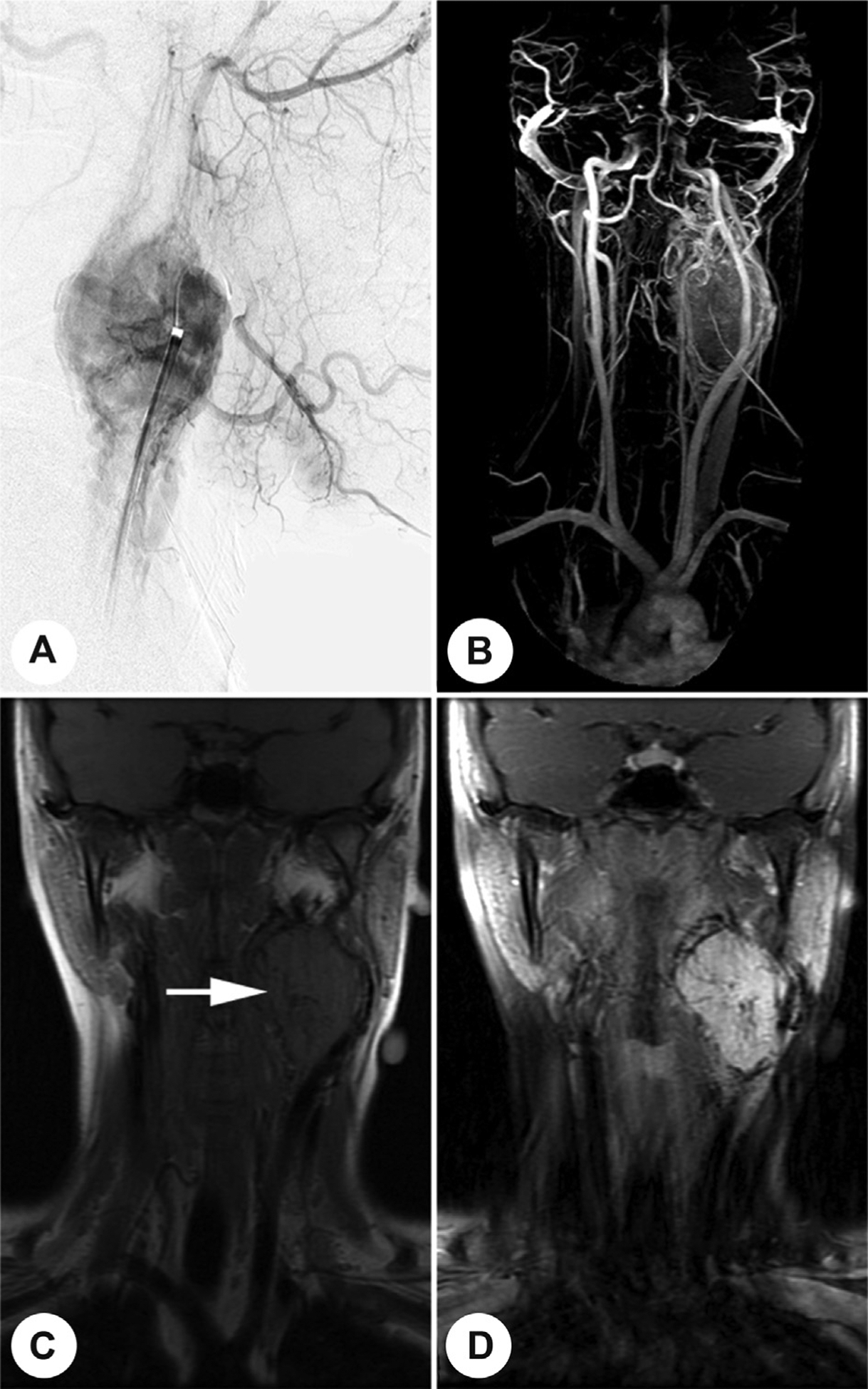
A carotid body paraganglioma at the bifurcation of the carotid artery is highlighted by angiography (A), vascular magnetic resonance (MR) imaging after contrast administration (B), T1-weighted MR imaging (C), or MR imaging after contrast administration (D).
5. Core elements
A summary of the core elements is outlined in Table 1.
Table 1.
Core and non-core elements for pathology reporting of pheochromocytoma and paraganglioma.
| Core elements | Non-core elements |
|---|---|
| Clinical information | Tumor dimensions |
| Operative procedure | Additional dimensions (largest tumor) |
| Specimen(s) submitted | Margin status |
| Tumor focality | Distance of the tumor to the closest margin |
| Tumor site | Closest margin, specify if possible |
| Specimen integrity | Lymph node status |
| Tumor dimensions | Extranodal extension (ENE) |
| Medullary hyperplasia | Adverse features |
| Histological tumor type | Ancillary studies |
| Extent of invasion | |
| Lymphovascular invasion | |
| Margin status | |
| Proliferative fraction | |
| Lymph node status | |
| Histologically confirmed distant metastases | |
| Pathological staging |
5.1. Clinical information
Clinical data in paraganglioma/pheochromocytoma are uniquely important for two reasons. First, there are distinctive correlations between genotype, biochemical phenotype [6], tumor distribution, prognosis, and syndromic associations [7,8]. Second, up to 50% of tumors are hereditary, making them the most genetically determined of all human tumors, with more than 20 hereditary susceptibility genes associated with their development [9]. Most pheochromocytomas and sympathetic paragangliomas are associated with clinical signs and symptoms related to catecholamine excess (Fig. 3). In contrast, parasympathetic paragangliomas are rarely symptomatic, and while some produce dopamine, others often lack tyrosine hydroxylase (TH), the enzyme required for catecholamine synthesis, making them biochemically and clinically silent [10]. Biochemical testing for pheochromocytoma/paraganglioma should include measuring the level of metabolites of norepinephrine and epinephrine, such as metanephrines and/or methoxytyramine, measured either in plasma or urine, as these are superior to measurements of the catecholamines themselves [11,12]. Many clinically silent paragangliomas, particularly of the sympathoadrenal type, will produce normetanephrines and/or methoxytyramine and thus are amenable to biochemical testing [6,9]. Similar to other neuroendocrine neoplasms, pheochromocytomas and extra-adrenal paragangliomas may produce and secrete peptides that can cause other clinical syndromes [13]. While not an exhaustive list, production of adrenocorticotropic hormone, β-endorphin, corticotropin-releasing hormone, calcitonin gene-related peptide, vasoactive intestinal peptide, growth hormone–releasing hormone, neuropeptide Y, peptide YY, insulin-like growth factor 1, galanin, adrenomedullin, serotonin, somatostatin, and gastrin-like neuropeptide has all been reported [7]. Thus, information on biochemical function, individual and family history, multiple tumors (Fig. 4a), and the presence of additional endocrine or nonendocrine tumors that may be components of a syndrome must be included in the data set [14,15]. If germ line mutation or familial syndrome testing has been performed, documenting the specific mutation if it is known, aids in providing a complete report.
Fig. 3.
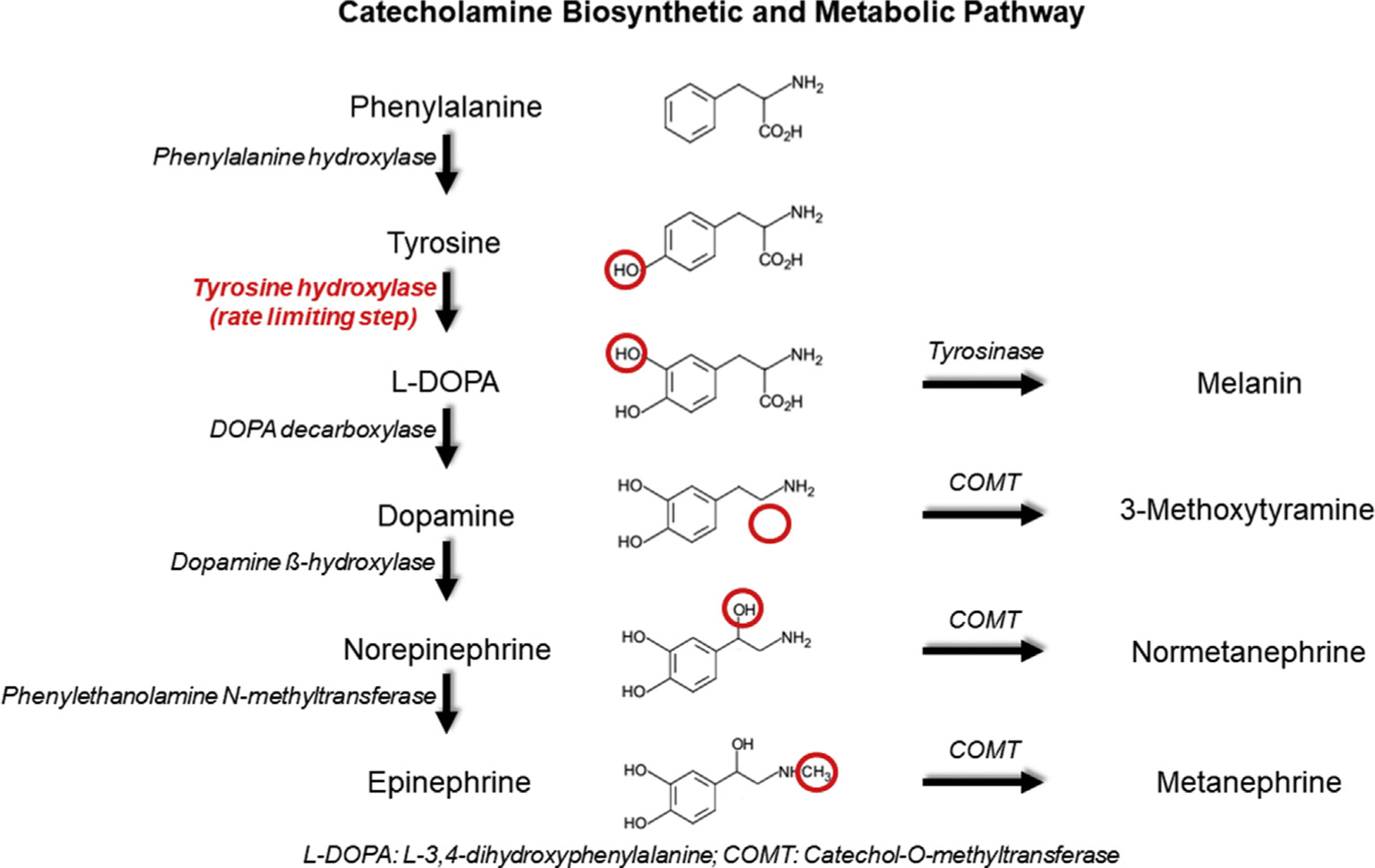
The biosynthetic pathway of catecholamines is demonstrated, including the enzymes necessary for synthesis. Metabolites are also shown as these are frequently used in testing. Not all pathways are illustrated, and not all intermediate steps are included. Tyrosinase is not expressed in the adrenal medulla or paraganglia, but is shown to illustrate the parallel utilization of tyrosine to produce melanin in melanocytes. Red circles show the structural region altered by the preceding enzyme’s action.
