

Abstract
Introduction
As a multifactorial polygenic disorder, Alzheimer's disease (AD) can be associated with complex haplotypes or compound genotypes.
Methods
We examined associations of 4960 single nucleotide polymorphism (SNP) triples, comprising 32 SNPs from five genes in the apolipoprotein E gene (APOE) region with AD in a sample of 2789 AD‐affected and 16,334 unaffected subjects.
Results
We identified a large number of 1127 AD‐associated triples, comprising SNPs from all five genes, in support of definitive roles of complex haplotypes in predisposition to AD. These haplotypes may not include the APOE ε4 and ε2 alleles. For triples with rs429358 or rs7412, which encode these alleles, AD is characterized mainly by strengthening connections of the ε4 allele and weakening connections of the ε2 allele with the other alleles in this region.
Discussion
Dissecting heterogeneity attributed to AD‐associated complex haplotypes in the APOE region will target more homogeneous polygenic profiles of people at high risk of AD.
Keywords: age‐related phenotypes, Alzheimer's disease, APOE polymorphism, linkage disequilibrium
1. BACKGROUND
Late‐onset Alzheimer's disease (AD), referred hereto as AD, is commonly considered as a multifactorial polygenic disorder. 1 Despite relatively high heritability of AD of 58% to 79% estimated in the most extensive study to date using all twins in the Swedish Twin Registry aged 65 years and older, 2 no one gene appears to be causative of AD. 3 , 4 , 5 , 6 , 7 In contrast, early onset AD can be caused by highly penetrant mutations in the APP gene (chromosome 21) and two homologous genes, PSEN1 (chromosome 14) and PSEN2 (chromosome 1). 8 , 9 , 10 , 11
The ε4 allele from the apolipoprotein E (APOE) gene ε2/ε3/ε4 polymorphism has been known for decades as the strongest single genetic risk factor for AD in various populations, 12 whereas the ε2 allele can be protective. 13 , 14 Studies also advocate for shifting “category of the APOE gene from ‘risk factor’ to ‘major gene.’” 15 Nevertheless, even the role of the strongest genetic risk factor for AD is still controversial. Indeed, although most researchers believe that the APOE ε4 allele itself is a risk factor for AD; 12 the others argue that the association between this allele and AD can be modulated by variants from nearby genes in this region. For example, Roses et al. advocate that variants from the nearby TOMM40 gene, such as tightly correlated with ε4 allele long poly thymine repeat polymorphism tagged by rs10524523, can increase susceptibility to AD either independently or in cis combination with the ε4 allele. 16 , 17 Franceschi et al. identified that a haplotype, including rs405509_T and ε4 alleles, increases the risk of AD when they are both in cis position. 18 Furthermore, a more complex haplotype in the APOE region may increase susceptibility to AD independently of the ε4 allele. 19
To better understand the complex role of genetic variants in AD pathogenesis, more comprehensive methods can be used. For example, Nielsen et al. reviewed and extended methods based on testing deviations from Hardy‐Weinberg equilibrium (HWE) in the affected subjects to localize disease‐susceptibility loci. 20 Zaykin et al. developed a method based on contrasting linkage‐disequilibrium (LD) patterns between the affected and unaffected subjects to map patterns of single nucleotide polymorphisms (SNPs) to disease. 21 Some studies attempted to access the differences in LD structures in the APOE region between AD‐affected and unaffected subjects qualitatively. 22 , 23 Recently we reported significant associations of both the entire SNP patterns and specific SNPs pairs in the APOE region with AD in populations of different ancestries. 24 , 25 , 26
In this study, we use the standardized third mixed moment, or co‐skewness, to generalize LD between pairs of SNPs to triples of SNPs, and then use this metric to study the relationship of triples of SNPs in the APOE region (19q13.3) to AD. This region is represented by 32 SNPs from the BCAM, NECTIN2, TOMM40, APOE, and APOC1 genes, which include rs429358 and rs7412 SNPs coding the ε2/ε3/ε4 polymorphism. We performed the analysis using a mega sample of 2789 AD‐affected and 16,334 unaffected subjects from four studies. Our findings include heterogeneous patterns of triples of SNPs associated with AD led by the triple comprising rs2075650 (TOMM40), rs12721046 (APOC1), and the APOE ε4‐coding rs429358 SNP. Our results support the definitive roles of complex haplotypes in predisposition to AD in the APOE region.
RESEARCH IN CONTEXT
Systematic review: A literature review (PubMed and Google Scholar) identified interest in complex haplotypes/genotypes predisposing to Alzheimer's disease (AD), particularly in the apolipoprotein E gene (APOE) region. To better understand the role of such variants in AD pathogenesis, more comprehensive methods can be used. These relevant citations are appropriately cited.
Interpretation: We leverage a new method to map triples of single nucleotide polymorphisms (SNPs) in the APOE region to AD. The analysis supports the definitive role of heterogeneous AD‐related haplotypes in AD, which include SNPs from five genes in the APOE region. AD is characterized mainly by strengthening connections of the ε4 allele and weakening connections of the ε2 allele with other SNPs in this region.
Future directions: This work presents an approach to examine associations of haplotypes/genotypes comprising triples of SNPs with diseases such as AD. Extension to a larger number of SNPs would suggest more homogeneous genetic profiles of AD risk and protection.
2. METHODS
2.1. Study cohorts and phenotypes
We used data from the Framingham Heart Study (FHS) original and offspring cohorts, 27 Cardiovascular Health Study (CHS), 28 Health and Retirement Study (HRS), 29 and the National Institute on Aging Late‐Onset Alzheimer's Disease Family Study (LOADFS) 30 for individuals of European ancestry. LOADFS and FHS released information on AD defined using diagnoses made according to the National Institute of Neurological and Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association. A diagnosis of AD in HRS and CHS was defined based on ICD‐9:331.0x codes in Medicare service use files. There were N = 2799 AD‐affected subjects (cases) and N = 16,354 AD‐unaffected subjects (non‐cases) (Table 1).
TABLE 1.
