

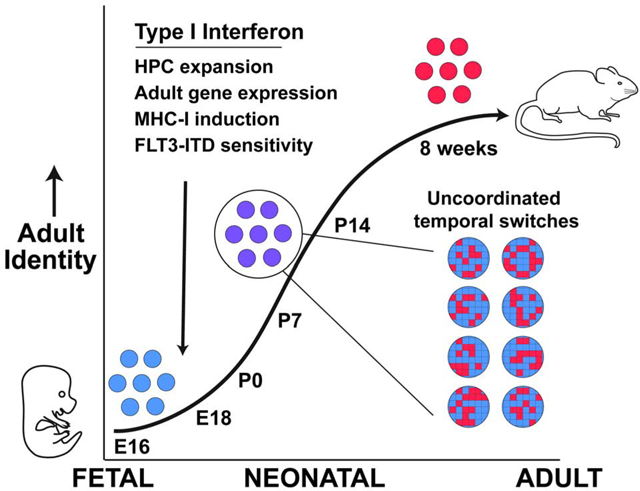
Summary
Fetal and adult hematopoietic stem cells (HSCs) have distinct proliferation rates, lineage biases, gene expression profiles and gene dependencies. While these differences are widely recognized, it is not clear how the transition from fetal to adult identity is coordinated. Here we show that murine HSCs and committed hematopoietic progenitors (HPCs) undergo a gradual, rather than precipitous, transition from fetal to adult transcriptional states. The transition begins prior to birth and is punctuated by a late prenatal spike in type I interferon signaling that promotes perinatal HPC expansion and sensitizes progenitors to the leukemogenic FLT3ITD mutation. Most other changes in gene expression and enhancer activation are imprecisely timed and poorly coordinated. Thus, heterochronic enhancer elements, and their associated transcripts, are activated independently of one another rather than as part of a robust network. This simplifies the regulatory programs that guide neonatal HSC/HPC ontogeny, but it creates heterogeneity within these populations.
Graphical Abstract
eTOC blurb
Li et al. have used single cell RNA-seq to understand how hematopoietic stem cells transition from fetal to adult states. They show that the shift is gradual and uncoordinated, but it begins with a precisely timed interferon pulse during late fetal development. Neonatal interferon modulates normal hematopoiesis and leukemia initiation.
Introduction
Hematopoiesis is sustained by multipotent hematopoietic stem cells (HSCs) and lineage committed hematopoietic progenitor cells (HPCs) (He et al., 2009; Laurenti and Gottgens, 2018). Definitive HSCs arise during mid-gestation from the aorta-gonad-mesonephros (AGM) (Medvinsky and Dzierzak, 1996; North et al., 2002). They migrate to the fetal liver and then, ultimately, the bone marrow where they persist throughout life (Mikkola and Orkin, 2006; Waas and Maillard, 2017). Fetal and adult HSCs have several fundamentally different properties. Fetal HSCs divide frequently and exhibit extensive self-renewal capacity (Copley and Eaves, 2013; He et al., 2011; Kim et al., 2007; Morrison et al., 1995). Adult HSCs divide infrequently and exhibit reduced self-renewal capacity relative to fetal HSCs (He et al., 2009; Pietras and Passegue, 2013). Fetal and adult HSCs have distinct lymphoid, myeloid and erythroid lineage biases (Benz et al., 2012; Mold et al., 2010; Rowe et al., 2016), with some innate immune cells (e.g. peritoneal B1a cells) arising almost exclusively from fetal HSCs (Barber et al., 2011; Beaudin et al., 2016; Ghosn et al., 2012; Hardy and Hayakawa, 1991). Thus, HSC self-renewal and differentiation mechanisms change across ontogeny.
As evidence of these changes, fetal and adult HSCs have different gene expression profiles, and they require different transcription factors and epigenetic regulators to maintain self-renewal capacity. For example, the transcription factor SOX17 is required to maintain fetal HSCs, but not adult HSCs, whereas ETV6 and GFI1 are required to maintain adult, but not fetal HSCs (Hock et al., 2004a; Hock et al., 2004b; Kim et al., 2007). Likewise, the epigenetic regulators BMI1, EED and ASH1L are required to maintain adult, but not fetal HSCs (Jones et al., 2015; Park et al., 2003; Xie et al., 2014). Fate mapping experiments have shown that adult HSCs arise from a sub-population of fetal HSCs rather than an independent, non-HSC precursor (Beaudin et al., 2016; Gothert et al., 2005; Samokhvalov et al., 2007). Thus, fetal HSCs must undergo transcriptional and epigenetic reprogramming as they transition to adult-like states.
Murine HSCs are generally thought to transition from fetal- to adult-like states between 3 and 4 weeks after birth, based on evidence showing that they become quiescent during this time period (Bowie et al., 2007; Bowie et al., 2006). The transition is thought to be regulated by a cell-intrinsic program driven, at least in part, by increasing C/EBPA expression (Ye et al., 2013). Around 3–4 weeks after birth, HSCs also become dependent on ASH1L and the tumor suppressor, PTEN, to enforce quiescence and maintain long-term self-renewal capacity (Jones et al., 2015; Magee et al., 2012). Prior studies have shown that, at the population level, neonatal HSCs gradually downregulate fetal gene expression programs, and they gradually upregulate adult gene expression programs (Copley et al., 2013; Porter et al., 2016). However, it is not clear whether, at a single cell level, the transition is bi-modal, like a switch, or gradual, like a rheostat. Furthermore, it is not clear how HSCs record developmental time so that they can transition from fetal to adult states on cue.
Single cell RNA-sequencing (scRNA-seq) provides an effective tool for mapping state changes in HSCs and other progenitors. The technique has been used previously to map the differentiation trajectories of adult hematopoietic progenitors (Giladi et al., 2018; Nestorowa et al., 2016; Olsson et al., 2016; Paul et al., 2015; Velten et al., 2017). These studies showed that the transcriptomes of differentiating progenitors change continuously, as cells become more lineage restricted, rather than precipitously at specified branch points of the hematopoietic hierarchy. The epigenomes of differentiating blood cells also evolve in a continuous manner (Buenrostro et al., 2018; Lara-Astiaso et al., 2014). By analogy, these observations raise the question of whether the transcriptomes of developing HSCs change continuously, over time, or precipitously at specified ages.
We used scRNA-seq, ATAC-seq and ChIP-seq to characterize the transcriptional and epigenomic landscapes of HSCs and HPCs as they transit the neonatal period. We found that the transition from fetal to adult identity is gradual rather than bi-modal, and individual neonatal HSCs/HPCs co-express fetal and adult transcripts. Both HSCs and HPCs begin transitioning to an adult-like transcriptional state even before birth, well before HSCs become quiescent. Furthermore, temporal changes in gene expression appear to be driven by imprecisely timed, uncoordinated changes in enhancer activity rather than a robust, precisely timed gene regulatory network. As a consequence, at any given neonatal age, individual HSCs (and HPCs) express different repertoires of fetal and adult transcripts.
Against this backdrop of uncoordinated transcriptional changes, we did observe a precisely timed spike in type I interferon (IFN) target gene activation just before birth. Interferons alpha and beta (IFNα and IFNβ) emanate from the skin, and potentially other distal organs, rather than local sources in the liver or bone marrow, and they are expressed even under germ-free conditions. IFN signaling promotes perinatal expansion of the HPC population while sparing HSCs. It reinforces an adult transcriptional program in both HSCs and HPCs, and it sensitizes postnatal progenitors to the leukemogenic FLT3-Internal Tandem Duplication (FLT3ITD) mutation. Altogether, these observations provide a timeline, and an overarching framework, that describes how HSCs and HPCs transition from fetal to adult transcriptional states. Furthermore, they implicate type I IFN signaling as a key developmental switch during perinatal and postnatal HSC/HPC ontogeny.
Results
Neonatal HSCs and HPCs undergo a gradual, synchronous transition from fetal to adult transcriptional states
To test whether the transition from fetal to adult identity is gradual or bi-modal, we performed scRNA-seq on HSCs and HPCs harvested at embryonic day (E)16.5, postnatal day (P)7, P14 and 8 weeks after birth (Figure 1A, Figure S1A). We expected that P7 and P14 progenitors would cluster with either the fetal or adult progenitors if the transition is bi-modal, or they would cluster separately if the transition is gradual (Figure 1B). Cells were clustered with Seurat, after regressing out unwanted variables such as cell cycle stage, and visualized by t-distributed Stochastic Neighbor Embedding (t-SNE) (Figure 1C, D; Figure S1B–E; Table S1). Fetal, postnatal and adult HSCs coalesced into 3 discrete clusters, as did the HPCs (Figure 1C, D). These patterns were most consistent with a graded transition.
Figure 1. The transition from fetal to adult HSC/HPC identity is gradual.
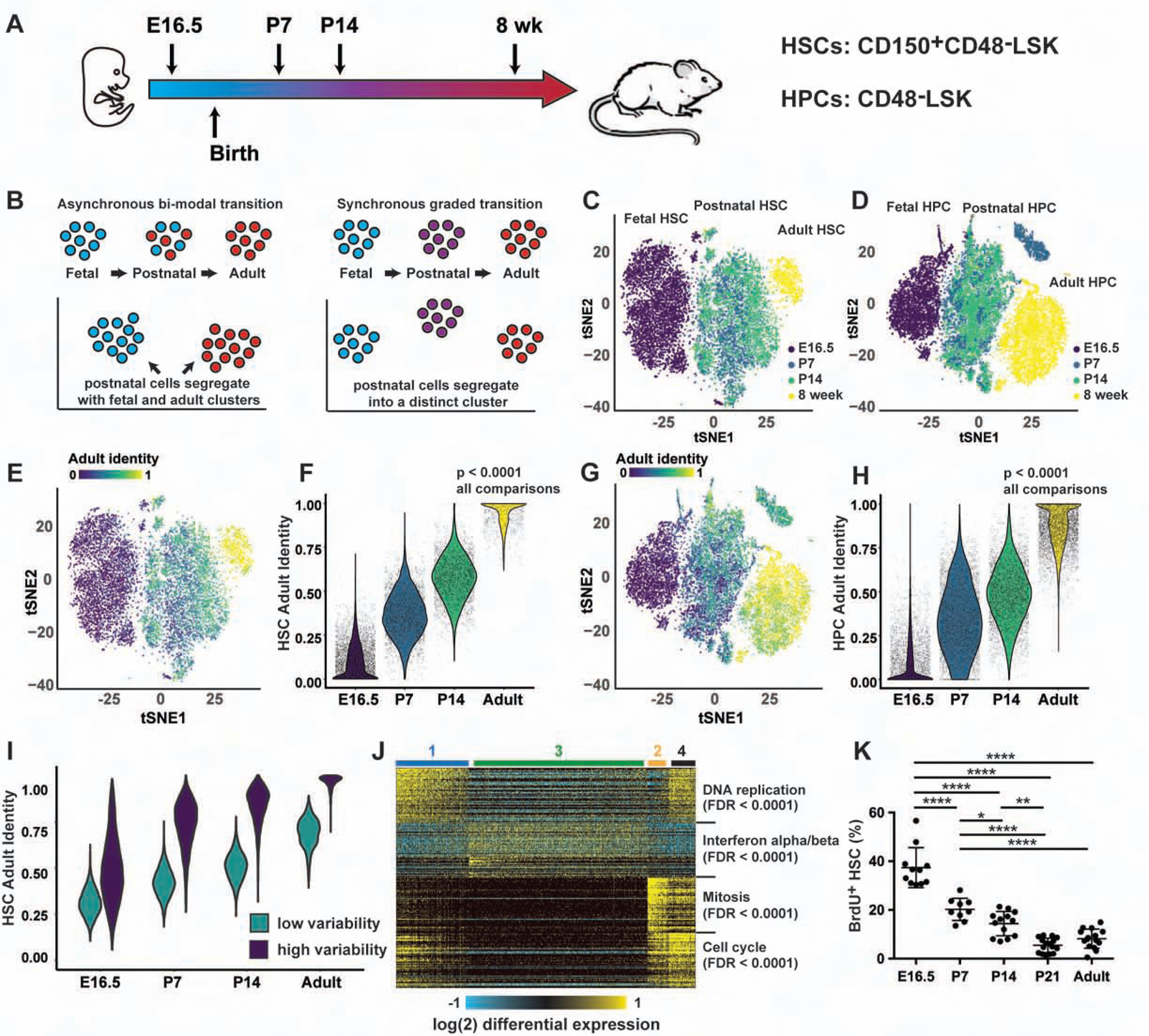
(A) Overview of scRNA-seq experiments. (B) scRNA-seq can resolve an asynchronous, bimodal transition from a synchronous, graded transition. (C, D) Postnatal HSCs/HPCs cluster separately from fetal and adult HSCS/HPCs. (E-H) Adult identity scores for HSCs (E, F) and HPCs (G, H); p<0.0001 for all comparisons. (I) Adult identity scores based on randomly selected genes from groups that exhibited low or high variability. (J) ICGS analysis of the HSC scRNA-seq timecourse. The heatmap illustrates four clusters of cells with Reactome pathway analysis of the guide genes shown to the right. (K) Twenty-four-hour BrdU incorporation in HSCs at the indicated ages. Error bars show standard deviation. *p<0.05, **p<0.01, ****p<0.0001 by one-way ANOVA and Holm-Sidak post-hoc test. See also Figures S1 and S2, and Table S1, S2 and S3.
To better quantify the extent to which P7 and P14 progenitors resemble fetal or adult progenitors, we used quadratic programming to assign adult identity scores to each cell. The output from this computational method was a fraction, between 0 and 1, that represented the degree to which each cell resembled either the initial (in this case fetal) or final (in this case adult) stage of the transition (Biddy et al., 2018; Kong et al., 2020; Treutlein et al., 2016). Scores were initially based on genes that distinguished fetal and adult progenitors in both the current data set and previously published, whole population data (McKinney-Freeman et al., 2012; Porter et al., 2016) (Tables S2 and S3). Adult identity genes were significantly enriched for type I IFN targets (Figure S1F). Many fetal identity genes promoted self-renewal in prior studies or in our own transplantation assays (e.g. Hmga2, Igf2bp1, Igf2bp2, Igf2bp3, Mecom; Figure S1G, H). When we used the curated lists to compute adult identity scores for each cell, we found that at each age, HSCs and HPCs were progressively more adult-like, and less fetal-like, than cells at the preceding ages (Figure 1E–H). There was no apparent bi-modal switch. Identity scores did not predict the cell cycle status of HSCs at any given age (data not shown), consistent with the fact that cell cycle genes were explicitly regressed out of the analysis.