Fig. 4.
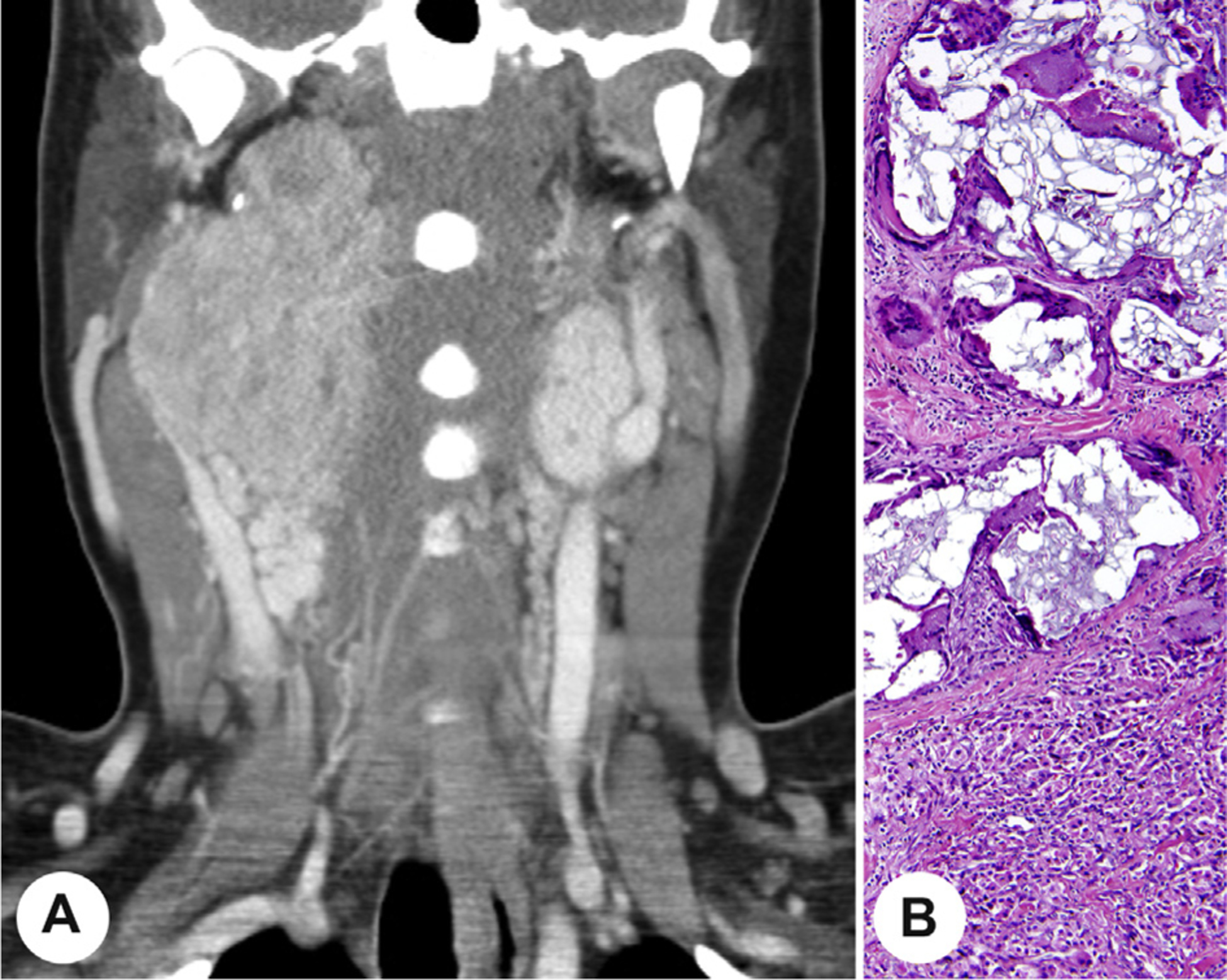
A, A coronal computed tomography image demonstrating bilateral carotid body paragangliomas in a syndrome-associated patient. B, A neck paraganglioma containing embolic material with associated foreign body giant cell reaction.
Previous therapy (chemotherapy, radiotherapy, embolization, targeted therapy, and/or immunotherapy) along with previous procedures (such as fine-needle aspiration [FNA] or core needle biopsy) may alter the microscopic appearance of a tumor, resulting in tumor infarction, or may interfere with assessment of invasion. It is generally not prudent to perform FNA or core needle biopsy on paraganglioma, especially the sympathetic type or pheochromocytoma, as this may cause catecholamine crisis or severe bleeding, in addition to usually producing a bloody smear with limited diagnostic yield [16–19]. Furthermore, FNA alone cannot reliably document a primary versus metastatic tumor and thus does not aid in final interpretation [20]. Preoperative embolization (Fig. 4b) is an established cause of necrosis in head and neck paragangliomas [10] and should not be interpreted as an adverse prognostic sign. In fact, it is in head and neck paragangliomas that FNA is most likely to be performed, especially in patients who are at poor surgical risk. Partial adrenalectomy, which is increasingly used in treating patients with pheochromocytomas, particularly those that are familial and likely to be or become bilateral [21], might also be expected to cause long-term changes in histology of the residual adrenal. Thus, including this information in the data set allows for a better understanding of the overall tumor. The data set does include an information not provided option, but as the information in these elements is vital to a comprehensive, clinically relevant pathology report that guides further adjuvant therapy, the not provided option should only be used in rare instances after all good faith efforts to obtain the information have been thoroughly exhausted. In many countries, an electronic medical record has been implemented, which allows for easy access to many of these results that may otherwise be challenging to report.
5.2. Operative procedure
Laparoscopic surgery may lead to disruption or fragmentation of the gland and/or tumor, potentially making it difficult to assess tumor size, integrity of the tumor capsule, and completeness of excision, and may also cause distortion of vascular channels, making assessment of lymphovascular invasion difficult. In rare cases wherein the specimen has been morcellated, tumor size should be obtained from either the surgeon or from preoperative cross-sectional imaging studies.
5.3. Specimen(s) submitted
All anatomical structures removed or biopsied as part of the procedure should be identified. Examples of other specimens may include additional tissues or organs (eg, kidney, larynx, lymph node) or deposits of recurrent or metastatic tumor. Laterality information is needed for correct identification of specimens, including right, left, or midline.
5.4. Tumor focality
A single tumor (unifocal) is easily captured, but the presence of multiple tumors is an important clue to potential hereditary disease [22]. Multiple tumors encompasses multifocality, defined as separate foci of tumor in the same organ (Fig. 5), whereas multicentric is used for more than one tumor identified in separate organs (eg, two or three topographically separate paragangliomas or a paraganglioma and a pheochromocytoma). These designations apply to primary tumors, not metastases, and require histologic confirmation that tumor is present. In some cases, it may not be possible to determine whether a specimen represents a metastasis or a separate primary (eg, a suspected lymph node with no residual lymph node architecture or a solitary pulmonary nodule [23]). Similarly, it may not be possible to determine whether a fragmented specimen contains multifocal tumors. When presented with these cases, the indeterminate category should be used. Specimens should be carefully examined both macroscopically and microscopically to determine whether multiple or multifocal tumors are present (Fig. 5). In most cases, multifocality applies to the adrenal gland specifically, but multicentric tumors may be identified in adrenalectomy which contains a pheochromocytoma and an additional, nearby extra-adrenal paraganglioma.
Fig. 5.
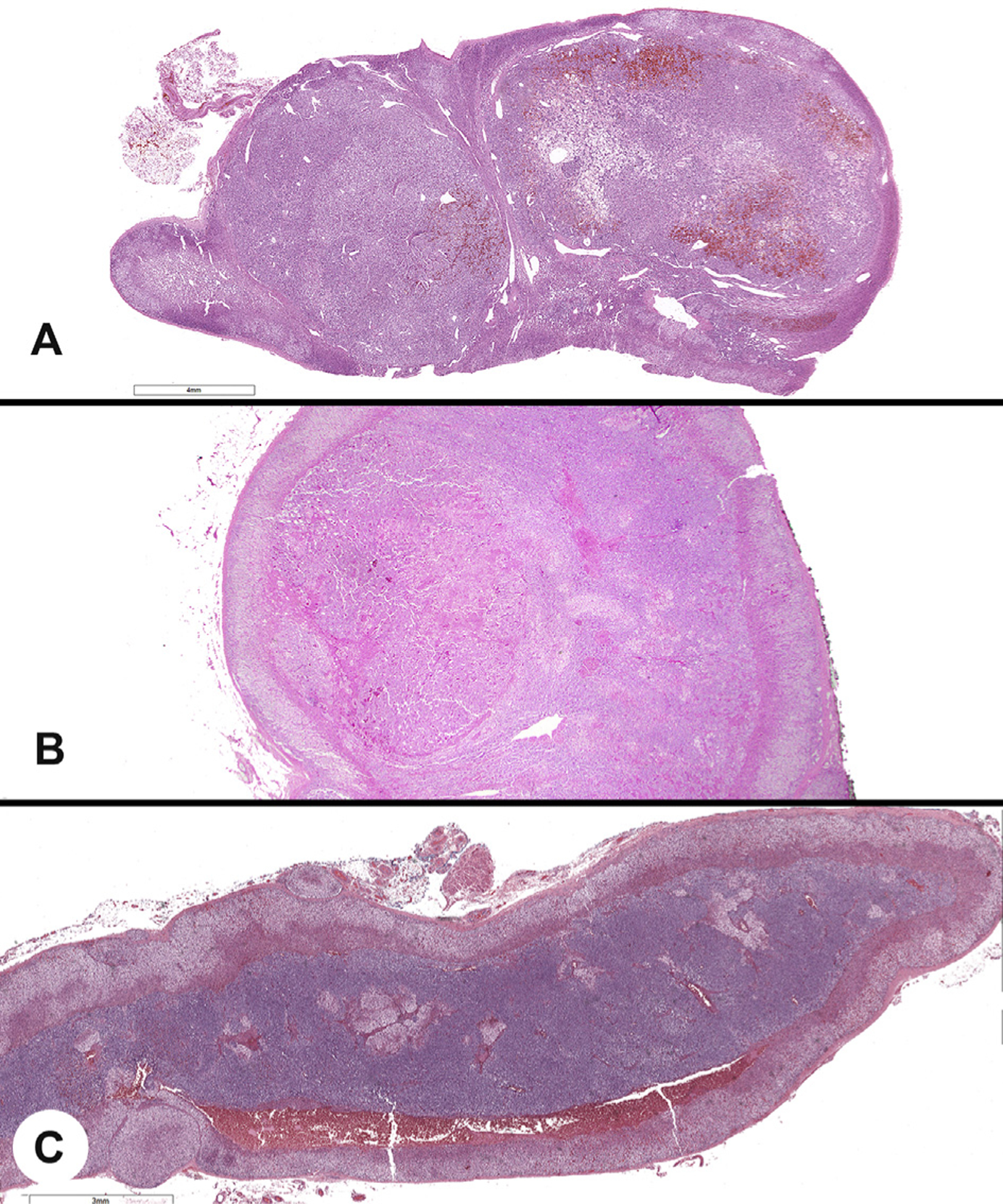
A, Multifocal tumors showing two topographically separate pheochromocytomas in the same adrenal gland. B, A microscopic pheochromocytoma identified in a background of diffuse hyperplasia. C, Diffuse adrenal medullary hyperplasia, with the medullary zone much greater than one-third of the gland thickness.