Basic characteristics of the genotyped participants in the selected studies
| Sample | Ntotal | AD cases (%) | Men (%) | Birth year mean (SD) | Age at the end of follow‐up mean (SD), years | Follow‐up through |
|---|---|---|---|---|---|---|
| LOADFS | 3999 | 1973(49.3) | 1491(37.3) | 1928.3 (12.5) | 76.7 (12.5) | 2015 |
| HRS | 7226 | 263 (3.6) | 3129 (43.3) | 1934.2 (8.4) | 79.1 (8.1) | 2012 |
| CHS | 4273 | 247 (5.8) | 1864 (43.6) | 1914.1 (5.7) | 83.5 (5.4) | 2002 |
| FHS | 3625 | 306 (8.4) | 1862 (51.4) | 1931.6 (12.7) | 76.2 (11.1) | 2012 |
Ntotal is total number of subjects in the analysis; AD cases: the number of Alzheimer's disease cases.
Abbreviations: SD, standard deviation; LOADFS, the National Institute on Aging Late‐Onset Alzheimer's Disease Family Study; HRS, Health and Retirement Study; CHS, Cardiovascular Health Study; FHS, Framingham Heart Study parental and offspring cohorts.
LOADFS is a study with a case‐control design that explains a large proportion of AD cases. The other studies are of longitudinal design following the study participants for long periods of time. HRS is a population‐based study, whereas FHS and CHS are community‐based studies. All studies included AD‐affected and unaffected subjects and, therefore, they could be separated into the samples according to the affection status to be used in the comparative analyses of co‐skewness.
2.2. Genotypes
We used genotypes from genome‐wide and custom SNP arrays available for the selected studies, including the same customized Illumina iSelect array (the IBC‐chip, ≈50K single nucleotide polymorphisms [SNPs]) in the FHS and CHS cohorts, Affymetrix 500K in the FHS, Illumina HumanCNV370v1 chip (370K SNPs) in the CHS, Illumina HumanOmni 2.5 Quad chip (≈2.5 M SNPs) in the HRS, and Illumina Human 610Quadv1_B Beadchip (≈610K SNPs) in the LOADFS.
The analyses focused on the same 32 SNPs representing the BCAM‐NECTIN2‐TOMM40‐APOE‐APOC1 (19q13.3) region (Table S1) as in previous studies. 26 We selected SNPs available from common GWAS arrays, which were genotyped directly in at least two cohorts and which were not in strong LD in the mega sample of all studies (r2 <0.8). We excluded subjects with missingness >5%. Genotypes in these cohorts were phased and imputed using the Michigan imputation server 31 with a reference panel from the Haplotype Reference Consortium (HRC) (version r1.1 2016). Only SNPs with high imputation quality were selected for the analysis (Table S1).
2.3. Co‐skewness metrics
We used the standardized third mixed moment, or co‐skewness, to generalize LD between pairs of SNPs to triples of SNPs (see Note S1). Then we used this metric to study the associations of triples of SNPs in the APOE region with AD by computing the difference in co‐skewness between AD‐affected and unaffected subjects. We derived two metrics based on genotype () and haplotype () counts. Haplotypes were inferred under the assumption of Hardy‐Weinberg equilibrium (or HWE) using the EM algorithm (the R package haplo.stats). HWE was interpreted in the broad sense that the gametes are probabilistically independent. Therefore, as in the case of pairwise LD, 20 , 21 the difference between and , that is., , characterizes deviation from HWE at the haplotype level, which, otherwise, may be difficult to detect. Because all SNPs were selected to be in HWE in the sample of cases and non‐cases combined, this deviation is unlikely to be an artifact and warrants biological interpretation. Haplotype‐based co‐skewness also provided convenient decomposition of the metric into two fractions () measuring (1) how far the triple is from mutual independence () and (2) a weighted sum of the pairwise LD values (). Co‐skewness is interpreted as joint deviation of the distributions of random variables from the normal distribution, that is, that larger values of or imply stronger connections between SNPs in a triple (Figure 1). This is consistent with the interpretation of the coefficient of pairwise LD. As in the case of pairwise LD, individually, the and coefficients are invariant under the change of the sign, whereas their signs have to be used consistently in comparative analyses, for example, when evaluating the difference .
FIGURE 1.

Illustration of co‐skewness in the AD‐affected and unaffected subjects. The diagram shows the joint distributions of the compound genotypes for the SNPs rs2075650, rs12721046, and rs429358 as proportions. The left (blue) column shows proportions of the compound genotypes among Alzheimer's disease (AD) non‐cases, and the right (red) displays proportions among AD cases. Larger dot size indicates a larger proportion. Rows keep track of the number of rs429358 minor alleles coding the ε4 allele. Observe that the distribution for cases is considerably more clustered along the diagonal (0,0,0), (1,1,1), (2,2,2) than for non‐cases. This phenomenon visually confirms our result that the mutual term increases substantially from non‐cases to cases for this triple. Thus we can think of the mutual term as a direct generalization of linkage disequilibrium and view co‐skewness as a more subtle metric as it contains additional pairwise information
A permutation test was employed to compute the significance of the effects defined by the differences in co‐skewness between the AD‐affected and unaffected subjects. We resampled the entire set of subjects into two groups with the same sizes as those of the affected and unaffected groups to estimate the permutation distribution of χ² = (g₁‐g₀)², where g₁ and g₀ are the co‐skewness estimates for the resampled groups for a fixed triple of SNPs. Using quantile‐quantile plots and Shapiro‐Wilk tests, we checked that the resulting permutation distribution of g₁‐g₀ is approximately normal and, therefore, that the permutation distribution of χ² indeed follows a chi‐squared distribution. We then calculated , where and s are the sample mean and standard deviation of the permutation distribution of g₁‐g₀, respectively. G₁ is the estimate of the co‐skewness for the original (non‐permuted) affected and unaffected groups. Afterward, we compared z² to a chi‐squared distribution with one degree of freedom to obtain P‐values.
