

Abstract
Pharmacophore-directed retrosynthesis applied to ophiobolin A led to bicyclic derivatives that were synthesized and display anticancer activity. Key features of the ultimate defensive synthetic strategy include a Michael addition/facially-selective protonation sequence to set the critical C6 stereocenter and a ring-closing metathesis to form the cyclooctene. Cytotoxicity assays toward a breast cancer cell line (MDA-MB-231) confirm the anticipated importance of structural complexity for selectivity (vs. MCF10A cells) while C3 variations modulate stability.
Graphical Abstract
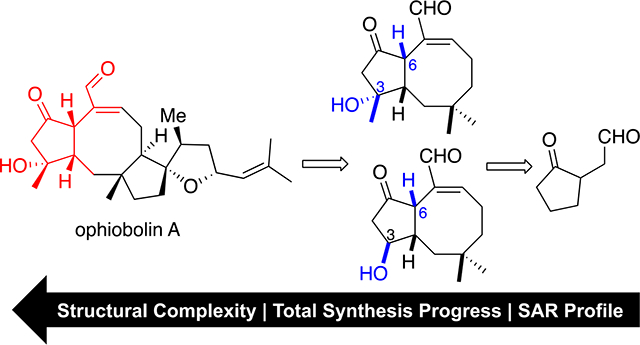
The ophiobolins are fungal-derived sesterterpenoids consisting of over 50 members,1, 2 with ophiobolin A (1, OpA, Figure 1) being the first isolated and also one of the most bioactive congeners (Figure 1).3–5 This family of natural products is drawing increasing interest2, 6, 7 not only because of their high potency against various cancer cell lines1, 8 (e.g. chronic lymphocytic leukemia (CLL), IC50 = 1 nM;8 P335, IC50 = 63 nM;6 glioblastoma Hs683, IC50 = 620 nM9, 10) for OpA, but also their unique cellular effects and selectivity observed. For example, both OpA (1) and B11 (2) show selectivity towards cancer stem cells (CSCs) by significantly reducing the CD44/CD24-positive population in a MDA-MB-231 cell line.12 Moreover, the ability of certain ophiobolins to induce paraptosis7, 9, 13 or autophagy14 makes these natural products valuable leads toward apoptosis-resistant cancer cells. Our interest in the ophiobolins stems from their complex structures, unique bioactivities and unknown cellular targets, and was further enhanced by our finding that OpA, but not 3-deoxy OpA, specifically induces cell death in stem-like cells with epithelial-mesenchymal transition (EMT) properties.15
Figure 1.

Structure of selected ophiobolins (A-C) and proposed minimal pharmacophore (yellow).
Several structure-activity relationship (SAR) studies of the anticancer effects of ophiobolins, derived mainly from derivatization of OpA or congeners, led us to apply our recently described pharmacophore-directed retrosynthesis (PDR) strategy16, 17 to elaborate on SAR information with respect to effects on stem-like cancer cells. In particular, the anticancer effects of various ophiobolins1, 2, 6 and synthetic derivatives,10 in conjunction with our own work15 point to the importance of the A/B-rings as an initial minimal pharmacophore consisting of a 1,4- keto unsaturated aldehyde moiety, the C5/C6-relative stereochemistry, the C3-hydroxyl, and syn-fused cyclooctene (Figure 1, highlighted in yellow). The potential reactivity of the 1,4-ketoaldehyde to primary amines including cellular protein lysines through a potential Paal-Knorr pyrrole- reaction has been reported.10, 18 Isolation of a phosphatidylethanolamine-adduct from HEK293T cells treated with OpA supports this notion.19 The natural C6-stereochemistry appears vital for the observed bioactivity across multiple cell lines with significant loss of activity observed for the C6-epimer of OpA.1, 2 Molecular modeling studies8 suggests one rationale for significant loss of bioactivity of 6-epi-ophiobolin congeners, namely significant alteration of the eight-membered B ring conformation. This altered conformation changes the orientation of the C21 aldehyde, thus impacting any potential Paal-Knorr condensation with the ketoaldehyde and also possibly impacting potential Michael additions to the unsaturated aldehyde.8 Therefore, we incorporate the cyclooctene into our current proposed pharmacophore (Figure 1). Efforts toward a broader, deep-seated SAR profile of the ophiobolins is precluded by structural convergence of the natural ophiobolins. Thus, we envisioned that application of PDR would enable a greater dissection of minimal structural requirements for anticancer activity through synthesis of simplified derivatives of OpA that retain structural features known to be critical for activity. Our initial PDR-guided studies toward this goal are described herein.
