

Abstract
Objective:
Epithelial cell death is an important innate mechanism at mucosal surfaces, which enables the elimination of pathogens and modulates immunoinflammatory responses. Based on the antimicrobial and anti-inflammatory properties of cell death, we hypothesized that oral epithelial cell (OECs) death is differentially modulated by oral bacteria.
Material and Methods:
We evaluated the effect of oral commensals Streptococcus gordonii (Sg), Streptococcus sanguinis (Ss), and Veillonella parvula (Vp), and pathogens Porphyromonas gingivalis (Pg), Tannerella forsythia (Tf), and Fusobacterium nucleatum (Fn) on OEC death. Apoptosis and necrosis were evaluated by flow cytometry using FITC Annexin-V and Propidium Iodide staining. Caspase-3/7 and caspase-1 activities were determined as markers of apoptosis and pyroptosis, respectively. IL-1β and IL-8 protein levels were determined in supernatants by ELISA.
Results:
Significant increases in apoptosis and necrosis were induced by Sg and Ss. Pg also induced apoptosis, although at a substantially lower level than the commensals. Vp, Tf, and Fn showed negligible effects on cell viability. These results were consistent with Sg, Ss, and Pg activating caspase-3/7. Only Ss significantly increased the levels of activated caspase-1, which correlated to IL-1β over-expression.
Conclusions:
OEC death processes were differentially induced by oral commensal and pathogenic bacteria, with Sg and Ss being more pro-apoptotic and pro-pyroptotic than pathogenic bacteria. Oral commensal-induced cell death may be a physiological mechanism to manage the extent of bacterial colonization of the outer layers of mucosal epithelial surfaces. Dysbiosis-related reduction or elimination of pro-apoptotic oral bacterial species could contribute to the risk for persistent inflammation and tissue destruction.
Keywords: Oral epithelial cells, oral commensal bacteria, oral pathogenic bacteria, apoptosis, pyroptosis
1. Introduction
The oral epithelium is colonized by a diverse and high number of microorganisms including both commensal and pathogenic bacteria. Fluctuations in the numbers of these bacterial groups are reflected by a continuum ranging from health (symbiosis) to disease (dysbiosis) (Hajishengallis et al., 2011). Among several mechanisms involved in maintaining host-microbe homeostatic interactions at mucosal surfaces, controlled mechanisms of epithelial cell viability and death play a critical role, not only in the physiological and permanent renewal of epithelial surfaces during health and wound healing, but also as an intrinsic immune defense mechanism in response to microbial infections controlling invasive bacteria and viruses (Carvalho-Filho et al., 2013).
Importantly, evidence indicates that strategies used by various bacterial pathogens can manipulate host cell death pathways to enhance their capacity to replicate, survive and disseminate (Lamkanfi & Dixit, 2010). The molecular pathways by which a cell dies (i.e., apoptosis, necrosis, pyroptosis) determine the level of collateral damage and inflammation inflicted on surrounding healthy tissues (Westman et al., 2019). For example, necrosis is considered to be a drastic and uncontrolled form of cell death which necrotic debris are potent inducers of inflammation, delaying the regeneration required after injury and sustaining collateral inflammatory damage (Pasparakis & Vandenabeele, 2015). Pyroptosis is a type of cell death caused by extensive inflammasome activation, marked by caspase-1 activation, and IL-1β and IL-18 secretion (Bergsbaken et al., 2009). Apoptosis, the prototypical form of programmed cell death, is morphologically described as cell shrinkage and chromatin condensation, followed by fragmentation of the entire cell into smaller, sealed apoptotic bodies. These apoptotic bodies are promptly cleared by neighboring phagocytes, importantly without initiating a pro-inflammatory response or disturbing tissue homeostasis (Westman et al., 2019).
It has been shown that apoptotic cell death is an essential mechanism that regulates the immunoinflammatory response against pathogens, through the generation of anti-inflammatory signals affecting phagocytes at the site of the infection (e.g., TGF-β), as well as contributing to the characteristics of the T helper and T regulatory (Treg) cell responses (Henson & Bratton, 2013; Nakahashi-Oda et al., 2016). Therefore, the potential for inhibition of apoptosis at epithelial surfaces would increase the likelihood for persistent infection and inflammation, which are central features of periodontal disease. Overall, most of the existing studies involving cell death are primarily focused on periodontopathogenic bacteria, and the role of apoptosis in the pathogenesis of periodontal disease remains uncertain (Gamonal et al., 2001; Jarnbring et al., 2002; Tsuda et al., 2012). Studies suggest that apoptosis may be one mechanism underlying the pathophysiology of periodontal disease progression (Abuhussein et al., 2014; Dabiri et al., 2016). Although the ability to manipulate cell death in oral epithelial cells has been shown for some periodontopathogens such as P. gingivalis (Mao et al., 2007; Nakayama et al., 2015; Yao et al., 2010), the impact of other oral bacteria including commensals in oral epithelial cell viability remains limited (Zhao et al., 2019). Moreover, whether oral bacteria selectively and differentially modulate other types of cell death, such as pyroptosis remains unknown.
The historical emphasis on late colonizers of the biofilm overlooks the initial changes and the potential impact of commensal bacteria that occurs earlier in the process of inflammation and infection associated with periodontal disease. A better understanding of the mechanisms by which oral bacteria modulate oral epithelial cells (OECs) death may help to develop new preventive strategies aimed at minimizing the persistent infection and inflammation in the oral mucosa as two central early events in the pathogenesis of periodontitis. Herein, we evaluated the effect of representative oral commensal and periodontopathogenic bacterial species on human OECs death including apoptosis, pyroptosis, and necrosis.
2. Materials & Methods
2.1. Oral epithelial cell cultures
The effect of both oral commensal and periodontopathogenic bacteria in cell death was tested in vitro. The immortalized keratinocyte cell line OKF6/hTERT-2 (OECs), established by ectopic expression of the telomerase catalytic subunit in cells from normal oral mucosal epithelium, obtained from Dr. James Rheinwald, Harvard Medical School (Dickson et al., 2000), was used in this study as previously described.(Al-Attar et al., 2018; Gonzalez et al., 2013). These cells have normal growth and differentiation characteristics and represent a broadly used model of oral epithelial cell biology allowing for improved reproducibility relative to variation between sources and short lifespan of primary cultures of epithelial cells. OECs were grown in Keratinocyte serum-free media supplemented with 25 μg/ml Pituitary extract and 0.2 ng/ml epidermal growth factor (Fisher Scientific Company, MA).
2.2. Bacterial cultures
The commensal Streptococcus sanguinis ATCC10556, Streptococcus gordonii ATCC10558 and Veillonella parvula ATCC10790, and pathogenic Porphyromonas gingivalis 381, Tannerella forsythia ATCC43037, and Fusobacterium nucleatum ATCC 25586 bacterial strains were used in the study. All bacterial strains were grown in 5 mL of Brain Heart infusion culture media (BD, Sparks, MD, USA) at appropriate aerobic and anaerobic atmospheric conditions as previously reported (Al-Attar et al., 2018; Gonzalez et al., 2013; Gonzalez et al., 2010; Huang et al., 2009). T. forsythia was grown in ATCC Medium: 1921 NAM medium. Each bacterial strain was grown in 2–3 ml of appropriate broth media to reach logarithmic growth, centrifuged, and resuspended in OEC media (Ker-SFM). Bacterial numbers were determined by count of the cell suspensions using a Petroff Hausser chamber and adjusted to the amounts needed for the corresponding OEC: bacteria ratios to be used in cell death experiments as described in the following section.
