

Abstract
The picoloyl ester (Pico) has proven to be a versatile protecting group in carbohydrate chemistry. It can be used for the purpose of stereocontrolling glycosylations via an H-bond-mediated Aglycone Delivery (HAD) method. It can also be used as a temporary protecting group that can be efficiently introduced and chemoselectively cleaved in the presence of practically all other common protecting groups used in synthesis. Herein, we will describe a new method for rapid, catalytic, and highly chemoselective removal of the picoloyl group using inexpensive copper(ii) or iron(iii) salts.
Introduction
The chemical synthesis of glycans is a difficult task that typically involves manipulation of a variety of protecting groups to obtain selectively protected building blocks.1–4 As once stated by Fraser-Reid, “protecting groups do more than protect”:5 they are also known to control all types of selectivity: regio-, stereo-, and chemoselectivity in glycosylation. Protecting groups may also have a powerful effect on the building block reactivity in glycosylation.6 During the synthesis of carbohydrates and other complex biomolecules, protecting groups often need to be chemoselectively removed over other protecting and functional groups present in the molecule. Some reaction conditions used for chemoselective protecting group removal are harsh or rely on using toxic reagents. Others lead to only marginal chemoselectivity and hence require careful refinement of reaction conditions to avoid undesired removal of other protecting groups. Dedicated studies in this area led to the discovery of a few sets of orthogonal protecting group. Orthogonal combinations identified by Boons: levulinoyl (Lev), acetyl, fluorenylmethoxycarbonyl (Fmoc), tert-butyldiphenylsilyl (TBDPS);7 Fmoc, naphthyl, Lev, and allyloxycarbonyl (Alloc);8 Schmidt: Fmoc, phenoxyacetyl, Lev, Alloc;9 Seeberger: naphthyl, Lev, Fmoc, 2-(azidomethyl)benzoyl;10–13 and others13,14 offer excellent flexibility for selective liberation of particular hydroxyl groups. These protecting group strategies are commonly employed in glycan assembly using reactions in solution and on solid supports.15 Nevertheless, identifying other stable and selectively removable protecting groups that can be selectively removed under mild and/or unique reaction conditions is always a desirable direction of research in the field of polyfunctional compound synthesis and modification. In particular, new protecting groups that would easily fit into existing schemes and orthogonal combinations are of particular interest.
Recently, our group16–25 and others26–36 have done extensive studies on the use of the picoloyl (Pico) protecting group. In particular, the Pico group assisted H-bond-mediated Aglycone Delivery (HAD) glycosylation reaction provided high facial α- or β-stereoselectivity that was always syn in respect to the Pico group. The stereoselectivity was only one advantage of using the Pico protecting group. The Pico group can be cleaved in traditional Zemplén conditions37 using sodium methoxide in methanol.23 It was also found that Pico could be selectively cleaved off in the presence of practically all other known protecting groups using zinc(ii) acetate27 or copper(ii) acetate.17,18,20,23 This reaction, however, is slow with reported times of 16 h,17,21 and typically requires stoichiometric amount of Cu(OAc)2 (1–1.3 equiv.). Reported herein are new reaction conditions that allow for entirely chemoselective removal of Pico using catalytic (30 mol%) ferric chloride or Cu (OAc)2. The developed conditions are directly compatible with all other protecting groups used in orthogonal glycan synthesis.
Results and discussion
After preliminary screening of potential reagents, we discovered that iron(iii) chloride provides a much faster removal of Pico under the same reaction conditions to those previously reported for Cu(OAc)2. We have purposefully chosen compounds equipped with Pico at the C-4 position that was particularly resistant towards removal in our previous studies.17,21 Thus, deprotection of 4-Pico in a series of linear and branched glycans required excess Cu(OAc)2 and prolonged reaction time (16 h). Deprotection of thioglycoside 116 equipped with 4-Pico group with Cu(OAc)2 (1.3 equiv.) in MeOH–DCM (1/9) was more rapid, but still required 3 h to complete (Table 1, entry 1). As a result, the deprotected derivative 2 was obtained in 99% yield. Performing the reaction with FeCl3 (1.3 equiv.) under similar reaction conditions afforded compound 2 in 99% yield in 3.5 h (entry 2).
Table 1.
Optimization of the Pico group removal under catalytic conditions
 | ||
|---|---|---|
| Entry | Catalyst, solvent, time | Yield |
| 1 | Cu(OAc)2 (130), MeOH/DCM (1/9), 3 h | 99% |
| 2 | FeCl3 (130), MeOH/DCM (1/9), 3.5 h | 98% |
| 3 | FeCl3 (30), MeOH/DCM (1/9), 48 h | 75% |
| 4 | FeCl3 (30), MeOH/DCM (1/1), 18 h | 87% |
| 5 | FeCl3 (30), MeOH/DCM (9/1), 5 h | 91% |
| 6 | FeCl3 (30), MeOH (neat), 5 h | 89% |
| 7 | FeCl3 (15), MeOH/DCM (9/1), 10 h | 99% |
| 8 | FeCl3 (5), MeOH/DCM (9/1), 28 h | 92% |
| 9 | Cu(OAc)2 (30), MeOH/DCM (9/1), 1.5 h | 99% |
After recording these promising results, we endeavored to optimizing the reaction condition to determine whether sub-stoichiometric amounts of metal salts would be sufficient for driving the Pico deprotection to completion. We first found that upon reducing the amount of FeCl3 to 30 mol%, the reaction still occurred. However, this reaction was significantly slower, and required 48 h to obtain compound 2 in 75% yield (entry 3). In a further attempt to refine the reaction conditions, we investigated the effect of the solvent. Increasing the amount of MeOH in respect to DCM gave us the desired outcome. Thus, deprotection in MeOH–DCM (1/1) produced compound 2 in 87% in 18 h (entry 4). Furthermore, deprotection in MeOH–DCM (9/1) afforded compound 2 in 91% yield in 5 h (entry 5). Using neat methanol showed no further improvement (entry 6). Reactions using even lower amounts of FeCl3, 15 and even 5 mol%, could still be driven to completion, but required longer reaction time, 10 and 28 h, respectively (entries 7 and 8). Nevertheless, compound 2 was obtained in excellent yields of 99% and 92%, respectively. We also wanted to see how well Cu(OAc)2 worked under these new reaction conditions. As depicted in entry 9, this reaction was even faster, and compound 2 was produced in 99% yield in only 1.5 h. It should be noted that the reaction did not proceed when performed in the presence of methanol, without the addition of a catalyst.