2.4. Analysis
Co‐skewness was first evaluated in LOADFS and the mega sample of non‐LOADFS studies (created by pooling FHS, CHS, and HRS data sets) to examine the consistency of the directions of the effects in independent samples that is widely regarded as replication. 32 Then we used the mega sample of all studies combined to increase statistical power. This study did not examine the roles of sex and age as we showed their trivial effect for the same SNPs. 25
3. RESULTS
3.1. Co‐skewness for triples of SNPs
We evaluated LD between triples of SNPs using the standardized third mixed moment, or co‐skewness (see Methods). For 32 SNPs representing the BCAM‐NECTIN2‐TOMM40‐APOE‐APOC1 (19q13.3) region (see Methods and Table S1), there were 4960 triples. We examined associations of these triples with AD by evaluating differences in co‐skewness between the AD‐affected (cases, N = 2,789) and unaffected (non‐cases, N = 16,334) subjects using data from four studies (Table 1). To perform the analysis, we used genotype‐based, , and haplotype‐based, , co‐skewness metrics.
The genotype‐based method identified 51 triples of SNPs associated with AD at a Bonferroni‐adjusted locus‐wide significance level P < 10−5 = 0.05/4960 in LOADFS (Table S2). For most of these triples, 47 of 51 (92.2%), the effect direction, that is, the difference in co‐skewness between the AD‐affected () and unaffected () subjects , was replicated in the non‐LOADFS mega sample. The analysis of the mega sample of all studies revealed 302 significant associations at P < 10−5. For 269 of them (89.1%), the effect directions were consistent in LOADFS and non‐LOADFS.
The haplotype‐based method identified significant effects at P < 10−5 for 313 triples in LOADFS. The effect directions were replicated for 277 (88.5%) triples in non‐LOADFS studies (Table S3). The analysis of the mega sample of all studies combined identified 1127 significant differences, of which consistent effect directions in LOADFS and non‐LOADFS were for 999 triples. The haplotype‐based estimates of the associations of with AD were consistent with those from the genotype‐based method for 989 of 999 triples.
The top difference in the magnitude of co‐skewness was observed for the triple of rs2075650 (TOMM40), rs12721046 (APOC1), and rs429358 (minor allele of this SNP encodes the APOE ε4 allele) SNPs, , P = 7.38×10−178. The effect for this triple was much more extreme than the effects for the other triples, followed by the rs17561351 (NECTIN2), rs4081918, (NECTIN2), and rs405509 (APOE) triple with a 2.5‐fold smaller effect , P = 1.59×10−15 (Table 2, RΔG). The effect for the first triple was also the most significant, followed by , P = 1.24×10−167 for the rs157580 (TOMM40), rs440446 (APOE), and rs429358 (APOE) triple (Table 2, Rp‐val). Notably, the top 20 most significant triples included SNPs only from the TOMM40‐APOE‐APOC1 locus.
TABLE 2.
Top differences in co‐skewness between Alzheimer's disease (AD) affected and unaffected subjects ranked using two metrics
| ID | Rp‐val | RΔG | SNP1 | SNP2 | SNP3 | E4 | E2 |
|
|
|
p_value |
|
|
|
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4922 | 1 | 1 | rs2075650 | rs429358 | rs12721046 | e4 | no | 0.414 | 1.112 | –0.698 | 7.38E‐178 | 0.439 | 0.276 | –0.025 | 0.836 | |||||||
| 4895 | 2 | 12 | rs157580 | rs440446 | rs429358 | e4 | no | 0.258 | 0.058 | 0.199 | 1.24E‐167 | –0.551 | –0.267 | 0.809 | 0.325 | |||||||
| 4952 | 3 | rs440446 | rs429358 | rs439401 | e4 | no | 0.222 | 0.044 | 0.178 | 3.70E‐148 | 0.210 | –0.285 | 0.012 | 0.329 | ||||||||
| 4956 | 4 | rs440446 | rs439401 | rs12721046 | no | no | 0.195 | 0.043 | 0.151 | 2.00E‐127 | –0.511 | –0.290 | 0.706 | 0.333 | ||||||||
| 4921 | 5 | 18 | rs2075650 | rs429358 | rs439401 | e4 | no | 0.155 | 0.347 | –0.192 | 5.05E‐124 | –0.501 | –0.552 | 0.656 | 0.899 | |||||||
| 4898 | 6 | rs157580 | rs440446 | rs12721046 | no | no | 0.229 | 0.058 | 0.171 | 2.32E‐123 | –0.494 | –0.270 | 0.724 | 0.328 | ||||||||
| 4900 | 7 | rs157580 | rs429358 | rs439401 | e4 | no | –0.155 | 0.006 | –0.161 | 4.35E‐119 | –0.212 | –0.174 | 0.057 | 0.179 | ||||||||
| 4937 | 8 | 7 | rs8106922 | rs429358 | rs12721046 | e4 | no | 0.174 | 0.383 | –0.209 | 8.96E‐115 | 0.251 | 0.086 | –0.078 | 0.297 | |||||||
| 4907 | 9 | 5 | rs2075650 | rs8106922 | rs429358 | e4 | no | 0.118 | 0.336 | –0.218 | 3.88E‐105 | 0.249 | 0.085 | –0.132 | 0.251 | |||||||
| 4932 | 10 | rs8106922 | rs440446 | rs7412 | no | e2 | 0.106 | 0.249 | –0.143 | 1.01E‐104 | 0.130 | 0.193 | –0.024 | 0.057 | ||||||||
| 4880 | 11 | 6 | rs157580 | rs2075650 | rs429358 | e4 | no | 0.108 | 0.325 | –0.217 | 2.87E‐102 | –0.382 | –0.557 | 0.490 | 0.882 | |||||||
| 4904 | 12 | rs157580 | rs439401 | rs12721046 | no | no | –0.139 | 0.008 | –0.146 | 2.87E‐98 | 0.509 | 0.275 | –0.648 | –0.267 | ||||||||
| 4947 | 13 | 19 | rs405509 | rs429358 | rs12721046 | e4 | no | 0.334 | 0.525 | –0.191 | 3.08E‐93 | 0.326 | 0.119 | 0.008 | 0.406 | |||||||
| 4920 | 14 | rs2075650 | rs429358 | rs7412 | e4 | e2 | 0.031 | 0.120 | –0.088 | 1.42E‐89 | –0.159 | –0.213 | 0.190 | 0.332 | ||||||||