We next tested whether an unsupervised analysis would similarly reveal gradual, linear changes in HSC/HPC identity. We calculated adult identity scores using the entire list of expressed genes, rather than a select list. This recapitulated the linear transitions observed with the select identity genes (Figure S2A, B). We next calculated predicted expression values for each gene based on the identity score for each cell, and we measured the degree to which actual gene expression varied from the predicted value. We identified two major groups of genes, one that was highly variable over time and drove the change in adult identity scoring, and another that was relatively static (Figure S2C, D). From each group, we randomly selected 214 genes (HSCs) or 115 genes (HPCs) to match the size of the non-randomly selected gene sets. We then calculated adult identity scores, and repeated the process 100 times, averaging the scores. We observed a gradual transition toward adult HSC identity over time, particularly when highly variable genes were sampled (Figure 1I). The transition was more subtle than was evident with non-randomly selected fetal and adult identity genes, but it nevertheless demonstrated gradual, linear, global reprogramming of HSC transcriptomes with age. We observed a similar trend for HPCs (Figure S2E). The selected fetal and adult identity gene lists therefore reflect more extensive temporal changes in neonatal HSC/HPC transcriptomes.
One limitation of quadratic programming is that it assumes a linear transition, so it could overlook transient, neonate-specific transcriptional programs. To address this possibility, we used Iterative Clustering and Guide-gene Selection (ICGS) to perform an unsupervised analysis of fetal, neonatal and adult HSC gene expression (Olsson et al., 2016). ICGS should capture clusters of transiently expressed genes, yet HSCs did not cluster according to age. Instead, the clustering was guided by expression of genes associated with DNA replication and cell division (Figure 1J – clusters 1, 2 and 4) and type I IFN target gene expression (cluster 3). This clustering pattern argues against a discrete transition (or a series of discreet transitions) from fetal to adult identity. It did implicate type I IFN signaling as a potential temporal regulator, but there was no one specific age at which the clustered genes appeared to become active or inactive. Changes in gene expression coincided with a gradual decline in HSC proliferation (Figure 1K). Altogether, the data show that HSCs undergo a gradual, sustained transition from fetal to adult transcriptional states rather than an abrupt bi-modal transition. This raises the question of how such a transition could be coordinated and timed.
Temporal changes in HSC/HPC gene expression are imprecisely timed and poorly coordinated at the single cell level
Gradual changes in temporal identity could arise via a few different mechanisms. One possibility is that precise regulatory networks allow cells to mark time intrinsically and execute coordinated, stage-specific changes in gene expression (Figure 2A). A second possibility is that extrinsic cues guide temporal changes in gene expression, again in a coordinated manner. A third possibility is that HSCs and HPCs activate adult identity genes, and inactivate fetal identity genes, in an uncoordinated manner (Figure 2B). In this model, the pace of development is determined by the stochastic likelihood that individual genes will convert from a fetal- to an adult-like state rather than sequential temporal cues. The circuit is simpler because it does not require precisely timed interactions between the constituent genes, but it is noisy.
Figure 2. Fetal identity genes are inactivated, and adult genes are activated, in an uncoordinated manner.
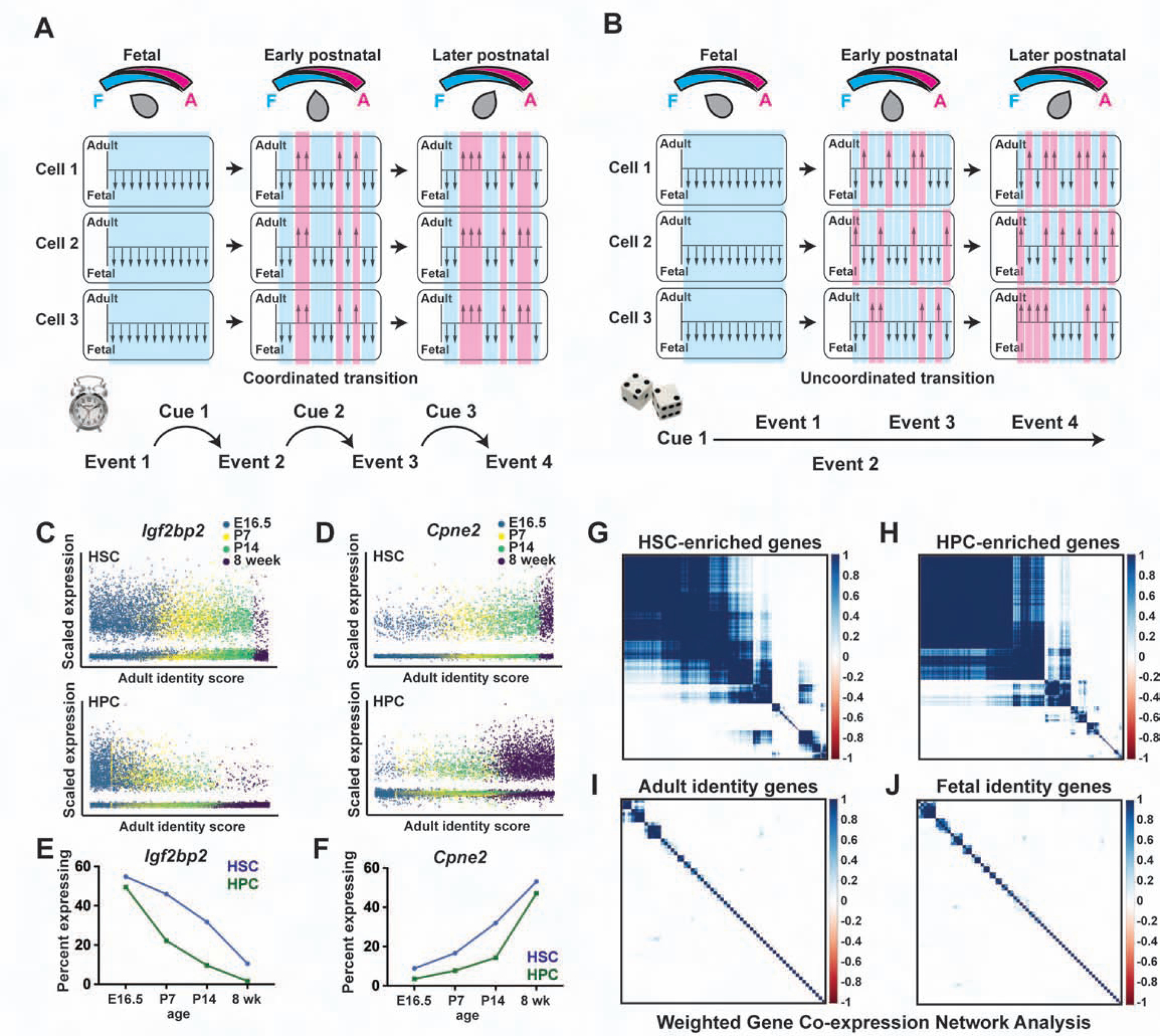
(A) Schematic overview of a coordinated transition from fetal to adult identity. As cells become older, individual genes or enhancers (shown as arrows) convert from a fetal-like to an adult-like state. The changes are uniform and precisely timed. (B) Schematic overview of an uncoordinated transition. As cells become more adult-like, individual genes convert from a fetal-like to an adult-like state non-uniformly. (C, D) Expression of Igf2bp2 and Cpne2 as a function of adult identity scores. (E, F) The fraction of Igf2bp2-expressing HSCs/HPCs declines with age, and the fraction Cpne2-expressing HSCs/HPCs increases with age. (G, H) WGCNA for genes that are differentially expressed during HSC to HPC differentiation, with correlation co-efficients indicating modular patterns of co-expression. (I, J) Fetal and adult identity genes do not exhibit patterns of co-expression. See also Figure S3.
To distinguish between these potential models, we evaluated expression of individual fetal and adult identity genes as a function of the adult identity score. We found that changes in gene expression were not precisely timed in individual cells. Fetal genes, such as Igf2bp2, Hmga2 and Arid3a, were expressed not only in fetal HSCs/HPCs, but also in a subset of cells with high adult identity scores (Figure 2C; Figure S3A, B). Likewise, adult genes, such as Cpne2, H2-Q7 and Sdsl, were expressed not only in adult HSCs/HPCs, but also in a subset of cells with low adult identity scores (Figure 2D; Figure S3C, D). At each tested age, individual HSCs (and HPCs) expressed different combinations of fetal and adult identity genes, but the ratios of adult to fetal transcripts were similar (Figure 2E, F; Figure S3E–I). These patterns are consistent with a model in which a majority of fetal identity genes are turned off, and a majority of adult identity genes are turned on, independently of one another rather than as part of a tightly coordinated, precisely timed developmental program.
To further assess the degree to which heterochronic genes are coordinately regulated, we adapted Weighted Gene Co-expression Network Analysis (WGCNA) for use with scRNA-seq data. WGCNA can identify modular patterns of gene co-expression that are most evident when cells undergo highly coordinated state changes. For example, WGCNA successfully identified modules of genes that were co-expressed during HSC to HPC differentiation (Figure 2G, H). This indicated that the technique is sensitive enough to detect correlations among transcripts within the single cell data set. However, in contrast to the differentiation paradigm, WGCNA failed to identify strong correlations among fetal or adult identity genes during postnatal HSC/HPC development (Figure 2I, J). This suggests that gradual, temporal changes in HSC/HPC gene expression are less tightly coordinated at the single cell level than the changes that accompany differentiation.
Neonatal epigenome reprogramming is gradual and discordant
We next tested whether HSC and HPC epigenomes also undergo gradual, discordant changes during neonatal development. We used ATAC-seq (Buenrostro et al., 2015) and ChIPmentation (Schmidl et al., 2015) to identify chromatin regions, and enhancer elements, that gain or lose accessibility between fetal and adult stages of HSC/HPC development. We then evaluated ATAC-seq peak heights at P7, P14 and P21. We observed a gradual transition from fetal- to adult-like chromatin accessibility patterns that was apparent even by P7 (Figure 3A, B). We identified 1230 putative enhancers – ATAC-seq peaks with overlapping histone 3, lysine 4 monomethylation (H3K4me1) peaks – that mapped within 100 kB of adult identity genes. Of these, 271 enhancers were active in adult HSCs/HPCs based on histone 3, lysine 27 acetylation (H3K27ac) (Figure 3C–E), and they converted gradually from fetal to adult ATAC-seq profiles (Figure 3F). The adult enhancers were enriched for ETS, RUNX, AP-1 and IRF binding motifs (Figure 3G). Only ~20% met criteria as super-enhancers (Figure 3H). Analysis of previously described ChIP-seq data (Komorowska et al., 2017; Wilson et al., 2016) showed that the enhancers bind to well-characterized hematopoietic transcription factors (Figure 3I), which were expressed, in most cases, at similar levels at each age (data not shown). One explanation for this pattern is that adult enhancers may harbor low affinity binding sites for a multitude of hematopoietic transcription factors. Rare, low affinity interactions between transcription factors could gradually, and non-uniformly, activate individual enhancers.
Figure 3. Neonatal HSC/HPC epigenome remodeling is gradual and uncoordinated.
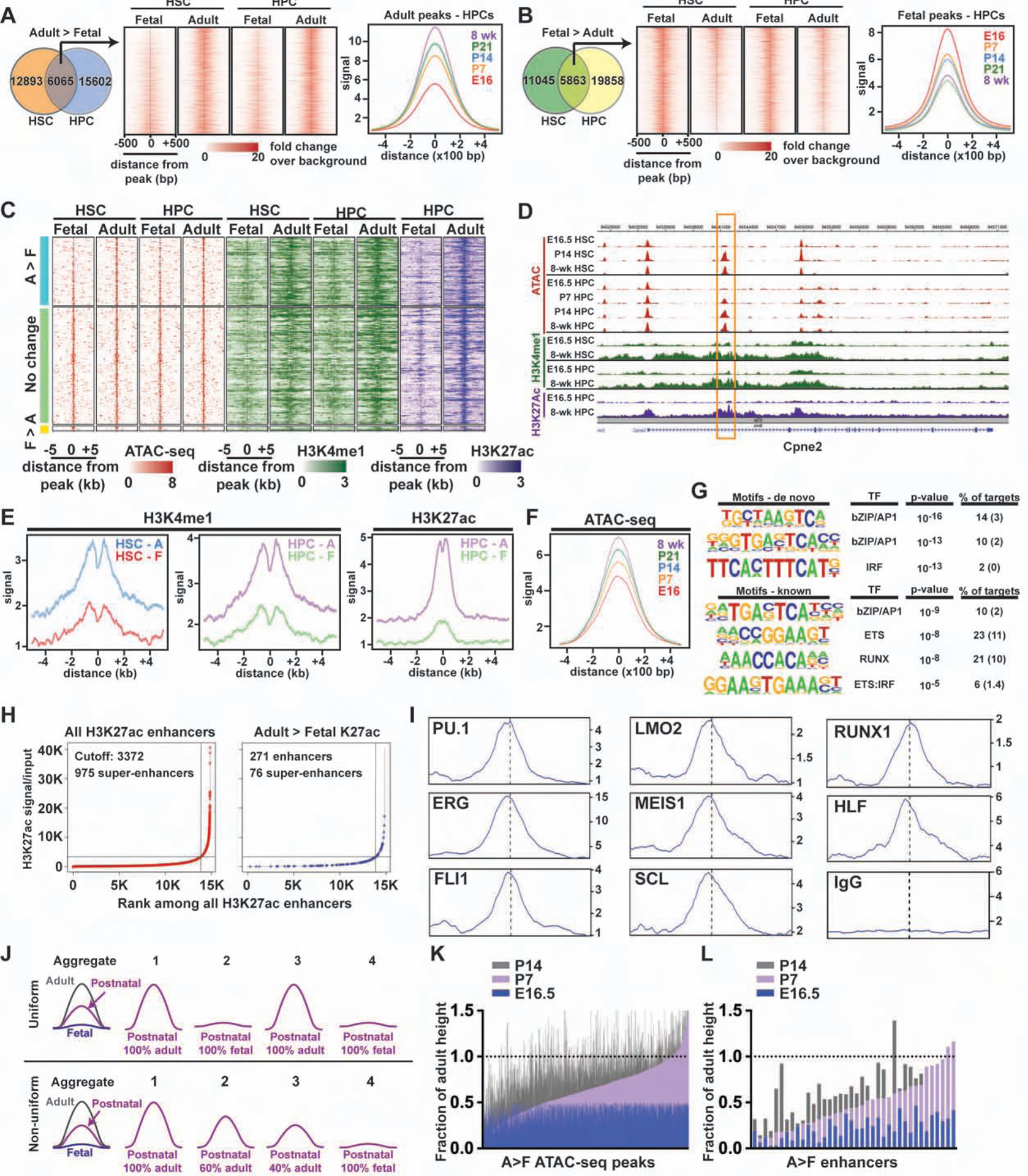
(A, B) ATAC-seq analysis of HSCs and HPCs at the indicated ages. Temporal changes in aggregate peak heights were gradual for both adult (A) and fetal (B) peaks. (C) ATAC-seq (red), H3K4me1 (green) and H3K27ac (purple) peaks for enhancers that map within 100 kB of adult identity genes. Most enhancers were either more accessible in adult HSCs/HPCs relative to fetal HSCs/HPCs (A>F) or exhibited no change in accessibility. (D) Representative tracks for the adult identity gene Cpne2, including a putative intron 1 enhancer (box). (E) Aggregate H3K4me1 and H3K27ac peaks for adult enhancers at the indicated ages. (F) Histogram showing that accessibility of A>F enhancers increased gradually with age. (G) HOMER analysis of adult enhancer elements. Parentheses indicate background. (H) ROSE analysis of all enhancer elements and adult-specific enhancer elements, as determined by adult > fetal H3K27ac. (I) Histograms indicating hematopoietic transcription factor binding at adult enhancer elements. (J) A model for how aggregate ATAC-seq profiles can reflect uniform or non-uniform changes in chromatin remodeling. (K, L) Heights for individual adult > fetal ATAC-seq peaks (K), or adult enhancers (L) with at least a 2-fold dynamic range, are shown normalized to the adult peak height.