From these optimizations, we carried out subsequent deprotection reactions using 30 mol% of the catalyst in MeOH–DCM (9/1). First, we wanted to investigate other regioisomers of 1 wherein Pico was present at C-2, C-3, and C-6 positions, compounds 3, 5,24,25 and 7,16 respectively (Scheme 1). Interestingly, removing Pico from the C-2 position in 3 with FeCl3 was very sluggish, which resulted in a much longer and incomplete reaction giving compound 4 in only 71% yield after 3 days. We are unsure of the rationale for this result that seems to stand out from the general trend. In contrast, a similar reaction in the presence Cu(OAc)2 rapidly produced 2-OH derivative 4 in 91% yield in 2 h. Deprotection of 3-Pico in 5 with FeCl3 afforded 3-OH derivative 6 in 91% yield in 5 h. The removal of 6-Pico in 7 was very rapid and efficient in the presence of either catalyst, and 6-OH derivative 8 was obtained in 99% in 10–20 min.
Scheme 1.
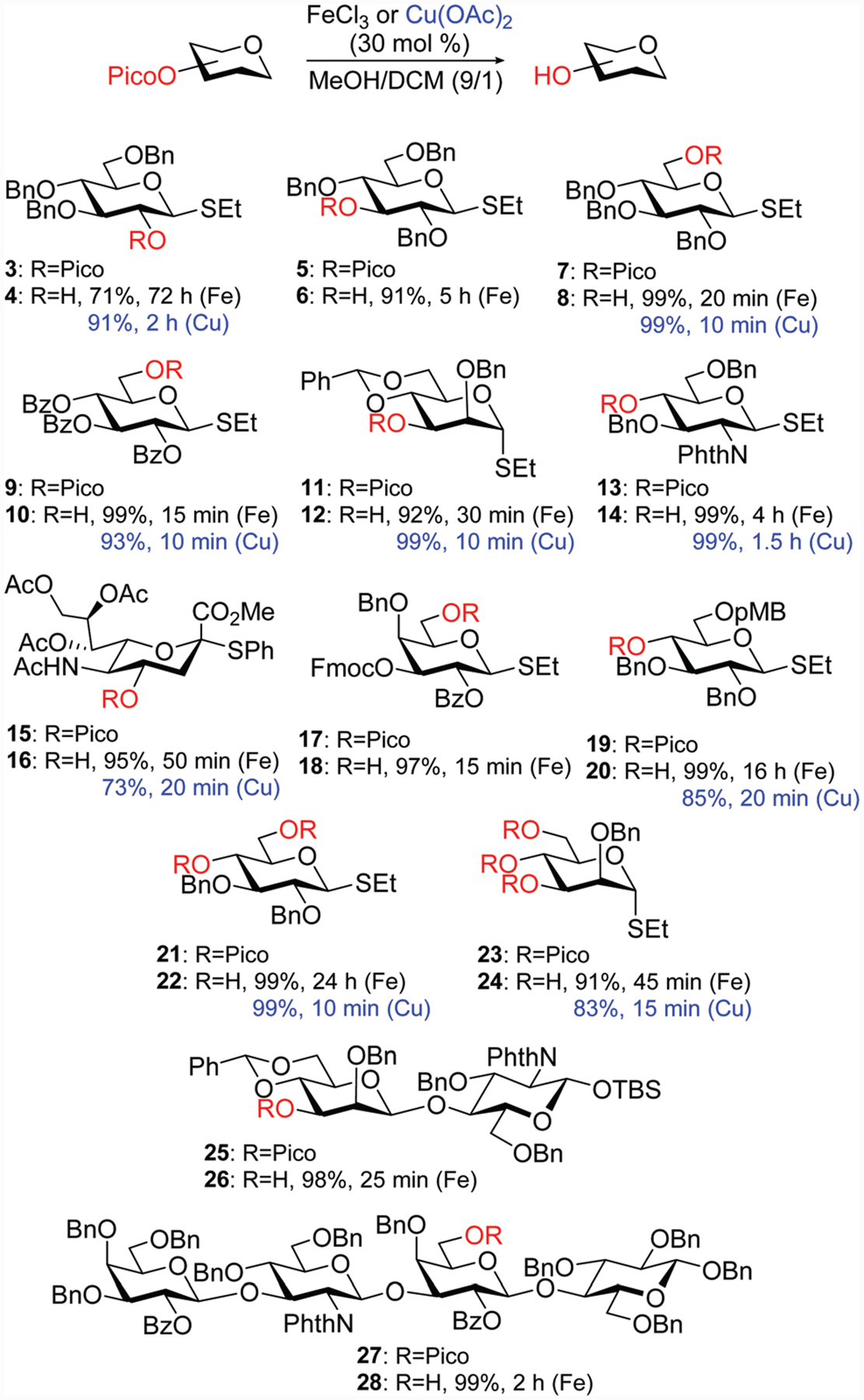
Broadening the scope of the chemoselective Pico cleavage using Fe(iii) or Cu(ii) catalysts.
Following the success of our preliminary trials, we moved on to investigating the compatibility of the developed reaction conditions with other temporary protecting groups. Removing the 6-Pico group in benzoylated thioglycoside 9 was rapid and chemoselective with either catalyst. The desired 6-OH derivative 10 was obtained in 93–99% yield in 10–15 min (Scheme 1). This result demonstrates that Pico can be chemoselectively removed in the presence of benzoyl groups. Deprotection of the 3-Pico group in benzylidene-protected thiomannoside 1118 was also rapid and efficient. 3-OH derivative 12 was rapidly produced (10–30 min) in the presence of either catalyst. The yields for the formation of 12 were also excellent (92–99%), which confirms compatibility of the acid-labile benzylidene acetal group with the developed reaction conditions. The removal of 4-Pico in glucosamine derivative 13 was also very efficient, and the resulting 4-OH derivative 14 was obtained in 99% yield in 1.5–4 h. This result indicated the efficiency of the developed method in application to aminosugars and compatibility of the phthalimido group with these reaction conditions.
The method also proved successful in chemoselective removal of the 4-Pico group in acetylated sialic acid derivative 15.32 4-OH Sialoside 16 was rapidly produced in the presence of FeCl3 in 95% yield in 50 min. A similar reaction in the presence of copper(ii) acetate was even faster (20 min), but this translated in a somewhat lower yield of compound 16 (73%). Deprotection of 6-Pico with FeCl3 in differentially protected thioglycoside 1738 was very rapid (15 min) affording 6-OH derivative 18 in 97% yield. This result indicated excellent compatibility of the Fmoc group that is commonly used as a temporary protecting group in iterative oligosaccharide synthesis. The removal of 4-Pico in compound 19 was somewhat slow with FeCl3, but the desired 4-OH derivative 20 was smoothly produced in an excellent yield (99%). This result ultimately confirms the compatibility of p-methoxybenzyl group with the developed reaction conditions. The 4-Pico group removal in 19 in the presence of copper(ii) acetate was significantly faster (20 min), but the yield of product 20 was somewhat lower (85%).
We also wanted to evaluate whether these reaction conditions are capable of concomitant removal of multiple Pico groups. When 4,6-di-Pico derivative 21 was treated with 30 mol% of iron(iii) chloride the desired diol 22 was produced in 99% yield. This reaction required 24 h to go to completion. As in a number of previous cases deprotection in the presence of copper(ii) acetate was much more rapid (10 min) without affecting the efficiency: diol 22 was produced in 99% yield. Even tri-Pico compound 2323 could be efficiently deprotected using only 30 mol% of either catalyst. As a result, triol 24 was isolated in 83–91% yield in 15–45 min.