5.5. Tumor site
This element is defined as the site from which the surgeon removed tumor tissue and requires histologic confirmation that the tumor is present. The sites include groupings in the abdominal or pelvic region, thorax, and head and neck as an aid in documenting the location, with an open entry box allowing for the number of tumor(s) in each site to be included. As stated previously, the anatomic location of a paraganglioma has important clinical correlations with respect to predictive values concerning genotype, hormonal function, likelihood of additional and syndromically associated tumors, and risk of metastasis [24]. When metastases are sampled, the site (bone, lymph node, and so on) should specifically indicate which bone(s), and which lymph node(s), and to include the number of tumors independently for each sampled anatomic site.
5.6. Specimen integrity
This element becomes important when tumor fragmentation is present as this may cause difficulties in determining the completeness of excision, overall tumor size, the integrity of the tumor capsule, and whether there is capsular and/or lymphovascular invasion present. As such, this element helps to explain responses in other categories that have used cannot be assessed.
5.7. Tumor dimensions
A maximum single tumor dimension of the largest tumor is considered a core reporting element (Table 1) and may have to be assessed by gross and microscopic means, recognizing that additional dimensions may not be easily documented (and are thus non-core elements). Tumor measurements must exclude adjacent fat or other non-neoplastic tissue (Fig. 6). The assessment often depends on the specimen type and extent of disease, with straightforward documentation in a single, localized tumor in a well-oriented specimen, while nearly impossible in a curettage or debulking specimen. In this type of setting, obtaining a tumor size from the surgeon or from preoperative cross-sectional imaging studies may yield the most accurate information. Tumor size (≥50 mm) is used in staging [25,26], although with mixed results, as an independent prognostic criterion [27–29].
Fig. 6.
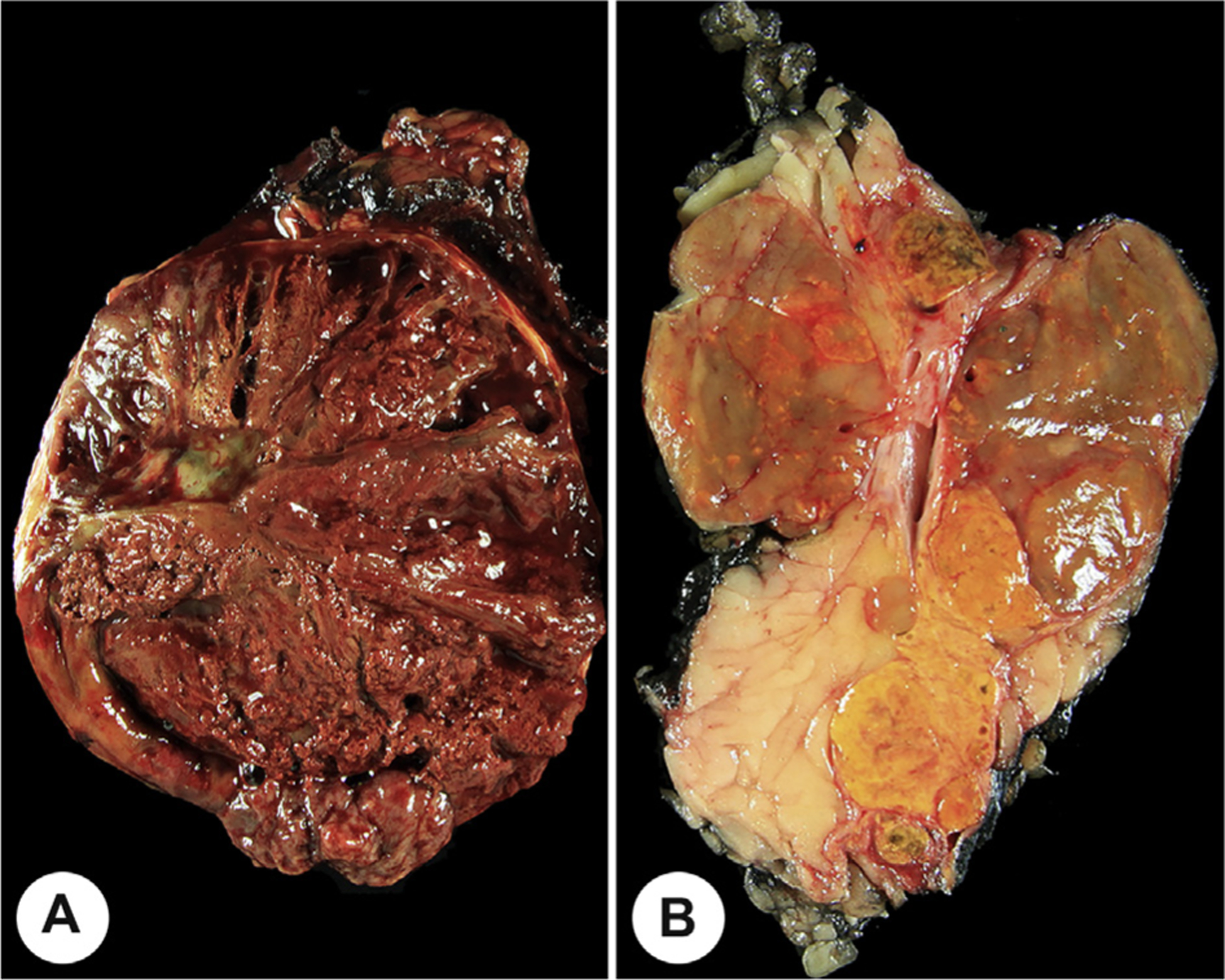
A, The overall size of this pheochromocytoma is large as a consequence of hemorrhage and necrosis. Fat has been removed to aid in accurate measuring. B, This is a gross photograph of a composite tumor (yellow) and medullary hyperplasia (brown), with measurements from each separate component documented.
Tumor sampling for microscopy should represent all variations in gross appearance and consistency of the tumor (Fig. 6), as well as margins and other specific features of interest. The general guideline of at least 1 section per cm of tumor should be used.
5.8. Medullary hyperplasia
It has been well documented that hereditary disease may be associated with adrenal medullary nodules either coexisting with pheochromocytoma/paraganglioma or in a background of diffuse adrenal medullary expansion [22]. They are most often associated with multiple endocrine neoplasia type 2 (MEN2), but have recently been described in other disorders [30]. Historically, nodules <10 mm have been arbitrarily called hyperplastic nodules or nodular adrenal medullary hyperplasia. However, molecular evidence supports calling them micropheochromocytomas [31].
The adrenal gland(s) received for diagnosis of possible micropheochromocytoma or adrenal medullary hyperplasia (Fig. 5b and c) should be oriented and dissected clean of as much fat/connective tissue as possible and then accurately weighed. This should not be done in cases wherein invasive tumor is a consideration clinically because this would preclude evaluation of the fat for microscopic involvement by a tumor. Sequential serial sections of roughly equal thickness are made to display the distribution and amount of medullary tissue in the lateral wings and tail of the gland (the coronal plane divides the gland into anterior and posterior portions, whereas the transverse plane divides the gland into superior and inferior/cranial and caudal portions) [32]. Medullary tissue is normally present only in the head and body of the gland, with extension into the wings but only minimally into the tail; the normal medulla represents up to one-third of the gland thickness, with cortex on each side comprising the other two-thirds. The presence of substantial adrenal medullary tissue in the tail or thickened medullary tissue comprising more than one-third of the thickness of the wings strongly suggests adrenal medullary hyperplasia. However, anatomic variation exists, and definitive diagnosis of medullary hyperplasia in the absence of nodules may require quantitative morphometric analysis [33].
Although it is sometimes difficult to define the tail of an adrenal gland distorted by a pheochromocytoma, it should be remembered that adrenal medullary nodules [33] and pheochromocytomas can occur in adrenals in MEN2 syndrome without an obvious background of diffuse hyperplasia. The adrenal gland adjacent to an apparently sporadic pheochromocytoma should therefore be sectioned as mentioned previously and carefully examined for small nodules [7].
5.9. Histological tumor type
All tumors of the adrenal medulla and extra-adrenal paraganglia should be assigned a type based on the most recent edition of the WHO Classification of Tumours of Endocrine Organs (see Table 2) [14,15]. A composite tumor is defined as a tumor that combines morphological features of paraganglioma or pheochromocytoma with those of a developmentally related neurogenic tumor, including ganglioneuroma (Fig. 7), ganglioneuroblastoma, neuroblastoma, or malignant peripheral nerve sheath tumor [14,15], listed in the reporting guide from more primitive to mature. There is no specified percentage of the second tumor type required, and as such, an estimation of the percentage of tumor present is documented. However, complete histoarchitecture of the second tumor type is required. Scattered neuron-like cells often seen in pheochromocytomas are insufficient. The composite designation is unique from mixed corticomedullary neoplasms, which would be included in the other, specify selection box.
Table 2.