To test whether adult enhancers are commissioned uniformly or non-uniformly, we evaluated the heights of individual ATAC-seq peaks in HSCs/HPCs at E16.5, P7 and P14, relative to adult heights. A uniform transition should yield bi-modal changes in individual peak heights – i.e. if each enhancer changes uniformly in all cells (Figure 2A), then neonatal peaks will either be completely fetal-like or completely adult-like (Figure 3J). A non-uniform transition should yield varying peak heights that trend incrementally toward adult heights over time – i.e. if each enhancer is accessible in some cells, but not others (Figure 2B), then neonatal peaks will range between fetal and adult heights (Figure 3J). The observed peak height changes were incremental rather than bi-modal (Figure 3K, L). This observation is most consistent with non-uniform, continuous epigenome remodeling rather than uniform, precisely coordinated epigenomic changes. Altogether, the epigenetic analyses reinforce our scRNA-seq observations. They favor a model in which adult identity genes, and their associated enhancers, are activated independently of one another rather than as part of a precisely timed circuit.
Fetal-specific enhancers remain accessible and epigenetically active in adulthood
We next tested whether fetal enhancers are deactivated gradually, and non-uniformly, much as adult enhancers are activated. Interestingly, enhancers near fetal identity genes remained accessible in adulthood, and they exhibited only modest decreases in H3K4me1 and H3K27ac levels (Figure 4A–E). Several fetal identity genes, such as Hmga2 and Igf2bp2, had qualitatively higher H3K4me1 levels throughout the gene bodies at E16.5, as compared to 8-weeks after birth (Figure 4F, G), but the profiles of distal enhancer elements were remarkably similar at all ages. This raised the question of whether other epigenetic mechanisms exist to suppress fetal gene expression in the adult HSCs. To test this, we used ChIPmentation to assess repressive histone 3, lysine 27 trimethylation (H3K27me3) at the fetal and adult promoters and enhancers. Adult promoters and enhancers had elevated H3K27me3 in fetal HSCs/HPCs, relative to adult HSCs/HPCs, consistent with lower expression levels (Figure 4H). In contrast, fetal promoters and enhancers had identical levels of H3K27me3 at both ages (Figure 4H). Again, the data suggest that fetal enhancers and promoters remain epigenetically competent to drive gene expression even in the adult stage of development. We did observe strong enrichment for BCL11A sites within the fetal promoters (Figure 4I). BCL11A directly represses fetal hemoglobin promoters after birth (Liu et al., 2018), and it may perform a similar function at other fetal-specific loci. Regardless of the exact transcription factors involved, the data show that neonatal HSC development is accompanied by widespread commissioning of adult enhancer elements but not concomitant decommissioning of fetal enhancer elements.
Figure 4. Fetal identity gene enhancers remain accessible and primed in adult HSCs and HPCs.
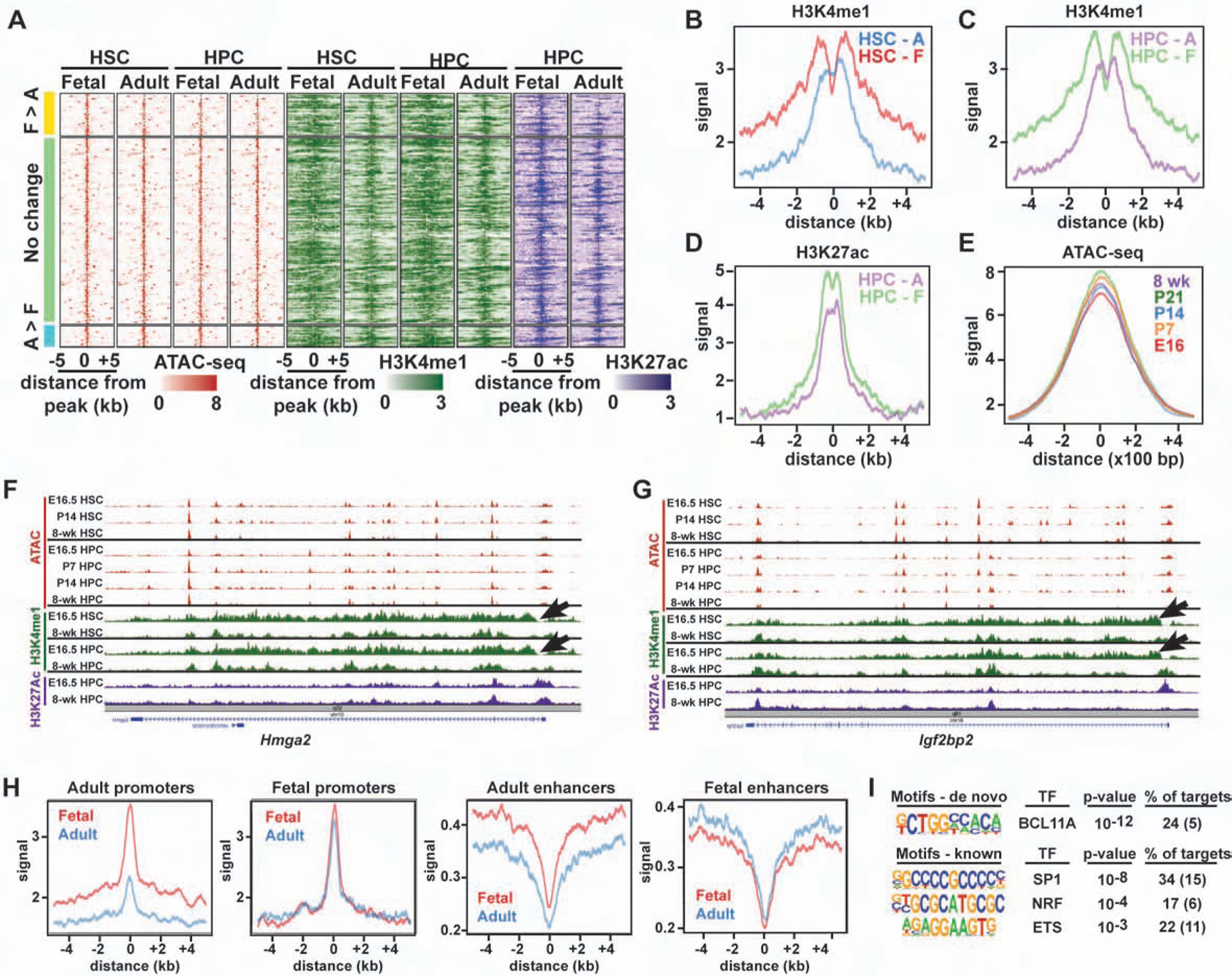
(A) ATAC-seq (red), H3K4me1 (green) and H3K27ac (purple) peaks for enhancers that map within 100 kB of fetal identity genes. (B-D) Aggregate H3K4me1 and H3K27ac levels in adult HSCs and HPCs. (E) Aggregate ATAC-seq profiles for the same enhancers. (F, G) The gene bodies of Hmga2 and Igf2bp2 have qualitatively higher H3K4me1 in fetal HSCs and HPCs (arrows). (H) Aggregate histograms of H3K27me3 at promoters and enhancers associated with fetal and adult identity genes. (I) HOMER analysis of fetal identity gene promoters.
The transition to adult transcriptional programs begins prior to birth
We considered that the neonatal transition might commence when HSCs migrate from the fetal liver to the bone marrow. To test this possibility, we compared single cell transcriptomes of P0 liver and bone marrow HSCs to one another and to HSCs harvested from the E16.5 liver and P7 bone marrow (Figure 5A, Figure S4A, B). If localization to the bone marrow were to coincide with the onset of fetal to adult identity changes, then P0 liver and bone marrow HSCs would cluster according to their location rather than their age (Figure 5A). Instead, clustering and t-SNE visualization showed that P0 bone marrow and liver HSCs formed a single cluster that was distinct from the E16.5 and P7 HSCs (Figure 5B, C). We performed the same analysis on E16.5, P0 and P7 HPCs (Figure 5D, E; Figure S4C–H). Again, the P0 bone marrow and liver HPCs clustered with one another rather than with the E16.5 and P7 HPCs (Figure 5D, E). Both P0 liver and bone marrow HSCs (and HPCs) had adult identity scores that closely resembled P7 HSCs (and HPCs) (Figure 5F, G). Thus, HSCs and HPCs begin transitioning from fetal to adult transcriptional programs before birth, irrespective of whether they are located in the liver or bone marrow.
Figure 5. The transition from fetal to adult identity begins prior to birth and coincides with a transient pulse in type I IFN target gene expression.
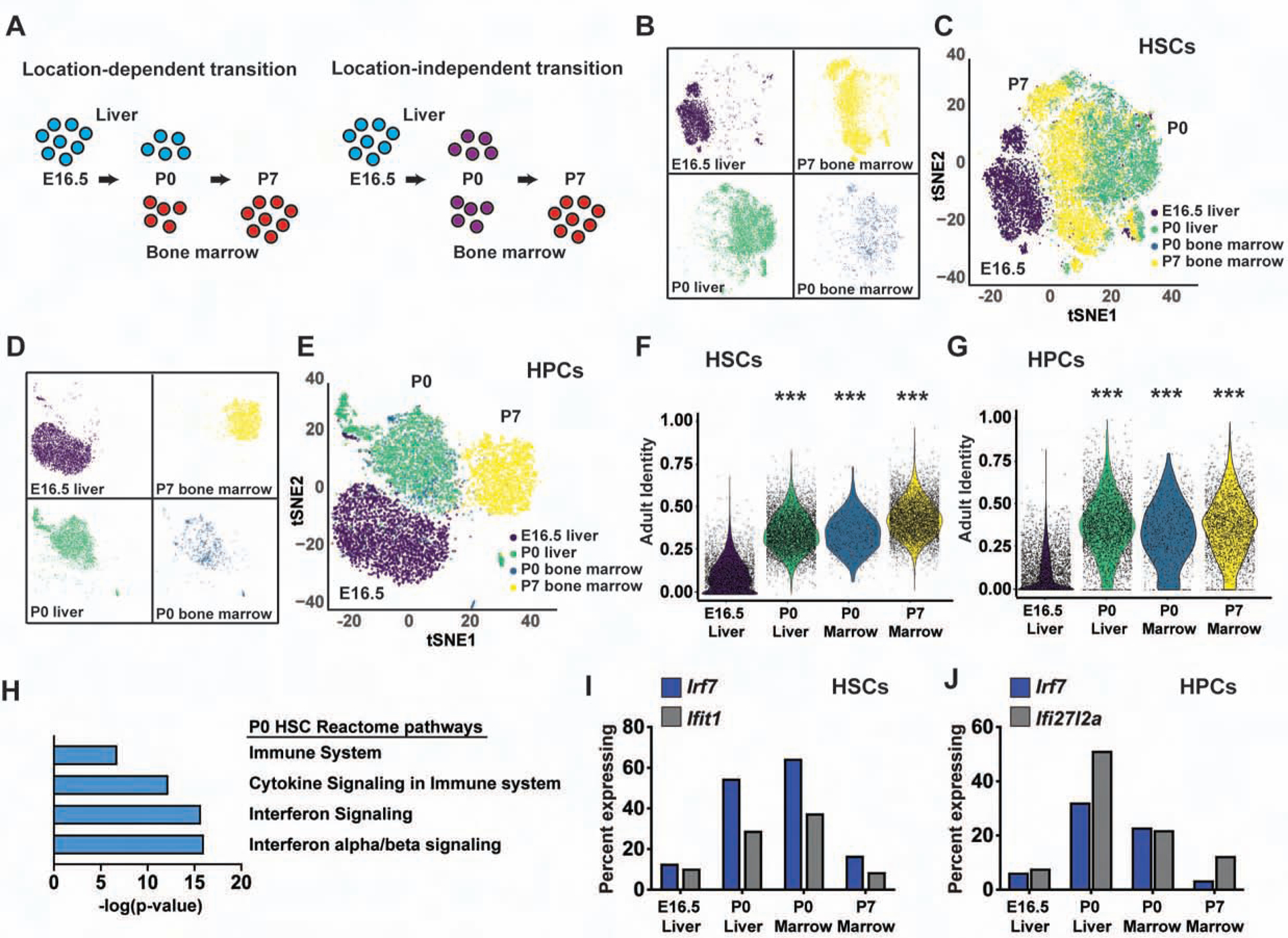
(A) Overview of scRNA-seq profiles for location-dependent and location-independent transitions. (B-E) P0 liver and bone marrow HSCs (B, C) and HPCs (D, E) cluster together, and separate from both E16.5 and P7 HSCs/HPCs. (F, G) Adult identity scores for E16.5 liver, P0 liver, P0 bone marrow and P7 bone marrow HSCs and HPCs. ***p<0.0001 relative to E16.5; two-tailed Student’s t-test. (H) Reactome pathway analysis of genes expressed higher in P0 HSCs than E16.5 and P7 HSCs (FDR <0.001). (I, J) Percentages of HSCs and HPCs at each age with detectable Irf7, Ifit1 and Ifi27l2a expression. See also Figure S4, Tables S1 and S4.