To date, three completed total syntheses of various ophiobolin and derivatives have been reported including the synthesis of OpA by the Nakada group who employed a late-stage ring closing metathesis (RCM) to form the 8-membered B ring20, 21 and ophiobolin C by the Kishi group who employed a Nozaki-Hiyama-Kishi coupling to generate the C-ring.22 Recently, Maimone reported an elegant 9-step radical cascade process to fashion the BC ring system of (–)-6-epi-OpN,23 and more recently, a 15-step synthesis of (+)-6-epi-OpA employing a similar synthetic strategy.24
Herein, we report the two initial stages of our PDR approach towards OpA toward gaining further SAR information of this complex terpene. To address questions of the C6-stereochemistry, the C3-hydroxy group, and required ring systems for bioactivity, we applied PDR to arrive at simplified monocyclic (e.g. known ketoaldehyde (±)-1125) and bicyclic (e.g. (±)-10) derivatives (Figure 2), all bearing our current hypothesized minimal pharmacophore (Figure 2, yellow highlight).
Figure 2.

(a) Pharmacophore-directed retrosynthesis applied to OpA (b) Staged syntheses of OpA derivatives including monocyclic (±)-11) and bicyclic (±)-10 derivatives bearing minimal pharmacophore described herein.
Our synthetic strategy placed an emphasis on installing the critical C6-stereocenter while retaining flexibility at certain positions such as the C3-hydroxyl bearing center to invert stereochemistry or alter substitution. For synthesis of the challenging 8-membered B-ring, we relied on the strategy of Nakada employing a RCM reaction.20, 21 Michael addition of a cuprate derived from iodide 6 to cyclopentenone 5 to form the C1-C4 bond followed by facially-selective protonation would enable control of the key C6-stereocenter. A protected secondary alcohol at C3 would enable variations in substitution and also stereochemistry at this center. The described synthetic strategy sets the stage for more elaborate tricyclic congeners or the natural product itself. Importantly, application of PDR enables the exploration of key strategic bond disconnections that typically would only be considered model studies in a total synthesis effort however, these studies can be utilized to access simplified derivatives and provide important SAR information as described herein.
We initiated our synthetic efforts by studying the conjugate addition of cuprate reagents derived from known neopentyl iodide 12a26 to enone (±)-5, to mimic the eventual cyclopentyl cuprate that would be required for tricyclic analogs (Scheme 1). However, several problems quickly surfaced including the anticipated low reactivity of the neopentyl cuprate reagent which could be overcome by forming the more reactive higher order organocuprate using the method of Lipshutz.27 Unfortunately, the resulting cuprate was prone to 5-exo-trig cyclization (see inset, Scheme 1), 26, 28 likely accelerated by the Thorpe-Ingold (gem-dimethyl) effect. This side reaction is unavoidable even at −78 °C unless the olefin is trisubstituted, directing us toward a defensive strategy29 and the use of iodide 1330 bearing a pendant trisubstituted alkene known to lower the rate of intramolecular cyclization.31 Lithium-halogen exchange32 followed by addition of (2-thienyl)Cu(CN)Li27 converted iodide 13 into a reactive organocuprate species leading to conjugate addition without detectable 5-exo-trig cyclization at −78 °C (Scheme 1). Use of TMSCl33, 34 was also beneficial for this conjugate addition but led to a TMS enol ether that was too labile for isolation. This problem was circumvented by treating the crude, intermediate silyl enol ether (±)-15 directly with MeLi followed by addition of Ac2O. This led to in situ conversion to the more stable and readily purified enol acetate (±)-16.35 Overall, this process entailed a one-pot conjugate addition/double-enolate trapping for the key C1-C5 bond construction. However, enol acetate (±)-16 failed to undergo the subsequent desired RCM reaction36–39 (not shown) to deliver the targeted cyclooctene despite trying a variety of conditions.
Scheme 1.