Finally, we also investigated the removal of the Pico group from oligosaccharides. When 3′-Pico protected disaccharide 25 was treated with FeCl3 compound 26 was efficiently produced in 98% yield in 25 min. This result signified compatibility of the developed conditions with tert-butyldimethylsilyl (TBS), benzylidene, and phthalimido groups, all in one platform. Lacto-N-tetraose 27,39,40 a common core human milk tetrasaccharide, equipped with 6′-Pico group could also be efficiently deprotected with FeCl3 to afford compound 28 in 99% in 2 h.
Mechanistically, we hypothesize that when a picoloylated derivative A is used, iron(iii) chloride (or copper(ii) acetate) coordinate between both the carbonyl oxygen and the nitrogen atoms of the Pico group as shown in Scheme 2 for intermediate B. This pulls electron density away from the carbonyl carbon allowing for a nucleophile to attack, in our case methanol via tetrahedral intermediate C. The subsequent proton exchange leads to intermediate D, and the tetrahedral intermediate collapse the transesterification products, unprotected alcohol E and methyl picolinate. Iron(iii) chloride is released and is available for the next catalytic cycle. To reinforce this reaction mechanism, we also investigated whether other positional isomers of the Pico group could be removed accordingly. For this purpose we obtained 3-niconoyl and 3-O-iso-niconoyl protected compounds 29 and 30 (Scheme 2).24,25 No deprotection took place even in 24 h under the established reaction conditions. This outcome ultimately proves the complexation mode of metal salts that leads to swift deprotection of Pico groups.
Scheme 2.
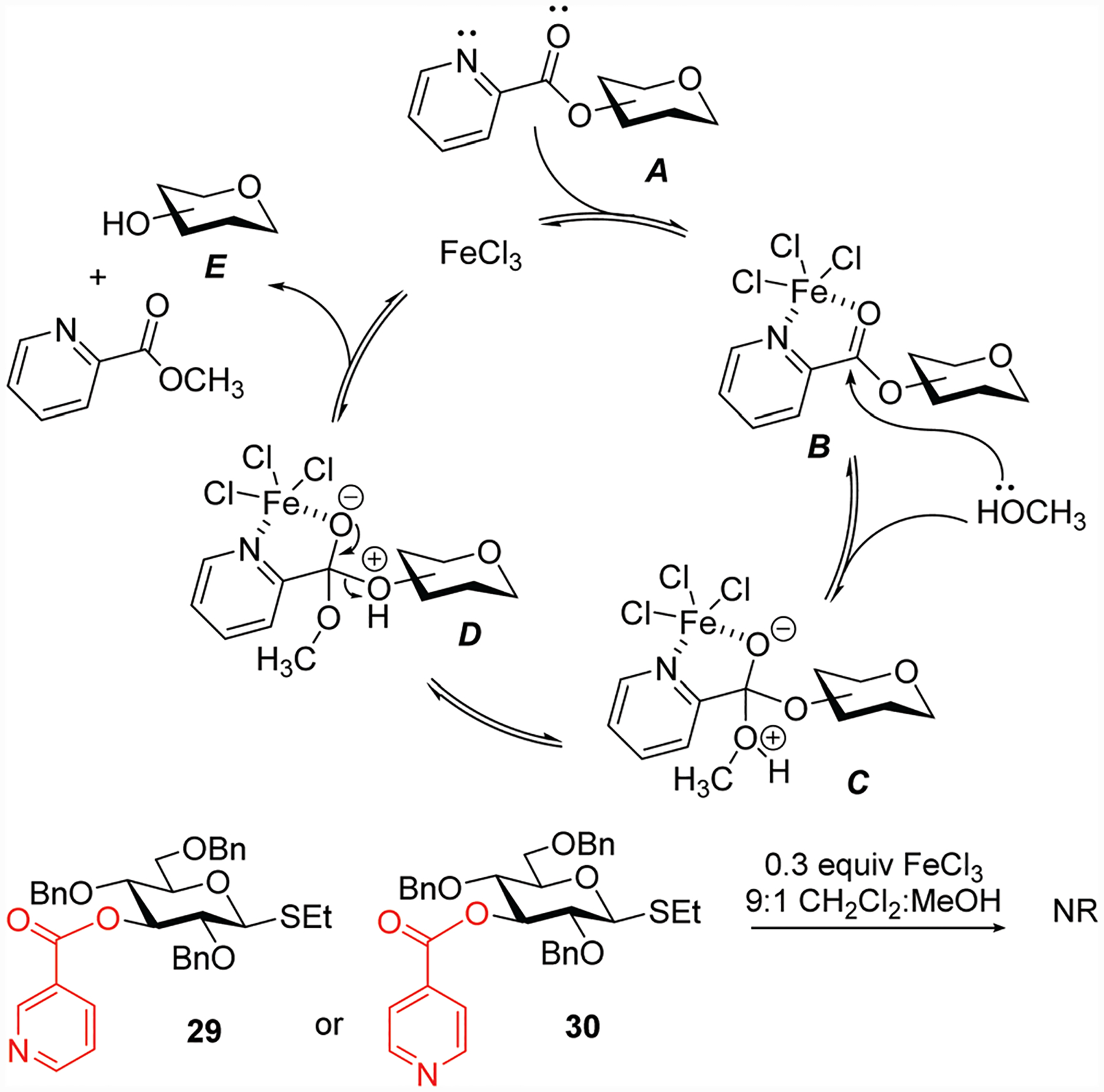
Proposed mechanism of picoloyl cleavage.
Conclusions
The Pico group can be used as an effective temporary protecting group. The Pico group can be cleaved using traditional Zemplén conditions using sodium methoxide in methanol, and its stability is similar to that of benzoyl ester group. In contrast to previous reports dealing with the chemoselective removal of the Pico group in the presence of other esters that employed stoichiometric reagents, this study demonstrated that the Pico group can be removed in a catalytic manner using 30 mol% of iron(iii) chloride or copper(ii) acetate. These conditions are also capable of chemoselective removal of even multiple Pico groups. Reactions performed with Cu(OAc)2 were generally faster, but on a number of occasions FeCl3-catalyzed reactions provided better yields. The developed conditions are directly compatible with all other protecting groups used in other orthogonal protection schemes. Hence, it is to be expected that the Pico group can directly supplement the arsenal of existing orthogonal group combinations used for glycan synthesis.
Experimental section
General
Column chromatography was performed on silica gel 60 (70–230 mesh), reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. Optical rotations were measured at ‘Jasco P-2000’ polarimeter. Unless noted otherwise, 1H NMR spectra were recorded in CDCl3 at 300, 13C NMR spectra were recorded in CDCl3 at 75 MHz. Accurate mass spectrometry determinations were performed using Agilent 6230 ESI TOF LCMS.