2.3. Determination of oral epithelial cell death
OECs (2 × 105 cells/well) were incubated in 6-wells plates at 37°C and 5% CO2 for 24 hours with either Ker-SFM alone or containing oral bacteria growing at the mid-log phase. Different epithelial cell bacteria ratios (1:10, 1:50, and 1:100) and times (4, 8, 16, and 24 hours) were tested. After bacterial challenge, OECs were harvested by trypsinization and combined with cells rescued from supernatants, washed with PBS and centrifuged at 1100 RPM for 5 minutes. Then, 1×105 cells were reconstituted in PBS and labeled with 5 μL of FITC-Annexin V and 5 μL Propidium iodide (BD Pharmingen, San Jose, CA) for 15 minutes at room temperature followed by flow cytometry analysis (FACS). OECs incubated with 16μM staurosporine (Sigma, St. Louis, MO) or only media (Mock) were used as a positive and negative control for apoptosis, respectively. In cells undergoing death process the membrane phospholipid, phosphatidylserine (PS), is translocated from the inner to the outer leaflet, exposing PS to the external cellular environment. With a high affinity for PS, FITC-Annexin V will bind to cells with membrane exposure. Propidium iodide (PI) was added in conjunction with FITC-Annexin V to enable identification of necrotic cells, since PI permeates membranes that are not intact. Annexin V positive but PI negative cells were defined as early apoptotic cells, whereas Annexin V positive and PI positive cells were defined as late apoptotic cells. For each sample 10,000 events were read in a flow cytometer FACSCalibur (Becton Dickinson, San Jose, CA).
2.4. Determination of levels of activated caspases
Activation of caspase-3 is an essential event during apoptosis. Levels of activated caspase-3/7 were determined in OECs by FACS using the Vybrant FAM FLICA Caspase-3/7 Assay Kit (Molecular Probes, Eugene, OR). Levels of activated caspase-1 (pyroptosis marker) were similarly determined using the FAM FLICA Caspase-1 Assay kit (Immunochemistry Technologies, LLC. Bloomington, MN). These systems detect active caspases by use of a fluorescent affinity label which binds covalently with a cysteine that has a specific caspase-amino acid sequence. The fluorescent signal allows for a direct measure of caspase activity (Wlodkowic et al., 2009). After bacterial challenge, cells (attached and from supernatants) were harvested and resuspended in Ker-SFM at a concentration of 1×106 cells/ml. Then, 300 μL of cell suspension was transferred to FACS tubes and incubated for 60 minutes with 10 μL FLICA reagent for either activated Caspase 3/7 or activated Caspase 1. Tubes were swirled during incubation for an even distribution of the reagent among the cells. Cells were then washed twice with 1X wash buffer, centrifuged at 1100 RPM for 5 minutes, and pellets resuspended in wash buffer for FACS analysis.
2.5. Determination of IL-1β and IL-8
Levels of IL-1β and IL-8 in cell supernatants from unstimulated cells and bacterial-stimulated cells were determined by ELISA (R&D Systems, Minneapolis, MN). The reaction was evaluated at 450 nm in the SpectraMax M2 Spectrophotometer (Molecular Devices, Sunnyvale, CA).
2.6. Statistical Analysis
Statistical analysis included descriptive statistics for the effect of oral bacteria on death of oral epithelial cells. At least 2 independent experiments were done, with each experimental condition performed in duplicate or triplicate. Variations in these continuous variables were compared under normality and equal variance assumptions in un-stimulated cells vs. bacterial-stimulated cells using an Analysis of variance (ANOVA). For significant overall tests, post hoc Fisher’s least significant difference (LSD) was performed. Statistical significance was considered at p<0.05. Graphs were made using GraphPad Prism version 8 for Windows (GraphPad Software, La Jolla California).
3. Results
The ability of different amounts of oral commensal and pathogenic bacterial species to induce OEC apoptosis and necrosis was first evaluated by FACS using Annexin V and PI staining. Figure 1A shows a representative flow cytometry dot plot for the effects of each bacteria, as well as positive (staurosporine) and negative controls (mock/unstimulated cells). Cells that were double negative (lower left quadrant) represent the percentage of viable cells, Annexin V positive cells (lower right quadrant) represent cells undergoing early apoptosis, double positive cells for Annexin V and PI (upper right quadrant) represents cells in late apoptosis, and cells positive for PI (upper left) denote necrotic cells. The overall effect of different doses of oral bacteria is shown in Figure 1B. The oral Gram-positive commensals S. sanguinis and S. gordonii induced a significant decrease in the percentage of viable cells along with significant increases in apoptosis and necrosis. In contrast, the oral commensal Gram-negative bacterium V. parvula did not affect OEC viability. Among the evaluated pathogens, P. gingivalis was the only one that induced any increase in apoptotic cells, although at a significantly lower level compared with the outcomes observed with both oral streptococci. Time course experiments (Figure 2) confirmed the above experiments and showed that only S. sanguinis and S. gordonii consistently decreased cell viability as early as 4–8h post-challenge, which correlated with significant increases in cells undergoing early and late apoptosis, as well as necrosis (Figure 2A). Likewise, P. gingivalis was the only pathogen eliciting any apoptotic events with the major effect seen at 16h post-challenge.
Figure 1.