WHO Classification of Tumours of Endocrine Organs (2017): Tumours of the adrenal medulla and extra-adrenal paraganglia.
| Descriptor | ICD-O codes |
|---|---|
| Phaeochromocytoma | 8700/3 |
| Extra-adrenal paragangliomas | |
| Head and neck paragangliomas | |
| Carotid body paraganglioma | 8692/3a |
| Jugulotympanic paraganglioma | 8690/3a |
| Vagal paraganglioma | 8693/3 |
| Laryngeal paraganglioma | 8693/3 |
| Sympathetic paragangliomas | 8693/3 |
| Neuroblastic tumours of the adrenal gland | |
| Neuroblastoma | 9500/3 |
| Ganglioneuroblastoma, nodular | 9490/3 |
| Ganglioneuroblastoma, intermixed | 9490/0 |
| Ganglioneuroma | 9490/0 |
| Composite phaeochromocytoma | 8700/3 |
| Composite paraganglioma | 8693/3 |
WHO, World Health Organization.
These new codes were approved by the International Agency for Research on Cancer/WHO Committee for ICD-O. © WHO/IARC. Reproduced with permission.
Fig. 7.
A, The classical appearance of a paraganglioma with well-developed zellballen architecture. B, A composite ganglioneuroma and pheochromocytoma.
The most common second component of composite tumors is ganglioneuroma (70–80% of cases), followed by ganglioneuroblastoma (15–20%). Although the latter is morphologically comparable with pediatric ganglioneuroblastoma, it differs in molecular and clinical perspectives and confers only a low risk of metastases [14,15,32].
5.10. Extent of invasion
Invasion is a risk factor for development of metastases when evaluated in conjunction with other adverse features. However, invasion is currently categorized and weighted inconsistently [22]. Toward a more reproducible approach, precise descriptions of the nature and extent of invasion are included, with microscopic transgression of the tumor capsule (if one is present), the organ capsule, extension into periadrenal soft tissues (Fig. 8), or other organs included. As pheochromocytomas usually do not have a capsule [32], the adrenal gland capsule becomes the capsule of the tumor in most cases. If a tumor capsule is present, invasion of the organ capsule and tumor capsule should be documented separately. Capsular invasion is not assessed in a biopsy.
Fig. 8.
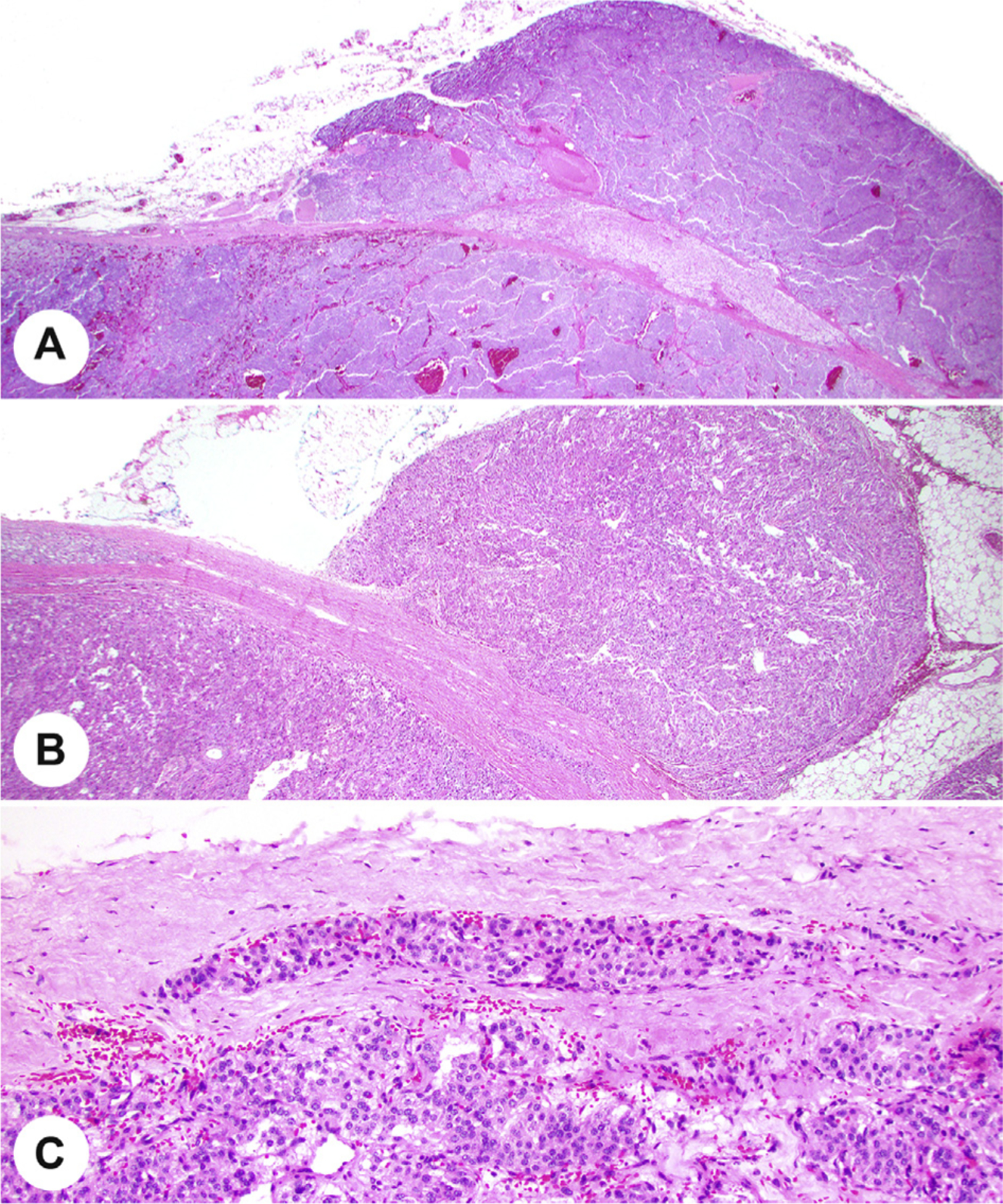
A, The pheochromocytoma invades through the capsule of the adrenal and expands into the adjacent adipose tissue. B, There is a well-developed capsule with tumor in the adjacent adipose tissue. C, Tumor cells are noted within an oval lymphatic endothelium-lined space in the capsule of a paraganglioma.
5.11. Lymphovascular invasion
Vessel invasion is a risk factor for development of metastases [22,27,34], but no specific data are available to separate between lymphatics, capillaries, veins, or arteries, and thus, separation between lymphatic and vascular invasion has not yet been advocated for these tumors. Precise descriptions of the nature and extent of vascular invasion are required in conjunction with other adverse factors to optimally guide patient management [22]. While the presence of thrombus associated with tumor in an endothelial lined space is unquestionable vessel invasion, thrombus and disrupted endothelium may not be seen in adrenal or paraganglioma tumors (Fig. 8). Vessel invasion should be documented at the periphery of the tumor or near the advancing edge as capsular vessels may not be present. Intratumoral vessels are generally not considered when evaluating lymphovascular invasion. Furthermore, definitive documentation of increased metastatic risk progressively with involvement of small to larger vessels is not available, although extrapolation from other tumors would suggest that is the case [35–38]. In the adrenal gland, invasion of one or more tributaries of the central vein (perhaps facilitated by the discontinuous arcades of smooth muscle in the wall of the central vein) may be an important event leading to involvement of the adrenal vein and the vena cava.
5.12. Margin status
Adrenalectomy specimens may be distorted and irregular. There are no data indicating a distance to the closest margin as being predictive of outcomes, and thus, the closest margin distance when not involved is not a core element (Table 1). Incomplete excision has been associated with local recurrence [39]. Positive margins are defined both grossly, as tumor obviously transected, and microscopically, as tumor on ink, if the surface is inked. Tumors in morcellated adrenalectomy specimens or curetted lesions (eg, bladder) cannot be assessed for margin involvement. In these cases, the margins cannot be assessed, and a comment about the reason can be entered.
5.13. Proliferative index
Mitotic count and/or the Ki-67 proliferation index is now widely used in risk stratification for neuroendocrine tumors as a whole. A high proliferative index based on either mitotic count [27,40] or Ki-67 labeling [34,41] is a well-documented risk factor for development of metastases for pheochromocytoma and paraganglioma and thus is considered an essential reporting criterion (Fig. 9). Mitotic count should be performed in a minimum area of 2 mm2, which is equivalent to approximately 10 high-power fields (HPFs) in many microscopes [42]. There is currently no standard approach to scoring a Ki-67 proliferation index in pheochromocytoma and paraganglioma, and this is emphasized. Using criteria established for other neuroendocrine tumors [14,15,43], it is recommended that the Ki-67 proliferation index should be reported as the percentage of positive tumor cells in the area of highest nuclear labeling (so called hot spot) [7,34]. Counts should ideally be based on manual counts of printed images or appropriately validated automated image analysis; visual estimates are unreliable and are not recommended [14,15,43].
Fig. 9.