Synthesis of picoloyl containing compounds
General procedure for the Pico group introduction.
Picolinic acid (2–3 equiv. per OH), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, 2–3 equiv. per OH), and 4-dimethylaminopyridine (DMAP, 0.2–0.5 equiv. per OH) were added to a solution of a starting material containing at least one OH group in CH2Cl2, and the resulting mixture was stirred under argon for 16 h at rt. After that, the reaction mixture was diluted with CH2Cl2 and washed with water (twice). The organic phase was separated, dried with magnesium sulfate, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane gradient elution) to give the corresponding compound containing one or more Pico groups.
Ethyl 3,4,6-tri-O-benzyl-2-O-picoloyl-1-thio-β-d-glucopyranoside (3).
The title compound was prepared from ethyl 3,4,6-tri-O-benzyl-1-thio-β-d-glucopyranoside41 (4, 34.6 mg, 0.07 mmol) in CH2Cl2 (2.0 mL), picolinic acid (26.0 mg, 0.21 mmol), EDC (40.3 mg, 0.21 mmol), and DMAP (4.3 mg, 0.03 mmol) in accordance with the general procedure as a white amorphous solid in 87% yield (36.3 mg, 0.60 mmol). Analytical data for 3: Rf = 0.50 (ethyl acetate/toluene, 1/1, v/v); +22.3 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.77 (d, 1H, aromatic), 8.12 (d, 1H, aromatic), 7.83 (m, 1H, aromatic), 7.48 (m, 1H, aromatic), 7.40–7.24 (m, 8H, aromatic), 7.24–7.15 (m, 2H, aromatic), 7.15–7.05 (m, 5H, aromatic), 5.38 (dd, J2,3 = 9.6 Hz, 1H, H-2), 4.82 (d, 2J = 10.6, 1H, CHPh), 4.75 (m, 2J = 11.0 Hz, 2H, 2 × CHPh) 4.67 (d, J1,2 = 9.6 Hz, 1H, H-1) 4.68–4.51 (m, 3H, 3 × CHPh), 3.97 (dd, J3,4 = 9.1 Hz, 1H, H-3), 3.77 (dd, J4,5 = 9.6 Hz, 1H, H-4), 3.76 (m, 1H, H-6a), 3.75 (m, J6a,6b = 4.6 Hz, 1H, H-6b), 3.57 (dd, J5,6a = J5,6b = 9.1 Hz, 1H, H-5), 2.85–2.62 (m, 2H, SCH2CH3), 1.24 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 164.3, 150.0, 147.8, 138.3, 138.1, 138.0, 137.2, 128.6 (×2), 128.4, 128.3, 128.1, 127.9, 127.8, 127.7, 127.2, 126.0, 84.4, 83.4, 79.6 (×2), 78.1 (×2), 75.6 (×2), 75.3 (×2), 73.6 (×2), 73.4 (×2), 69.0 (×2), 24.1, 15.1 ppm; HRMS [M + Na]+ calcd for C35H37NO6SNa 622.2236 found 622.2244.
Ethyl 2,3,4-tri-O-benzoyl-6-O-picoloyl-1-thio-β-d-glucopyranoside (9).
The title compound was prepared from ethyl 2,3,4-tri-O-benzoyl-1-thio-β-d-glucopyranoside42 (10, 4.65 g, 8.66 mmol) in CH2Cl2 (100 mL), picolinic acid (2.15 g, 17.32 mmol), EDC (3.32 g, 17.32 mmol), and DMAP (0.21 g, 1.73 mmol) in accordance with the general procedure as a white amorphous solid in 99% yield (5.56 g, 8.65 mmol). Analytical data for 9: Rf = 0.30 (ethyl acetate/hexane, 1/1, v/v); +16.2 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.71 (m, 1H, aromatic), 8.10 (m, 1H, aromatic), 8.00–7.92 (m, 2H, aromatic), 7.92–7.85 (m, 2H, aromatic), 7.85–7.76 (m, 3H, aromatic), 7.55–7.23 (m, 10H, aromatic), 5.93 (dd, J3,4 = 9.5 Hz, 1H, H-3), 5.66 (dd, J4,5 = 9.8 Hz, 1H, H-4), 5.58 (dd, J2,3 = 9.7 Hz, 1H, H-2), 4.87 (dd, J1,2 = 10.0 Hz, 1H, H-1), 4.68 (dd, 1H, H-6a), 4.62 (dd, J6a,6b = 12.2 Hz, 1H, H-6b), 4.27 (dd, J5,6a = 3.4 Hz, J5,6b = 5.5 Hz, 1H, H-5), 2.86–2.65 (m, 2H, SCH2CH3), 1.23 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 165.9, 165.4 (×2), 164.7, 150.2, 147.6, 137.2, 133.7, 133.5 (×2), 130.1 (×2), 130.0 (×2), 129.9 (×2), 129.2, 128.9, 128.8, 128.6 (×4), 128.5 (×2), 127.2, 125.5, 84.1, 76.2, 74.2, 70.7, 69.9, 64.4, 24.6, 15.1 ppm; HRMS [M + Na]+ calcd for C35H31NO9SNa 664.1617 found 664.1626.
Ethyl 3,6-di-O-benzyl-2-deoxy-2-phthalimido-4-O-picoloyl-1-thio-β-d-glucopyranoside (13).
The title compound was prepared from ethyl 3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside43 (14, 1.20 g, 1.88 mmol) in CH2Cl2 (50 mL), picolinic acid (0.46 g, 3.76 mmol), EDC (0.58 g, 3.76 mmol), and DMAP (0.045 g, 0.37 mmol) in accordance with the general procedure as a white amorphous solid in 85% yield (0.86 g, 1.62 mmol). Analytical data for 13: Rf = 0.2 (ethyl acetate/hexane, 1/1, v/v); +59.4 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.77 (d, 1H, aromatic), 8.08 (d, 1H, aromatic), 7.88–7.76 (m, 2H, aromatic), 7.74–7.62 (m, 3H, aromatic), 7.53–7.45 (m, 1H, aromatic), 7.29–7.12 (m, 6H, aromatic), 6.93 (m, 2H, aromatic), 6.85–6.74 (m, 3H, aromatic), 5.52 (dd, 1H, H-4), 5.35 (d, J1,2 = 10.6 Hz, 1H, H-1), 4.74 (dd, 1H, H-3), 4.57 (d, 2J = 12.1 Hz, 1H, CHPh), 4.51 (s, 2H, CH2Ph), 4.45–4.33 (m, 2H, H-2, CHPh), 4.06 (m, J5,6a = J5,6b = 4.4 Hz, 1H, H-5), 3.71 (dd, 2H, H-6a, 6b), 2.68 (m, 2H, SCH2CH3), 1.19 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 168.1, 167.5, 164.3, 150.1, 147.6, 138.0, 137.7, 137.2, 134.1, 134.0, 131.7, 128.3 (×2), 128.1 (×4), 127.8 (×2), 127.6, 127.5, 127.3, 125.8, 123.7, 123.5, 81.3, 77.9, 77.6, 74.4, 74.1, 73.6, 69.8, 54.9, 24.2, 15.1 ppm; HRMS [M + Na]+ calcd for C36H34N2O7SNa 661.1984 found 661.1993.