Development of the Conjugate Addition Sequence Leading to Enol Acetate 16
We anticipated that the syn-substituted dienyl cyclopentane (±)-18 would undergo the RCM reaction more readily. Toward this end, we next attempted to introduce the C6 stereocenter prior to the RCM through a facially selective protonation of a derived enolate following treatment of the enol acetate with MeLi. However, enolate generation and protonation led to complete isomerization of the alkene to the more conjugated position leading to tetra-substituted alkene (±)-17 rather than the desired syn-substituted cyclopentane (±)-18. To preclude this olefin isomerization, we decided to again employ a defensive strategy29 by refining the structure of the substrates without altering the overall synthetic plan. After extensive exploration, we settled on introduction of a phenyl ring at C8 (OpA numbering) to preclude C7-C8 olefin isomerization (cf. enone (±)-26, Scheme 2) during installation of the critical C6 stereocenter (for further description of this defensive strategy see Scheme S1 in the ESI). Our revised synthesis began by developing an efficient route to cyclopentenone (±)-26 (Scheme 3). Sonogashira coupling40 of PMB-protected propargyl alcohol 19 with iodobenzene followed by regioselective hydroboration with pinacol diborane,41 both on multi-decagram scale, delivered vinyl borane 23. Hydrolysis to the more reactive boronic acid 24,42 followed by Suzuki-Miyaura coupling43–45 with racemic, known cyclopentenyl bromide (±)-2546 gave enone (±)-26 on a multi-gram scale in 83% yield.
Scheme 2.

Refined Scalable Synthesis of the Refined Enone Substrate 26
Scheme 3.

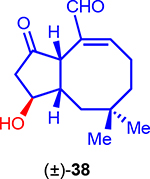
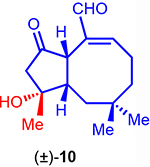
Synthesis of Bicyclic OpA Derivatives (±)-38 and (±)-10
Enone (±)-26 was then subjected to the one-pot cuprate addition silylation sequence developed previously, however a direct protonation of the intermediate enolate was also investigated to eliminate a step (Scheme 3). Following the same procedure developed previously for the conjugate addition-silylation/desilylation sequence beginning with iodide 27, a kinetic protonation delivered a single diastereomer of the trisubstituted cyclopentanone (±)-29 upon quenching with methyl acetoacetate (28) as the proton source.47 This sequence forged the required C1-C5 bond and also sets the C6 stereocenter with high diastereoselectivity. As expected, minimal olefin isomerization was observed due to the presence of the conjugated phenyl ring. This complex Michael addition/kinetic protonation sequence affording cyclopentanone (±)-29 was scalable (up to 8 g scale) and proceeded with high diastereoselectivity (>19:1) in 77% yield. Diastereoselective reduction of the ketone with L-selectride gave alcohol (±)-30 (>19:1, dr) and protection as the benzyl ether gave cyclopentane (±)-31. At this stage, the refined substrate served us well to prevent problematic issues encountered previously. However, in order to enable the proposed RCM reaction, terminal monosubstituted alkenes are required for optimal reactivity and efficiency which we verified through extensive experimentation.48 Thus, benzyl deprotection followed by ozonolysis revealed 1,8-ketoaldehyde (±)-32. While direct cyclization of ketoaldehydes by a McMurry reaction49, 50 to form 8-membered rings have been reported,51 several attempts were unsuccessful. Thus, toward applying RCM for cyclooctene formation with a substrate related to Nakada’s, we converted the ketoaldehyde (±)-32 to the bis-terminal alkene (±)-33. However, this bis-olefination required extensive optimization as the ketone was found to be inert to many common methylenation conditions.52, 53 After extensive studies, we were delighted to find that conditions developed by Takai54–57 delivered the desired bis-alkene (±)-33 in 52% yield. Following PMB ether deprotection to further reduce steric bulk around one alkene following the precedent of Nakada,20, 21 RCM of bis-olefin (±)-34 successfully established the 5/8 bicyclic core.58–60 Benzyl ether deprotection with Freeman’s reagent,61, 62 Swern oxidation,63, 64 and TBS-ether deprotection provided the first bicyclic OpA derivative (±)-38 which was best purified on Fluorosil®. This bicyclic OpA derivative shares the entire A/B ring system with the natural product OpA, differing only by having an epimeric, secondary alcohol at C3.