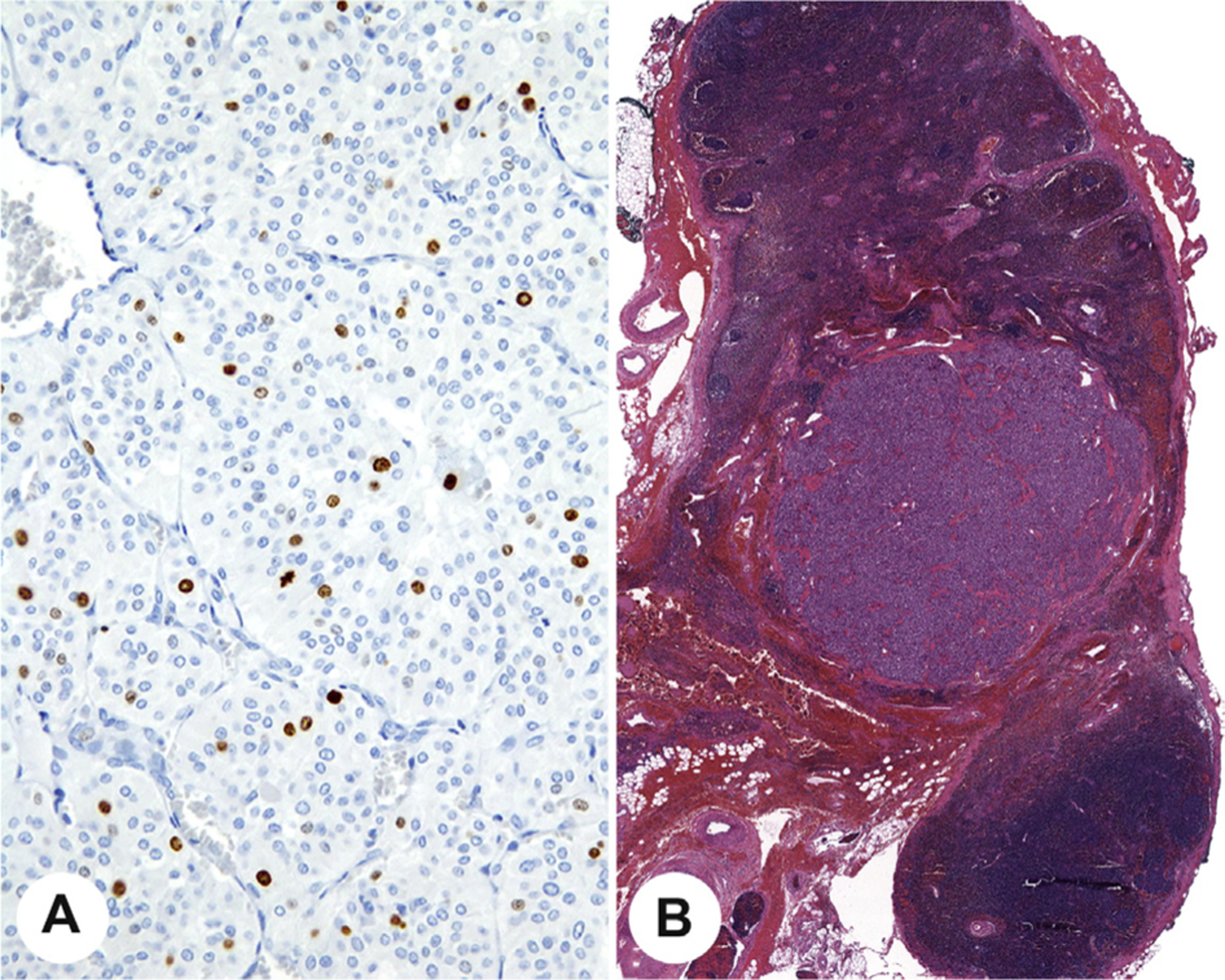
A, A Ki-67 proliferation index demonstrating >3% of the neoplastic nuclei are reactive. B, Histologic confirmation of lymph node metastasis from a paraganglioma.
5.14. Lymph node status
Regional lymph nodes are identified within the anatomic area in which a tumor is located and receive lymphatic drainage from that area. They are, therefore, anatomically related to the tumor and may be the earliest sites of metastases. It is important to recognize that multicentric tumors (multiple tumors in different anatomic sites) on imaging may mimic metastatic disease to a lymph node chain because the distribution of paraganglia closely mimics that of para-aortic lymph nodes. Thus, as shown in specimen(s) submitted, histologic evidence of tumor within a lymph node must be confirmed to verify nodal metastasis (Fig. 9). Similar to risk stratification for other organs, the pathology report should state the total number of lymph nodes examined and the total number of lymph nodes with metastases. Lymph node metastases are incorporated into tumor staging. Two additional features (size of tumor deposit and extranodal extension [ENE]) are included in the non-core elements (see Lymph node status below).
5.15. Histologically confirmed distant metastases
A diagnosis of metastasis is appropriate when pheohromocytoma or paraganglioma is present in a site where normal paraganglia do not exist. The only such sites a priori are bone and histologically confirmed lymph nodes. It is crucial to remember the normal anatomic distribution of paraganglia to consider the possibility of multiple primary tumors [44]. The assessment of distant metastasis can be particularly challenging in some cases because primary paragangliomas occur in the thyroid, pituitary, gallbladder, liver, and lung, to name a few rare sites. Therefore, tumor in these rare locations should not automatically be considered metastatic, but should be further evaluated to confirm primary versus metastatic paraganglioma. In addition, owing to the ease of performing needle core biopsies of various organs, metastatic disease is now increasingly seen histologically, and in many cases, biopsies may be the only tissue samples available owing to the advanced nature of the primary tumor or the comorbidities associated with surgical resection. In patients with germ line predisposition, the possibility of multiple primary tumors rather than metastases should be considered, depending on the exact anatomic sites evaluated.
5.16. Pathological staging
The American Joint Committee on Cancer staging system for pheochromocytomas and sympathetic paragangliomas was implemented in 2017 to guide clinicians in determining the therapies and follow-up that patients require [25]. Importantly, all sympathetic paragangliomas are of pT2 stage no matter the size, whereas paraganglioma of the head and neck (parasympathetic) is not staged. It is expected that staging and survival data collected will lead to increased understanding of these tumors and to future improvements in patient care [25,26].
6. Non-core elements
6.1. Lymph node status
The size of a tumor deposit within the lymph node may be correlated with outcomes, but this has not yet been widely validated and, as such, is a non-core element. It is included as a data point which may prove to be significant with more study. Similarly, ENE, also called extracapsular lymph node extension, whether microscopic (ENEmi) or macroscopic (ENEma), has been shown in many other organ system cancers to be a poor prognostic indicator (ie, patients do worsen) [45–51] and by extrapolation may be a useful prognostic marker for pheochromocytoma and paraganglioma metastatic foci also. However, validation studies of this empiric finding have yet to be performed, and thus, this element is a non-core data point. When lymph node dissections are received as multiple fragments, an accurate lymph node number may only be obtained from the surgeon or should otherwise be stated as undetermined.
6.2. Adverse features
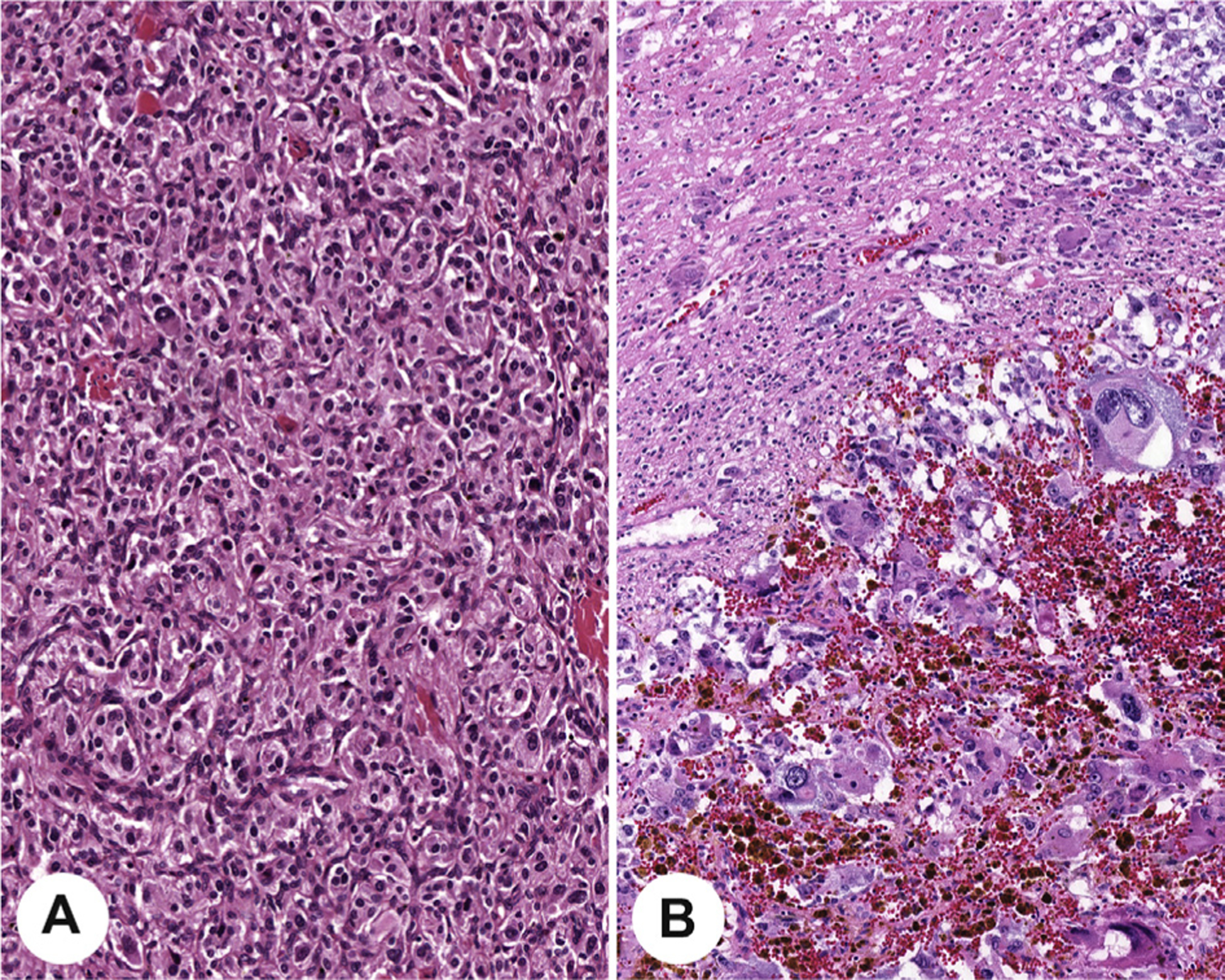
Currently, there is no universally adopted risk stratification for pheochromocytoma and paragangliomas, and thus, although the aggregate of adverse features is clinically beneficial, it is not yet required. Several histological features are putative risk factors for the development of metastases in proposed scoring systems for risk stratification: the Pheochromocytoma of the Adrenal gland Scoring Scale (PASS) [27] and Grading system for Adrenal Pheochromocytoma and Paraganglioma (GAPP) [34]. However, the individual parameters are assessed and weighted differently in these systems. The PASS was developed for adrenal tumors only and was developed using histology parameters only, whereas the GAPP incorporates findings for both pheochromocytoma and sympathetic paraganglioma, combining the catecholamine type, Ki-67 proliferation index, and histologic features to yield three progressive tumor grades (well, moderately, and poorly differentiated) which can be combined with succinate dehydrogenase B (SDHB) immunohistochemistry (IHC) to help predict metastasis. Variable concordance between expert pathologists has been reported [12,52], although a meta-analysis of published articles using the PASS or GAPP concluded that a low score with either histological system is a strong predictor of low metastatic risk, whereas high scores may not be predictive without additional features (such as genotype and biochemical testing) [53]. Comedonecrosis (Fig. 10), growth pattern (Fig. 10), and high proliferative index are the most readily recognized and possibly the most predictive parameters [8,41], whereas cellularity is much more subjective (Fig. 10). To reduce subjectivity, it has been recommended that cellularity be quantitated by counting the number of cells within an area (U) encompassed by a square grid in a ×10 ocular viewed with a ×40 HPF, corresponding to 0.0625 mm2 [9,34]. Although not required, reporting these histologic features may be considered in conjunction with other data for cumulative risk stratification to optimally guide patient management. There is presently no scoring system applied to head and neck paragangliomas, although individual parameters may provide useful information for those tumors [54].