Ethyl 2,3-di-O-benzyl-4-O-p-methoxybenzyl-6-O-picoloyl-1-thio-β-d-glucopyranoside (19).
NaH (60% in mineral oil, 703.4 mg 17.60 mmol) was added portionwise to a solution of ethyl 4,6-O-p-methoxybenzylidene-1-thio-β-d-glucopyranoside44 (31, 3.0 g, 8.79 mmol) in dimethylformamide (25 mL), and the resulting mixture was cooled to 0 °C. Benzyl bromide (4.5 g, 26.37 mmol) was added dropwise, a second batch of NaH (60% in mineral oil, 703.4 mg, 17.60 mmol) was then added portionwise, and the resulting mixture was stirred under argon for 5 h. After that, the reaction mixture was poured into ice water (50 mL) and stirred for 30 min. The aqueous phase was extracted with ethyl acetate/diethyl ether (1/1, v/v, 3 × 75 mL). The combined organic extract (~225 mL) was washed with cold water (3 × 30 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane gradient elution) to give ethyl 2,3-di-O-benzyl-4,6-O-p-methoxybenzylidene-1-thio-β-d-glucopyranoside (32, 4.42 g, 8.46 mmol) in 96% yield. Selected analytical data for 32: Rf = 0.65 (ethyl acetate/hexane, 2/3, v/v); 1H NMR (300 MHz, CDCl3): δ 7.47–7.18 (m, 12H, aromatic), 6.95–6.85 (m, 2H, aromatic), 5.54 (s, 1H, CHPh), 4.85 (dd, 2J = 11.3 Hz, 2H, CH2Ph), 4.84 (dd, 2J = 10.2 Hz, 2H, CH2Ph), 4.56 (d, J1,2 = 9.8 Hz, 1H, H-1), 4.33 (dd, J3,4 = 10.4, 1H, H-3), 3.81 (s, 3H, OCH3), 3.80–3.64 (m, 3H, H-4, 6a, 6b), 3.50–3.39 (m, 2H, H-2, 5), 2.76 (m, 2H, SCH2CH3), 1.32 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm.
A mixture containing compound 32 (4.42 g, 8.46 mmol), molecular sieves (4 Å, 3.0 g) in dimethylformamide (20 mL) was stirred under argon for 1 h at rt. The resulting mixture was cooled to 0 °C, sodium cyanoborohydride (2.66 g, 42.3 mmol) was added followed by slow dropwise addition of trifluoroacetic acid (9.65 g, 84.6 mmol), the reaction mixture was allowed to warm to ambient temperature and stirred for 16 h at rt. After that, the solids were filtered off through a pad of Celite and rinsed successively with DCM. The combined filtrate (~150 mL) was washed with sat. aq. NaHCO3 (3 × 40 mL). The layers were separated, and the aqueous phase was extracted with dichloromethane (2 × 150 mL). The combined organic phase was dried with magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane gradient elution) to give ethyl 2,3-di-O-benzyl-4-O-p-methoxybenzyl-1-thio-β-d-glucopyranoside 20 in 79% yield (3.51 g, 6.68 mmol). Analytical data for 20: Rf = 0.45 (ethyl acetate/hexane, 3/2, v/v); −32.5 (c = 1.86, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.50–7.19 (m, 12H, aromatic), 6.87 (d, 2H, aromatic), 4.85 (dd, 2J = 9.4 Hz, 2H, CH2Ph), 4.83 (dd, 2J = 10.2 Hz, 2H, CH2Ph), 4.50 (s, 2H, CH2Ph), 4.48 (d, J1,2 = 9.7 Hz, 1H, H-1), 3.80 (s, 3H, OCH3), 3.71 (d, 1H, H-6a), 3.70 (d, 1H, H-6b), 3.62 (dd, J4,5 = 9.1 Hz, 1H, H-4), 3.51 (dd, J3,4 = 8.6 Hz, 1H, H-3), 3.44 (dd, J5,6a = J5,6b = 4.8 Hz, 1H, H-5), 3.40 (dd, J2,3 = 9.0 Hz, 1H, H-2), 2.87–2.62 (m, 2H, SCH2CH3), 1.32 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 159.5, 138.7, 138.1, 130.0, 129.6, 128.8, 128.6 (×2), 128.2, 128.1 (×2), 114.0, 86.2, 85.3, 81.4, 77.9, 75.7, 75.6, 73.5, 72.5, 70.6, 55.5, 25.3, 15.4 ppm; HRMS [M + Na]+ calcd for C30H36O6SNa 547.2125 found 547.2131.
Compound 19 was prepared from 20 (1.48 g, 2.83 mmol) in CH2Cl2 (50 mL), picolinic acid (0.701 g, 5.65 mmol), EDC (1.08 g, 5.65 mmol), and DMAP (0.069 g, 0.57 mmol) in accordance with the general procedure as a white amorphous solid in 97% yield (1.72 g, 2.74 mmol). Analytical data for 19: Rf = 0.55 (acetone/hexane, 1/1, v/v); −27.6 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.78–8.69 (m, 1H, aromatic), 7.98 (m, 1H, aromatic), 7.78 (m, 1H, aromatic), 7.46 (m, 1H, aromatic), 7.41–7.24 (m, 5H, aromatic), 7.18–7.02 (m, 7H, aromatic), 6.77–6.66 (m, 2H, aromatic), 5.37 (dd, J4,5 = 9.7 Hz, 1H, H-4), 4.84 (dd, 2J = 10.2 Hz, 2H, CH2Ph), 4.73 (dd, 2J = 11.2 Hz, 2H, CH2Ph), 4.55 (d, J1,2 = 9.8 Hz, 1H, H-1), 4.39 (dd, 2H, H-6a, 6b), 3.90 (dd, J3,4 = 9.1 Hz, 1H, H-3), 3.82 (dd, 1H, H-5), 3.73 (s, 3H, OCH3), 3.60 (dd, 2J = 4.4 Hz, 2H, CH2Ph), 3.56 (dd, J2,3 = 8.9 Hz, 1H, H-2), 2.89–2.69 (m, 2H, SCH2CH3), 1.34 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 164.3, 159.1, 145.0, 147.7, 138.1, 138.0, 137.1, 130.1, 129.5 (×2), 128.6 (×4), 128.3 (×2), 128.1 (×3), 127.7, 127.1, 125.7, 113.7, 85.3, 83.8, 81.8, 77.4, 75.8, 75.6, 73.3, 72.5, 69.5, 55.3 (×2), 25.2, 15.4 ppm; HRMS [M + Na]+ calcd for C36H39NO7SNa 652.2345 found 652.2347.