Late prenatal type I IFN signaling drives adult HSC and HPC transcriptional programs
In comparing bone marrow and liver HSCs/HPCs, we noted higher expression of type I IFN target genes, including Irf7, Ifit1 and Ifi27l2a, at P0 relative to both E16.5 and P7 (Figure 5H–J, Table S4). This unexpected, coordinated spike in gene expression contrasted with the more gradual, uncoordinated changes that were observed for other fetal and adult identity genes. It raised questions as to the precise age at which IFN target gene expression increases in fetal HSCs and how IFN signaling influences perinatal and postnatal gene expression.
To determine when IFN target genes are induced, we performed bulk RNA-seq on E14.5, E16.5, E18.5, P0, P7, P14 and 8-week-old HSCs, and E16.5, P0 P7, P14 and 8-week-old HPCs. A large number of IFN- and inflammation-related genes were induced between E16.5 and E18.5 in HSCs (Figure 6A–C). Most of these genes spiked in expression at E18.5 and P0. Expression then persisted at slightly lower levels after birth. Similar patterns were observed for HPCs (Figure S5A, B). IFNα and IFN03B2 protein levels were elevated by approximately 3-fold in the E18.5 liver, relative to E14.5 and E16.5 livers (Figure 6D, E), but maternal blood IFNα levels were very low at both ages (Figure S5C). IFN target gene induction at E18 required the IFNα/β receptor, IFNAR (Figure 6F, G). Altogether, these data show that there is a spike in type I IFN signaling between E16 and E18 that alters gene expression in both HSCs and HPCs.
Figure 6. Type I IFN signaling spikes at E18 and promotes adult identity gene expression.
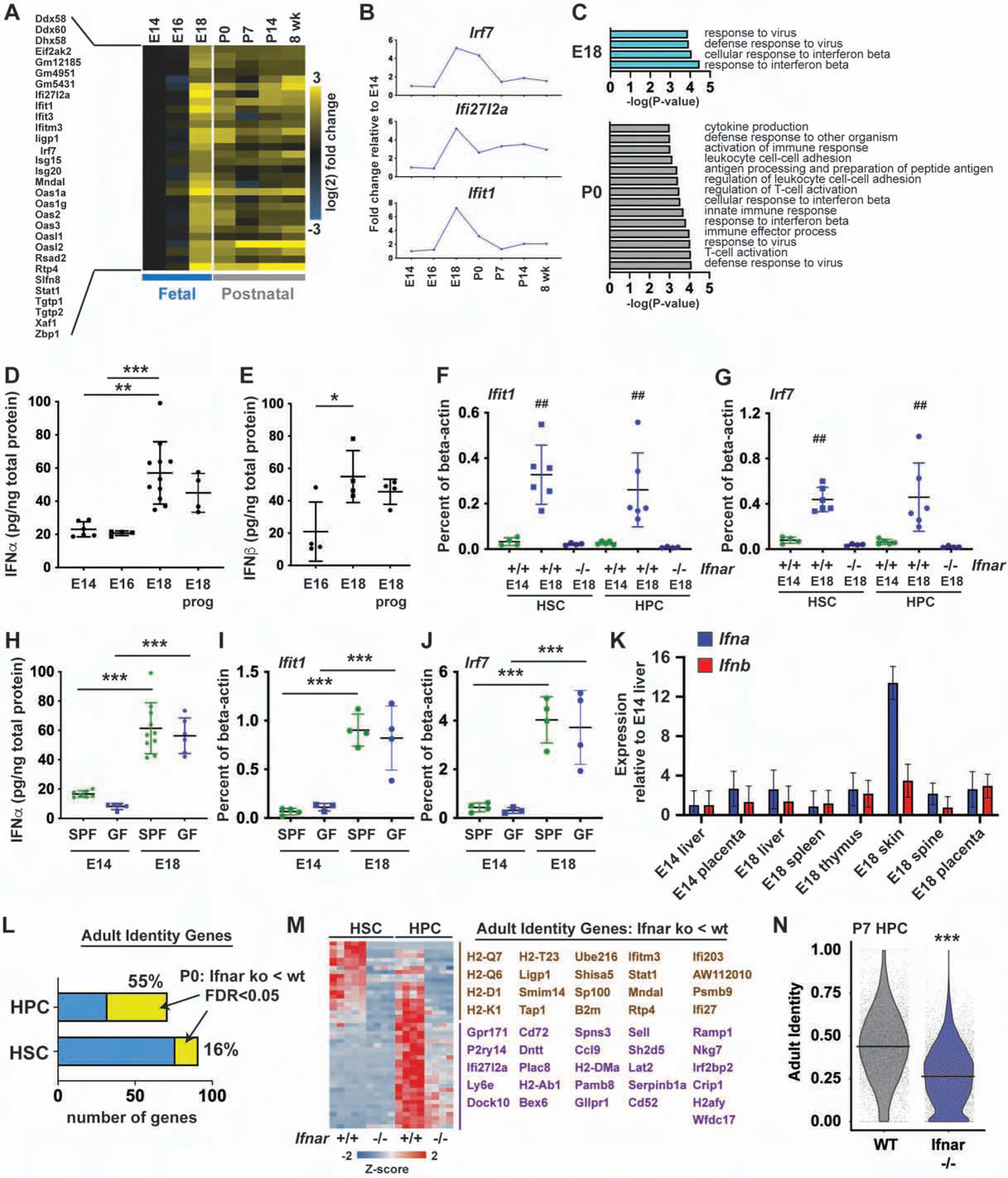
(A) IFN target gene expression in HSCs at indicated ages. n=4. (B) Irf7, Ifi27l2a and Ifit1 expression. (C) Biological process genes sets up in E18.5 or P0 HSCs relative to E16.5 (FDR<0.001). (D, E) IFNα and IFNβ levels in fetal livers at E14.5, E16.5 and E18.5 (+/− progesterone). n=4–10. (F, G) Ifit1 and Irf7 expression in wild type and Ifnar1−/− HSCs/HPCs at E14.5 and E18.5. n=4–6. (H) IFNα expression in E14 and E18 fetal livers from standard pathogen free “SPF” or germ free “GF” mothers. n=5–10. (I, J) Ifit1 and Irf7 expression in HSCs/HPCs from SPF and GF mice. n=3–4. (K) Expression of pan-Ifna and Ifnb1 in indicated tissues, normalized to E14 liver. n=3 (L) Percent of Ifnar-dependent adult identity genes in P0 HSCs and HPCs. (M) Ifnar-dependent adult identity genes (HPC-specific genes in purple, HSC/HPC genes in brown). (N) Adult identity scores for P7 wild type and Ifnar1−/− HPCs. All error bars indicate standard deviations. *p<0.05, **p<0.01 and ***p<0.0001; for panels F and G, ## reflects p<0.001 for E18.5 wild type HSCs/HPCs, relative to E14.5 wild type and E18.5 Ifnar−/− HSCs/HPCs. All comparisons were made by one-way ANOVA with Holm-Sidak post-hoc testing (panels D-J) or a two-tailed Student’s t-test (panel N). See also Figure S5 and Tables S1 and S5.
We next sought to identify the IFN source and stimulus. Progesterone signaling has been shown to suppress inflammation in some contexts, and progesterone levels drop during late gestation (Shah et al., 2019; Virgo and Bellward, 1974), but exogenous progesterone did not suppress INFα and IFNβ levels at E18 (Figure 6D, E). Fetal gut flora diversity expands considerably between E16 and 18, raising the possibility that this diversification stimulates IFN expression (Younge et al., 2019). To test this possibility, we measured IFNα and IFNβ levels in germ free mice at E14 and E18. We found that expression increased at E18 even under germ free conditions (Figure 6H, I). Ifit1 and Irf7 expression increased in HSCs/HPCs harvested from germ free mice (Figure 6J), and we did not observe changes in HSC or HPC numbers under germ free conditions (Figure S5D, E). Altogether, the data show that late gestation IFN signaling arises as part of a normal, sterile developmental program rather than in response to changes in the microbiome.
IFNα/β expression has previously been observed in the skin, thymus and spleen in murine and human fetuses, including under germ-free conditions (Colantonio et al., 2011; Duc-Goiran et al., 1994; Tovey et al., 1987). To identify a potential source, we performed quantitative RT-PCR for pan-Ifna or Ifnb1 transcripts, using cDNA from a variety of organs harvested at E14 and E18. We detected low level expression of Ifna and Ifnb in the E18 placenta and thymus, relative to the E14 liver. We detected a high level of Ifna expression in the E18 skin (Figure 6K). We were unable to detect Ifna/b expression in isolated hematopoietic populations (dendritic cells, liver monocytes, liver lymphocytes, thymocytes or splenocytes (data not shown). The data implicate the skin, and possibly other organs, as a non-local IFN source.
Since several lines of evidence have identified type I IFN signaling as a developmental switch in neonatal HSCs, we tested whether it regulates of adult identity gene expression. We performed RNA-seq on P0 HSCs and HPCs from wild type and Ifnar−/− mice. Many adult identity genes were Ifnar-dependent, particularly in HPCs, but also to a lesser extent in HSCs (Figure 6L, M; Figure S5F; Table S5). We therefore used scRNA-seq to test whether Ifnar deletion could blunt adult transcriptional programs in P7 HPCs (Figure 6N, Figure S5G, H). Identity scores were indeed more fetal-like, and less adult-like, in Ifnar1−/− HPCs relative to wild type HPCs (Figure 6N). ATAC-seq profiles of Ifnar−/− HPCs were also qualitatively less adult-like than those of wild type HPCs at P7 (Figure S5I), consistent with a delayed transition to adult identity.
Type I IFN signaling drives HPC expansion, and it enhances Major Histocompatibility I gene expression, from late gestation onward
We tested whether the prenatal IFN spike regulates perinatal and postnatal hematopoiesis. We first measured HSC and HPC numbers at E16 and P0. HSC numbers were similar in wild type and Ifnar−/− mice, both at E16 and P0 (Figure 7A). In contrast, HPC numbers increased ~3–4-fold between E16 and P0 in wild type mice, but not in Ifnar−/− mice (Figure 7B). Ifnar−/− HSCs were functional at both P0 and P14, as determined by competitive transplants with 20 sorted HSCs (Figure 7C, Figure S6A–D). Bromo-deoxyuridine (BrdU) incorporation was similar in wild type and Ifnar−/− HSCs at P7, indicating that the transition to quiescence is not delayed in the absence of IFN signaling (Figure S6E). Furthermore, Ifnar deletion did not cause postnatal HSCs to retain fetal-like lineage biases, such as peritoneal B1a B-cell potential (Figure S6F). Thus, the late prenatal IFN pulse promotes HPC expansion, but it does not alter HSC numbers, proliferation or function.
Figure 7. Ifnar deletion impairs perinatal HPC expansion and MHC-I expression, and it prevents FLT3ITD-driven HPC expansion.
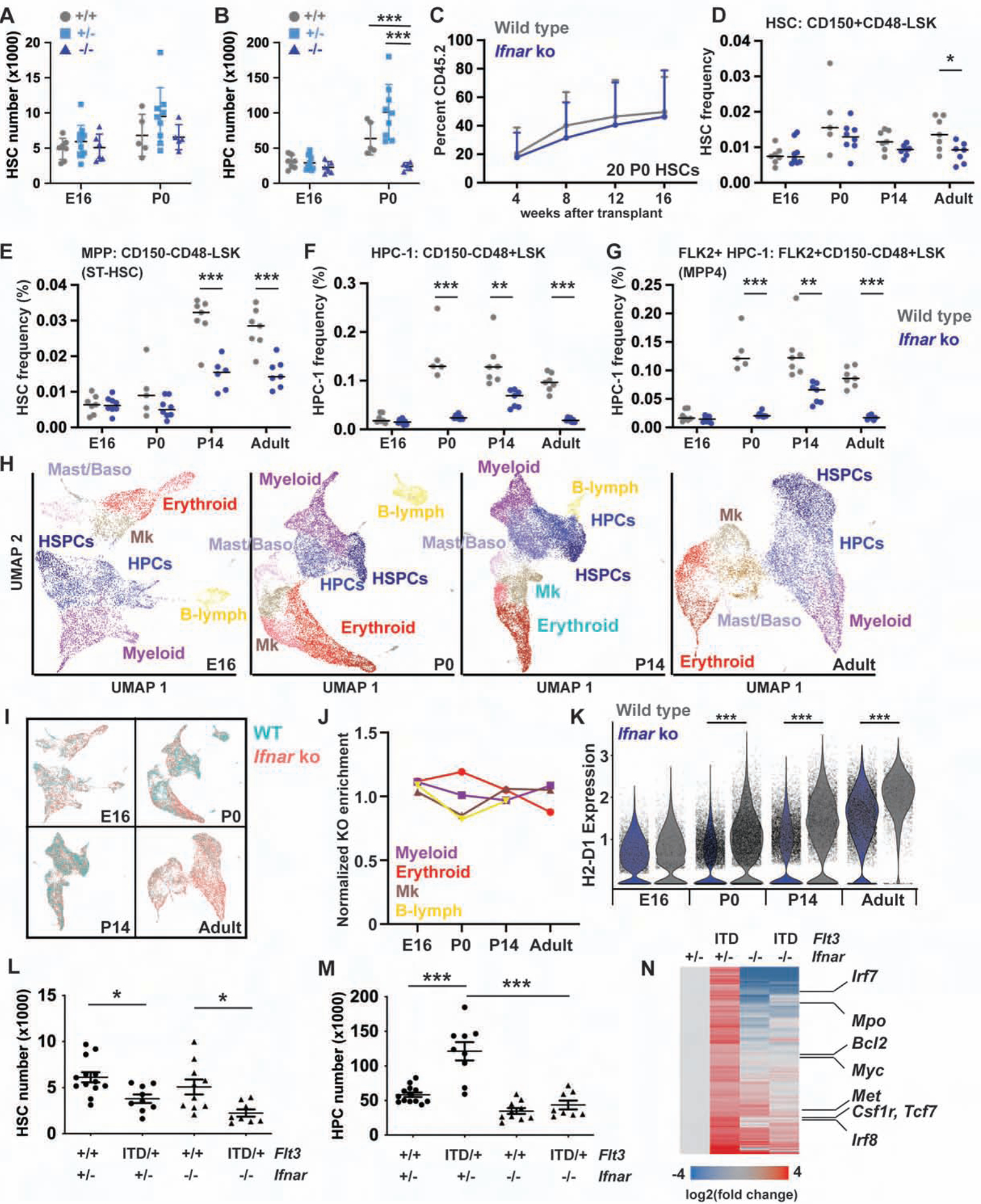
(A, B) HSC and HPC numbers in E16 and P0, wild type, Ifnar+/− and Ifnar−/− livers. n=5–9. (C) Peripheral blood reconstitution from P0 wild type and Ifnar−/− HSCs. n=12–14. (D-G) HSC, MPP, HPC-1 and FLK2+HPC-1 frequencies in wild type and Ifnar−/− mice at E16, P0, P14 and 8-weeks old. n=5–8. (H) UMAP plots of Lineage−c-kit+ cells based on scRNA-seq transcriptomes at E16, P0, P14 and 8-weeks old. Clusters are color coded based on expression of known lineage markers (see Figure S7C for the adult example). (I) UMAP plots, as in H, with color coding to indicate wild type and Ifnar−/− cells. (J) Normalized frequencies of Ifnar−/− cells in each lineage biased population. (K) H2-D1 expression in wild type and Ifnar−/− Lineage−c-kit+ cells at indicated ages. (L, M) Hindlimb HSC/HPC-1 numbers in P14 Flt3 and Ifnar mutant mice. n=8–13. (N) Gene expression changes for Ifnar-dependent FLT3ITD targets. Genes involved in AML pathogenesis are indicated to the right. All error bars indicate standard deviations. *p<0.05, **p<0.01 and ***p<0.001; comparisons by one-way ANOVA (A, B) or a two-tailed Student’s t-test (D-G, K) with Holm-Sidak post-hoc testing. See also Figures S6 and S7, and Tables S1, S6 and S7.