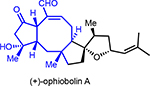
Toward gaining more information regarding the importance of the C3-teriary alcohol, we also targeted derivative (±)-10 bearing a tertiary alcohol with natural configuration at C3 (Scheme 3b). While use of a later stage, common intermediate such as benzyl ether (±)-35, would appear prudent, attempts to selectively manipulate the C3 alcohol required extensive protecting group manipulations or led to extensive β-elimination. We therefore opted to begin with alcohol (±)-33 toward C3-derivatives and an abbreviated synthetic scheme is shown (Scheme 3b; for the detailed synthetic sequence, see Scheme S2 in ESI). Methyl Grignard addition to the derived ketone (±)-39 proceeded diastereoselectively (dr > 20:1) to deliver tertiary alcohol (±)-40. Subsequent synthetic steps toward this bicyclic derivative followed a similar strategy to that described above for OpA derivative (±)-38, however the final step, a bis-oxidation step to afford the ketoaldehyde, was found to be quite challenging so is described briefly here. This bis-oxidation was plagued by C6-epimerization or C3-dehydration through β-elimination under a variety of oxidative conditions screened. Ultimately, we found that Hünig’s base in lieu of the more commonly employed Et3N was key to a clean Swern bis-oxidation but still afforded a modest yield (33%). Furthermore, it should be noted that the resulting ketoaldehyde (±)-10, while stable once purified, was quite sensitive to many stationary phases for purification. Silica gel deactivated with Et3N, basic alumina, and Fluorosil® all resulted in extensive decomposition. Use of reversed-phase semi-prep HPLC was uniquely successful in enabling purification of OpA derivative (±)-10. This is in stark contrast to the stability observed for the C3-secondary alcohol-bearing derivative (±)-38 and points to the importance of the C3-substitution pattern for stability of OpA derivatives. While OpA itself is more stable to silica gel than (±)-38 and (±)-10, it is also prone to C6 epimerization and C3 dehydration under acidic or basic conditions leading to significant loss in bioactivity.1, 2, 6
We then assayed the cytotoxicity of the racemic, simplified OpA derivatives synthesized toward a breast cancer cell line (MDA-MB-231) and a non-cancerous cell line (MCF-10A), to probe selectivity, if any, in comparison to OpA and a few naturally-occurring OpA congeners (Table 1). While significantly less potent than OpA, simplified bicyclic OpA derivatives (±)-10 and (±)-38 bearing the proposed pharmacophore displayed cytotoxicity (~7 and 11 μM, respectively) against MDA-MB-231 cells. The activity of the highly simplified and unstable ketoaldehyde (±)-11 was unsurprisingly less active (~44 μM). The selectivity observed with OpA between a cancer vs non-cancer cell line (~69 vs 200 nM, ~3-fold difference) is erased with the structurally simplified bicyclic derivatives (±)-10 and (±)-38. The similar potency of these simplified bicyclic derivatives is noteworthy and suggests that the C3-stereochemistry as either a secondary or tertiary alcohol has little effect on bioactivity. Overall, the described SAR data with racemic bicyclic derivatives of OpA suggests that cytotoxicity and selectivity increase in parallel with increasing complexity as expected.
Table 1.
Cell Viability Data of Synthesized, Simplified Analogs of Ophiobolin A and Natural Congenersa
| Cell lines (IC50, μM)╲OpA & derivatives | 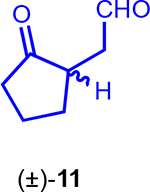 |
 |
 |
 |
 |
 |
 |
|---|---|---|---|---|---|---|---|
| MCF10A | 197 ± 14.4b | 13.4 ± 3.10 | 6.36 ± 1.99 | 0.198 ± 0.076 | 1.05 ± 0.076 | 1.74 ± 0.614 | 4.16 ± 0.763 |
| MDA-MB-231 | 44.2 ± 16.1b | 11.0 ± 0.935 | 7.09 ± 2.20 | 0.069 ± 0.024 | 0.80 ± 0.28 | 0.552 ± 0.124 | 4.94 ± 0.921 |
IC50 values are denoted in mean ± standard deviation of at least 3 independent replicates.
Ketoaldehyde (± )-11 was unstable in DMSO and thus a freshly prepared sample was required to preclude inconsistencies observed when using stored DMSO stock solutions.
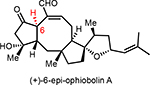
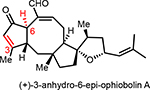
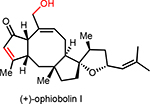
A few closely related congeners of OpA were also assayed. Direct comparison of OpA and (+)-6-epi-OpA suggest the importance of the C6 relative stereochemistry for cytotoxicity toward this breast cancer cell line leading to a ~12-fold drop in activity (~69 nM vs ~0.8 μM, Table 1). Other structural changes also impact cytotoxicity including loss of the C3-hydroxyl through β-elimination in conjunction with epimerization at C6 leads to an 8-fold drop in activity (cf. (+)-3-anhydro-6-epi-OpA). Retaining the C6-stereochemistry but dehydration in combination with reduction of the aldehyde to the primary alcohol as found in (+)-OpI leads to a significant loss in cytotoxicity (~10-fold drop vs OpA) with insignificant selectivity between the cancer vs non-cancer cell line, ~4.9 vs 4.2 μM, respectively.