Ethyl 2,3-di-O-benzyl-4,6-di-O-picoloyl-1-thio-β-d-glucopyranoside (21).
The title compound was prepared from ethyl 2,3-di-O-benzyl-1-thio-β-d-glucopyranoside45 (22, 237.3 mg, 0.59 mmol) in CH2Cl2 (10 mL), picolinic acid (364.0 mg, 2.90 mmol), EDC (555.9 mg, 2.90 mmol), and DMAP (36.1 mg, 0.30 mmol) in accordance with the general procedure as a white amorphous solid in 86% yield (311.1 mg, 0.51 mmol). Analytical data for 21: Rf = 0.30 (acetone/hexane, 1/3, v/v); −6.40 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.77–8.64 (m, 2H, aromatic), 8.15–8.06 (m, 1H, aromatic), 8.06–7.98 (m, 1H, aromatic), 7.86–7.74 (m, 2H, aromatic), 7.52–7.29 (m, 7H, aromatic), 7.08 (s, 5H, aromatic), 5.52 (dd, J4,5 = 9.8 Hz, 1H, H-4), 4.85 (dd, 2J = 10.2 Hz, 2H, CH2Ph), 4.76 (dd, 2J = 11.2 Hz, 2H, CH2Ph),4.61 (d, J1,2 = 9.8 Hz, 1H, H-1), 4.56 (d, 2H, H-6a, 6b), 4.06 (dd, J5,6a = 4.3 Hz, J5,6b = 4.4 Hz, 1H, H-5), 3.97 (dd, J3,4 = 9.1 Hz, 1H, H-3), 3.61 (dd, J2,3 = 8.9 Hz, 1H, H-2), 2.89–2.65 (m, 2H, SCH2CH3), 1.29 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 164.6, 164.3, 150.1, 150.0, 147.7, 147.4, 137.9, 137.8, 137.2, 137.1, 128.6 (×3), 128.4 (×2), 128.2 (×2), 128.1 (×2), 127.8, 127.3, 127.1, 125.9, 125.6, 85.4, 83.7, 81.7, 75.9, 75.8, 75.5, 71.9, 64.3, 25.2, 15.3 ppm; HRMS [M + Na]+ calcd for C34H34N2O7SNa 637.1984 found 637.1988.
Ethyl 2-O-benzyl-3,4,6-tri-O-picoloyl-1-thio-α-d-mannopyranoside (23).
p-Toluenesulfonic acid (3.1 mg, 0.016 mmol) and ethanethiol (12.3 mg, 0.198 mmol) were added to a solution of ethyl 2-O-benzyl-4,6-O-benzylidene-3-picoloyl-1-thio-α-d-mannopyranoside18 (33, 16.6 mg, 0.033 mmol) in DCM (0.5 mL), and the resulting solution was stirred under argon for 2 h at rt. The reaction mixture was then neutralized with triethylamine, the volatiles were removed under reduced pressure, and the residue containing ethyl 2-O-benzyl-1-thio-α-d-mannopyranoside (34) was dried in vacuo for 2 h. The title compound was then obtained from crude 34 (0.033 mmol) in dichloromethane (0.5 mL), picolinic acid (24.6 mg, 0.198 mmol), EDC (38.0 mg, 0.198 mmol), and DMAP (0.81 mg, 0.007 mmol) in accordance with the general procedure as a white amorphous solid in 86% yield (17.7 mg, 0.028 mmol). Analytical data for 23: Rf = 0.4 (acetone/toluene, 1/1, v/v); +52.5 (c = 2.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.70 (d, 3H, aromatic), 8.17–7.95 (m, 3H, aromatic), 7.84–7.66 (m, 3H, aromatic), 7.51–7.38 (m, 3H, aromatic), 7.38–7.25 (m, 2H, aromatic), 7.25–7.10 (m, 3H, aromatic), 6.17 (dd, J4,5 = 10.0 Hz, 1H, H-4), 5.70 (dd, J3,4 = 10.0 Hz, 1H, H-3), 5.51 (dd, 1H, H-1), 4.87 (dd, 1H, H-5), 4.68 (dd, 2J = 12.0 Hz, 2H, CH2Ph), 4.67–4.64 (m, 2H, H-6a, 6b), 4.27 (dd, J2,3 = 1.5 Hz, 1H, H-2), 2.84–2.47 (m, 2H, SCH2CH3), 1.29 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 164.5, 164.0, 163.7, 150.2, 150.1 (×2), 147.6, 147.1, 147.0, 137.5, 137.2 (×2), 137.1, 128.4 (×2), 127.9 (×3), 127.3, 127.1, 126.9, 125.7, 125.6, 125.4, 82.0, 77.1, 73.1, 72.8, 68.8, 68.3, 64.0, 25.4, 14.9 ppm; HRMS [M + Na]+ calcd for C33H31N3O8SNa 652.1724 found 652.1738.
Selective deprotection of the Pico group
General procedure for Pico removal.
Iron(iii) chloride (0.017 mmol) or copper(ii) acetate (0.017 mmol) was added to a solution of a Pico derivative (0.051 mmol) in MeOH–DCM (1.0 mL, 1.0/1, v/v), and the resulting mixture was stirred under argon at rt. Upon completion (see the reaction time listed in Scheme 1), the volatiles were removed under reduced pressure. The residue was diluted with DCM (~5 mL) and washed with sat. aq. NaHCO3 (5 mL) and water (2 × 5 mL). The organic phase was separated, dried using magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate – hexane gradient elution) to give the corresponding deprotected derivative in yields listed in Scheme 1.
Ethyl 2,4,6-tri-O-benzyl-1-thio-β-d-glucopyranoside (2).
The title compound was obtained from ethyl 2,3,6-tri-O-benzyl-4-O-picoloyl-1-thio-β-d-glucopyranoside16 (1, 29.1 mg, 0.052 mmol) and iron(iii) trichloride (2.5 mg, 0.016 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 5 h in 91% yield (23.3 mg, 0.047 mmol). Alternatively, the title compound was obtained from thioglycoside 1 (29.6 mg, 0.049 mmol) and copper(ii) acetate (3.0 mg, 0.015 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 1.5 h in 99% yield (24.8 mg, 0.048 mmol). Analytical data for 2 was in accordance with that reported previously.46
Ethyl 3,4,6-tri-O-benzyl-1-thio-β-d-glucopyranoside (4).
The title compound was obtained from precursor 3 (29.0 mg, 0.048 mmol) and iron(iii) trichloride (2.4 mg, 0.015 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 48 h in 71% yield (17.0 mg, 0.034 mmol). Alternatively, the title compound was obtained from thioglycoside 3 (33.1 mg, 0.055 mmol) and copper(ii) acetate (3.3 mg, 0.017 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 2 h in 91% yield (15.2 mg, 0.03 mmol). Analytical data for 4 was in accordance with that reported previously.41
Ethyl 2,4,6-tri-O-benzyl-1-thio-β-d-glucopyranoside (6).