| 4959 | 15 | 11 | rs429358 | rs439401 | rs12721046 | e4 | no | 0.116 | 0.317 | –0.201 | 2.85E‐89 | 0.243 | 0.088 | –0.127 | 0.229 | |||||||
| 4916 | 16 | rs2075650 | rs440446 | rs429358 | e4 | no | 0.161 | 0.347 | –0.187 | 2.99E‐88 | 0.233 | 0.084 | –0.072 | 0.264 | ||||||||
| 4953 | 17 | 13 | rs440446 | rs429358 | rs12721046 | e4 | no | 0.105 | 0.303 | –0.198 | 4.15E‐76 | –0.479 | –0.523 | 0.585 | 0.826 | |||||||
| 4901 | 18 | 8 | rs157580 | rs429358 | rs12721046 | e4 | no | 0.108 | 0.315 | –0.207 | 4.61E‐75 | –0.476 | –0.547 | 0.583 | 0.862 | |||||||
| 4958 | 19 | rs429358 | rs7412 | rs12721046 | e4 | e2 | 0.036 | 0.121 | –0.085 | 1.31E‐69 | –0.126 | –0.205 | 0.162 | 0.326 | ||||||||
| 4938 | 20 | rs8106922 | rs7412 | rs439401 | no | e2 | 0.084 | 0.199 | –0.115 | 6.53E‐69 | 0.135 | –0.132 | –0.051 | 0.331 | ||||||||
| 4019 | 2 | rs17561351 | rs4081918 | rs405509 | no | no | 0.102 | 0.380 | –0.278 | 1.59E‐15 | 0.680 | 0.542 | –0.579 | –0.162 | ||||||||
| 4015 | 3 | rs17561351 | rs4081918 | rs283813 | no | no | 0.270 | 0.014 | 0.255 | 1.27E‐09 | 0.182 | 0.162 | 0.087 | –0.147 | ||||||||
| 4897 | 4 | rs157580 | rs440446 | rs439401 | no | no | 0.457 | 0.222 | 0.235 | 6.72E‐53 | –1.114 | –0.865 | 1.571 | 1.087 | ||||||||
| 1361 | 9 | rs10402271 | rs4803763 | rs3852856 | no | no | 0.151 | 0.354 | –0.203 | 1.82E‐30 | –1.231 | –1.152 | 1.382 | 1.506 | ||||||||
| 4422 | 10 | rs519113 | rs387976 | rs405509 | no | no | 0.036 | 0.239 | –0.203 | 1.74E‐26 | 0.494 | 0.413 | –0.458 | –0.174 | ||||||||
| 4012 | 14 | rs17561351 | rs4081918 | rs11667640 | no | no | 1.397 | 1.595 | –0.198 | 9.75E‐04 | 0.145 | 0.155 | 1.252 | 1.440 | ||||||||
| 4912 | 15 | rs2075650 | rs405509 | rs429358 | e4 | no | 0.271 | 0.467 | –0.196 | 2.06E‐61 | 0.309 | 0.107 | –0.038 | 0.360 | ||||||||
| 4771 | 16 | rs6859 | rs2075650 | rs429358 | e4 | no | 0.172 | 0.367 | –0.195 | 1.18E‐23 | –0.600 | –0.805 | 0.772 | 1.172 | ||||||||
| 4915 | 17 | rs2075650 | rs405509 | rs12721046 | no | no | 0.268 | 0.461 | –0.193 | 3.76E‐62 | 0.288 | 0.114 | –0.020 | 0.347 | ||||||||
| 2657 | 20 | rs4803763 | rs429358 | rs12721046 | e4 | no | 0.110 | 0.301 | –0.191 | 4.71E‐16 | 0.406 | 0.292 | –0.295 | 0.009 |
The data are from the pooled sample of the National Institute on Aging Late‐Onset Alzheimer's Disease Family Study, the Health and Retirement Study, the Cardiovascular Health Study, and the Framingham Heart Study parental and offspring cohorts.
ID in this column corresponds to IDs in Table S3.
Rp‐val and RΔG show ranking based on P‐value and effect size, respectively. Blank cells indicate ranking outside of top 20 triples for each metric.
E4 or E2 indicates presence of the APOE ε4‐coding rs429358 SNP or ε2‐coding rs7412 SNP in the triple.
is the effect defined as difference in co‐skewness between the AD‐affected () and unaffected () subjects using the haplotype‐based method.
and denote partitions of the and metrics into two fractions of mutual independence () and a weighted sum of the pairwise linkage disequilibrium () values (see Methods).
Italic shows SNPs from the NECTIN2 gene. Underlining denotes BCAM SNP. The other SNPs are in the TOMM40‐APOE‐APOC1 locus (see more details in Table S3).
The top 30 differences in co‐skewness between AD‐affected and unaffected subjects ranked using and P‐value‐based metrics included 18 triples with rs429358 SNP and four triples with rs7412 (minor allele of this SNP encodes the APOE ε2 allele) (Table 2). Two of these four triples included both rs429358 and rs7412 SNPs, and either rs2075650 (TOMM40) or rs12721046 (APOC1). LD between rs2075650 and rs12721046 was moderate (r 2 = 0.48).
Co‐skewness for the top triple of rs2075650, rs12721046, and rs429358 SNPs in cases ( ) appears to be smaller than in non‐cases ( ); that should indicate weaker connections between SNPs in this triple in the AD‐affected subjects than in the AD‐unaffected subjects. Partitioning these haplotype‐based metrics into two fractions of mutual independence () and a weighted sum of the pairwise LD values () (see Methods) shows, however, that the component increases, whereas the component decreases in cases (Table 2). This change in co‐skewness between AD‐affected and unaffected subjects is consistent with stronger connections between all three SNPs in cases (Figure 1).
3.2. Co‐skewness for triples harboring the ε2 or ε4 coding SNP
We further characterized differences in co‐skewness for 465 triples, which included rs429358, and 465 triples, which included rs7412. In the rs429358‐tailored set, there were 116 triples for which the difference in co‐skewness attained the locus‐wide significance (P < 10−5) and 105 such triples in the rs7412‐tailored set (Figure 2A and Table S3). These triples included SNPs from all selected genes in this region. We found seven triples, which had both rs429358 and rs7412, and one of the following SNPs: rs4803763, rs440277, rs6859, rs283813 (all four are from NECTIN2), rs2075650 (TOMM40), rs405509 (APOE), and rs12721046 (APOC1).