In summary, we completed the first two stages of a PDR approach toward OpA to access bicyclic analogs bearing the AB ring system. To circumvent synthetic challenges including an inherent 5-exo-trig cyclization of a neopentyl alkyllithium and an undesired olefin isomerization, we adopted a defensive strategy by careful refinement of intermediates that ultimately enabled an RCM to form the required cyclooctene ring and maintained the critical C6-stereochemistry. Cell viability assays toward MDA-MB-231 breast cancer cells confirms our hypothesis that significant cytotoxicity observed for the ophiobolins originates from structural features of the A/B ring system. In addition to the SAR studies of the ophiobolins, the work described represent model studies of key reactions envisioned toward more elaborate OpA derivatives, a useful by-product of the PDR approach. This study further highlights the advantage of PDR to access SAR information at the early stages of a total synthesis effort and inform the design of more elaborate derivatives in the later stages.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge support by the National Institute of General Medical Sciences of the National Institutes of Health (R37 GM052964 and R35 GM134910, DR), the Robert A. Welch Foundation (AA-1280, DR), and the Cancer Prevention & Research Institute of Texas (RP180771, JHT). We thank Prof. Alexander Kornienko (Texas State University) for continued fruitful discussions and collaboration on the ophiobolins and Dr. Maurizio Vurro, Istituto di Scienze delle Produzioni Alimentari, CNR, Bari, Italy for a supply of culture filtrates of Dreschslera gigantea.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
Experimental procedures and compound characterization data (1H and 13C and select 2D (NOESY, HMBC, HSQC) NMR spectra) for all new compounds reported (PDF).
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- 1.Masi M; Dasari R; Evidente A; Mathieu V; Kornienko A, Chemistry and biology of ophiobolin A and its congeners. Bioorg. Med. Chem. Lett 2019, 29 (7), 859–869. [DOI] [PubMed] [Google Scholar]
- 2.Tian W; Deng Z; Hong K, The biological activities of sesterterpenoid-type ophiobolins. Mar. Drugs 2017, 15 (7), 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canonica L; Fiecchi A; Kienle MG; Scala A, The constitution of cochliobolin. Tetrahedron Lett. 1966, 7 (11), 1211–1218. [Google Scholar]
- 4.Nozoe S; Morisaki M; Tsuda K; Iitaka Y; Takahashi N; Tamura S; Ishibashi K; Shirasaka M, The structure of ophiobolin, a C25 terpenoid having a novel skeleton. J. Am. Chem. Soc 1965, 87 (21), 4968–4970. [DOI] [PubMed] [Google Scholar]
- 5.Ohkawa H; Tamura T, Studies on the Metabolites of Cochliobolus miyabeanus: Part I. Ophiobolosin A and Ophiobolosin B. Agr. Biol. Chem 1966, 30 (3), 285–291. [Google Scholar]
- 6.Au T; Chick WS; Leung P, The biology of ophiobolins. Life Sci. 2000, 67 (7), 733–742. [DOI] [PubMed] [Google Scholar]
- 7.Morrison R; Lodge T; Evidente A; Kiss R; Townley H, Ophiobolin A, a sesterpenoid fungal phytotoxin, displays different mechanisms of cell death in mammalian cells depending upon the cancer cell origin. Int. J. Oncol 2017, 50 (3), 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bladt TT; Dürr C; Knudsen PB; Kildgaard S; Frisvad JC; Gotfredsen CH; Seiffert M; Larsen TO, Bioactivity and dereplication-based discovery of ophiobolins and other fungal secondary metabolites targeting leukemia cells. Molecules 2013, 18 (12), 14629–14650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim IY; Kwon M; Choi M-K; Lee D; Lee DM; Seo MJ; Choi KS, Ophiobolin A kills human glioblastoma cells by inducing endoplasmic reticulum stress via disruption of thiol proteostasis. Oncotarget 2017, 8 (63), 106740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dasari R; Masi M; Lisy R; Ferdérin M; English LR; Cimmino A; Mathieu V; Brenner AJ; Kuhn JG; Whitten ST, Fungal metabolite ophiobolin A as a promising anti-glioma agent: In vivo evaluation, structure–activity relationship and unique pyrrolylation of primary amines. Bioorg. Med. Chem. Lett 2015, 25 (20), 4544–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Canonica L; Friecchi A; Kienle UG; Scala A, Isolation and constitution of cochliobolin B. Tetrahedron Lett. 1966, 7 (13), 1329–1333. [Google Scholar]