Since type I IFN signaling promotes HPC expansion, we tested whether it also promotes expansion of other committed progenitor populations and whether the effects were age-specific. We analyzed HSC, multipotent progenitor (MPP), HPC-1, HPC-2, pre-granulocyte-monocyte progenitors (pGM) and granulocyte-monocyte progenitor frequencies using previously described surface marker phenotypes (Oguro et al., 2013; Pronk et al., 2007). MPPs and HPC-1s were significantly depleted in Ifnar−/− mice from P0 onward (Figure 7E, F), as was the lymphoid-biased FLK2+ HPC-1 (sometimes called MPP4) subset (Figure 7G). HPC-2s were less severely affected, and pGMs and GMPs were either marginally expanded or not affected at all (Figure S6G–I). Thus, Ifnar deletion selectively impaired MPP and HPC-1 expansion.
To further test whether IFN signaling regulates commitment and gene expression in more committed progenitors, we performed scRNA-seq on Lineage−c-kit+ progenitors from E16, P0, P14 and adult wild type and Ifnar−/− mice (Figure 7H, Figure S7A, B). Cells were clustered with Seurat and visualized with Uniform Manifold Approximation and Projection (UMAP) (Becht et al., 2018). We annotated clusters based on expression of known lineage commitment genes (Figure 7H, Figure S7C). ICGS yielded very similar clustering results (Figure S7D–F). Ifnar−/− progenitors were represented within all lineage biased clusters (Figure 7I, Figure S7G, H). In fact, Ifnar deletion did not enrich or deplete these populations relative to the overall percentage of Ifnar−/− cells in each age group (Figure 7J). Thus, Ifnar deletion selectively depleted immature MPPs/HPCs. One notable consequence of Ifnar deletion was reduced expression of genes associated with Major Histocompatibility type I (MHC-I) antigen expression (e.g. H2-K1, H2-D1, B2m) across all progenitor clusters (Figure 7K, Figure S7I–L, Table S6). MHC-I gene expression increases with age in wild type progenitors, but Ifnar deletion blunts the change and potentially masks these progenitors from T-cell mediated destruction. Altogether, the data show that type I IFN signaling promotes adult transcriptional programs in both HSCs and HPCs, it promotes late prenatal and postnatal HPC expansion, and it promotes MHC-I expression that increases during ontogeny.
Type I IFN signaling sensitizes postnatal HPCs to FLT3ITD
Mechanisms of leukemogenesis change through the course of neonatal developmental (Lopez et al., 2019; Okeyo-Owuor et al., 2019; Porter et al., 2016), raising the question of whether neonatal type I IFN signaling can modulate sensitivity to leukemogenic mutations. We have previously shown that the Flt3ITD mutation induces HPC expansion and HSC depletion after birth, but not before (Porter et al., 2016). To test whether neonatal IFN signaling sensitizes HPCs to FLT3ITD, we generated compound mutant Flt3ITD/+;Ifnar−/− mice and analyzed HSC and HPC numbers at P14. Flt3ITD caused HSC depletion irrespective of Ifnar genotype, but FLT3ITD-driven HPC-1 expansion was completely rescued in Ifnar−/− mice (Figure 7L, M). Prior studies have shown that HPC-1s are the cell of origin for FLT3ITD-driven AML (Mead et al., 2013; Shih et al., 2015), and we therefore tested whether Ifnar deletion altered FLT3ITD target gene expression in these cells. Ifnar deletion rescued expression of a subset of FLT3ITD target genes, including many that are not canonical IFN targets (Figure 7N, Table S7). Several of these genes have established roles in AML pathogenesis (e.g. Bcl2, Myc, Met, Csf1r and Irf8). These findings establish a role for type I IFN signaling not only in normal hematopoiesis, but also in FLT3ITD-driven malignant hematopoiesis. Normal temporal changes in IFN expression can determine when in ontogeny HPCs become competent to transform.
Discussion
Our data provide a model, and a timeline, that describes how HSCs and HPCs transition from fetal to adult identities. The transition begins before birth, and it coincides with a spike in type I IFN signaling. Interestingly, the transition does not require migration to the bone marrow, as neonatal liver and bone marrow HSCs have indistinguishable single cell transcriptomes. After birth, heterochronic genes convert, slowly and non-uniformly, from fetal- to adult-like expression patterns. In aggregate, this causes a gradual shift toward adult HSC/HPC identities, but it also results in extensive epigenetic and transcriptional heterogeneity during the neonatal transition period. We propose that the uncoordinated nature of the circuit allows HSC development to progress gradually with only limited guidance from sequential, precisely timed cues.
The only precisely timed switch that we identified was a late prenatal IFN pulse at E18.5. IFN signaling appears to function differently in neonates than at other ages. During the earliest stages of hematopoietic ontogeny, IFN promotes emergence of HSCs from the AGM (He et al., 2015; Kim et al., 2016; Li et al., 2014). During adulthood, acute type I IFN stimulation promotes HSC proliferation and megakaryocyte activation, whereas chronic stimulation promotes HSC quiescence (Essers et al., 2009; Haas et al., 2015; Pietras et al., 2014; Walter et al., 2015). In neonates, IFN signaling had no effect on HSC numbers or function. Instead, it promoted perinatal and postnatal expansion of the HPC population, it reinforced an adult-like transcriptional program within the HPCs (and to a lesser extent the HSCs), and it drove expression of the MHC-I complex in all progenitor populations. The latter finding suggests that fetal and neonatal progenitors may be masked from T-cell mediated destruction relative to older progenitors. This has potential implications for idiopathic aplastic anemia, as it suggests that IFN signaling could potentially be targeted to mitigate T-cell-mediated HSC and progenitor cell losses.
One intriguing finding from our epigenomic studies was that most fetal enhancers are not decommissioned in adult progenitors. Thus, the adult enhancer landscape appears to superimpose itself on the fetal landscape, rather than replace it. Efforts to reprogram adult progenitors to fetal-like states could benefit from the fact that fetal enhancers remain primed and accessible. However, the permissive chromatin state may also allow fetal programs to be reactivated in adult hematopoietic malignancies. For example, Dnmt3a mutant murine progenitors can ectopically activate a fetal gene expression signature (Guryanova et al., 2016). Likewise, IGF2BP1, 2 and 3 are ectopically expressed and functionally important in some human leukemias (Elcheva et al., 2020; Palanichamy et al., 2016). While it is not clear how these programs are activated in leukemia cells, our data raise the possibility that leukemias may co-opt fetal regulatory elements.
Our findings have implications for childhood leukemogenesis. Mutations have age-specific effects on HSC/HPC fates, suggesting that normal developmental switches can make cells more, or less, competent to transform (Lopez et al., 2019; Magee et al., 2012; Okeyo-Owuor et al., 2019; Porter et al., 2016). We have now identified neonatal IFN signaling as a switch that sensitizes HPCs to FLT3ITD. It will be interesting to test whether IFNα/β signaling modulates age-specific responses to other leukemogenic mutations, as well.
Limitations of the study
Our data raise the question of why additional developmental switches were not identified and whether they would have been identified if we could have surveilled single cell transcriptomes in their entirety. It is possible that with a deeper sampling of the single cell transcriptomes, additional switches and correlations among heterochronic genes would emerge. However, a lack of depth cannot, by itself, explain the patterns that we observed. Essentially every adult HSC identity gene was expressed in a percentage of fetal HSCs, and essentially every fetal identity gene was expressed in a percentage of adult HSCs (Figure 2, Figure S3). This pattern generalized to a large percentage of expressed genes (Figure 1I, Figure S2). This is not compatible with a precise developmental circuit wherein well-defined inductive signals activate super-enhancers and feed-forward loops to enforce robust, uniform changes in HSC identity. The data are more consistent with a model in which neonatal HSCs and HPCs are exposed to chronic intrinsic and/or extrinsic stimuli that gradually recruit and activate adult-specific enhancers. The latter model predicts that, over time, all HSCs/HPCs will arrive at an adult-like state, but the path that each cell takes to get to that point will vary.
In addition, several questions need to be addressed in future studies. For example, our data predict that adult enhancers could be calibrated to activate earlier, or later, by altering affinity for the transcription factors shown in Figure 3I. This has yet to be tested. Likewise, we have not yet identified the transcription factors that limit fetal gene expression to the fetal stage despite epigenetic competence in adulthood. Finally, the putative link between skin IFNα/β expression and HSC/HPC gene expression changes needs to be functionally tested. These will be addressed in ongoing studies.
STAR METHODS
RESOURCE AVAILABILTY
Lead Contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Jeffrey Magee (mageej@wustl.edu).
Materials Availability
This study did not generate new unique reagents.
Data availability
The datasets generated during this study are available at Gene Expression Omnibus: GSE128758 (ATAC-seq), GSE128759 (RNA-seq), GSE128760 (ChIPmentation) and GSE128761 (scRNA-seq).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were performed with C57Bl/6 strain mice purchased from the Jackson Laboratory or bred (with Jackson Laboratory founders) in the Magee lab colony. Ages are indicated in the text and figures; both sexes were used equivalently. For fetal studies, timed pregnant C57Bl/6 female mice were purchased from the Jackson Laboratory. Deliveries were coordinated so that all cells in each independent experiment were harvested simultaneously, and the pregnant females were given several days to acclimate to the colony before they were euthanized. Ifnar1−/− – RRID:IMSR_JAX:028288 (Prigge et al., 2015), Flt3ITD – RRID:IMSR_JAX:011112 (Lee et al., 2007), and Rosa26M2−rTTA – RRID:IMSR_JAX:006965 (Hochedlinger et al., 2005) mice were obtained from the Jackson Laboratory. Mice were housed in a standard pathogen free barrier facility, except for the germ free studies as indicated in the text. For comparisons of germ free and standard pathogen-free housed mice, in vitro fertilizations were performed under sterile conditions. Fertilized embryos were then implanted into pseudopregnant germ free or standard mothers, on the same days with the same embryo sources. Fetal livers were then analyzed at the indicated time points, in parallel. All procedures were performed according to an IACUC approved protocol at Washington University School of Medicine. All mice were healthy and immune competent. All mice were naïve to prior procedures, drugs or tests. Cell lines were not used in these studies.
METHOD DETAILS
Flow cytometry
Bone marrow cells were obtained by flushing the long bones (tibias and femurs) or by crushing long bones, pelvic bones and vertebrae with a mortar and pestle in calcium and magnesium-free Hank’s buffered salt solution (HBSS), supplemented with 2% heat inactivated bovine serum (Gibco). Fetal and P0 liver cells were obtained by macerating livers with frosted slides. Single cell suspensions were filtered through a 40 μm cell strainer (Fisher). The cells were then stained sequentially, for 20 minutes each, with biotin conjugated anti-CD117 (c-kit; 2B8) and then Streptavidin-conjugated paramagnetic beads (Biolegend). c-kit+ cells were enriched by magnetic selection with an Automacs or LS magnetic columns (Miltenyi Biotec). Cells were stained for flow cytometry using the following antibodies, all of which were from Biolegend except as indicated: CD150 (TC15–12F12.2), CD48 (HM48–1), Sca1 (D7), c-kit (2B8), Ter119 (Ter-119), CD3 (17A2), CD11b (M1/70), Gr1 (RB6–8C5), B220 (RA3–6B2), CD4 (GK1.5), CD8a (53–6.7), CD2 (RM2–5), CD45.1 (A20), CD45.2 (104), surface IgM (RMM-1), CD5 (53–7.3), CD21 (7E9), CD23 (B3B4), CD135 (eBioscience; A2F10), CD105 (MJ7/18), CD41 (MWReg30), CD16/32 (93). Lineage stains for all experiments included CD2, CD3, CD8a, Ter119, B220 and Gr1. Antibodies to CD4 and CD11b were omitted from the lineage stains because they are expressed on fetal HSCs at low levels. The following surface marker phenotypes were used to define cell populations at all ages: HSCs (CD150+, CD48−, Lineage−, Sca1+, c-kit+), HPCs (CD48+, Lineage−, Sca1+, c-kit+), MPP (CD150−, CD48−, Lineage−, Sca1+, c-kit+), HPC-1 (CD150−, CD48+, Lineage−, Sca1+, c-kit+), HPC-2 (CD150+, CD48+, Lineage−, Sca1+, c-kit+), FLK2+ HPC-1 (CD135+, CD150−, CD48+, Lineage−, Sca1+, c-kit+), pGM (Lineage−, Sca1−, c-kit+, CD150−, CD105−, CD16/32−) and GMP (Lineage−, Sca1−, c-kit+, CD150−, CD105−, CD16/32+). Non-viable cells were excluded from analyses by 4’,6-diamidino-2-phenylindone (DAPI) staining (1 μg/ml). Flow cytometry was performed on a BD FACSAria Fusion flow cytometer (BD Biosciences). Cells were double sorted to ensure purity.