FIGURE 2.
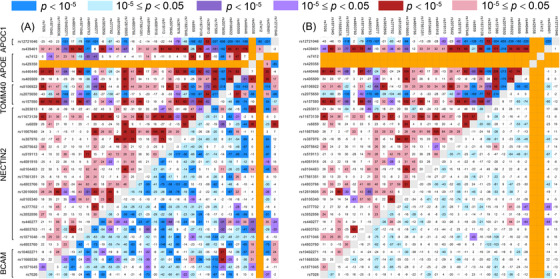
Heat maps for triples harboring ε4‐coding rs429358 (upper‐left triangle) or ε2‐coding rs7412 (lower‐right triangle). Heat maps for triples in the samples with (A) no exclusions and (B) exclusion of either carriers of the ε2 (upper‐left) or ε4 (lower‐right) allele. Numbers show the effect multiplied by 103 defined as the difference in co‐skewness between the AD‐affected () and unaffected () subjects. Red (blue) shows a positive (negative) difference that indicates strengthening (weakening) connections between SNPs in the triple. Purple shows triples for which the effect directions are of opposite signs in AD cases and non‐cases. Color shades denote significance levels, as shown in the upper inset. No color shows P > 0.05. Orange indicates undefined values where either rs429358 or rs7412 would occur twice in the same triple (A and B), or the value is undefined due to one of rs429358 or rs7412 being constant on the strata (B)
3.3. Co‐skewness for triples in the sample with no either the ε4 or ε2 allele carriers
Next, we examined the roles of the ε4 and ε2 alleles in the associations of with AD identified in Section 3.2. In the sample with no carriers of the ε4 allele (ie, excluding subjects carrying a minor allele of rs429358), there were five significant associations of with AD at P < 10−5 and 529 significant associations at 10−5 ≤ P < .05 (Table S4). In contrast, in the sample with no carriers of the ε2 allele (ie, excluding subjects carrying a minor allele of rs7412), there were 899 significant associations at P < 10−5 and 1474 significant associations at 10−5 ≤ P < .05 (Table S4). In part, this difference is due to a 2.5‐fold smaller sample of the AD cases with no carriers of the ε4 allele (maximum of cases is Ncase = 1035) compared to that with no carriers of the ε2 allele (maximum of cases is Ncase = 2578).
To examine potential connections of the ε2 allele with alleles from other SNPs in the triples, we focused on all 435 triples with rs7412 in the sample with no carriers of the ε4 allele. The heat maps in Figure 2 (lower‐right triangle) show that the significance of the associations was substantially decreased for most triples in this sample; that was due to reduced sample size and/or effect size (Table S4). For two triples, was associated with AD at P < 10−5. Both of them included rs7412 and rs8106922 (TOMM40), and either rs440446 (APOE) or rs405509 (APOE). LD between rs440446 and rs405509 was modest (r 2 = 0.62). For 85 triples, the association of with AD attained significance at 10−5 < P < 0.05.
The ε4 allele was more extensively involved in the associations of with AD than the ε2 allele as we observed 95 of 435 associations of with AD at P < 10−5 in the sample with the exclusion of the ε2 allele carriers (Figure 2, upper‐left triangles, and Table S4).
Figure 2 shows prevailing patterns of decreased co‐skewness in the ε2‐bearing triples and increased co‐skewness in the ε4‐bearing triples in AD‐affected subjects.
4. DISCUSSION
Here we leverage a new method to map triples of SNPs in the APOE region to AD. This method uses haplotype‐ and genotype‐based metrics of co‐skewness and generalizes LD between pairs of SNPs to SNP triples (see Methods and Note S1). By analogy with pairwise LD, 24 the associations have been assessed by evaluating the difference in co‐skewness between the AD‐affected and unaffected subjects of European ancestry.
The haplotype‐based method identified 1127 triples associated with AD at the locus‐wide significance P < 10−5, whereas the genotype‐based method identified 302 triples. This 3.7‐fold difference is driven mainly by the sensitivity of the haplotype‐based method to the deviation from HWE, as haplotypes are inferred under HWE, whereas the genotype‐based metric () does not require HWE. Because HWE for individual SNPs in full samples of LOADFS and non‐LOADFS (Table S1) does not guarantee HWE for subsamples (eg, as those stratified by the AD status) or at haplotype level (see Methods), consistently with the case of LD between SNP pairs, 20 , 21 , 25 the deviation from HWE is likely meaningful. This means that such a departure is unlikely an artifact but rather the result of some unobserved biological processes in an organism. Here this sensitivity is driven primarily by deviation from HWE at haplotype level in the AD‐affected subjects, as indicated by consistently larger differences in cases than non‐cases (Figure 3, Tables S2 and S3). Accordingly, the observed sensitivity indicates SNPs, which can be involved in the regulation of such unobserved biological processes specific to AD‐affected subjects. This information may be missed using the genotype‐based method alone as it is extracted from the difference of the results from the genotype‐ and haplotype‐based methods. These findings support a role of haplotypes comprising at least three alleles from different SNPs from the same or different genes in the APOE region in AD pathogenesis, rather than independent alleles (see the Introduction).
FIGURE 3.