- 12.Najumudeen A; Jaiswal A; Lectez B; Oetken-Lindholm C; Guzmán C; Siljamäki E; Posada I; Lacey E; Aittokallio T; Abankwa D, Cancer stem cell drugs target K-ras signaling in a stemness context. Oncogene 2016, 35 (40), 5248–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bury M; Girault A; Megalizzi V; Spiegl-Kreinecker S; Mathieu V; Berger W; Evidente A; Kornienko A; Gailly P; Vandier C, Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell death & disease 2013, 4 (3), e561–e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodolfo C; Rocco M; Cattaneo L; Tartaglia M; Sassi M; Aducci P; Scaloni A; Camoni L; Marra M, Ophiobolin A induces autophagy and activates the mitochondrial pathway of apoptosis in human melanoma cells. PloS one 2016, 11 (12), e0167672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reisenauer KN; Tao Y; Song S; Patel S; Ingros A; Sheesley P; Masi M; Boari A; Taube JH; Evidente A, Epithelial-mesenchymal transition sensitizes breast cancer cells to cell death via the fungus-derived sesterterpenoid ophiobolin A. BioRkiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbasov ME; Alvariño R; Chaheine CM; Alonso E; Sánchez JA; Conner ML; Alfonso A; Jaspars M; Botana LM; Romo D, Simplified immunosuppressive and neuroprotective agents based on gracilin A. Nat. Chem 2019, 11 (4), 342–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Truax NJ; Ayinde S; Van K; Liu JO; Romo D, Pharmacophore-Directed Retrosynthesis Applied to Rameswaralide: Synthesis and Bioactivity of Sinularia Natural Product Tricyclic Cores. Org. Lett 2019, 21 (18), 7394–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Au TK; Leung PC, Identification of the binding and inhibition sites in the calmodulin molecule for ophiobolin A by site-directed mutagenesis. Plant Physiol. 1998, 118 (3), 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chidley C; Trauger SA; Birsoy K; O’Shea EK, The anticancer natural product ophiobolin A induces cytotoxicity by covalent modification of phosphatidylethanolamine. Elife 2016, 5, e14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsuna K; Noguchi N; Nakada M, Convergent Total Synthesis of (+)-Ophiobolin A. Angew. Chem. Int. Ed 2011, 50 (40), 9452–9455. [DOI] [PubMed] [Google Scholar]
- 21.Tsuna K; Noguchi N; Nakada M, Enantioselective Total Synthesis of (+)-Ophiobolin A. Chem. Eur. J 2013, 19 (17), 5476–5486. [DOI] [PubMed] [Google Scholar]
- 22.Rowley M; Tsukamoto M; Kishi Y, Total synthesis of (+)-ophiobolin C. J. Am. Chem. Soc 1989, 111 (7), 2735–2737. [Google Scholar]
- 23.Brill ZG; Grover HK; Maimone TJ, Enantioselective synthesis of an ophiobolin sesterterpene via a programmed radical cascade. Science 2016, 352 (6289), 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thach DQ; Brill ZG; Grover HK; Esguerra KV; Thompson JK; Maimone TJ, Total Synthesis of (+)-6-epi-Ophiobolin A. Angew. Chem. Int. Ed 2020, 59 (4), 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molander GA; Cameron KO, Neighboring group participation in Lewis acid-promoted [3+ 4] and [3+ 5] annulations. The synthesis of oxabicyclo [3. n. 1] alkan-3-ones. J. Am. Chem. Soc 1993, 115, 830–846. [Google Scholar]