Single cell RNA-seq library construction
For each independent experiment, HSCs and HPCs for all groups were isolated by flow cytometry at the same time, using identical gating strategies, to minimize technical variation. For most groups, fifteen thousand HSCs or HPCs were double sorted into Phosphate Buffered Saline (PBS) with 0.1% Bovine Serum Albumin (BSA). Fewer cells were obtained from P0 bone marrow. Cell capture was performed immediately after cell isolation using the 10x Genomics platform. Kits included the Chromium Single Cell 3′ Library and Gel Bead Kit v2 (PN-120237), Chromium Single Cell 3′ Chip kit v2 (PN-120236) and Chromium i7 Multiplex Kit (PN-120262), and they were used according to the manufacturer’s instructions as per the Chromium Single Cell 3′ Reagents Kits V2 User Guide. The entire sorted population was loaded per lane of the chip with a goal of capturing 10,000 single-cell transcriptomes. All samples within a given experiment were processed in parallel, on the same day. Resulting cDNA libraries were quantified on an Agilent Tapestation and sequenced on an Illumina HiSeq 3000. scRNA-seq data can be accessed from Gene Expression Omnibus (GSE128761).
ATAC-seq and Chipmentation
For ATAC-seq, HSCs or HPCs were double sorted into PBS + 0.1% BSA. For most assays, 50,000 cells were isolated, though only 30,000 cells could be obtained for P7 HPCs. At least 3 independent replicates were analyzed per time point and population. Cells were processed, and libraries were generated, using the method described by Corces et al. (Corces et al., 2017). Library amplification was performed using barcoded primers as described by Buenrostro et al. (Buenrostro et al., 2013). After library amplification, fragments were size selected with SPRI beads (Beckman Coulter) with ratios of 0.9x and 1.8x for right- and left-sided selection. Sequencing was performed on a HiSeq2500.
For Chipmentation assays, 30,000 HSCs or HPCs were double sorted into PBS + 0.1% BSA. Chipmentation was performed as described by Schmidl et al. (Schmidl et al., 2015). Sonication was performed with a Covaris E220 using empirically defined setting (Peak incident power 105, duty factor 2%, cycles per burst 200, duration 840 seconds). ChIP-seq grade H3K4me1, H3K27ac and H3K27me3 antibodies were purchased from Diagenode. Library amplification was performed using barcoded primers as described by Buenrostro et al. (Buenrostro et al., 2013). After library amplification, fragments were size selected with SPRI beads with ratios of 0.65x and 0.9x for right- and left-sided selection. Sequencing was performed on a HiSeq2500.
RNA-seq
For each replicate, ten thousand HSCs or HPCs were double sorted into PBS with 0.1% BSA. Cells were pelleted by centrifugation and resuspended in RLT-plus RNA lysis buffer (Qiagen). RNA was isolated with RNAeasy micro-plus columns (Qiagen). RNAseq libraries were generated with Clontech SMRTer kits, and sequencing was performed on a HiSeq3000.
IFNα and IFNβ ELISA
IFNα and IFNβ levels were measured in fetal liver and maternal serum by ELISA (PBL assay science, Mouse Interferon alpha and beta ELISA kits). Fetal liver lysates were obtained by homogenizing liver fragments in a buffer of 100 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton-X100, 0.5% sodium deoxycholate, 1 mM EDTA, 1 mM EGTA. IFNα/β concentrations were normalized to total protein concentrations that were measured by Bradford assay (Coomassie Plus Bradford kit, Thermo Fisher).
Quantitative RT-PCR
For analysis of IFN target genes by quantitative RT-PCR, total RNA was isolated as described above using the Qiagen RNAeasy micro plus kit. For analysis of Ifna and Ifnb expression, the indicated organs were dissected and homogenized in Trizol reagent (Thermo Fisher) with a tissue homogenizer. RNA was then isolated according to manufacturer protocol. cDNA was generated with Superscript III reverse transcriptase (Thermo Fisher). Quantitative RT-PCR was performed with taqman probes specific to each indicated target gene (Thermo Fisher) or with pan-Ifna primers (Marro et al., 2019) using iTaq Universal Prober or SYBR green master mixes (Bio Rad). Expression values were normalized to Actb using the ΔΔCT method.
Long-term repopulation assays and IGF2BP1/2 overexpression assays
Eight- to ten-week old C57BL/6Ka-Thy-1.2 (CD45.1) recipient mice were given two doses of 550 rad delivered at least 3 hours apart. Wild type or Ifnar−/− HSCs were isolated from the livers of P0 mice or the bone marrow of P14 mice (CD45.2). Twenty HSCs were mixed with 300,000 competitor bone marrow cells per recipient and injected via the retroorbital sinus. To assess donor chimerism, peripheral blood was obtained from the submandibular veins of recipient mice at the indicated times after transplantation. Blood was subjected to ammonium-chloride lysis of the red blood cells and leukocytes were stained with antibodies to CD45.2, CD45.1, B220, CD3, CD11b and Gr1 to assess multilineage engraftment. For IGF2BP-1 and −2 overexpression assays, coding sequences for each protein were fused to EGFP with a P2A linker and cloned in place of the Oct4 cDNA in the TetO-FUW-oct4 lentivirus backbone (Brambrink et al., 2008). HSCs were isolated from Rosa26M2-rTTA mice (Jackson Labs strain 006965). HSCs were transduced with lentiviruses to express EGFP, IGF2B1–2A-EGFP or IGF2BP2–2A-EGFP proteins in the presence of doxycycline. Transduced cells were transplanted with 300,000 wild type competitor bone marrow cells as described above. Recipient mice were placed on doxycycline chow prior to transplantation. HSC and HPC chimerism levels (%GFP+ cells) were measured in recipient bone marrow at 16 weeks after transplantation.
BrdU incorporation assays
Bromo-deoxyuridine (BrdU; Sigma) was administered by IP injections (100 mg/kg/dose) given every 8 hours beginning 24 hours prior to bone marrow or fetal liver harvest. HSCs were stained and enriched by c-kit selection as described above. BrdU incorporation was measured by flow cytometry using the APC BrdU Flow Kit (BD Biosciences).
Assessment of B1a potential
P14 bone marrow cells were isolated from wild type and Ifnar−/− littermates. The cells were stained for HSCs as described above, and 100 HSCs were sorted into separate wells of a 96 well plate. Each well also contained 300,000 wild type CD45.1 bone marrow cells. The HSCs and competitor cells were transplanted into lethally irradiated CD45.1 recipient mice (two doses of 550 cGy separated by 3 hours). The recipients were then euthanized to assess CD45.2+ peritoneal B1a chimerism at 8 weeks after transplantation. Peritoneal washes were performed with staining media. Spleens and bone marrow were then isolated and stained as described above. B1a B-cells were defined within the peritoneal cell population as IgM+CD5+CD11b+. Spleen follicular B-cells were defined as B220+IgM+CD21+CD23−.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Comparisons
Group sizes and statistical tests are indicated in the text, with the exception of the bioinformatic analyses that are described below. In all figures, error bars indicate standard deviations. Sample sizes and numbers of replicates are indicated in the figure legends. In all cases in which multiple comparisons were made, a Holm-Sidak posthoc test was used to correct for multiple comparisons. A majority of the comparisons of wild type and Ifnar−/− progenitors were made prior to weaning/genotyping and were therefore performed in a blinded fashion. All other comparisons (e.g. age-related comparisons) were unblinded.
Alignment, digital gene expression matrix generation and filtering
The Cell Ranger v.2.1.0 and v3.0.1 pipelines (10x Genomics) were used to process data generated from the 10x Chromium platform. Digital gene expression (DGE) files were then filtered, as follows, for quality control. We first removed cells with a low number (<200) of unique detected genes. We then removed cells for which the total number of unique molecular identifiers (UMIs) (after log transformation) was not within three standard deviations of the mean. This was followed by removal of outlying cells with an unusually high or low number of UMIs (greater than 3 standard deviations from the mean after log transformation). The removal step was performed by fitting a LOESS curve (span = 0.5, degree = 2) to the number of UMIs with number of reads as the predictor. The same process was used to remove cells with unusually high or low number of genes, relative to the UMI counts. Finally, we removed cells in which the proportion of the UMI count attributable to mitochondrial genes was greater than 20%. Reads per cell, genes per cell and UMI per cell are shown in Table S1.
Seurat and quadratic programming analyses
After filtering, the R package Seurat was used to normalize the DGE matrices, and to cluster and visualize cells (Butler et al., 2018). Cell cycle scores were calculated for each cell using the CellCycleScoring function in Seurat. Metadata, such as cell cycle phase and time point, were collected for each cell in addition to the expression data. We regressed out differences based on the number of UMIs, proportion of mitochondrial UMIs, and cell cycle scores. Highly variable genes were then identified and used as input for dimensionality reduction via principal component analysis. The resulting principal components, and the correlated genes, were examined to determine the number of components to include in downstream analysis. These principal components were then used as input to generate and visualize clusters using t-SNE and UMAP. When we compared data from independent experiments to assess reproducibility, the data were integrated using canonical correlation analysis and mutual nearest neighbors with the Seurat V3 pipeline (Butler et al., 2018; Stuart et al., 2019). Quadratic programming was used to calculate adult identity scores based on expression of fetal and adult identity genes in each cell (Table S3). The identity genes were differentially expressed between the fetal and adult HSC or HPC populations (Table S2), and they were also found to be differentially expressed in fetal-adult comparisons described by either Porter et al. or McKinney-Freeman et al. (McKinney-Freeman et al., 2012; Porter et al., 2016). The R package QuadProg was used to generate cell identity scores using methods that we have previously reported (Biddy et al., 2018; Kong et al., 2020; Treutlein et al., 2016).
Random Gene Selection QP Analysis
The underlying assumption of quadratic programming is that each single-cell transcriptome is a linear combination of the reference cell types, where the estimated coefficients labels the fractional identities toward each cell type (Biddy et al., 2018; Kong et al., 2020; Treutlein et al., 2016). Let f = [ffetal, fadult] be the identity scores for each cell; X = [xfetal, xadult] represented the reference transcriptome matrix; and denoted the single-cell transcriptome. Then, we denoted the underlying model as
The linear model enabled us to calculate a “predicted” transcriptome profile based on the reference as . Thus, the deviation of each gene for each cell between the real and predicted expression was calculated as . The total absolute deviation of each gene was computed as . Here, we first used the entire list of expressed genes to calculate the fractional identity scores via quadratic programming, from which the “predicted” expression for each cell were then computed. We further calculated the total absolute deviation for each gene. The distribution of the total absolute deviation was observed to be bi-modal (Figure S2C, D). We divided the genes into two groups by manual selection of thresholds such that the peaks in the distribution could be captured and separated (HSC: Group 1: total absolute deviation > 3500 (high variability); Group 2: total absolute deviation < 2000 (low variability); HPC: Group 1: total absolute deviation > 5000 (high variability); Group 2: total absolute deviation < 2000 (low variability). From each group, we randomly sampled 214 genes HSC and 115 genes for HPC and calculated the adult identity scores using quadratic programming. We repeated this random sampling process 100 times and averaged the identity scores.
Network construction via WGCNA
We performed weighted correlation network analysis using the R package WGCNA (Langfelder and Horvath, 2008). Though WGCNA was initially developed for analysis of bulk expression data, we adapted the approach for single cell analysis (https://hms-dbmi.github.io/scw/WGCNA.html). For each age and cell type, differentially expressed genes were identified in Seurat. Pairwise correlation, soft threshold, and adjacency matrices were computed and clustered using Ward’s method to identify modules of highly correlated genes. The correlation heatmap was generated based on Pearson’s correlation matrix.
ICGS analysis
We performed Iterative Clustering and Guide-gene Selection (ICGS, version 2) analysis with the AltAnalyze toolkit (Venkatasubramanian et al., 2020) following provided documentation (https://altanalyze.readthedocs.io/en/latest/ICGS/). In brief, with the 10x filtered data matrix as input, ICGS (version 1) was used to iteratively identify guide genes. Dimensional reduction and initial clustering were then performed using sparse non-negative matrix factorization (SNMF). Each cell cluster was re-evaluated based on the weight matrix from SNMF, and highly correlated cluster-specific genes were identified. Clusters were refined using cluster fitness scores and re-assigned using support vector machines. Comparisons of wild type and Ifnar−/− progenitors were performed using ICGS2 in conjunction with CellHarmony (DePasquale et al., 2019) via the AltAnalyze graphical interface. CellHarmony was used to integrate and compare independent datasets from different treatments (wild-type and Ifnar−/−). Cells from the query were aligned/projected to the reference through k-nearest neighbor graph and community clustering within each dataset. With the alignment, cells from the query were merged with the reference based on their correlation with their projecting cells. Integrated data was further projected and visualized using UMAP.
RNA-seq analysis
Sequences were aligned to the mouse genome (Ensembl release 76 top-level assembly) using STAR version 2.0.4b (Dobin et al., 2013). Linear modeling (limma/voom) was used to compare gene expression across samples (Ritchie et al., 2015). False discovery rates were calculated using the Benjamini and Hochberg method. Pathway analysis was performed with Generally Applicable Gene-set Enrichment (Luo et al., 2009) or Reactome (Joshi-Tope et al., 2005). Heatmaps for visualizing RNA-seq data were generated with the Spotfire Omics package. RNA-seq data can be accessed from Gene Expression Omnibus (GSE128759).