Fig. 10.
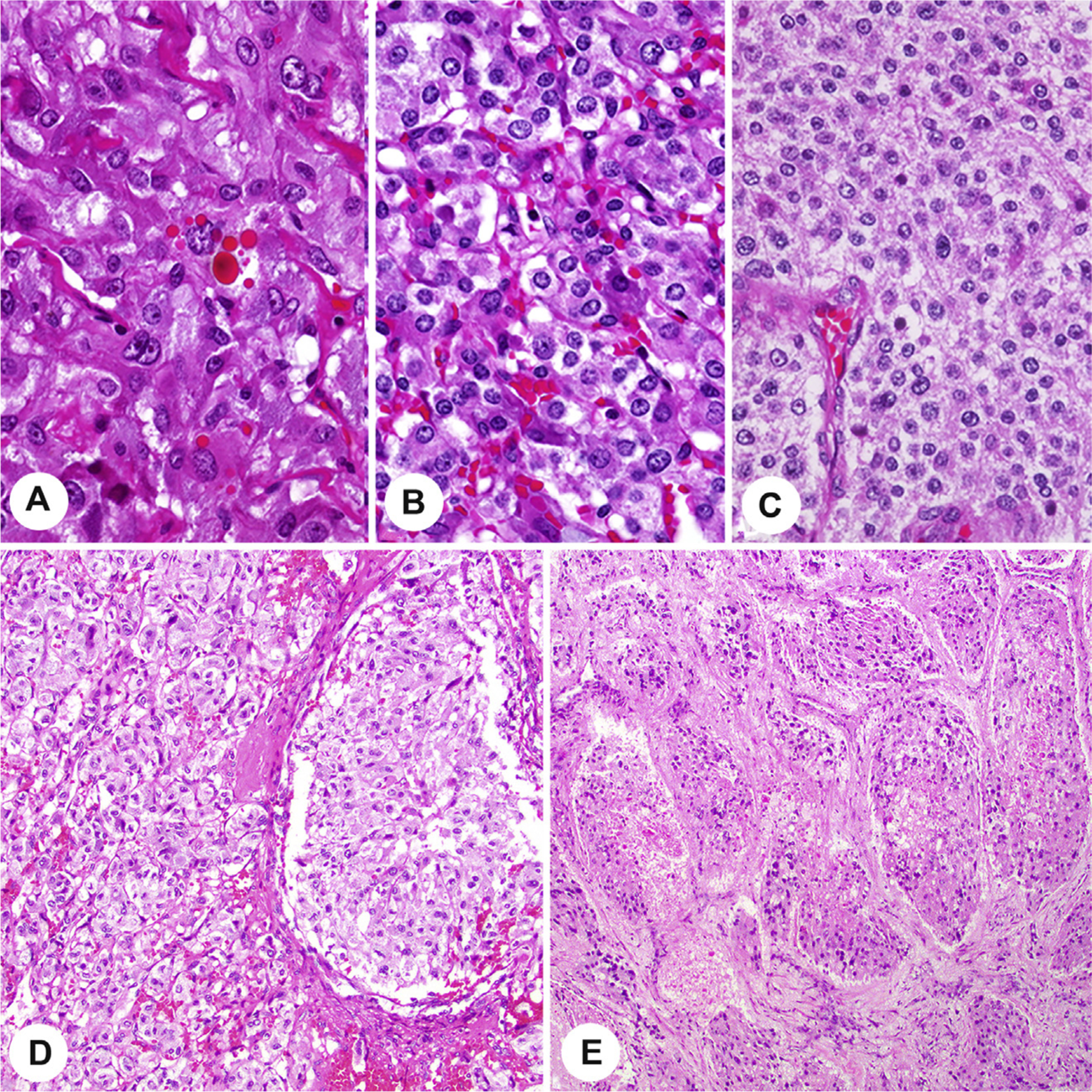
The top row shows increasing cellularity, from low cellularity (A) to intermediate cellularity (B) to high cellularity (C) taken at the same magnification. The lower row demonstrates large nest size in comparison with usual smaller nests of paraganglioma (D) and tumor comedonecrosis (E).
6.3. Ancillary studies
The differential diagnosis of pheochromocytoma or paraganglioma often requires use of IHC markers to establish the neuroendocrine nature of a tumor together with additional more specific markers to confirm the diagnosis or exclude other entities, especially other neuroendocrine neoplasms [10,44,55]. The most frequently used positive markers in most contexts are chromogranin A (CGA) and synaptophysin; synaptophysin, however, can be expressed in the normal adrenal cortex and adrenal cortical tumors and thus may not be reliable in distinguishing pheochromocytomas from cortical neoplasms. Thus, CGA is one of the most reliable markers of neuroendocrine tumors because it has relative specificity for the matrix of dense core granules, although it is not the most sensitive marker [56]. An additional marker that can define a lesion as a member of the neuroendocrine tumor family is the transcription factor INSM1 (Fig. 11) [57]. Additional potentially useful positive markers include GATA3 (Fig. 11) [44,58], a transcription factor that helps to distinguish paragangliomas and pheochromocytomas from most epithelial neuroendocrine tumors; tyrosine hydroxylase (TH) and dopamine β-hydroxylase (DBH) (Fig. 11) demonstrate capacity for catecholamine synthesis [12] and can clarify functional status [59]. S100 protein or SOX10 may be used to demonstrate sustentacular cells (Fig. 11); decrease or loss of sustentacular cells is associated with more biologically aggressive tumors [27,34,60]. An important feature is the lack of expression of various keratins that can be used to distinguish these tumors from epithelial neuroendocrine tumors. HMB45 can be used to exclude a melanoma in a tumor with cytologic atypia. Inhibin has been traditionally used to distinguish the adrenal cortex from medulla; however, a recent study has shown that inhibin may be expressed in pheochromocytomas/paragangliomas arising in patients with von Hippel-Lindau (VHL) and SDHx-driven pseudohypoxic pathway disease [61], and tumors from patients with von Hippel-Lindau syndrome are the tumors that can have a clear cytoplasm and mimic cortical lesions [62,63]. It is noteworthy that head and neck paragangliomas may be focally reactive to completely negative for TH and also negative or only focally positive for CGA and synaptophysin [10,64]. In those cases that show low expression of catecholamine enzymes, GATA3 reactivity is of value and may be superior to TH or DBH in this setting [65], but keratin and parathyroid hormone immunohistochemistry would be needed to exclude the differential diagnosis of parathyroid carcinoma. The presence of sustentacular cells can also be found in other neuroendocrine tumors and is therefore not considered to have great diagnostic value.
Fig. 11.
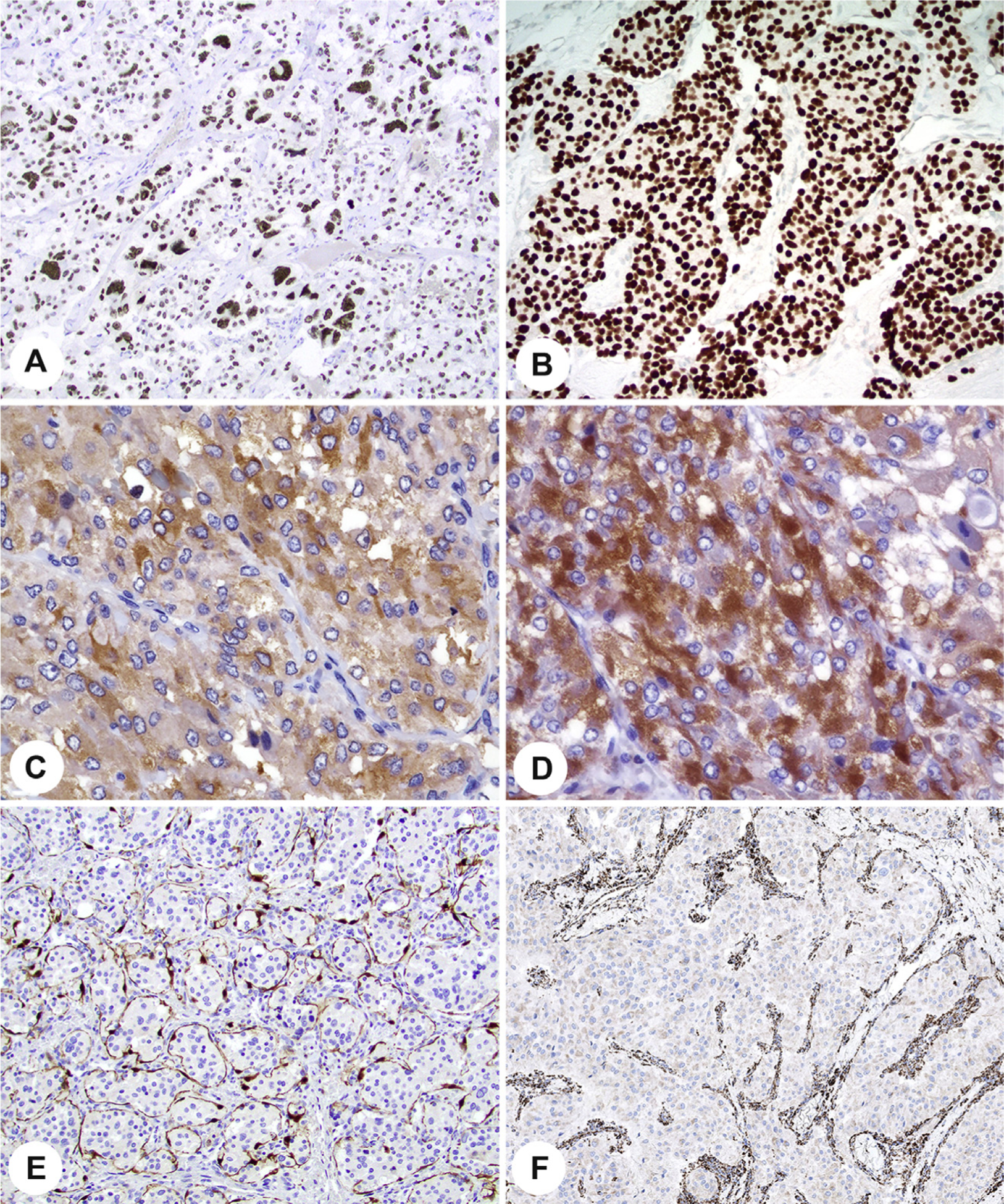
Various ancillary studies aid in the diagnosis and evaluation of paraganglioma and pheochromocytoma. A, Strong, diffuse, nuclear INSM1. B, Strong, diffuse, nuclear reaction with GATA3. C, Tyrosine hydroxylase in a cytoplasmic distribution. D, Dopamine beta-hydroxylase with a cytoplasmic distribution. E, Sustentacular supporting S100 proteinepositive cells. F, Loss of SDHB in the neoplastic cells, with a strong internal control. SDHB, succinate dehydrogenase B.