- 26.Ashby E; Pham T; Amrollah-Madjdabadi A, Concerning the mechanism of reaction of lithium aluminum hydride with alkyl halides. J. Org. Chem 1991, 56 (4), 1596–1603. [Google Scholar]
- 27.Lipshutz BH; Kozlowski JA; Parker DA; Nguyen SL; McCarthy KE, More highly mixed, higher order cyanocuprates “RT (2-thienyl) Cu (CN) Li2”. Efficient reagents which promote selective ligand transfer. J. Organomet. Chem 1985, 285 (1–3), 437–447. [Google Scholar]
- 28.Ashby E; Goel A; DePriest R, Evidence for single electron transfer in the reations of alkali metal amides and alkoxides with alkyl halides and polynuclear hydrocarbons. J. Org. Chem. 1981, 46 (11), 2429–2431. [Google Scholar]
- 29.Wyatt P; Warren S, Organic synthesis: strategy and control. John Wiley & Sons: 2007. [Google Scholar]
- 30.Ashby E; Pham T; Madjdabadi AA, Another challenge to the validity of the use of cyclizable probes as evidence for single-electron transfer in nucleophilic aliphatic substitution. The reaction of lithium aluminum hydride with alkyl iodides. J. Org. Chem 1988, 53 (26), 6156–6158. [Google Scholar]
- 31.Trost BM; Pattenden G; Fleming I, Carbon-Carbon Sigma-Bond Formation. Pergamon: 1991; Vol. 3. [Google Scholar]
- 32.Negishi E; Swanson DR; Rousset CJ, Clean and convenient procedure for converting primary alkyl iodides and. alpha.,. omega.-diiodoalkanes into the corresponding alkyllithium derivatives by treatment with tert-butyllithium. J. Org. Chem 1990, 55 (19), 5406–5409. [Google Scholar]
- 33.Lipshutz BH; Dimock SH; James B, The role of trimethylsilyl chloride in Gilman cuprate 1, 4-addition reactions. J. Am. Chem. Soc 1993, 115 (20), 9283–9284. [Google Scholar]
- 34.Bertz SH; Miao G; Rossiter BE; Snyder JP, New Copper Chemistry. 25. Effect of TMSCl on the Conjugate Addition of Organocuprates to. alpha.-Enones: A New Mechanism. J. Am. Chem. Soc 1995, 117 (44), 11023–11024. [Google Scholar]
- 35.Lange GL; Otulakowski JA, Improved preparation of methyl 3-oxo-1-cyclohexene-1-carboxylate and its use in the synthesis of substituted 1, 5-cyclodecadienes. J. Org. Chem 1982, 47 (26), 5093–5096. [Google Scholar]
- 36.Fu GC; Nguyen ST; Grubbs RH, Catalytic ring-closing metathesis of functionalized dienes by a ruthenium carbene complex. J. Am. Chem. Soc 1993, 115 (21), 9856–9857. [Google Scholar]
- 37.Grubbs RH; Miller SJ; Fu GC, Ring-closing metathesis and related processes in organic synthesis. Acc. Chem. Res 1995, 28 (11), 446–452. [Google Scholar]
- 38.Maier ME, Synthesis of Medium-Sized Rings by the Ring-Closing Metathesis Reaction. Angew. Chem. Int. Ed 2000, 39 (12), 2073–2077. [DOI] [PubMed] [Google Scholar]
- 39.Hoye TR; Jeffrey CS; Tennakoon MA; Wang J; Zhao H, Relay ring-closing metathesis (RRCM): A strategy for directing metal movement throughout olefin metathesis sequences. J. Am. Chem. Soc 2004, 126, 10210–10211. [DOI] [PubMed] [Google Scholar]
- 40.Sonogashira K, Development of Pd–Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem 2002, 653 (1–2), 46–49. [Google Scholar]
- 41.Zhou Y; You W; Smith KB; Brown MK, Copper-Catalyzed Cross-Coupling of Boronic Esters with Aryl Iodides and Application to the Carboboration of Alkynes and Allenes. Angew. Chem. Int. Ed 2014, 53 (13), 3475–3479. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura H; Fujiwara M; Yamamoto Y, A practical method for the synthesis of enantiomerically pure 4-borono-L-phenylalanine. Bull. Chem. Soc. Jpn 2000, 73 (1), 231–235. [Google Scholar]
- 43.Miyaura N; Yamada K; Suzuki A, A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20 (36), 3437–3440. [Google Scholar]
- 44.Miyaura N; Suzuki A, Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev 1995, 95 (7), 2457–2483. [Google Scholar]
- 45.Molander GA; Felix LA, Stereoselective Suzuki− Miyaura Cross-Coupling Reactions of Potassium Alkenyltrifluoroborates with Alkenyl Bromides. J. Org. Chem 2005, 70, 3950–3956. [DOI] [PubMed] [Google Scholar]