ATAC-seq and ChIPmentation analysis
ATAC-seq reads were demultiplexed and mapped to mm10 using bowtie2. Peaks were identified and ChIP-seq coverage tracks were generated with MACS2 using the ATAC-seq and DNase-seq processing pipeline developed by the Kundaje lab (https://github.com/kundajelab/atac_dnase_pipelines, Version 0.3.3) (Roadmap Epigenomics et al., 2015). Consistency among replicates was assessed based on Irreproducible Discovery Rates (Li et al., 2011). Differential binding peaks were identified with the R package DiffBind. Signal tracks were visualized with the WashU Epigenome browser (Zhou et al., 2013).
ChIPmentation Reads were demultiplexed and mapped to mm10. Peaks were identified and ChIP-seq coverage tracks were generated with MACS2 using the AQUAS TF and histone ChIP-seq pipeline with the “histone” option (https://github.com/kundajelab/chipseq_pipeline, version 0.3.3) (Roadmap Epigenomics et al., 2015). Analysis of transcription factor binding sites was performed by lifting previously published tracks from HPC-7 cells (Wilson et al., 2016) onto mm10. Analysis of super enhancers was performed with Rank Ordering of Super Enhancers (Whyte et al., 2013). Transcription factor motif analysis was performed with HOMER (Heinz et al., 2010).
ATAC-seq and Chipmentation data can be accessed from Gene Expression Omnibus (GSE128758 and GSE128760, respectively).
Supplementary Material
Table S1. Quality control data for single cell RNA-seq (related to Figures 1, 5, 6 and 7). Metadata are shown for each single cell RNA-seq experiment.
Table S2. Heterochronic genes as determined by single cell RNA-seq (related to Figure 1). Differentially expressed genes are shown for comparisons of E16.5 fetal and 8-week-old adult HSCs and HPCs by single cell RNA-seq. Positive values indicate genes that are more highly expressed in fetal HSCs or HPCs.
Table S3. Fetal and adult identity genes for HSCs and HPCs (related to Figure 1). These identity genes were used to compute adult identity scores for HSCs and HPCs.
Table S4. Differential gene expression in E16, P0 and P7 HSCs (related to Figure 5). Differential expression was calculated from the scRNA-seq experiment in Figure 5.
Table S5. IFN target genes in HSCs and HPCs (related to Figure 6). Significantly differentially expressed genes (FDR<0.05) in Ifnar−/− HSCs and HPCs as compared to wild type.
Table S6. Cluster specific IFN target genes (related to Figure 7). For each age, genes that are differentially expressed between wild type and Ifnar−/− Lineage−c-kit+ cells are indicated for each annotated cluster, based on scRNA-seq analysis.
Table S7. Ifnar-dependent, FLT3-ITD target genes (related to Figure 7). Significantly differentially expressed genes (FDR<0.05) in Flt3ITD/+; Ifnar+/− HPCs as compared to Ifnar+/− that were then rescued in Flt3ITD/+; Ifnar−/− HPCs.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD150-PE | Biolegend | 115904; RRID: AB_313683 |
| CD48-APC | Biolegend | 103412; RRID: AB_571997 |
| Sca-1-PercpCy5.5 | Biolegend | 108124; RRID: AB_893615 |
| CD117-PE-Cy7 | Biolegend | 105814; RRID: AB_313223 |
| CD117-APC-Cy7 | Biolegend | 105826; RRID: AB_1626278 |
| CD2-FITC | Biolegend | 100105; RRID: AB_312652 |
| CD3-FITC | Biolegend | 100204; RRID: AB_312661 |
| CD8a-FITC | Biolegend | 100706; RRID: AB_312745 |
| B220-FITC | Biolegend | 103206; RRID: AB_312991 |
| Gr-1-FITC | Biolegend | 108406; RRID: AB_313371 |
| Ter119-FITC | Biolegend | 116206; RRID: AB_313707 |
| CD48-PE-Cy7 | Biolegend | 103424; RRID: AB_2075049 |
| CD16/32-BV711 | Biolegend | 101337; RRID: AB_2565637 |
| CD105-APC | Biolegend | 120414; RRID: AB_2277914 |
| CD45.1-APC-Cy7 | Biolegend | 110716; RRID: AB_313505 |
| CD45.2-FITC | Biolegend | 109806; RRID: AB_313443 |
| CD45.2-AF700 | Biolegend | 109822; RRID: AB_493731 |
| CD11b-APC | Biolegend | 101212; RRID: AB_312795 |
| Gr-1-PE-Cy7 | Biolegend | 108416; RRID: AB_313381 |
| B220-PercpCy5.5 | Biolegend | 103236; RRID: AB_893354 |
| CD3-PE | Biolegend | 100206; RRID: AB_312663 |
| CD117-Biotin | Biolegend | 135129; RRID: AB_2566721 |
| CD117-Biotin | Biolegend | 105804; RRID: AB_313213 |
| CD135-Biotin | Invitrogen | 13-1351-82; RRID: AB_466599 |
| H3K4me1 | Diagenode | C15410194; RRID: AB_2637078 |
| H3K4me3 | Diagenode | C15410003; RRID: AB_2616052 |
| H3K27ac | Diagenode | C15410196; RRID: AB_2637079 |
| H3K27me3 | Diagenode | C15410069; RRID: AB_2814977 |
| Bacterial and Virus Strains | ||
| TetO-FUW-oct4 | Brambrink et al, 2008 | Addgene: #20323 |
| Biological Samples | ||
| None | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Streptavidin-PE-Cy7 | Biolegend | 405206 |
| Streptavidin-APC-Cy7 | Biolegend | 405208 |
| Mojosort Streptavidin Nanobeads | Biolegend | 76447 |
| Critical Commercial Assays | ||
| APC BrdU Flow Kit | BD | 51–9000019AK |
| RNeasy Plus Micro Kit | QIAGEN | 74034 |
| MinElute Reaction Cleanup Kit | QIAGEN | 28204 |
| Illumina Tagment DNA Enzyme and Buffer (Small Kit) | Illumina | 20034210 |
| Sera-Mag Speedbeads | Fisher | 09-981-123 |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | M0541S |
| VeriKine-HS Mouse IFN-a All Subtype ELISA Kit | PBL | 42115 |
| VeriKine-HS Mouse IFN Beta Serum ELISA Kit | PBL | 42410 |
| Coomassie Plus (Bradford) Assay Kit | Thermo | 23236 |
| Ampure XP SPRI Beads | Beckman | B23318 |
| Chromium Single Cell 3’ Library and Gel Bead Kit v2 | 10x Genomics | PN-120237 |
| Chromium Single Cell 3’ Chip kit v2 | 10x Genomics | PN-120236 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| Deposited Data | ||
| ATAC-seq | This paper | GEO: GSE128758 |
| RNA-seq | This paper | GEO: GSE128759 |
| ChIPmentation | This paper | GEO: GSE128760 |
| scRNA-seq | This paper | GEO: GSE128761 |
| Experimental Models: Cell Lines | ||
| None | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:006965 |
| Mouse: B6(Cg)-Ifnar1tm1.2Ees/J | The Jackson Laboratory | RRID:IMSR_JAX:028288 |
| Mouse: B6.129-Flt3tm1Dgg/J | The Jackson Laboratory | RRID:IMSR_JAX:011112 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | RRID:IMSR_JAX:002014 |
| Oligonucleotides | ||
| Primer for Ifnar1 genotyping: Forward (Common): ACTCAGGTTCGCTCCATCAG | https://www.jax.org/Protocol?stockNumber=028256&protocolID=27369 | N/A |
| Primer for Ifnar1 genotyping: WT Reverse: CTTTTAACCACTTCGCCTCGT | https://www.jax.org/Protocol?stockNumber=028256&protocolID=27369 | N/A |
| Primer for Ifnar1 genotyping: MUT Reverse: GAACCTGAGGCTGTCGAAGG | https://www.jax.org/Protocol?stockNumber=028256&protocolID=27369 | N/A |
| Primer for qPCR: Ifna Forward: CCCTCCTAGACTCATTCTGCA | Marro et al., 2019 | N/A |
| Primer for qPCR: Ifna Reverse: AGGCACAGGGGCTGTGTTTC | Marro et al., 2019 | N/A |
| Primer for Flt3-ITD genotyping: Forward (Common): TCTGGTTCCATCCATCTTCC | https://www.jax.org/Protocol?stockNumber=011112&protocolID=27300 | N/A |
| Primer for Flt3-ITD genotyping: WT Reverse: AGGAAGTCGATGTTGGCACT | https://www.jax.org/Protocol?stockNumber=011112&protocolID=27300 | N/A |
| Primer for Flt3-ITD genotyping: MUT Reverse: TGGCTACCCGTGATATTGCT | https://www.jax.org/Protocol?stockNumber=011112&protocolID=27300 | N/A |
| Actb | Thermo Fisher | Cat#4352663 |
| Ifna (Mm03030145_Gh) | Thermo Fisher | Cat#4331182 |
| Ifnb1 (Mm00439546_s1) | Thermo Fisher | Cat#4331182 |
| Irf7 (Mm00516793_g1) | Thermo Fisher | Cat#4331182 |
| Ifit1 (Mm00515153_m1) | Thermo Fisher | Cat#4331182 |
| ATAC-seq and ChIPmentation primers | Buenrostro et el., 2013 | N/A |
| Recombinant DNA | ||
| TetO-FUW-EGFP | This study | N/A |
| TetO-FUW-IGFP2B1–2A-EGFP | This study | N/A |
| TetO-FUW- IGFP2B2–2A-EGFP | This study | N/A |
| Software and Algorithms | ||
| RStudio | RStudio | https://rstudio.com |
| Seurat v2/v3 | (Butler et al., 2018, Stuart et al., 2019) | https://satijalab.org/seurat/ |
| Quadratic Programming | (Treutlein et al., 2016; Biddy et al., 2018) | https://cran.r-project.org/web/packages/quadprog/index.html |
| ICGS2 | (Venkatasubramanian et al., 2020) | https://altanalyze.readthedocs.io/en/latest/ICGS |
| CellHarmony | (DePasquale et al., 2019) | https://github.com/nsalomonis/altanalyze/wiki/cellHarmony |
| CellRanger v2.1.0/v3.0.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Weighted correlation network analysis (WGCNA) | (Langfelder and Horvath, 2008) | https://hms-dbmi.github.io/scw/WGCNA.html |
| STAR version 2.0.4b | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| GAGE (Generally Applicable Gene-set Enrichment) | (Luo et al., 2009) | https://bioconductor.org/packages/release/bioc/html/gage.html |
| Reactome | (Joshi-Tope et al., 2005) | https://reactome.org |
| limma/voom | (Ritchie et al. 2015) | https://www.bioconductor.org/packages/release/bioc/html/limma.html |
| Spotfire Omics package | Spotfire Omics package | https://www.tibco.com/products/tibco-spotfire |
| ATAC-seq and DNase-seq processing pipeline, v0.3.3 | (Roadmap Epigenomics et al., 2015) | https://github.com/kundajelab/atac_dnase_pipelines |
| AQUAS TF and histone ChIP-seq pipeline, v0.3.3 | (Roadmap Epigenomics et al., 2015) | https://github.com/kundajelab/chipseq_pipeline |
| Bowtie2 | Bowtie, v2.2.26 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| DiffBind | DiffBind | http://www.bioconductor.org/packages/2.13/bioc/html/DiffBind.html |
| Irreproducible Discovery Rate (IDR) | (Li et al, 2011) | https://www.encodeproject.org/software/idr |
| MACS2 | MACS2, V2.1.0 | https://github.com/macs3-project/MACS |
| Rank Ordering of Super Enhancers (ROSE) | (Whyte et al., 2013) | http://younglab.wi.mit.edu/super_enhancer_code.html |
| HOMER | (Heinz et al. 2010) | http://homer.ucsd.edu/homer/motif |
| BD FACSDiva | BD | N/A |
| Flowjo v.10 | Treestar | N/A |
| GraphPad Prism v.8 | GraphPad Software | N/A |
| WashU Epigenome browser | (Zhou et al, 2013) | https://epigenomegateway.wustl.edu |
Highlights.
Murine HSCs and HPCs transition gradually from fetal to adult transcriptional states
Regulatory networks are uncoordinated, causing extensive neonatal HSC heterogeneity
Fetal to neonatal transitions coincide with a late gestational type I interferon pulse
The pulse promotes HPC expansion and sensitivity to leukemogenic FLT3-ITD signaling
Acknowledgements
This work was supported by grants to J.A.M. from the NHLBI (R01 HL136504 and R01 HL152180), Alex’s Lemonade Stand Foundation (‘A’ Award), Gabrielle’s Angel Foundation, The V Foundation, the American Society of Hematology and the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital. It was also supported by grants to S.A.M. from the NIGMS (R01 GM126112), Silicon Valley Community Foundation and the Chan Zuckerberg Initiative (grant HCA2-A-1708-02799). S.A.M. is supported by an Allen Distinguished Investigator Award (through the Paul G. Allen Frontiers Group), a Vallee Scholar Award, and a Sloan Research Fellowship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
Supplemental Information
Supplemental figures 1–7 and corresponding legends are provided as supplemental materials.