The title compound was obtained from ethyl 2,4,6-tri-O-benzyl-3-O-picoloyl-1-thio-β-d-glucopyranoside24,25 (5, 9.5 mg, 0.017 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 91% yield (8.1 mg, 0.016 mmol). Analytical data for 6 was in accordance with that reported previously.47
Ethyl 2,3,4-tri-O-benzyl-1-thio-β-d-glucopyranoside (8).
The title compound was obtained from 2,3,4-tri-O-benzyl-6-O-picoloyl-1-thio-β-d-glucopyranoside16 (7, 35.4 mg, 0.055 mmol) and iron(iii) trichloride (2.7 mg, 0.016 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 20 min in 98% yield (28.8 mg, 0.054 mmol). Alternatively, the title compound was obtained from thioglycoside 7 (18.1 mg, 0.03 mmol) and copper(ii) acetate (1.8 mg, 0.009 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 10 min in 99% yield (15.2 mg, 0.03 mmol). Analytical data for 8 was in accordance with that reported previously.48
Ethyl 2,3,4-tri-O-benzoyl-1-thio-β-d-glucopyranoside (10).
The title compound was obtained from substrate 9 (31.9 mg, 0.050 mmol) and iron(iii) trichloride (2.4 mg, 0.015 mmol) in methanol (0.9 mL) and dichloromethane (0.10 mL) in accordance with the general procedure as a colorless syrup in 15 min in 96% yield (25.7 mg, 0.0048 mmol). Alternatively, the title compound was obtained from thioglycoside 9 (32.3 mg, 0.050 mmol) and copper(ii) acetate (3.0 mg, 0.015 mmol) in methanol (0.9 mL) and dichloromethane (0.10 mL) in accordance with the general procedure as a colorless syrup in 10 min in 93% yield (25.1 mg, 0.047 mmol). Analytical data for 10 was essentially the same as reported previously.42
Ethyl 2-O-benzyl-4,6-O-benzylidene-1-thio-α-d-mannopyranoside (12).
The title compound was obtained from ethyl 2-O-benzyl-4,6-O-benzylidene-3-O-picoloyl-1-thio-α-d-mannopyranoside18 (11, 31.7 mg, 0.062 mmol) and iron(iii) trichloride (3.0 mg, 0.019 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 30 min in 88% yield (22.2 mg, 0.055 mmol). Alternatively, the title compound was obtained from 11 (14.6 mg, 0.029 mmol) and copper(ii) acetate (1.8 mg, 0.0087 mmol) in methanol (0.45 mL) and dichloromethane (0.05 mL) in accordance with the general procedure as a colorless syrup in 10 min in 93% yield (25.1 mg, 0.047 mmol). Analytical data for 12 was essentially the same as reported previously.49
Ethyl 3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-d-glucopyranoside (14).
The title compound was obtained from compound 13 (29.9 mg, 0.047 mmol) and iron(iii) trichloride (2.3 mg, 0.014 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 5 h in 99% yield (25.2 mg, 0.047 mmol). Alternatively, the title compound was obtained from thioglycoside 13 (36.4 mg, 0.057 mmol) and copper(ii) acetate (1.8 mg, 0.017 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 1.5 h in 99% yield (30.0 mg, 0.056 mmol). Analytical data for 14 was essentially the same as reported previously.43
Methyl (phenyl 5-acetamido-7,8,9-tri-O-acetyl-3,5-dideoxy-2-thio-d-glycero-α-d-galacto-non-2-ulopyranosid)onate (16).
The title compound was obtained from methyl (phenyl 5-acetamido-7,8,9-tri-O-acetyl-3,5-dideoxy-4-O-picoloyl-2-thio-d-glycero-α-d-galacto-non-2-ulopyranosid)onate32 (15, 22.9 mg, 0.035 mmol) and iron(iii) trichloride (1.7 mg, 0.01 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 50 min in 95% yield (17.9 mg, 0.033 mmol). Alternatively, the title compound was obtained from thioglycoside 15 (26.8 mg, 0.041 mmol) and copper(ii) acetate (2.5 mg, 0.012 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 20 min in 73% yield (16.2 mg, 0.030 mmol). Analytical data for 16: Rf = 0.40 (methanol/dichloromethane, 1/9, v/v); +25.3 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.53–7.46 (m, 2H, aromatic), 7.43–7.29 (m, 3H, aromatic), 5.99 (d, J = 8.4 Hz, 1H, NH), 5.32 (dd, J7,8 = 7.3 Hz, 1H, H-7), 5.27 (dt, J8,9a = 5.0 Hz, J8,9b = 2.4 Hz 1H, H-8), 4.39 (dd, J9a,9b = 12.6 Hz, 1H, H-9a), 4.26 (dd, 1H, H-9b), 3.90 (dd, J6,7 = 10.4 Hz, 1H, H-6), 3.86 (s, 1H, 4-OH), 3.64 (dd, J4,5 = 10.6, 1H, H-4), 3.57 (s, 3H, OCH3), 3.50 (dd, J5,6 = 8.5 Hz, 1H, H-5), 2.89 (dd, J3eq,3ax = 13.0, J3eq,4 = 4.4 Hz, 1H, H-3eq), 2.15, 2.06, 2.03, 1.96 (4 s, 12H, NCOCH3, 3 × OCOCH3), 1.86 (dd, J3ax,4 = 11.4 Hz, 1H, H-3ax) ppm; 13C NMR (75 MHz, CDCl3): δ 172.4 (×2), 170.9 (×2), 170.5 (×2), 170.3 (×2), 168.3 (×2), 136.5 (×3), 130.0, 129.0 (×4), 88.0, 74.1, 70.1, 69.2, 68.1, 62.1, 52.8 (×3), 41.4, 29.5, 23.7, 21.2 (×2), 21.0 (×3) ppm; HRMS [M + Na]+ calcd for C24H31NO11SNa 564.1510 found 564.1521.
Ethyl 2-O-benzoyl-4-O-benzyl-3-O-(9-fluorenylmethoxycarbonyl)-1-thio-β-d-galactopyranoside (18).