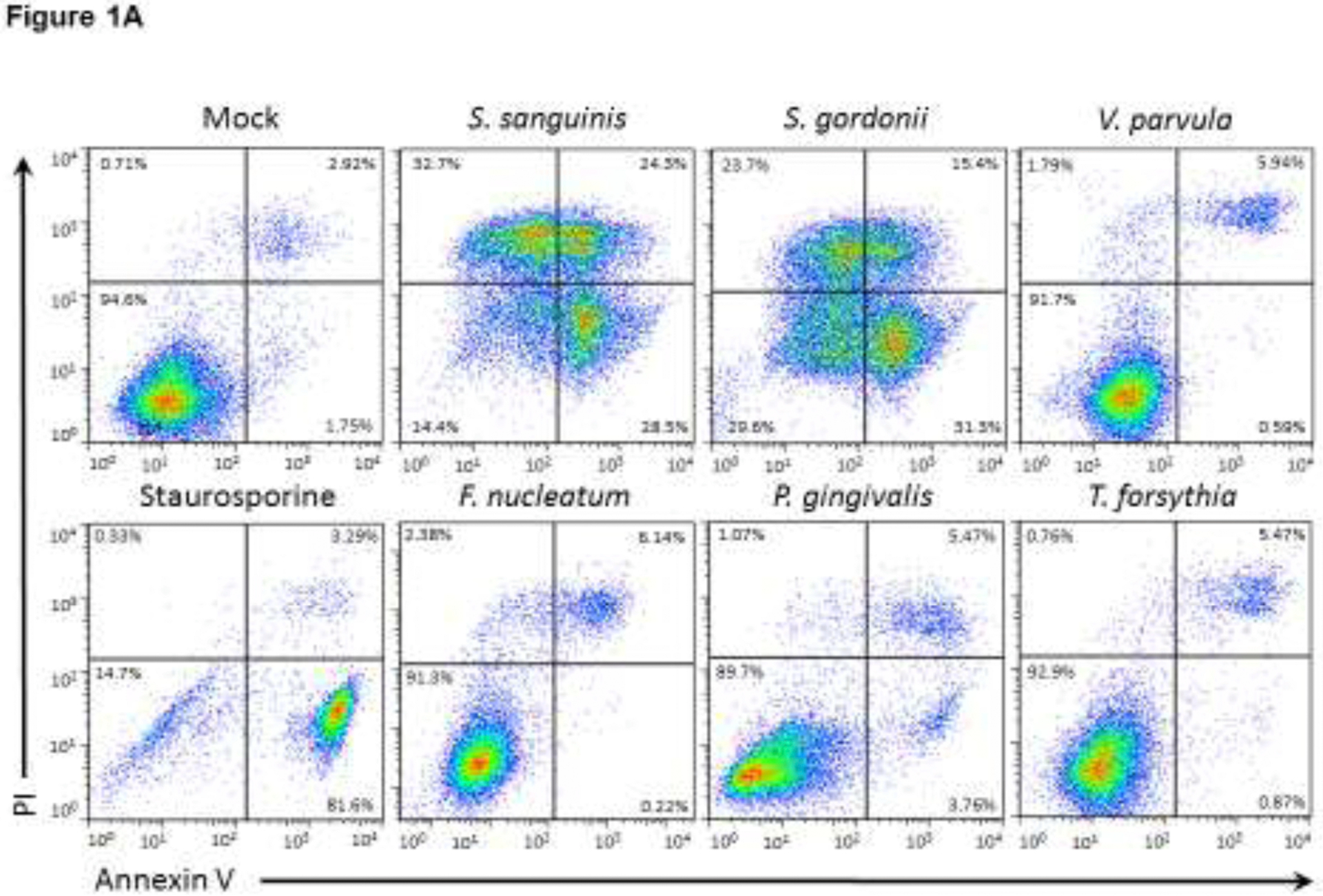
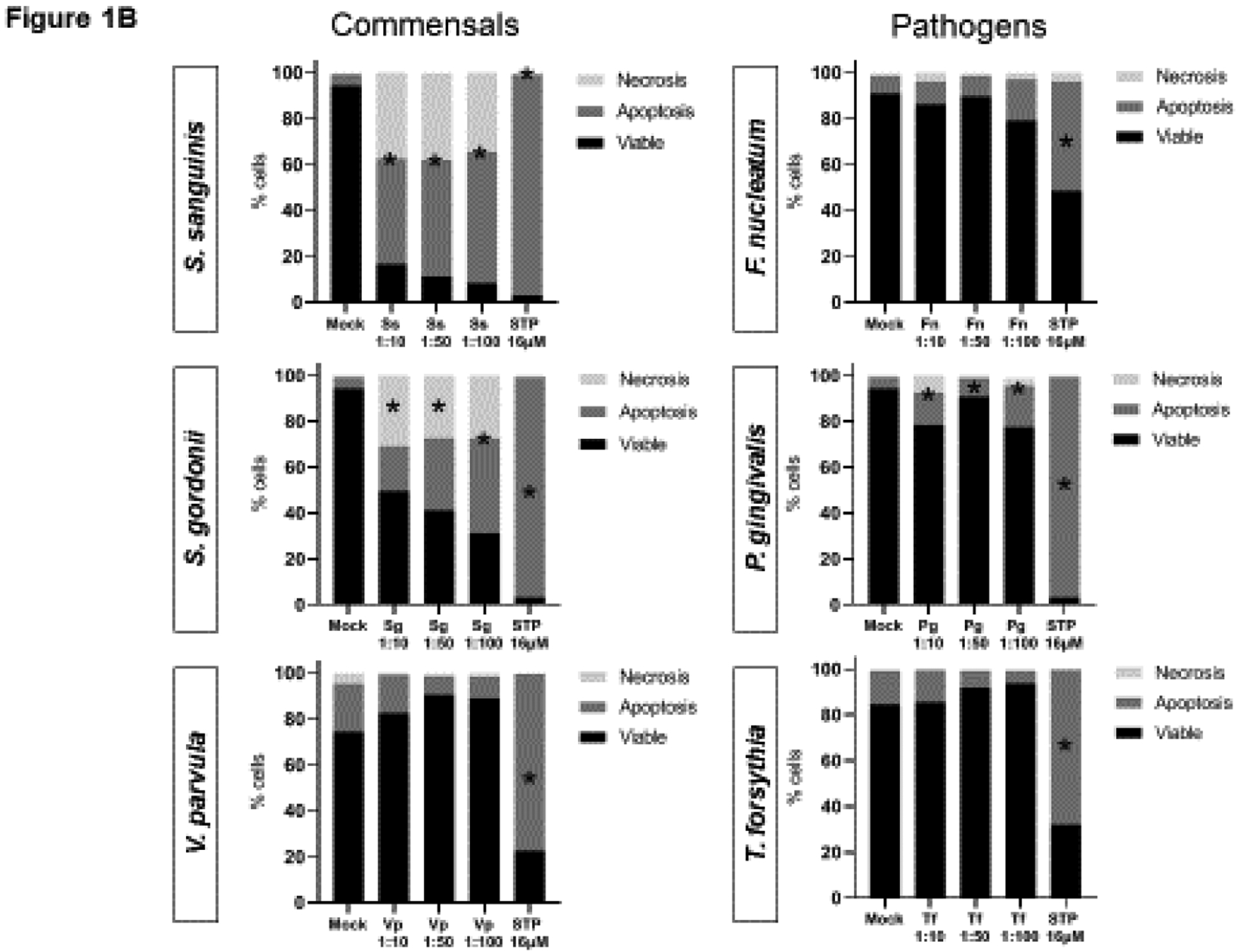
Effect of oral commensal and pathogenic bacteria on epithelial cell death (Dose-response). (A) Representative dot plots and (B) mean of percentages for viable cells or undergoing cell death (apoptosis or necrosis) after 24h bacterial challenge of two independent experiments analyzing at least 10,000 events by FACS for each condition is shown. Viable cells (Annexin V−/PI−), early apoptosis (Annexin V+), late apoptosis (Annexin V+/PI+), and Necrosis (PI+). STP: 16 μM Staurosporine was used as a positive control. *Treated cells vs. Unstimulated (Mock) cells p<0.05 (ANOVA and post hoc Fisher LSD).
Figure 2.