References
- Barber CL, Montecino-Rodriguez E, and Dorshkind K (2011). Reduced production of B-1-specified common lymphoid progenitors results in diminished potential of adult marrow to generate B-1 cells. Proc Natl Acad Sci U S A 108, 13700–13704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin AE, Boyer SW, Perez-Cunningham J, Hernandez GE, Derderian SC, Jujjavarapu C, Aaserude E, MacKenzie T, and Forsberg EC (2016). A Transient Developmental Hematopoietic Stem Cell Gives Rise to Innate-like B and T Cells. Cell Stem Cell 19, 768–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. [DOI] [PubMed] [Google Scholar]
- Benz C, Copley MR, Kent DG, Wohrer S, Cortes A, Aghaeepour N, Ma E, Mader H, Rowe K, Day C, et al. (2012). Hematopoietic stem cell subtypes expand differentially during development and display distinct lymphopoietic programs. Cell Stem Cell 10, 273–283. [DOI] [PubMed] [Google Scholar]
- Biddy BA, Kong W, Kamimoto K, Guo C, Waye SE, Sun T, and Morris SA (2018). Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie MB, Kent DG, Dykstra B, McKnight KD, McCaffrey L, Hoodless PA, and Eaves CJ (2007). Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proc Natl Acad Sci U S A 104, 5878–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, and Eaves CJ (2006). Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest 116, 2808–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambrink T, Foreman R, Welstead GG, Lengner CJ, Wernig M, Suh H, and Jaenisch R (2008). Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY, and Greenleaf WJ (2018). Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 173, 1535–1548 e1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantonio AD, Epeldegui M, Jesiak M, Jachimowski L, Blom B, and Uittenbogaart CH (2011). IFN-alpha is constitutively expressed in the human thymus, but not in peripheral lymphoid organs. PLoS One 6, e24252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley MR, Babovic S, Benz C, Knapp DJ, Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, et al. (2013). The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol 15, 916–925. [DOI] [PubMed] [Google Scholar]
- Copley MR, and Eaves CJ (2013). Developmental changes in hematopoietic stem cell properties. Exp Mol Med 45, e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePasquale EAK, Schnell D, Dexheimer P, Ferchen K, Hay S, Chetal K, Valiente-Alandi I, Blaxall BC, Grimes HL, and Salomonis N (2019). cellHarmony: cell-level matching and holistic comparison of single-cell transcriptomes. Nucleic Acids Res 47, e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duc-Goiran P, Robert B, Navarro S, Civas A, Cerutti I, Rudant C, Maury M, Condamine H, and Doly J (1994). Developmental control of IFN-alpha expression in murine embryos. Exp Cell Res 214, 570–583. [DOI] [PubMed] [Google Scholar]
- Elcheva IA, Wood T, Chiarolanzio K, Chim B, Wong M, Singh V, Gowda CP, Lu Q, Hafner M, Dovat S, et al. (2020). RNA-binding protein IGF2BP1 maintains leukemia stem cell properties by regulating HOXB4, MYB, and ALDH1A1. Leukemia 34, 1354–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, and Trumpp A (2009). IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 458, 904–908. [DOI] [PubMed] [Google Scholar]
- Ghosn EE, Yamamoto R, Hamanaka S, Yang Y, Herzenberg LA, Nakauchi H, and Herzenberg LA (2012). Distinct B-cell lineage commitment distinguishes adult bone marrow hematopoietic stem cells. Proc Natl Acad Sci U S A 109, 5394–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi A, Paul F, Herzog Y, Lubling Y, Weiner A, Yofe I, Jaitin D, Cabezas-Wallscheid N, Dress R, Ginhoux F, et al. (2018). Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat Cell Biol 20, 836–846. [DOI] [PubMed] [Google Scholar]
- Gothert JR, Gustin SE, Hall MA, Green AR, Gottgens B, Izon DJ, and Begley CG (2005). In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood 105, 2724–2732. [DOI] [PubMed] [Google Scholar]
- Guryanova OA, Lieu YK, Garrett-Bakelman FE, Spitzer B, Glass JL, Shank K, Martinez AB, Rivera SA, Durham BH, Rapaport F, et al. (2016). Dnmt3a regulates myeloproliferation and liver-specific expansion of hematopoietic stem and progenitor cells. Leukemia 30, 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, Wurzer S, Prendergast AM, Schnell A, Hexel K, et al. (2015). Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 17, 422–434. [DOI] [PubMed] [Google Scholar]
- Hardy RR, and Hayakawa K (1991). A developmental switch in B lymphopoiesis. Proc Natl Acad Sci U S A 88, 11550–11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Zhang C, Wang L, Zhang P, Ma D, Lv J, and Liu F (2015). Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood 125, 1098–1106. [DOI] [PubMed] [Google Scholar]
- He S, Kim I, Lim MS, and Morrison SJ (2011). Sox17 expression confers self-renewal potential and fetal stem cell characteristics upon adult hematopoietic progenitors. Genes Dev 25, 1613–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Nakada D, and Morrison SJ (2009). Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol 25, 377–406. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, and Jaenisch R (2005). Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 121, 465–477. [DOI] [PubMed] [Google Scholar]
- Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, and Orkin SH (2004a). Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431, 1002–1007. [DOI] [PubMed] [Google Scholar]
- Hock H, Meade E, Medeiros S, Schindler JW, Valk PJ, Fujiwara Y, and Orkin SH (2004b). Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev 18, 2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M, Chase J, Brinkmeier M, Xu J, Weinberg DN, Schira J, Friedman A, Malek S, Grembecka J, Cierpicki T, et al. (2015). Ash1l controls quiescence and self-renewal potential in hematopoietic stem cells. J Clin Invest 125, 2007–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Tope G, Gillespie M, Vastrik I, D’Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, et al. (2005). Reactome: a knowledgebase of biological pathways. Nucleic Acids Res 33, D428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Saunders TL, and Morrison SJ (2007). Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell 130, 470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PG, Canver MC, Rhee C, Ross SJ, Harriss JV, Tu HC, Orkin SH, Tucker HO, and Daley GQ (2016). Interferon-alpha signaling promotes embryonic HSC maturation. Blood 128, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komorowska K, Doyle A, Wahlestedt M, Subramaniam A, Debnath S, Chen J, Soneji S, Van Handel B, Mikkola HKA, Miharada K, et al. (2017). Hepatic Leukemia Factor Maintains Quiescence of Hematopoietic Stem Cells and Protects the Stem Cell Pool during Regeneration. Cell reports 21, 3514–3523. [DOI] [PubMed] [Google Scholar]
- Kong W, Fu Y, and Morris SA (2020). Capybara: A computational tool to measure cell identity and fate transitions. BioRxiv, 10.1101/2020.1102.1117.947390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. (2014). Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurenti E, and Gottgens B (2018). From haematopoietic stem cells to complex differentiation landscapes. Nature 553, 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Tothova Z, Levine RL, Anderson K, Buza-Vidas N, Cullen DE, McDowell EP, Adelsperger J, Frohling S, Huntly BJ, et al. (2007). FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell 12, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. The Annals of Applied Statistics 5, 1752–1779. [Google Scholar]
- Li Y, Esain V, Teng L, Xu J, Kwan W, Frost IM, Yzaguirre AD, Cai X, Cortes M, Maijenburg MW, et al. (2014). Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev 28, 2597–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. (2018). Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442 e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CK, Noguera E, Stavropoulou V, Robert E, Aid Z, Ballerini P, Bilhou-Nabera C, Lapillonne H, Boudia F, Thirant C, et al. (2019). Ontogenic Changes in Hematopoietic Hierarchy Determine Pediatric Specificity and Disease Phenotype in Fusion Oncogene-Driven Myeloid Leukemia. Cancer Discov 9, 1736–1753. [DOI] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, and Woolf PJ (2009). GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JA, Ikenoue T, Nakada D, Lee JY, Guan KL, and Morrison SJ (2012). Temporal changes in PTEN and mTORC2 regulation of hematopoietic stem cell self-renewal and leukemia suppression. Cell Stem Cell 11, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marro BS, Legrain S, Ware BC, and Oldstone MB (2019). Macrophage IFN-I signaling promotes autoreactive T cell infiltration into islets in type 1 diabetes model. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney-Freeman S, Cahan P, Li H, Lacadie SA, Huang HT, Curran M, Loewer S, Naveiras O, Kathrein KL, Konantz M, et al. (2012). The transcriptional landscape of hematopoietic stem cell ontogeny. Cell Stem Cell 11, 701–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead AJ, Kharazi S, Atkinson D, Macaulay I, Pecquet C, Loughran S, Lutteropp M, Woll P, Chowdhury O, Luc S, et al. (2013). FLT3-ITDs instruct a myeloid differentiation and transformation bias in lymphomyeloid multipotent progenitors. Cell reports 3, 1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvinsky A, and Dzierzak E (1996). Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 86, 897–906. [DOI] [PubMed] [Google Scholar]
- Mikkola HK, and Orkin SH (2006). The journey of developing hematopoietic stem cells. Development 133, 3733–3744. [DOI] [PubMed] [Google Scholar]
- Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, Weinberg K, Stoddart CA, and McCune JM (2010). Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science 330, 1695–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Hemmati HD, Wandycz AM, and Weissman IL (1995). The purification and characterization of fetal liver hematopoietic stem cells. Proc Natl Acad Sci U S A 92, 10302–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, and Gottgens B (2016). A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North TE, de Bruijn MF, Stacy T, Talebian L, Lind E, Robin C, Binder M, Dzierzak E, and Speck NA (2002). Runx1 expression marks long-term repopulating hematopoietic stem cells in the midgestation mouse embryo. Immunity 16, 661–672. [DOI] [PubMed] [Google Scholar]
- Oguro H, Ding L, and Morrison SJ (2013). SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeyo-Owuor T, Li Y, Patel RM, Yang W, Casey EB, Cluster AS, Porter SN, Bryder D, and Magee JA (2019). The efficiency of murine MLL-ENL-driven leukemia initiation changes with age and peaks during neonatal development. Blood Adv 3, 2388–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, and Grimes HL (2016). Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature 537, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanichamy JK, Tran TM, Howard JM, Contreras JR, Fernando TR, Sterne-Weiler T, Katzman S, Toloue M, Yan W, Basso G, et al. (2016). RNA-binding protein IGF2BP3 targeting of oncogenic transcripts promotes hematopoietic progenitor proliferation. J Clin Invest 126, 1495–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, and Clarke MF (2003). Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423, 302–305. [DOI] [PubMed] [Google Scholar]
- Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, et al. (2015). Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell 163, 1663–1677. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M, and Passegue E (2014). Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med 211, 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, and Passegue E (2013). Linking HSCs to their youth. Nat Cell Biol 15, 885–887. [DOI] [PubMed] [Google Scholar]
- Porter SN, Cluster AS, Yang W, Busken KA, Patel RM, Ryoo J, and Magee JA (2016). Fetal and neonatal hematopoietic progenitors are functionally and transcriptionally resistant to Flt3-ITD mutations. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge JR, Hoyt TR, Dobrinen E, Capecchi MR, Schmidt EE, and Meissner N (2015). Type I IFNs Act upon Hematopoietic Progenitors To Protect and Maintain Hematopoiesis during Pneumocystis Lung Infection in Mice. J Immunol 195, 5347–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk CJ, Rossi DJ, Mansson R, Attema JL, Norddahl GL, Chan CK, Sigvardsson M, Weissman IL, and Bryder D (2007). Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 1, 428–442. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RG, Wang LD, Coma S, Han A, Mathieu R, Pearson DS, Ross S, Sousa P, Nguyen PT, Rodriguez A, et al. (2016). Developmental regulation of myeloerythroid progenitor function by the Lin28b-let-7-Hmga2 axis. J Exp Med 213, 1497–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samokhvalov IM, Samokhvalova NI, and Nishikawa S (2007). Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 446, 1056–1061. [DOI] [PubMed] [Google Scholar]
- Schmidl C, Rendeiro AF, Sheffield NC, and Bock C (2015). ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat Methods 12, 963–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NM, Lai PF, Imami N, and Johnson MR (2019). Progesterone-Related Immune Modulation of Pregnancy and Labor. Front Endocrinol (Lausanne) 10, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, Antony-Debre I, Viale A, Socci N, Sun Y, et al. (2015). Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 27, 502–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovey MG, Streuli M, Gresser I, Gugenheim J, Blanchard B, Guymarho J, Vignaux F, and Gigou M (1987). Interferon messenger RNA is produced constitutively in the organs of normal individuals. Proc Natl Acad Sci U S A 84, 5038–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SA, Sim S, Neff NF, Skotheim JM, Wernig M, et al. (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, Hirche C, Lutz C, Buss EC, Nowak D, et al. (2017). Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol 19, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubramanian M, Chetal K, Schnell D, Atluri G, and Salomonis N (2020). Resolving single-cell heterogeneity from hundreds of thousands of cells through sequential hybrid clustering and NMF. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgo BB, and Bellward GD (1974). Serum progesterone levels in the pregnant and postpartum laboratory mouse. Endocrinology 95, 1486–1490. [DOI] [PubMed] [Google Scholar]
- Waas B, and Maillard I (2017). Fetal hematopoietic stem cells are making waves. Stem Cell Investig 4, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle B, Brocks D, Bayindir I, Kaschutnig P, et al. (2015). Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, Schoenfelder S, Hannah R, Sanchez Castillo M, Schutte J, Ladopoulos V, Mitchelmore J, Goode DK, Calero-Nieto FJ, Moignard V, et al. (2016). Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood 127, e12–23. [DOI] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, and Orkin SH (2014). Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14, 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Zhang H, Amabile G, Yang H, Staber PB, Zhang P, Levantini E, Alberich-Jorda M, Zhang J, Kawasaki A, et al. (2013). C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat Cell Biol 15, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younge N, McCann JR, Ballard J, Plunkett C, Akhtar S, Araujo-Perez F, Murtha A, Brandon D, and Seed PC (2019). Fetal exposure to the maternal microbiota in humans and mice. JCI Insight 4, e127806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lowdon RF, Li D, Lawson HA, Madden PA, Costello JF, and Wang T (2013). Exploring long-range genome interactions using the WashU Epigenome Browser. Nat Methods 10, 375–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Quality control data for single cell RNA-seq (related to Figures 1, 5, 6 and 7). Metadata are shown for each single cell RNA-seq experiment.
Table S2. Heterochronic genes as determined by single cell RNA-seq (related to Figure 1). Differentially expressed genes are shown for comparisons of E16.5 fetal and 8-week-old adult HSCs and HPCs by single cell RNA-seq. Positive values indicate genes that are more highly expressed in fetal HSCs or HPCs.
Table S3. Fetal and adult identity genes for HSCs and HPCs (related to Figure 1). These identity genes were used to compute adult identity scores for HSCs and HPCs.
Table S4. Differential gene expression in E16, P0 and P7 HSCs (related to Figure 5). Differential expression was calculated from the scRNA-seq experiment in Figure 5.
Table S5. IFN target genes in HSCs and HPCs (related to Figure 6). Significantly differentially expressed genes (FDR<0.05) in Ifnar−/− HSCs and HPCs as compared to wild type.
Table S6. Cluster specific IFN target genes (related to Figure 7). For each age, genes that are differentially expressed between wild type and Ifnar−/− Lineage−c-kit+ cells are indicated for each annotated cluster, based on scRNA-seq analysis.
Table S7. Ifnar-dependent, FLT3-ITD target genes (related to Figure 7). Significantly differentially expressed genes (FDR<0.05) in Flt3ITD/+; Ifnar+/− HPCs as compared to Ifnar+/− that were then rescued in Flt3ITD/+; Ifnar−/− HPCs.
Data Availability Statement
The datasets generated during this study are available at Gene Expression Omnibus: GSE128758 (ATAC-seq), GSE128759 (RNA-seq), GSE128760 (ChIPmentation) and GSE128761 (scRNA-seq).