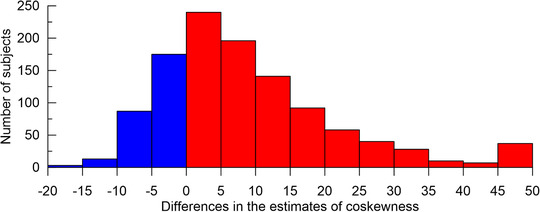
Differences in the estimates of co‐skewness using haplotype‐ and genotype‐based methods. The histogram shows the differences between the estimates of the differences in co‐skewness in cases and non‐cases multiplied by 103, that is, . Red (blue) denotes negative (positive) differences. Red shows dominant skewness driven by deviation from the Hardy‐Weinberg equilibrium at the haplotype level in the AD‐affected subjects. The differences in magnitude, which are larger than the x‐axis limits, are included in the flanking bins
Despite understanding that AD is a highly heterogeneous, genetically complex disorder, 33 current research often pursues the logic of medical genetics, assuming the existence of variants causing a complex trait. For example, a fundamental concept of genome‐wide association studies (GWAS) is that SNPs discovered by GWAS are merely proxies for actual causal variants for a given trait. 3 This logic is inherited from studies of monogenic diseases when a single highly penetrant mutation can cause a specific disease, which seems to be the case of APP, PSEN1, and PSEN2 genes and the autosomal dominant form of early onset AD. 8 , 9 , 10 , 11 For complex (ie, non‐Mendelian) diseases, such as late‐onset AD, GWAS report only small or moderate statistical effects. 5 , 6 , 7 These findings inspire studies of combined action of small‐effect variants. One common strategy is to aggregate the effects of many small‐effect variants spread through the entire genome into a polygenic risk score. 34
Another approach is to identify AD risks attributed to haplotypes comprising specific alleles. 17 , 18 , 19 , 22 , 23 An advantage of co‐skewness is that it highlights SNPs whose alleles can naturally define extended haplotypes. This is important in the framework of polygenic predisposition to complex diseases, as such haplotypes represent more accurate polygenic disease profiles. The complexity of the co‐skewness pattern in the APOE region is unlikely caused by pairwise LD between SNPs comprising different triples, as these SNPs have been selected not to be in strong LD. 25 This complexity supports genetic heterogeneity in susceptibility to AD beyond that related to race/ethnic differences. 26 Accordingly, the co‐skewness approach is also useful to highlight such genetic heterogeneity.
Our results show that AD is characterized mainly by the decrease and increase of co‐skewness in the ε2‐ and ε4‐bearing triples, respectively, in AD‐affected subjects. These changes indicate strengthening connections (ie, a higher rate of co‐occurrence) of the ε4 allele and weakening connections (ie, a smaller rate of co‐occurrence) of the ε2 allele with alleles from the other SNPs in this region (Figure 2). For example, for the top triple, which includes the ε4 coding rs429358 SNP and SNPs from the TOMM40 (rs2075650) and APOC1 (rs12721046) genes, we observe strengthening connections between all SNPs in the triple in the AD‐affected subjects (Table 2, Figure 1). Notably, we found that three triples include rs405509 (APOE) (Table 2), which is linked to all SNPs from the top triple, and that all these triples show the same type of associations with AD as the top triple, that is, stronger connections between all SNPs in the triple in the AD‐affected subjects. This result provides firm support to previous reports on the association of a haplotype comprising the rs405509_T and ε4 alleles with the risk of AD 18 and a reason for the dose‐dependent association of rs405509_T with AD in carriers of the ε4/ε4 homozygotes. 35 This result, however, suggests that the rs405509_T and ε4 haplotype should include a higher number of the risk alleles, which may be from the same or different genes, 19 that concurs with. 22 The functional role of NECTIN2, TOMM40, and APOC1 also suggests that they could contribute to the pathogenesis of AD in addition to APOE. 22
In contrast, four top rs7412‐bearing triples (Table 2) show that AD is associated with weakening connections of the ε2 allele with alleles from TOMM40 (rs2075650, rs8106922), APOE (rs429358, rs440446), and APOC1 (rs439401, rs12721046). The same prevailing pattern of weakening connections of rs7412 with other SNPs in the APOE region is observed in AD‐affected subjects who do not carry the ε4 allele (Figure 2B). Two triples, which include rs7412 and rs8106922 (TOMM40) and either rs440446 (APOE) or rs405509 (APOE), were associated with AD at P < 10−5. Thus better protection of the ε2 allele against AD may require haplotypes, including the ε2 allele and alleles from neighboring genes.
Figure 2 shows that the ε4 allele is associated with a more complex AD‐related pattern of co‐skewness than that for the ε2 allele. Therefore, although these alleles can be involved in haplotypes affecting the AD risks, there should be a higher number of the AD‐associated haplotypes with ε4 than ε2 in this region, and the ε4‐bearing haplotypes should be more sophisticated, including a more sizeable number of alleles from the other SNPs from the same or different genes.
Associations of triples without rs429358 and rs7412 with AD support the existence of AD‐predisposing haplotypes in this region, which may not include ε4 or ε2 alleles. 19 However, the lack of rs429358 and rs7412 SNPs in top triples (Table 2) should be interpreted with caution given that haplotypes may include more genetic variants.
Our results are supported by prior findings of a complex transcriptional regulatory structure in the APOE region, which includes multiple enhancers modulating gene expression. 36 Bekris et al. also showed that regional enhancers in cis could functionally influence the cell‐specific expression of TOMM40 and APOE, and suggested the biological role of promoter‐enhancer haplotypes in AD pathogenesis. 37 Given that the function of enhancers can be modulated by nearby and distant non‐coding variants, 38 the role of extended haplotypes harboring local and distant alleles in AD pathogenesis is feasible.
Our results raise a fundamental issue of a driving force of AD‐related haplotypes. By contrasting pairwise LD in younger and older subjects, we recently showed that LD structures observed in older AD‐free subjects and the younger subjects who were not under noticeable mortality risk were the same. 25 This finding favors the role of recent and specific (eg, within families or communities, a divergence of ancestral groups) selection, which can be indirectly relevant to AD. Such selection could be driven by exogenous factors supporting, thus, the concept of the AD exposome. 39 Trumble and Finch provided solid arguments supporting the role of exposures to environmental toxins such as air pollution and tobacco smoking in recent human evolution. 40 It was shown that the ε4 allele increases the risk of dementia from air pollution. 41 Thus environmental toxins, the prevalence of which increases over time, can be a driver of the adaptive haplotypes in the APOE region, which may become deleterious for cognition in late life. Another factor driving the adaptation of haplotypes in the APOE region could be health protection in infected environments in early life. 42 , 43 , 44 Of interest, such protection can be even extended to adult life, as was shown in the Tsimané indigenous population living in the highly infectious environment. 45
Despite the rigor of this study, we acknowledge the limitation that the AD diagnoses based on ICD‐9 codes in HRS and CHS can be less accurate than those in LOADFS and FHS. Although this inaccuracy may affect the precision of the co‐skewness estimates, the consistency of the directions of differences in the AD‐affected and unaffected subjects in independent samples partly offsets this problem.