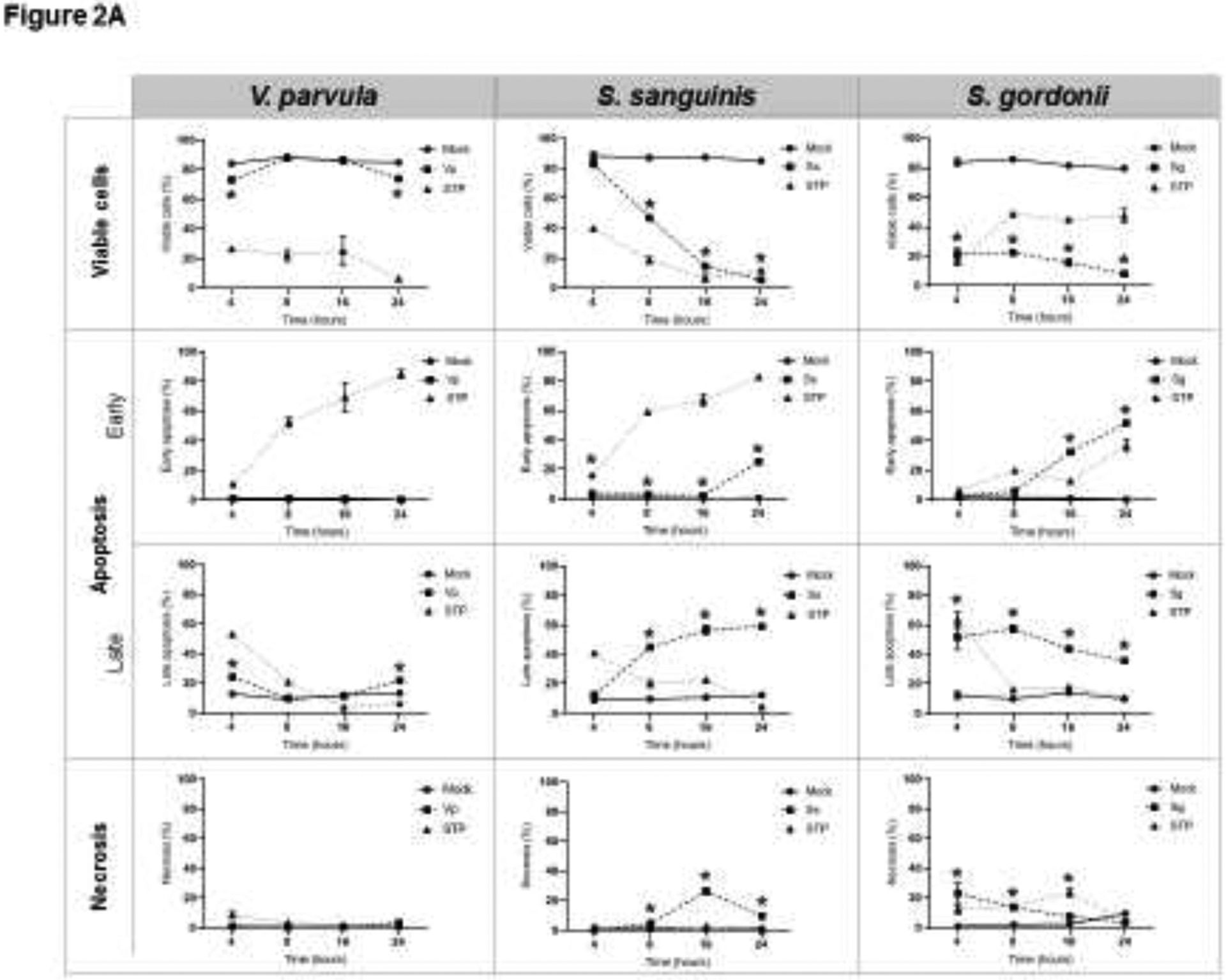
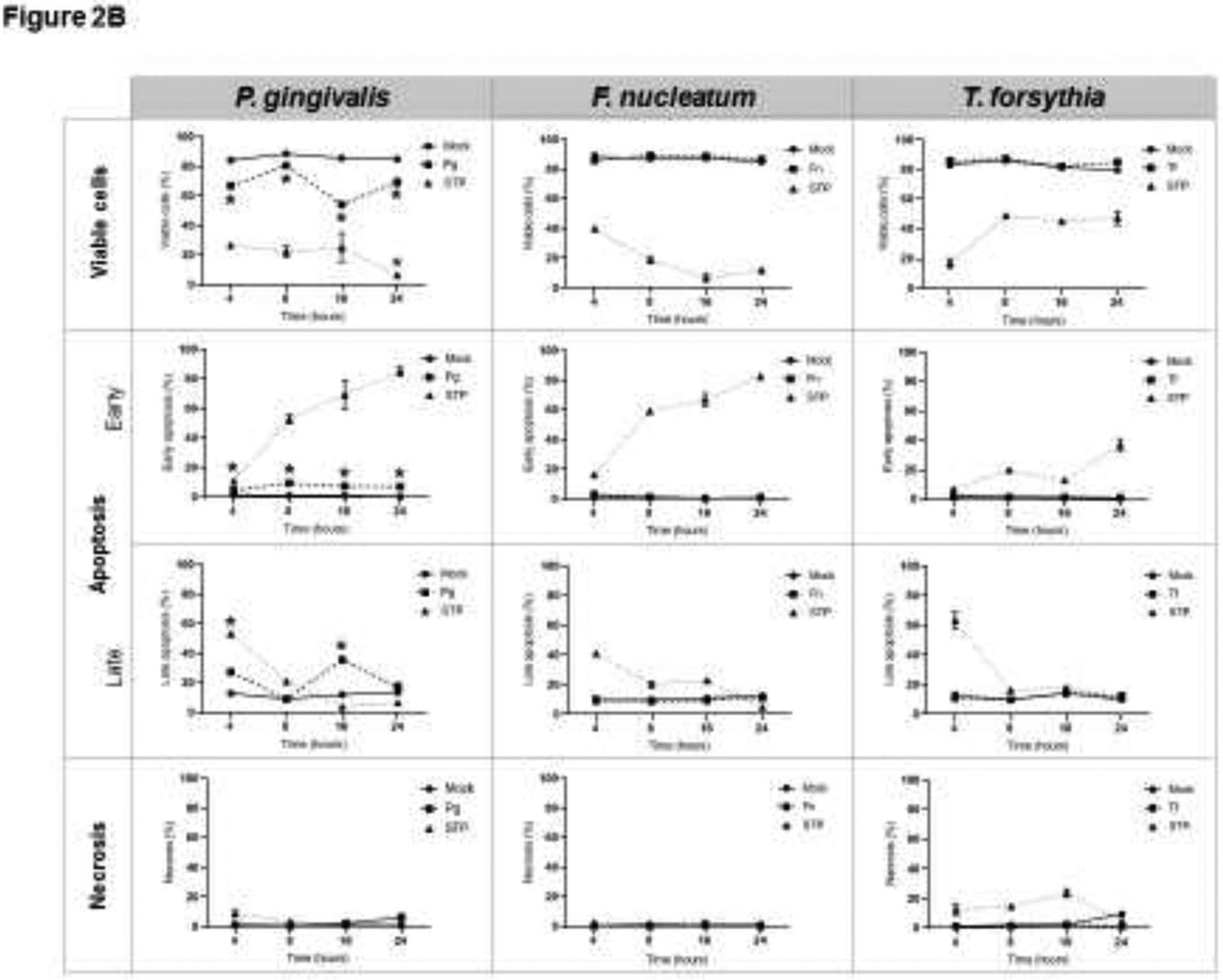
Effect of oral (A) commensal, and (B) pathogenic bacteria on epithelial cell death (Time-response). MOI [1:50] was used for all bacteria. For each condition, at least 10,000 events were analyzed by FACS to identify viable cells (Annexin V−/PI−), or undergoing early apoptosis (Annexin V+), late apoptosis (Annexin V+/PI+), and Necrosis (PI+). STP: 16 μM Staurosporine was used as a positive control. The mean ± standard deviation from two independent experiments with each condition in duplicate is shown. *Treated cells vs. Unstimulated (Mock) cells p<0.05 (ANOVA and post hoc Fisher LSD).
To determine if death of OECs induced by S. gordonii, S. sanguinis, and P. gingivalis was indeed through apoptosis, or perhaps could involve pyroptotic cell death, determination of the levels of activated caspase-3/7 (apoptosis) and caspase-1 (pyroptosis) were evaluated by FACS in OECs exposed to these three bacteria (Figure 3). Consistently, the percentage of OECs with elevated activated caspase-3/7 was increased in OECs exposed to all three bacteria, but not by F. nucleatum used as a negative control. Increases in the levels of activated caspase-1 were seen in OECs exposed to S. sanguinis and P. gingivalis; however, these increases reached significance only in cells challenged with S. sanguinis.