In addition to aiding in diagnosis, IHC is increasingly used as a genetic screen. When any component of mitochondrial respiratory chain complex 2 is completely inactivated, the entire complex becomes unstable, resulting in degradation of the SDHB subunit. Nearly all SDH mutations are germ line. Thus, there is loss of SDHB by IHC (Fig. 11) if there is complete inactivation of SDHA, SDHAF2, SDHB, SDHC, or SDHD (collectively “SDHx”), as would be seen in a germ line pathogenic variant accompanied by inactivation of the normal allele [66–68]. SDHC and SDHD form the anchoring component, and SDHA and SDHB form the catalytic component of the complex. An immunohistochemical loss is interpreted as no mitochondrial (granular cytoplasmic) staining in the presence of an appropriate internal control. Some potential pitfalls in interpretation must be taken into consideration during evaluation [69,70]. Germ line pathogenic variants in SDHA show loss of staining for SDHA, in addition to loss of staining for SDHB [71]. Pathogenic variants in VHL may contribute to false interpretation of SDHB IHC results when there is only a cytoplasmic blush or not true loss [67]. Despite these limitations, staining for SDHB should be performed in all cases to identify patients with any SDHx common germ line predisposition and also may serve as a prognostic marker [71–73]. Prognostically, paragangliomas associated with SDHB mutations have been associated with a high rate of metastasis compared with tumors without SDHB mutations. When preserved SDHB expression is seen, other genes (eg, RET, VHL) can be evaluated [71]. As indicated previously, expression of inhibin in SDHB-intact tumors suggests VHL-related disease [61]. Germ line fumarate hydratase (FH) mutations, which underlie hereditary leiomyomatosis and renal cell carcinoma syndrome, have been identified in a small subset of paragangliomas and pheochromocytomas [68,74,75] and can be detected by immunohistochemical loss of FH.
7. Conclusions
The goal of this data set developed to report pheochromocytoma and paraganglioma is to improve patient management worldwide, to advance national and international benchmarking, and to enable standardized data supporting research and tissue banking. This group of tumors is quite unique within the endocrine organs as they are classified along a risk stratification spectrum, making them more challenging to evaluate and study. There are limited guidelines regarding prognostic factors and patient outcomes of these tumors. By harmonizing reporting criteria that can be globally integrated into research, we hope to facilitate further research by using a single international data standard.
Acknowledgments
The authors appreciate the sponsoring societies and organizations and thank Fleur Webster for all her exceptional organizational and editing contributions. The views expressed are those of the authors solely and do not reflect the official policy of the United States Government, Army, Navy or Air Force, Department of Defense, or Department of Veterans Affairs nor of any other Sovereign Nation.
Disclosures: The authors did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. The authors declare no conflict of interest.
References
- [1].Srigley J, Lankshear S, Brierley J, McGowan T, Divaris D, Yurcan M, et al. Closing the quality loop: facilitating improvement in oncology practice through timely access to clinical performance indicators. J Oncol Pract 2013;9:e255–61. [DOI] [PubMed] [Google Scholar]
- [2].Ellis DW, Srigley J. Does standardised structured reporting contribute to quality in diagnostic pathology? The importance of evidence-based datasets. Virchows Arch 2016;468:51–9. [DOI] [PubMed] [Google Scholar]
- [3].Tischler AS, Asa SL, Clifton-Bligh R, de Krijger RR, Kimura N, Komminoth P, et al. Phaeochromocytoma and Paraganglioma Histopathology Reporting Guide. Sydney, Australia: International Collaboration on Cancer Reporting; 2019. [Google Scholar]
- [4].Merlin T, Weston A, Tooher R. Extending an evidence hierarchy to include topics other than treatment: revising the Australian ‘levels of evidence. BMC Med Res Methodol 2009;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Giordano T, Berney D, de Krijger R, Erickson LA, Fassnacht M, Mete O, et al. Carcinoma of the Adrenal Cortex Histopathology Reporting Guide. Sydney, Australia: International Collaboration on Cancer Reporting; 2019. [Google Scholar]
- [6].Eisenhofer G, Klink B, Richter S, Lenders JW, Robledo M. Metabologenomics of Phaeochromocytoma and Paraganglioma: An Integrated Approach for Personalised Biochemical and Genetic Testing. Clin Biochem Rev 2017;38:69–100. [PMC free article] [PubMed] [Google Scholar]
- [7].Mete O, Tischler AS, de Krijger R, McNicol AM, Eisenhofer G, Pacak K, et al. Protocol for the examination of specimens from patients with pheochromocytomas and extra-adrenal paragangliomas. Arch Pathol Lab Med 2014;138:182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Turchini J, Cheung VKY, Tischler AS, De Krijger RR, Gill AJ. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology 2018;72:97–105. [DOI] [PubMed] [Google Scholar]
- [9].Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, Welander J, et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocy- tomas and paragangliomas. Nat Rev Endocrinol 2017;13:233–47. [DOI] [PubMed] [Google Scholar]
- [10].Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: updates. Arch Pathol Lab Med 2008;132:1272–84. [DOI] [PubMed] [Google Scholar]
- [11].Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014;99:1915e42. [DOI] [PubMed] [Google Scholar]
- [12].Konosu-Fukaya S, Omata K, Tezuka Y, Ono Y, Aoyama Y, Satoh F, et al. Catecholamine-Synthesizing Enzymes in Pheochromocytoma and Extraadrenal Paraganglioma. Endocr Pathol 2018;29:302–9. [DOI] [PubMed] [Google Scholar]
- [13].Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and Paraganglioma. N Engl J Med 2019;381:552–65. [DOI] [PubMed] [Google Scholar]
- [14].Tischler AS, de Krijger RR, Gill A, Kawashima A, Kimura N, Komminoth P, et al. Tumours of the adrenal medulla and extra-adrenal paraganglia: Phaeochromocytoma. In: Lloyd RV, Osamura RY, Kloppel G, Rosai J, editors. Tumours of Endocrine Organs. Lyon, France: IARC; 2017. p. 183–9. [Google Scholar]
- [15].Kimura N, Capella C, DeLellis RA, Epstein JI, Gill A, Kawashima A, et al. Tumours of the adrenal medulla and extra-adrenal paraganglia: Extra-adrenal paragangliomas. In: Lloyd RV, Osamura RY, Kloppel G, Rosai J, editors. Tumours of Endocrine Organs. Lyon, France: IARC; 2017. p. 190–5. [Google Scholar]
- [16].Quayle FJ, Spitler JA, Pierce RA, Lairmore TC, Moley JF, Brunt LM. Needle biopsy of incidentally discovered adrenal masses is rarely informative and potentially hazardous. Surgery 2007;142:497–502. [DOI] [PubMed] [Google Scholar]
- [17].Casola G, Nicolet V, vanSonnenberg E, Withers C, Bretagnolle M, Saba RM, et al. Unsuspected pheochromocytoma: risk of blood-pressure alterations during percutaneous adrenal biopsy. Radiology 1986;159:733–5. [DOI] [PubMed] [Google Scholar]
- [18].Ford J, Rosenberg F, Chan N. Pheochromocytoma manifesting with shock presents a clinical paradox: a case report. CMAJ 1997;157: 923–5. [PMC free article] [PubMed] [Google Scholar]
- [19].de Vries AC, Poley JW. Hypertensive crisis after endoscopic ultra-sound-guided fine-needle aspiration of the right adrenal gland. Endoscopy 2014;46(Suppl 1 UCTN):E447–8. [DOI] [PubMed] [Google Scholar]
- [20].Ntanasis-Stathopoulos I, Tsilimigras DI, Klapsinou E, Daskalopoulou D, Vaida S, Arnogiannaki N, et al. Challenging differential diagnosis of an extra-adrenal paraganglioma; the role of fine needle aspiration cytology. Diagn Cytopathol 2017;45: 565–8. [DOI] [PubMed] [Google Scholar]
- [21].Asher KP, Gupta GN, Boris RS, Pinto PA, Linehan WM, Bratslavsky G. Robot-assisted laparoscopic partial adrenalectomy for pheochromocytoma: the National Cancer Institute technique. Eur Urol 2011;60:118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tischler AS, deKrijger RR. 15 years of paraganglioma: Pathology of pheochromocytoma and paraganglioma. Endocr Relat Cancer 2015; 22:T123–33. [DOI] [PubMed] [Google Scholar]
- [23].Aubertine CL, Flieder DB. Primary paraganglioma of the lung. Ann Diagn Pathol 2004;8:237–41. [DOI] [PubMed] [Google Scholar]
- [24].Benn DE, Robinson BG, Clifton-Bligh RJ. 15 years of paraganglioma: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr Relat Cancer 2015;22:T91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Amin MB, Edge SB, Greene FL, Byrd DR, Brookland RK, Washington MK, et al. AJCC Cancer Staging Manual. Chicago, USA: Springer; 2017. [Google Scholar]
- [26].Roman-Gonzalez A, Jimenez C. Malignant pheochromocytoma-paraganglioma: pathogenesis, TNM staging, and current clinical trials. Curr Opin Endocrinol Diabetes Obes 2017;24:174–83. [DOI] [PubMed] [Google Scholar]
- [27].Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002;26:551–66. [DOI] [PubMed] [Google Scholar]
- [28].Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium October 2005. Nat Clin Pract Endocrinol Metab 2007;3: 92–102. [DOI] [PubMed] [Google Scholar]
- [29].Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 2012;48:1739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Romanet P, Guerin C, Pedini P, Essamet W, Castinetti F, Sebag F, et al. Pathological and Genetic Characterization of Bilateral Adrenomedullary Hyperplasia in a Patient with Germline MAX Mutation. Endocr Pathol 2017;28:302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Korpershoek E, Petri BJ, Post E, van Eijck CH, Oldenburg RA, Belt EJ, et al. Adrenal medullary hyperplasia is a precursor lesion for pheochromocytoma in MEN2 syndrome. Neoplasia 2014;16: 868–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lack EE. Tumors of the Adrenal Glands and Extraadrenal Paraganglia. Washington DC: American Registry of Pathology; 2007. [Google Scholar]