- 46.Ren K; Zhao M; Hu B; Lu B; Xie X; Ratovelomanana-Vidal V; Zhang Z, An Enantioselective Approach to 4-O-Protected-2-cyclopentene-l, 4-diol Derivatives via a Rhodium-Catalyzed Redox-Isomerization Reaction. J. Org. Chem 2015, 80 (24), 12572–12579. [DOI] [PubMed] [Google Scholar]
- 47.Eames J, Diastereoselective protonation of chiral enolates derived from 2, 4-dimethyl-1-tetralone using carbonyl chelating proton donors. Tetrahedron Lett. 1999, 40 (31), 5787–5790. [Google Scholar]
- 48.Chatterjee AK; Choi T-L; Sanders DP; Grubbs RH, A general model for selectivity in olefin cross metathesis. J. Am. Chem. Soc 2003, 125 (37), 11360–11370. [DOI] [PubMed] [Google Scholar]
- 49.McMurry JE; Kees KL, Synthesis of cycloalkenes by intramolecular titanium-induced dicarbonyl coupling. J. Org. Chem 1977, 42 (15), 2655–2656. [Google Scholar]
- 50.McMurry JE; Lectka T; Rico JG, An optimized procedure for titanium-induced carbonyl coupling. J. Org. Chem 1989, 54 (15), 3748–3749. [Google Scholar]
- 51.Nicolaou K; Yang Z; Liu JJ; Ueno H; Nantermet P; Guy R; Claiborne C; Renaud J; Couladouros E; Paulvannan K, Total synthesis of taxol. Nature 1994, 367 (6464), 630–634. [DOI] [PubMed] [Google Scholar]
- 52.Greenwald R; Chaykovsky M; Corey E, The Wittig Reaction Using Methylsulfinyl Carbanion-Dimethyl Sulfoxide 1. J. Org. Chem 1963, 28 (4), 1128–1129. [Google Scholar]
- 53.Maercker A, The wittig reaction. Organic reactions 2004, 14, 270–490. [Google Scholar]
- 54.Hibino J.-i.; Okazoe T; Takai K; Nozaki H, Carbonyl methylenation of easily enolizable ketones. Tetrahedron Lett. 1985, 26 (45), 5579–5580. [Google Scholar]
- 55.Okazoe T; Hibino J -i.; Takai, K.; Nozakil, H., Chemoselective methylenation with a methylenedianion synthon. Tetrahedron Lett. 1985, 26 (45), 5581–5584. [Google Scholar]
- 56.Takai K; Kakiuchi T; Kataoka Y; Utimoto K, A novel catalytic effect of lead on the reduction of a zinc carbenoid with zinc metal leading to a geminal dizinc compound. Acceleration of the Wittig-type olefination with the RCHX2-TiCl4-Zn systems by addition of lead. J. Org. Chem 1994, 59 (10), 2668–2670. [Google Scholar]
- 57.Matsubara S; Oshima K; Utimoto K, Bis (iodozincio) methane—preparation, structure, and reaction. J. Organomet. Chem 2001, 617, 39–46. [Google Scholar]
- 58.Ackermann L; Fürstner A; Weskamp T; Kohl FJ; Herrmann WA, Ruthenium carbene complexes with imidazolin-2-ylidene ligands allow the formation of tetrasubstituted cycloalkenes by RCM. Tetrahedron Lett. 1999, 40 (26), 4787–4790. [Google Scholar]
- 59.Huang J; Stevens ED; Nolan SP; Petersen JL, Olefin metathesis-active ruthenium complexes bearing a nucleophilic carbene ligand. J. Am. Chem. Soc 1999, 121 (12), 2674–2678. [Google Scholar]
- 60.Scholl M; Trnka TM; Morgan JP; Grubbs RH, Increased ring closing metathesis activity of ruthenium-based olefin metathesis catalysts coordinated with imidazolin-2-ylidene ligands. Tetrahedron Lett. 1999, 40 (12), 2247–2250. [Google Scholar]
- 61.Freeman PK; Hutchinson LL, Organolithium reagents from alkyl halides and lithium DI-tert-butylbiphenyl. Tetrahedron Lett. 1976, 17 (22), 1849–1852. [Google Scholar]
- 62.Hill RR; Rychnovsky SD, Generation, stability, and utility of lithium 4, 4′-di-tert-butylbiphenylide (LiDBB). J. Org. Chem 2016, 81 (22), 10707–10714. [DOI] [PubMed] [Google Scholar]
- 63.Omura K; Swern D, Oxidation of alcohols by “activated” dimethyl sulfoxide. A preparative, steric and mechanistic study. Tetrahedron 1978, 34 (11), 1651–1660. [Google Scholar]
- 64.Mancuso AJ; Brownfain DS; Swern D, Structure of the dimethyl sulfoxide-oxalyl chloride reaction product. Oxidation of heteroaromatic and diverse alcohols to carbonyl compounds. J. Org. Chem 1979, 44 (23), 4148–4150. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.