Figure 3.
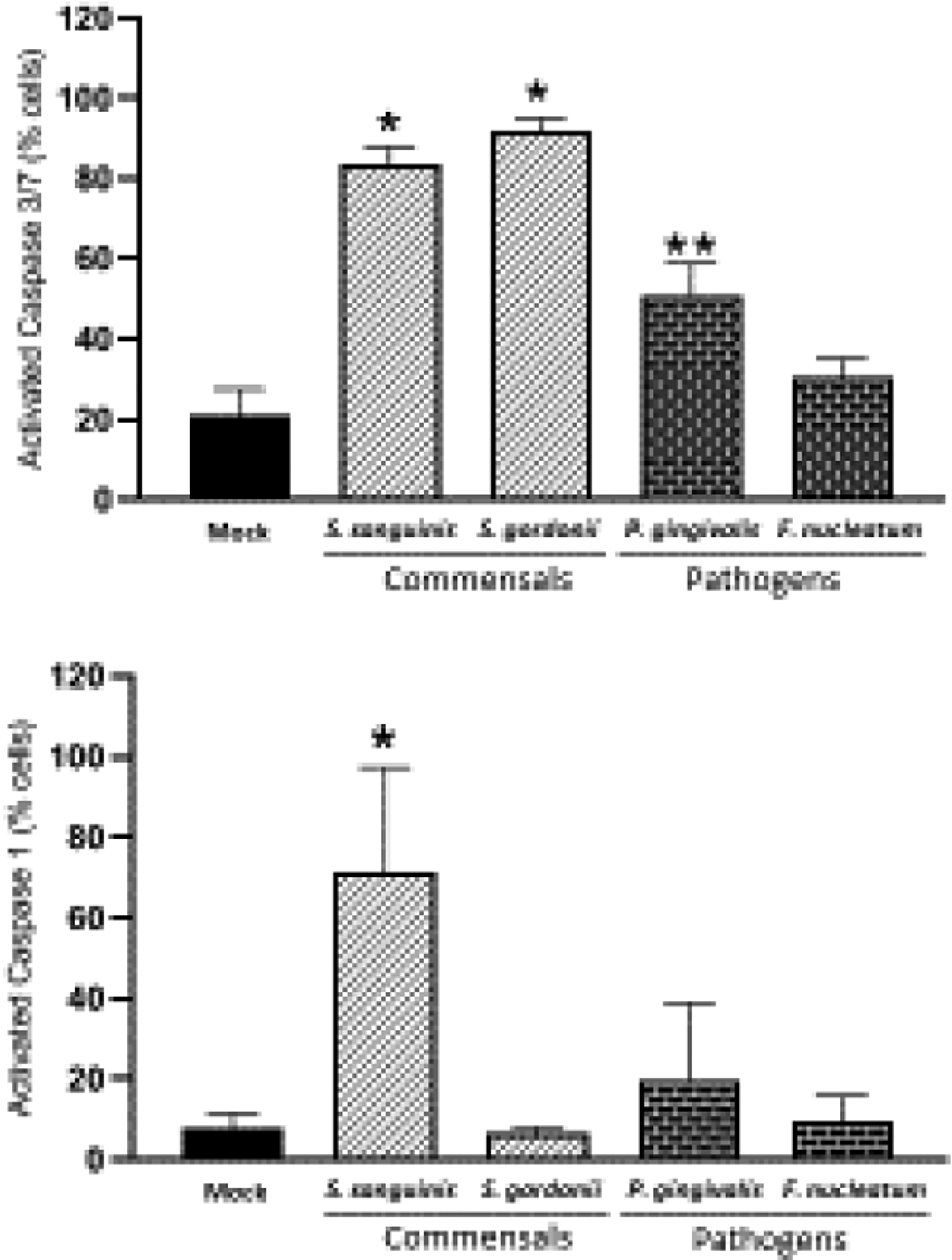
Effect of oral commensal and pathogenic bacteria on Caspase 3/7 (apoptosis) and Caspase-1 (Pyroptosis) activation in epithelial cells after 24h. Mean and standard deviation bars of percentage of cells expressing activated Caspase-3/7 or activated Caspase-1 for each condition are shown. For each condition 10,000 events were analyzed by FACS. The mean ± standard deviation from two independent experiments with each condition in duplicate is shown. MOI=1:50 *Treated cells vs. Unstimulated (Mock) cells *p<0.01; **p<0.05 (ANOVA and post hoc Fisher LSD).
Determination of IL-1β production by OECs in response to the oral bacteria indicated that only S. sanguinis, S. gordonii and P. gingivalis were able to stimulate the production of this pro-inflammatory cytokine (Figure 4). However, all oral bacteria stimulated the production of IL-8 in a time-dependent manner, which supported the fundamental biological response capacity of the OECs to the bacterial challenge (Figure 4).
Figure 4.
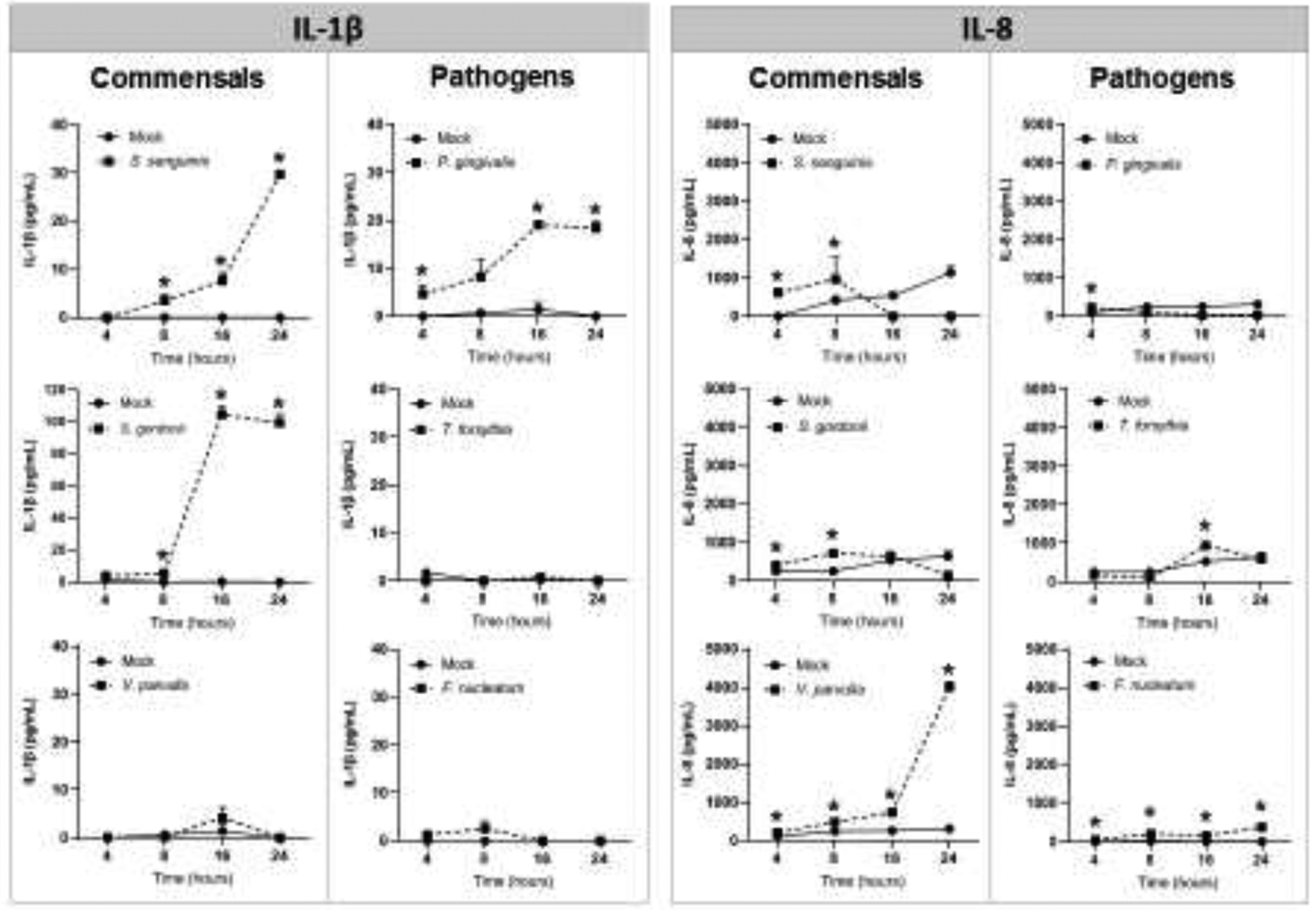
Effect of oral commensal and pathogenic bacteria [MOI=1:50] on IL-1β and IL-8 production in oral epithelial cells. Mean ± standard deviation bars of cytokine levels (pg/ml) detected by ELISA in cell culture supernatants from two independent experiments for each condition in duplicate is shown. *Treated cells vs. Unstimulated (Mock) cells p<0.05 (ANOVA and post hoc Fisher LSD).