- [33].DeLellis RA, Wolfe HJ, Gagel RF, Feldman ZT, Miller HH, Gang DL, et al. Adrenal medullary hyperplasia. A morphometric analysis in patients with familial medullary thyroid carcinoma. Am J Pathol 1976;83:177–96. [PMC free article] [PubMed] [Google Scholar]
- [34].Kimura N, Takayanagi R, Takizawa N, Itagaki E, Katabami T, Kakoi N, et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr Relat Cancer 2014; 21:405–14. [DOI] [PubMed] [Google Scholar]
- [35].Tripathi P, Rao SX, Zeng MS. Clinical value of MRI-detected extramural venous invasion in rectal cancer. J Dig Dis 2017;18:2–12. [DOI] [PubMed] [Google Scholar]
- [36].Tewari M Significance of pathological positive superior mesenteric/portal venous invasion in pancreatic cancer. Hepatobiliary Pancreat Dis Int 2016;15:572–8. [DOI] [PubMed] [Google Scholar]
- [37].Matsuda M, Suzuki T, Kono H, Fujii H. Predictors of hepatic venous trunk invasion and prognostic factors in patients with hepatocellular carcinomas that had come into contact with the trunk of major hepatic veins. J Hepatobiliary Pancreat Surg 2007;14:289–96. [DOI] [PubMed] [Google Scholar]
- [38].Bonsib SM. Renal veins and venous extension in clear cell renal cell carcinoma. Mod Pathol 2007;20:44–53. [DOI] [PubMed] [Google Scholar]
- [39].Li ML, Fitzgerald PA, Price DC, Norton JA. Iatrogenic pheochromocytomatosis: a previously unreported result of laparoscopic adrenalectomy. Surgery 2001;130:1072–7. [DOI] [PubMed] [Google Scholar]
- [40].Strong VE, Kennedy T, Al-Ahmadie H, Tang L, Coleman J, Fong Y, et al. Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery 2008;143:759–68. [DOI] [PubMed] [Google Scholar]
- [41].Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017;31: 181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yigit N, Gunal A, Kucukodaci Z, Karslioglu Y, Onguru O, Ozcan A. Are we counting mitoses correctly? Ann Diagn Pathol 2013;17: 536–9. [DOI] [PubMed] [Google Scholar]
- [43].Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod Pathol 2018;31:1770–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Asa SL, Ezzat S, Mete O. The Diagnosis and Clinical Significance of Paragangliomas in Unusual Locations. J Clin Med 2018;7:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Luchini C, Wood LD, Cheng L, Nottegar A, Stubbs B, Solmi M, et al. Extranodal extension of lymph node metastasis is a marker of poor prognosis in oesophageal cancer: a systematic review with meta-analysis. J Clin Pathol 2016;69:956–61. [DOI] [PubMed] [Google Scholar]
- [46].Suh S, Pak K, Seok JW, Kim IJ. Prognostic Value of Extranodal Extension in Thyroid Cancer: A Meta-Analysis. Yonsei Med J 2016; 57:1324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim CW, Kim J, Yeom SS, Lee JL, Yoon YS, Park IJ, et al. Extranodal extension status is a powerful prognostic factor in stage III colorectal cancer. Oncotarget 2017;8:61393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Invernizzi M, Corti C, Lopez G, Michelotti A, Despini L, Gambini D, et al. Lymphovascular invasion and extranodal tumour extension are risk indicators of breast cancer related lymphoedema: an observational retrospective study with long-term follow-up. BMC Cancer 2018;18:935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lop J, Rigo A, Codina A, de Juan J, Quer M, Leon X. Prognostic significance of extranodal extension in head and neck squamous cell carcinoma cN0 patients with occult metastatic neck nodes. Acta Otorrinolaringol Esp 2018;69:156–64. [DOI] [PubMed] [Google Scholar]
- [50].Luchini C, Veronese N, Nottegar A, Cheng M, Kaneko T, Pilati C, et al. Extranodal extension of nodal metastases is a poor prognostic moderator in non-small cell lung cancer: a meta-analysis. Virchows Arch 2018;472:939–47. [DOI] [PubMed] [Google Scholar]
- [51].Thompson LDR, Burchette R, Iganej S, Bhattasali O. Oropharyngeal Squamous Cell Carcinoma in 390 Patients: Analysis of Clinical and Histological Criteria Which Significantly Impact Outcome. Head Neck Pathol 2019. 10.1007/s12105-019-01096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wu D, Tischler AS, Lloyd RV, DeLellis RA, de Krijger R, van Nederveen F, et al. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am J Surg Pathol 2009;33:599–608. [DOI] [PubMed] [Google Scholar]
- [53].Stenman A, Zedenius J, Juhlin CC. The Value of Histological Algorithms to Predict the Malignancy Potential of Pheochromocytomas and Abdominal Paragangliomas-A Meta-Analysis and Systematic Review of the Literature. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ellis RJ, Patel D, Prodanov T, Nilubol N, Pacak K, Kebebew E. The presence of SDHB mutations should modify surgical indications for carotid body paragangliomas. Ann Surg 2014;260:158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kimura N, Takekoshi K, Naruse M. Risk Stratification on Pheochromocytoma and Paraganglioma from Laboratory and Clinical Medicine. J Clin Med 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Duan K, Mete O. Algorithmic approach to neuroendocrine tumors in targeted biopsies: Practical applications of immunohistochemical markers. Cancer Cytopathol 2016;124:871–84. [DOI] [PubMed] [Google Scholar]
- [57].Rooper LM, Bishop JA, Westra WH. INSM1 is a Sensitive and Specific Marker of Neuroendocrine Differentiation in Head and Neck Tumors. Am J Surg Pathol 2018;42:665–71. [DOI] [PubMed] [Google Scholar]
- [58].Miettinen M, McCue PA, Sarlomo-Rikala M, Rys J, Czapiewski P, Wazny K, et al. GATA3: a multispecific but potentially useful marker in surgical pathology: a systematic analysis of 2500 epithelial and nonepithelial tumors. Am J Surg Pathol 2014;38:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kimura N, Miura Y, Nagatsu I, Nagura H. Catecholamine synthesizing enzymes in 70 cases of functioning and non-functioning phaeochromocytoma and extra-adrenal paraganglioma. Virchows Arch A Pathol Anat Histopathol 1992;421:25–32. [DOI] [PubMed] [Google Scholar]
- [60].Fraga M, Garcia-Caballero T, Antunez J, Couce M, Beiras A, Forteza J. A comparative immunohistochemical study of phaeochromocytomas and paragangliomas. Histol Histopathol 1993;8: 429–36. [PubMed] [Google Scholar]
- [61].Pakbaz S, Asa SL, Mete O. Abstract #596: Alpha-inhibin Expression in Paragangliomas and Pheochromocytomas Shows Strong Correlation with VHL- and SDHx-driven Pseudohypoxic Pathway Disease. Mod Pathol 2020;33:591.31537895 [Google Scholar]
- [62].Ramsay JA, Asa SL, van Nostrand AW, Hassaram ST, de Harven EP. Lipid degeneration in pheochromocytomas mimicking adrenal cortical tumors. Am J Surg Pathol 1987;11:480–6. [DOI] [PubMed] [Google Scholar]
- [63].Lubensky IA, Pack S, Ault D, Vortmeyer AO, Libutti SK, Choyke PL, et al. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol 1998;153:223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Osinga TE, Korpershoek E, de Krijger RR, Kerstens MN, Dullaart RP, Kema IP, et al. Catecholamine-Synthesizing Enzymes Are Expressed in Parasympathetic Head and Neck Paraganglioma Tissue. Neuroendocrinology 2015;101:289–95. [DOI] [PubMed] [Google Scholar]
- [65].Kimura N, Shiga K, Kaneko K, Sugisawa C, Katabami T, Naruse M. The Diagnostic Dilemma of GATA3 Immunohistochemistry in Pheochromocytoma and Paraganglioma. Endocr Pathol 2020. [DOI] [PubMed] [Google Scholar]
- [66].Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, MeyerRochow GY, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol 2010;41:805–14. [DOI] [PubMed] [Google Scholar]
- [67].Pai R, Manipadam MT, Singh P, Ebenazer A, Samuel P, Rajaratnam S. Usefulness of Succinate dehydrogenase B (SDHB) immunohistochemistry in guiding mutational screening among patients with pheochromocytoma-paraganglioma syndromes. APMIS 2014;122:1130–5. [DOI] [PubMed] [Google Scholar]
- [68].Udager AM, Magers MJ, Goerke DM, Vinco ML, Siddiqui J, Cao X, et al. The utility of SDHB and FH immunohistochemistry in patients evaluated for hereditary paraganglioma-pheochromocytoma syndromes. Hum Pathol 2018;71:47–54. [DOI] [PubMed] [Google Scholar]
- [69].Santi R, Rapizzi E, Canu L, Ercolino T, Baroni G, Fucci R, et al. Potential Pitfalls of SDH Immunohistochemical Detection in Paragangliomas and Phaeochromocytomas Harbouring Germline SDHx Gene Mutation. Anticancer Res 2017;37:805–12. [DOI] [PubMed] [Google Scholar]
- [70].Papathomas TG, de Krijger RR, Tischler AS. Paragangliomas: update on differential diagnostic considerations, composite tumors, and recent genetic developments. Semin Diagn Pathol 2013;30:207–23. [DOI] [PubMed] [Google Scholar]
- [71].Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod Pathol 2015;28:807–21. [DOI] [PubMed] [Google Scholar]
- [72].van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol 2009;10:764–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Curras-Freixes M, Inglada-Perez L, Mancikova V, Montero-Conde C, Leton R, Comino-Mendez I, et al. Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J Med Genet 2015;52:647e56. [DOI] [PubMed] [Google Scholar]
- [74].Richter S, Gieldon L, Pang Y, Peitzsch M, Huynh T, Leton R, et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med 2019;21:705–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 2014;23:2440–6. [DOI] [PubMed] [Google Scholar]