In conclusion, our results on the 3.7‐fold excess in the estimates of the associations of triples of SNPs with AD using the haplotype‐based method compared to the genotype‐based method support the definitive roles of complex haplotypes in predisposition to AD in the APOE region. The complex structure of such haplotypes is supported by a large number of the AD‐associated triples, which include SNPs from all five genes. AD is characterized mainly by strengthening connections of the ε4 allele and weakening connections of the ε2 allele with the other alleles in this region. Finally, these results support a more extensive role of the ε4 allele than the ε2 allele in complex AD‐related haplotypes. However, the latter result should be interpreted with caution as the number of the ε2 carriers in this sample is substantially smaller than those of ε4.
CONFLICTS OF INTEREST
Nothing to report.
FUNDING INFORMATION
This research was supported by Grants No R01 AG047310, R01 AG061853, R01 AG065477, and R01 AG070488 from the National Institute on Aging. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
AUTHOR CONTRIBUTIONS
Alexander M Kulminski conceived and designed the experiment and wrote the paper. Ian Philipp developed co‐skewness metrics, wrote the paper, and performed statistical analyses. Irina Culminskaya wrote the paper. Yury Loika and Liang He prepared data.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGEMNTS
This manuscript was prepared using data sets obtained though dbGaP (accession numbers phs000007.v28.p10, phs000287.v5.p1, phs000428.v1.p1, phs000168.v2.p2) and the University of Michigan. Phenotypic Health and Retirement Study (HRS) data are available publicly and through restricted access from http://hrsonline.isr.umich.edu/index.php?p = data. The authors thank Arseniy P. Yashkin for help in preparation of phenotypes in HRS. The authors declare no competing interests.
The Framingham Heart Study (FHS) is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University (Contract No. N01‐HC‐25195 and HHSN268201500001I). This manuscript was not prepared in collaboration with investigators of the FHS and does not necessarily reflect the opinions or views of the FHS, Boston University, or NHLBI. Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract N02‐HL‐64278. SHARe Illumina genotyping was provided under an agreement between Illumina and Boston University. Funding for CARe genotyping was provided by NHLBI Contract N01‐HC‐65226. Funding support for the Framingham Dementia dataset was provided by NIH/NIA grant R01 AG08122.
The Cardiovascular Health Study (CHS) was supported by contracts HHSN268201200036C, HHSN268200800007C, N01‐HC‐85079, N01‐HC‐85080, N01‐HC‐85081, N01‐HC‐85082, N01‐HC‐85083, N01‐HC‐85084, N01‐HC‐85085, N01‐HC‐85086, N01‐HC‐35129, N01 HC‐15103, N01 HC‐55222, N01‐HC‐75150, N01‐HC‐45133, and N01‐HC‐85239; grant numbers U01 HL080295 and U01 HL130014 from the National Heart, Lung, and Blood Institute (NHLBI), and R01 AG‐023629 from the National Institute on Aging, with additional contribution from the National Institute of Neurological Disorders and Stroke. A full list of principal CHS investigators and institutions can be found at https://chs-nhlbi.org/pi. This manuscript was not prepared in collaboration with CHS investigators and does not necessarily reflect the opinions or views of CHS or the NHLBI. Additional support for infrastructure was provided by HL105756 and additional genotyping among the African American cohort was supported in part by HL085251. DNA handling and genotyping at Cedars‐Sinai Medical Center was supported in part by National Center for Research Resources grant UL1RR033176, now at the National Center for Advancing Translational Technologies CTSI grant UL1TR000124; in addition to the National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
The Health and Retirement Study (HRS) genetic data are sponsored by the Genetics Resource with HRS April 21, 2010, version G Page 5 of 7 National Institute on Aging (grant numbers U01AG009740, RC2AG036495, and RC4AG039029) and was conducted by the University of Michigan. This manuscript was not prepared in collaboration with HRS investigators and does not necessarily reflect the opinions or views of HRS.
Funding support for the Late Onset Alzheimer's Disease Family Study (LOADFS) was provided through the Division of Neuroscience, NIA. The LOADFS includes a genome‐wide association study funded as part of the Division of Neuroscience, NIA. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by Genetic Consortium for Late Onset Alzheimer's Disease. This manuscript was not prepared in collaboration with LOADFS investigators and does not necessarily reflect the opinions or views of LOADFS.
Kulminski AM, Philipp I, Loika Y, He L, Culminskaya I. Haplotype architecture of the Alzheimer's risk in the APOE region via co‐skewness. Alzheimer's Dement. 2020;12:e12129. 10.1002/dad2.12129
Alexander M. Kulminski and Ian Philipp contributed equally to this article.