4. Discussion
Our findings demonstrated that oral epithelial cell death is differentially induced by oral commensal and pathogenic bacteria, with oral commensals being more pro-apoptotic (S. sanguinis and S. gordonii) and pro-pyroptotic (S. sanguinis) than pathogenic bacteria (P. gingivalis, F. nucleatum, and Tannerella forsythia).
Interestingly, S. sanguinis induced a strong activation of caspase-1 consistent with IL-1β release into the supernatants suggesting that OECs exposed to this oral commensal could undergo a pyroptotic process. Pyroptosis has been considered an aggressive response of the innate immune system to pathogens, particularly due to its association with three major pro-inflammatory interleukins, IL-1β, IL-18, and IL-33 (Bortoluci & Medzhitov, 2010). Therefore, some selected oral commensal species could also be contributing to enhance inflammation through dysregulated innate immunity during the pathogenesis of periodontal disease linked to the ability to stimulate the production of pro-inflammatory cytokines, as has been shown in other streptococcal pathogenic species (LaRock & Nizet, 2015).
Caspase-3/7 was also activated by S. sanguinis. This is consistent with accumulating evidence suggesting that there seems to be cross-talk between cell death pathways where caspase-1 could proteolytically cleave (i.e. activate) other caspases involved in apoptosis (Miao et al., 2011).
Although S. gordonii did not activate caspase-1, the levels of IL-1β were elevated in OECs exposed to S. gordonii as previously shown by others in both OECs and monocytic cells (Ciabattini et al., 2006; Dickinson et al., 2011; Kesavalu et al., 2002). The mechanisms by which S. gordonii induces IL-1β production may be caspase-1 independent.
One mechanism by which apoptosis can function without inducing inflammation involves the production of TGF-β by apoptotic cells, which regulates the production of inflammatory mediators (Huynh et al., 2002). Most recently it has been shown that prostaglandin E2 (PGE2) is elevated in dying cells and PGE2 can exhibit significant anti-inflammatory properties(Hangai et al., 2016). Thus, increased levels of TGF-β and PGE2 produced by oral epithelial cells undergoing apoptosis after exposure to S. gordonii or S. sanguinis is a plausible mechanism by which pathologic inflammation could be modified by oral commensals. In addition, evidence indicates that both S. sanguinis and S. gordonii have the ability to produce hydrogen peroxide (H2O2), which has been shown to correlate with increased cell DNA fragmentation and caspase activity, both events that could be activating the intrinsic pathway of apoptosis (Li et al., 2016; Okahashi et al., 2014). The mechanisms by which these oral streptococci induce epithelial cell death require further study.
A recent study showed that inhibition of IL-8 up-regulated the expression of pro-apoptotic factors while downregulating anti-apoptotic factors simultaneously in endothelial cells (Choi et al., 2016). Consistently, IL-8 silencing decreased Bcl-2 expression in cancer cells with a consequent increase in apoptosis (Stronach et al., 2015). Similarly, herein S. sanguinis and S. gordonii significantly decreased the release of IL-8 after eight hours of infection, which was consistent with an increase in late or early apoptosis, respectively. The biological relevance and mechanisms involved in cell death associated with variations of chemokine signal expression would require future investigation.
Both apoptotic and pyroptotic cell death types contribute to removal of cells that have been infected by invasive bacteria such as P. gingivalis. Thus, a significant decrease in oral commensal species with the ability to induce cell death during dysbiosis, could improve the environment for pathogens like P. gingivalis to persist and orchestrate epithelial barrier damage and chronic inflammatory responses (Darveau, 2010). Oral commensal bacterial-induced OEC death could be an additional protective mechanism resulting from normal bacterial colonizers, such as S. gordonii, through which epithelial responses are modulated to limit inflammation and disease triggered by pathogens like P. gingivalis (Ohshima et al., 2019).
P. gingivalis enhanced apoptosis in OECs, although at a significant lower level compared with the effects found with commensal streptococci. These results are consistent with research by Stathopoulou et al. (Stathopoulou et al., 2009), where live P. gingivalis caused apoptosis in human epithelial cells using similar experimental conditions. Importantly, gingipains, the cysteine proteases produced by P. gingivalis, were directly involved in these responses. In addition, increased levels of IL-1β and caspase-1 were seen in OECs exposed to P. gingivalis, which indicates that this periodontopathogen could have the ability to induce oral epithelial cell death through pyroptosis, as has been shown in other cell types, i.e. monocytic cells (Park et al., 2014). This is also consistent with the ability of P. gingivalis to activate the inflammasome, a complex formed by a NOD-like receptor, an adaptor protein (ASC), and caspase-1 (Hung et al., 2013). These findings contrast with a previous report showing that IL-1β was not detected in OEC supernatants after P. gingivalis challenge (Dickinson et al., 2011). This variation could be related to differences in the experimental model and approaches used, since Dickinson’s group used organotypic cultures of primary human OECs.
The observation of apparent necrotic cell death in response to some of the oral bacteria (S. sanguinis, S. gordonii, and P. gingivalis) needs to be further defined since it could be secondary to apoptosis in the absence of phagocytosis (Krysko et al., 2008). Historically, necrosis is defined as a mode of cell death that resulted from abrupt changes in noxious stimuli, such as temperature and pressure. The lysed cellular contents would lead to inflammation through receptor activated release of cytokines, chemokines, and adhesion molecules (Yang et al., 2015). Normally, apoptotic cells within tissues are phagocytosed by macrophages without substantial inflammatory responses; however, since our experiments were developed using an in vitro system, the identified necrotic events could be secondary to the lack of physiologic phagocytic processes, and not reflecting a true bacterial-driven necrosis.
Of interest was that the oral bacterial species V. parvula, F. nucleatum and T. forsythia did not have a significant effect on oral epithelial cell viability. Previous studies have shown pro-cell death and pro-inflammatory properties of some of these bacteria in other cell types (e.g., neutrophils and mononuclear cells) (Bhattacharya et al., 2014; Jewett et al., 2000; Karlsson et al., 2002; Settem et al., 2012). Thus, cell death responses appear to be differentially induced and regulated by oral bacteria specifically related to the cell type. Moreover, enhancement of inflammatory responses may depend upon the need for direct contact with innate immune cells following epithelial barrier disruption. Accordingly, the preservation of the epithelial barrier integrity would be presumed to be critical in avoiding pathologic inflammatory responses induced by some oral bacterial species. Likewise, in situ synergistic cooperation among bacterial complexes may alter the presence or magnitude of a cell death response with a monoinfection challenge with these specific oral bacterial species (Inagaki et al., 2006).
Since oral commensal bacteria are more abundant in health and even disease microbial ecologies, oral epithelial cell death (either pyroptosis or apoptosis) could reflect a physiological mechanism, by which oral epithelial surfaces can more effectively manage the bacterial burden. The anti-inflammatory properties associated with apoptotic processes could also be critical for maintaining a balanced and homeostatic environment to avoid pathologic inflammation. Reduction or elimination of pro-apoptotic oral bacterial species during dysbiosis could lead to persistent inflammation and tissue destruction in the pathogenesis of periodontal disease. A better understanding of the mechanisms by which oral bacteria differentially modulate OEC death and survival will help to develop new disease management strategies aimed at controlling the persistent infection and inflammation in the oral mucosa, which are two central events involved in the pathogenesis of periodontal diseases. Future studies evaluating additional cell markers to distinguish specific death processes and in vitro and in vivo experimental strategies to validate these findings appear warranted as the field attempts to distinguish the molecular role of commensal bacteria as part of the innate immune capabilities.
Highlights.
Epithelial cell death is a regulatory process of inflammation and tissue homeostasis
Oral commensal bacteria differentially regulate epithelial cell death
Some oral commensal bacterial species likely contribute to oral inflammation
Oral epithelial cell death appears to be poorly induced by periodontopathogens
Acknowledgements
We thank the flow cytometry core from the University of Kentucky and Dr. Ahmad Al-Attar for their support with FACS analysis
Funding
These studies were funded by NIH/NIDCR DE024586 and Center for Oral Health Research from University of Kentucky (UK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare no potential conflicts of interest with respect to authorship and/or publication of this manuscript.
References
- Abuhussein H, Bashutski JD, Dabiri D, Halubai S, Layher M, Klausner C, Makhoul H, & Kapila Y (2014). The role of factors associated with apoptosis in assessing periodontal disease status. J Periodontol, 85(8), 1086–1095. 10.1902/jop.2013.130095 [DOI] [PubMed] [Google Scholar]
- Al-Attar A, Alimova Y, Kirakodu S, Kozal A, Novak MJ, Stromberg AJ, Orraca L, Gonzalez-Martinez J, Martinez M, Ebersole JL, & Gonzalez OA (2018). Activation of Notch-1 in oral epithelial cells by P. gingivalis triggers the expression of the antimicrobial protein PLA2-IIA. Mucosal Immunol, 11(4), 1047–1059. 10.1038/s41385-018-0014-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, & Cookson BT (2009). Pyroptosis: host cell death and inflammation. Nat Rev Microbiol, 7(2), 99–109. 10.1038/nrmicro2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R, Xu F, Dong G, Li S, Tian C, Ponugoti B, & Graves DT (2014). Effect of bacteria on the wound healing behavior of oral epithelial cells. PLoS One, 9(2), e89475 10.1371/journal.pone.0089475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortoluci KR, & Medzhitov R (2010). Control of infection by pyroptosis and autophagy: role of TLR and NLR. Cell Mol Life Sci, 67(10), 1643–1651. 10.1007/s00018-010-0335-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho-Filho PC, Trindade SC, Olczak T, Sampaio GP, Oliveira-Neto MG, Santos HA, Pereira BF, Moura-Costa L, Xavier MT, & Meyer R (2013). Porphyromonas gingivalis HmuY stimulates expression of Bcl-2 and Fas by human CD3+ T cells. BMC Microbiol, 13, 206 10.1186/1471-2180-13-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Park JY, Kang W, Kim SU, Kim do Y, Ahn SH, Ro SW, & Han KH (2016). Knockdown of HIF-1alpha and IL-8 induced apoptosis of hepatocellular carcinoma triggers apoptosis of vascular endothelial cells. Apoptosis, 21(1), 85–95. 10.1007/s10495-015-1185-2 [DOI] [PubMed] [Google Scholar]
- Ciabattini A, Cuppone AM, Pulimeno R, Iannelli F, Pozzi G, & Medaglini D (2006). Stimulation of human monocytes with the gram-positive vaccine vector Streptococcus gordonii. Clin Vaccine Immunol, 13(9), 1037–1043. 10.1128/CVI.00110-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabiri D, Halubai S, Layher M, Klausner C, Makhoul H, Lin GH, Eckert G, Abuhussein H, Kamarajan P, & Kapila Y (2016). The Role of Apoptotic Factors in Assessing Progression of Periodontal Disease. Int J Dent Oral Sci, 3(9), 318–325. 10.19070/2377-8075-1600064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveau RP (2010). Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol, 8(7), 481–490. 10.1038/nrmicro2337 [DOI] [PubMed] [Google Scholar]
- Dickinson BC, Moffatt CE, Hagerty D, Whitmore SE, Brown TA, Graves DT, & Lamont RJ (2011). Interaction of oral bacteria with gingival epithelial cell multilayers. Mol Oral Microbiol, 26(3), 210–220. 10.1111/j.2041-1014.2011.00609.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, & Rheinwald JG (2000). Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol, 20(4), 1436–1447. 10.1128/mcb.20.4.1436-1447.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamonal J, Bascones A, Acevedo A, Blanco E, & Silva A (2001). Apoptosis in chronic adult periodontitis analyzed by in situ DNA breaks, electron microscopy, and immunohistochemistry. J Periodontol, 72(4), 517–525. 10.1902/jop.2001.72.4.517 [DOI] [PubMed] [Google Scholar]