REFERENCES
- 1. Lutz MW, Crenshaw DG, Saunders AM, Roses AD. The importance of being connected. J Alzheimers Dis. 2011;24:247‐251. [DOI] [PubMed] [Google Scholar]
- 2. Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer's disease. Arch Gen Psychiatry. 2006;63:168‐174. [DOI] [PubMed] [Google Scholar]
- 3. Belloy ME, Napolioni V, Greicius MD. A quarter century of apoe and Alzheimer's disease: progress to date and the path forward. Neuron. 2019;101:820‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou X, Chen Y, Mok KY, et al. Identification of genetic risk factors in the chinese population implicates a role of immune system in Alzheimer's disease pathogenesis. Proc Natl Acad Sci U S A. 2018;115:1697‐1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goate A, Chartier‐Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704‐706. [DOI] [PubMed] [Google Scholar]
- 9. Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature. 1995;375:754‐760. [DOI] [PubMed] [Google Scholar]
- 10. Levy‐Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973‐977. [DOI] [PubMed] [Google Scholar]
- 11. Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775‐778. [DOI] [PubMed] [Google Scholar]
- 12. Raichlen DA, Alexander GE. Exercise, apoe genotype, and the evolution of the human lifespan. Trends Neurosci. 2014;37:247‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rebeck GW, Kindy M, LaDu MJ. Apolipoprotein e and alzheimer's disease: the protective effects of apoe2 and e3. J Alzheimers Dis. 2002;4:145‐154. [DOI] [PubMed] [Google Scholar]
- 14. Reiman EM, Arboleda‐Velasquez JF, Quiroz YT, et al. Exceptionally low likelihood of alzheimer's dementia in apoe2 homozygotes from a 5,000‐person neuropathological study. Nat Commun. 2020;11:667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Genin E, Hannequin D, Wallon D, et al. Apoe and Alzheimer disease: a major gene with semi‐dominant inheritance. Mol Psychiatry. 2011;16:903‐937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roses AD, Lutz MW, Amrine‐Madsen H, et al. A tomm40 variable‐length polymorphism predicts the age of late‐onset Alzheimer's disease. Pharmacogenomics J. 2010;10:375‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lutz MW, Crenshaw D, Welsh‐Bohmer KA, Burns DK, Roses AD. New genetic approaches to ad: lessons from apoe‐tomm40 phylogenetics. Curr Neurol Neurosci Rep. 2016;16:48. [DOI] [PubMed] [Google Scholar]
- 18. Lescai F, Chiamenti AM, Codemo A, et al. An apoe haplotype associated with decreased epsilon4 expression increases the risk of late onset Alzheimer's disease. J Alzheimers Dis. 2011;24:235‐245. [DOI] [PubMed] [Google Scholar]
- 19. Zhou X, Chen Y, Mok KY, et al. Non‐coding variability at the apoe locus contributes to the Alzheimer's risk. Nat Commun. 2019;10:3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nielsen DM, Ehm MG, Weir BS. Detecting marker‐disease association by testing for hardy‐weinberg disequilibrium at a marker locus. Am J Hum Genet. 1998;63:1531‐1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zaykin DV, Meng Z, Ehm MG. Contrasting linkage‐disequilibrium patterns between cases and controls as a novel association‐mapping method. Am J Hum Genet. 2006;78:737‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takei N, Miyashita A, Tsukie T, et al. Genetic association study on in and around the apoe in late‐onset Alzheimer disease in japanese. Genomics. 2009;93:441‐448. [DOI] [PubMed] [Google Scholar]
- 23. Yu CE, Seltman H, Peskind ER, et al. Comprehensive analysis of apoe and selected proximate markers for late‐onset Alzheimer's disease: patterns of linkage disequilibrium and disease/marker association. Genomics. 2007;89:655‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kulminski AM, Huang J, Wang J, He L, Loika Y, Culminskaya I. Apolipoprotein e region molecular signatures of Alzheimer's disease. Aging Cell. 2018;17:e12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kulminski AM, Shu L, Loika Y, et al. Genetic and regulatory architecture of Alzheimer's disease in the apoe region. Alzheimers Dement (Amst). 2020;12:e12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kulminski AM, Shu L, Loika Y, et al. Apoe region molecular signatures of Alzheimer's disease across races/ethnicities. Neurobiol Aging. 2020;87:141e1‐e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cupples LA, Heard‐Costa N, Lee M, Atwood LD. Genetics analysis workshop 16 problem 2: the framingham heart study data. BMC Proc. 2009;3(Suppl 7):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fried LP, Borhani NO, Enright P, et al. The cardiovascular health study: design and rationale. Ann Epidemiol. 1991;1:263‐176. [DOI] [PubMed] [Google Scholar]
- 29. Juster FT, Suzman R. An overview of the health and retirement study. J Human Res. 1995;30:S7‐S56. [Google Scholar]
- 30. Lee JH, Cheng R, Graff‐Radford N, Foroud T, Mayeux R. National Institute on Aging Late‐Onset Alzheimer's Disease Family Study G. Analyses of the national institute on aging late‐onset Alzheimer's disease family study: implication of additional loci. Arch Neurol. 2008;65:1518‐1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Das S, Forer L, Schonherr S, et al. Next‐generation genotype imputation service and methods. Nat Genet. 2016;48:1284‐1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marigorta UM, Rodriguez JA, Gibson G, Navarro A. Replicability and prediction: lessons and challenges from gwas. Trends Genet. 2018;34:504‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bertram L, Tanzi RE. Genomic mechanisms in Alzheimer's disease. Brain Pathol. 2020;30:966‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Del‐Aguila JL, Fernandez MV, Schindler S, et al. Assessment of the genetic architecture of Alzheimer's disease risk in rate of memory decline. J Alzheimers Dis. 2018;62:745‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi KY, Lee JJ, Gunasekaran TI, et al. Apoe promoter polymorphism‐219t/g is an effect modifier of the influence of apoe epsilon4 on Alzheimer's disease risk in a multiracial sample. J Clin Med. 2019;8:1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shao Y, Shaw M, Todd K, et al. DNA methylation of tomm40‐apoe‐apoc2 in Alzheimer's disease. J Hum Genet. 2018;63:459‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bekris LM, Lutz F, Yu CE. Functional analysis of apoe locus genetic variation implicates regional enhancers in the regulation of both tomm40 and apoe. J Hum Genet. 2012;57:18‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kikuchi M, Hara N, Hasegawa M, et al. Enhancer variants associated with Alzheimer's disease affect gene expression via chromatin looping. BMC Med Genomics. 2019;12:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Finch CE, Kulminski AM. The Alzheimer's disease exposome. Alzheimers Dement. 2019;15:1123‐1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Trumble BC, Finch CE. The exposome in human evolution: from dust to diesel. Q Rev Biol. 2019;94:333‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cacciottolo M, Wang X, Driscoll I, et al. Particulate air pollutants, apoe alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl Psychiatry. 2017;7:e1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oria RB, Patrick PD, Blackman JA, Lima AA, Guerrant RL. Role of apolipoprotein e4 in protecting children against early childhood diarrhea outcomes and implications for later development. Med Hypotheses. 2007;68:1099‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mitter SS, Oria RB, Kvalsund MP, et al. Apolipoprotein e4 influences growth and cognitive responses to micronutrient supplementation in shantytown children from northeast brazil. Clinics (Sao Paulo). 2012;67:11‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Exel E, Koopman JJE, Bodegom DV, et al. Effect of apoe epsilon4 allele on survival and fertility in an adverse environment. PLoS One. 2017;12:e0179497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vasunilashorn S, Finch CE, Crimmins EM, et al. Inflammatory gene variants in the tsimane, an indigenous bolivian population with a high infectious load. Biodemography Soc Biol. 2011;57:33‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information