- Gonzalez OA, Escamilla C, Danaher RJ, Dai J, Ebersole JL, Mumper RJ, & Miller CS (2013). Antibacterial effects of blackberry extract target periodontopathogens. J Periodontal Res, 48(1), 80–86. 10.1111/j.1600-0765.2012.01506.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez OA, Li M, Ebersole JL, & Huang CB (2010). HIV-1 reactivation induced by the periodontal pathogens Fusobacterium nucleatum and Porphyromonas gingivalis involves Toll-like receptor 2 [corrected] and 9 activation in monocytes/macrophages. Clin Vaccine Immunol, 17(9), 1417–1427. 10.1128/CVI.00009-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, & Curtis MA (2011). Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe, 10(5), 497–506. 10.1016/j.chom.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangai S, Ao T, Kimura Y, Matsuki K, Kawamura T, Negishi H, Nishio J, Kodama T, Taniguchi T, & Yanai H (2016). PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci U S A, 113(14), 3844–3849. 10.1073/pnas.1602023113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson PM, & Bratton DL (2013). Antiinflammatory effects of apoptotic cells. J Clin Invest, 123(7), 2773–2774. 10.1172/JCI69344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CB, Emerson KA, Gonzalez OA, & Ebersole JL (2009). Oral bacteria induce a differential activation of human immunodeficiency virus-1 promoter in T cells, macrophages and dendritic cells. Oral Microbiol Immunol, 24(5), 401–407. 10.1111/j.1399-302X.2009.00533.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung SC, Choi CH, Said-Sadier N, Johnson L, Atanasova KR, Sellami H, Yilmaz O, & Ojcius DM (2013). P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS One, 8(7), e70210 10.1371/journal.pone.0070210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh ML, Fadok VA, & Henson PM (2002). Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest, 109(1), 41–50. 10.1172/JCI11638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki S, Onishi S, Kuramitsu HK, & Sharma A (2006). Porphyromonas gingivalis vesicles enhance attachment, and the leucine-rich repeat BspA protein is required for invasion of epithelial cells by “Tannerella forsythia”. Infect Immun, 74(9), 5023–5028. 10.1128/IAI.00062-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarnbring F, Somogyi E, Dalton J, Gustafsson A, & Klinge B (2002). Quantitative assessment of apoptotic and proliferative gingival keratinocytes in oral and sulcular epithelium in patients with gingivitis and periodontitis. J Clin Periodontol, 29(12), 1065–1071. 10.1034/j.1600-051x.2002.291203.x [DOI] [PubMed] [Google Scholar]
- Jewett A, Hume WR, Le H, Huynh TN, Han YW, Cheng G, & Shi W (2000). Induction of apoptotic cell death in peripheral blood mononuclear and polymorphonuclear cells by an oral bacterium, Fusobacterium nucleatum. Infect Immun, 68(4), 1893–1898. 10.1128/iai.68.4.1893-1898.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson H, Hessle C, & Rudin A (2002). Innate immune responses of human neonatal cells to bacteria from the normal gastrointestinal flora. Infect Immun, 70(12), 6688–6696. 10.1128/iai.70.12.6688-6696.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesavalu L, Chandrasekar B, & Ebersole JL (2002). In vivo induction of proinflammatory cytokines in mouse tissue by Porphyromonas gingivalis and Actinobacillus actinomycetemcomitans. Oral Microbiol Immunol, 17(3), 177–180. 10.1034/j.1399-302x.2002.170307.x [DOI] [PubMed] [Google Scholar]
- Krysko DV, Vanden Berghe T, Parthoens E, D’Herde K, & Vandenabeele P (2008). Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol, 442, 307–341. 10.1016/S0076-6879(08)01416-X [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, & Dixit VM (2010). Manipulation of host cell death pathways during microbial infections. Cell Host Microbe, 8(1), 44–54. 10.1016/j.chom.2010.06.007 [DOI] [PubMed] [Google Scholar]
- LaRock CN, & Nizet V (2015). Inflammasome/IL-1beta Responses to Streptococcal Pathogens. Front Immunol, 6, 518 10.3389/fimmu.2015.00518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Zhai S, Xu M, Shang M, Gao Y, Liu G, Wang Q, & Zheng L (2016). SpxB-mediated H2 O2 induces programmed cell death in Streptococcus sanguinis. J Basic Microbiol, 56(7), 741–752. 10.1002/jobm.201500617 [DOI] [PubMed] [Google Scholar]
- Mao S, Park Y, Hasegawa Y, Tribble GD, James CE, Handfield M, Stavropoulos MF, Yilmaz O, & Lamont RJ (2007). Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol, 9(8), 1997–2007. 10.1111/j.1462-5822.2007.00931.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Rajan JV, & Aderem A (2011). Caspase-1-induced pyroptotic cell death. Immunol Rev, 243(1), 206–214. 10.1111/j.1600-065X.2011.01044.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahashi-Oda C, Udayanga KG, Nakamura Y, Nakazawa Y, Totsuka N, Miki H, Iino S, Tahara-Hanaoka S, Honda S, Shibuya K, & Shibuya A (2016). Apoptotic epithelial cells control the abundance of Treg cells at barrier surfaces. Nat Immunol, 17(4), 441–450. 10.1038/ni.3345 [DOI] [PubMed] [Google Scholar]
- Nakayama M, Inoue T, Naito M, Nakayama K, & Ohara N (2015). Attenuation of the phosphatidylinositol 3-kinase/Akt signaling pathway by Porphyromonas gingivalis gingipains RgpA, RgpB, and Kgp. J Biol Chem, 290(8), 5190–5202. 10.1074/jbc.M114.591610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima J, Wang Q, Fitzsimonds ZR, Miller DP, Sztukowska MN, Jung YJ, Hayashi M, Whiteley M, & Lamont RJ (2019). Streptococcus gordonii programs epithelial cells to resist ZEB2 induction by Porphyromonas gingivalis. Proc Natl Acad Sci U S A, 116(17), 8544–8553. 10.1073/pnas.1900101116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okahashi N, Sumitomo T, Nakata M, Sakurai A, Kuwata H, & Kawabata S (2014). Hydrogen peroxide contributes to the epithelial cell death induced by the oral mitis group of streptococci. PLoS One, 9(1), e88136 10.1371/journal.pone.0088136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Na HS, Song YR, Shin SY, Kim YM, & Chung J (2014). Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infect Immun, 82(1), 112–123. 10.1128/IAI.00862-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, & Vandenabeele P (2015). Necroptosis and its role in inflammation. Nature, 517(7534), 311–320. 10.1038/nature14191 [DOI] [PubMed] [Google Scholar]
- Settem RP, El-Hassan AT, Honma K, Stafford GP, & Sharma A (2012). Fusobacterium nucleatum and Tannerella forsythia induce synergistic alveolar bone loss in a mouse periodontitis model. Infect Immun, 80(7), 2436–2443. 10.1128/IAI.06276-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulou PG, Galicia JC, Benakanakere MR, Garcia CA, Potempa J, & Kinane DF (2009). Porphyromonas gingivalis induce apoptosis in human gingival epithelial cells through a gingipain-dependent mechanism. BMC Microbiol, 9, 107 10.1186/1471-2180-9-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stronach EA, Cunnea P, Turner C, Guney T, Aiyappa R, Jeyapalan S, de Sousa CH, Browne A, Magdy N, Studd JB, Sriraksa R, Gabra H, & El-Bahrawy M (2015). The role of interleukin-8 (IL-8) and IL-8 receptors in platinum response in high grade serous ovarian carcinoma. Oncotarget, 6(31), 31593–31603. 10.18632/oncotarget.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda H, Ning Z, Yamaguchi Y, & Suzuki N (2012). Programmed cell death and its possible relationship with periodontal disease. J Oral Sci, 54(2), 137–149. 10.2334/josnusd.54.137 [DOI] [PubMed] [Google Scholar]
- Westman J, Grinstein S, & Marques PE (2019). Phagocytosis of Necrotic Debris at Sites of Injury and Inflammation. Front Immunol, 10, 3030 10.3389/fimmu.2019.03030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Jiang G, Zhang P, & Fan J (2015). Programmed cell death and its role in inflammation. Mil Med Res, 2, 12 10.1186/s40779-015-0039-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, Jermanus C, Barbetta B, Choi C, Verbeke P, Ojcius DM, & Yilmaz O (2010). Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol Oral Microbiol, 25(2), 89–101. 10.1111/j.2041-1014.2010.00569.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JJ, Jiang L, Zhu YQ, & Feng XP (2019). Effect of Lactobacillus acidophilus and Porphyromonas gingivalis on proliferation and apoptosis of gingival epithelial cells. Adv Med Sci, 64(1), 54–57. 10.1016/j.advms.2018.04.008 [DOI] [PubMed] [Google Scholar]