The title compound was obtained from ethyl 2-O-benzoyl-4-O-benzyl-3-O-(9-fluorenylmethoxycarbonyl)-6-O-picoloyl-1-thio-β-d-galactopyranoside38 (17, 31.2 mg, 0.042 mmol) and iron(iii) trichloride (2.0 mg, 0.013 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 15 min in 91% yield (25.2 mg, 0.047 mmol). Analytical data for 18: Rf = 0.45 (ethyl acetate/hexane, 1/1, v/v); +25.7 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.05 (d, 2H, aromatic), 7.69 (dd, 2H, aromatic), 7.59–7.22 (m, 12H, aromatic), 7.19–7.04 (m, 2H, aromatic), 5.76 (dd, J2,3 = 10.0 Hz, 1H, H-2), 5.07 (dd, J3,4 = 2.8 Hz, 1H, H-3), 4.68 (dd, 2J = 11.6 Hz, 2H, CH2Ph), 4.60 (d, J1,2 = 9.9 Hz, 1H, H-1), 4.34 (dd, 1H, H-6a), 4.24 (dd, J6a,6b = 10.3 Hz, 1H, H-6b),4.08 (dd, J5,6a = 7.9 Hz, J5,6b = 7.1 Hz, 1H, H-5), 4.04 (dd, J4,5 = 5 Hz, 1H, H-4), 3.86 (dd, J = 11.1, 6.8 Hz, 1H, Fmoc), 3.67 (m, 1H, Fmoc), 3.56 (m, 1H, Fmoc), 2.88–2.60 (m, 2H, SCH2CH3), 1.23 (t, J = 7.3 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, CDCl3): δ 165.5, 154.7, 143.4, 142.9, 141.4, 141.3, 137.6, 133.5, 130.1 (×2), 129.6, 128.8 (×3), 128.7 (×3), 128.6 (×2), 128.4, 128.1 (×2), 127.3 (×2), 125.4, 125.1, 120.2, 84.0, 79.3, 79.1, 75.0, 73.5, 70.4, 62.0, 46.6, 24.1, 15.0 ppm; HRMS [M + Na]+ calcd for C37H36O8SNa 663.2029 found 663.2027
Ethyl 2,3-di-O-benzyl-4-O-p-methoxybenyl-1-thio-β-d-glucopyranoside (20).
The title compound was obtained from compound 19 (32.8 mg, 0.052 mmol) and iron(iii) trichloride (2.5 mg, 0.015 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 16 h in 99% yield (28.5 mg, 0.054 mmol). Alternatively, the title compound was obtained from 19 (29.4 mg, 0.047 mmol) and copper(ii) acetate (2.8 mg, 0.014 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 15 min in 85% yield (20.8 mg, 0.040 mmol).
Ethyl 2,3-di-O-benzyl-1-thio-β-d-glucopyranoside (22).
The title compound was obtained from 21 (57.0 mg, 0.093 mmol) and iron(iii) trichloride (4.5 mg, 0.028 mmol) in methanol (1.8 mL) and dichloromethane (0.2 mL) in accordance with the general procedure as a colorless syrup in 24 h in 99% yield (37.2 mg, 0.092 mmol). Alternatively, the title compound was obtained from thioglycoside 21 (30.4 mg, 0.049 mmol) and copper(ii) acetate (3.0 mg, 0.015 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 10 min in 99% yield (20.3 mg, 0.048 mmol). Analytical data for 22 was essentially the same as reported previously.45
Ethyl 2-O-benzyl-1-thio-α-d-mannopyranoside (24).
The title compound was obtained from 23 (36.5 mg, 0.058 mmol) and iron(iii) trichloride (2.8 mg, 0.017 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 45 min in 91% yield (16.6 mg, 0.052 mmol). Alternatively, the title compound was obtained from 23 (37.3 mg, 0.059 mmol) and copper(ii) acetate (3.6 mg, 0.018 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 15 min in 83% yield (15.5 mg, 0.049 mmol). Analytical data for 24: Rf = 0.50 (acetone/toluene, 1/1, v/v); +103.1 (c = 1.0, CHCl3); 1H NMR (300 MHz, MeOD): δ 7.49–7.22 (m, 5H, aromatic), 5.32 (s, 1H, H-1), 4.68 (dd, 2J = 11.9, 2H, CH2Ph), 3.97–3.60 (m, 6H, H-2, 3, 4, 5, 6a, 6b), 2.76–2.48 (m, 2H, SCH2CH3), 1.23 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C NMR (75 MHz, MeOD): δ 139.7 (×2), 129.5 (×2), 129.3, 128.9, 83.1, 81.5, 75.1, 73.8, 73.3, 69.4, 62.9, 26.0, 15.4 ppm; HRMS [M + Na]+ calcd for C15H22O5SNa 337.1080 found 337.1120.
tert-Butyldimethylsilyl O-(2-O-benzyl-4,6-O-benzylidene-β-d-mannopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside (26).
The title compound was obtained from tert-butyldimethylsilyl O-(2-O-benzyl-4,6-O-benzylidene-3-O-picoloyl-β-d-mannopyranosyl)-(1 → 4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranoside23 (25, 29.0 mg, 0.028 mmol) and iron(iii) trichloride (1.3 mg, 0.083 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 25 min in 91% yield (25.5 mg, 0.027 mmol). Analytical data for 26 was essentially the same as reported previously.23
Benzyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-d-galactopyranosyl)-(1 → 3)-O-(4,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 3)-O-(2-O-benzoyl-4-O-benzyl-β-d-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside (28).
The title compound was obtained from benzyl O-(2-O-benzoyl-3,4,6-tri-O-benzyl-β-d-galactopyranosyl)-(1 → 3)-O-(4,6-di-O-benzyl-2-deoxy-2-phthalimido-β-d-glucopyranosyl)-(1 → 3)-O-(2-O-benzoyl-4-O-benzyl-6-O-picoloyl-β-d-galactopyranosyl)-(1 → 4)-2,3,6-tri-O-benzyl-β-d-glucopyranoside39 (27, 9.9 mg, 0.0049 mmol) and iron(iii) trichloride (2.3 mg, 0.0015 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL) in accordance with the general procedure as a colorless syrup in 45 min in 99% yield (9.3 mg, 0.0049 mmol). Analytical data for 28 was essentially the same as reported previously.39
Attempted deprotection of Pico regioisomers
Iron(iii) chloride (1.3 mg, 0.0008 mmol) was added to a solution of ethyl 2,4,6-tri-O-benzyl-3-O-nicotinoyl-1-thio-β-d-glucopyranoside24,25 (29, 14.8 mg, 0.026 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL), and the resulting mixture was stirred under argon at rt. No reaction took place after 24 h.
Iron(iii) chloride (1.5 mg, 0.0009 mmol) was added to a solution of ethyl 2,4,6-tri-O-benzyl-3-O-iso-nicotinoyl-1-thio-β-d-glucopyranoside24,25 (30, 17.3 mg, 0.031 mmol) in methanol (0.9 mL) and dichloromethane (0.1 mL), and the resulting mixture was stirred under argon at rt. No reaction took place after 24 h.
Supplementary Material
Acknowledgements
This work was supported by awards from the NSF (CHE-1058112) and NIGMS (GM111835). We thank Dr Rensheng Luo (UM - St Louis) for aquiring spectral data using 600 MHz NMR spectrometer that was purchased thanks to the NSF (award CHE-0959360).
Footnotes
Dedicated to the memory of Bert Fraser-Reid (1934–2020) who was among the first to recognize the effect of protecting groups in carbohydrate chemistry “Protecting groups do more than protect”.
Electronic supplementary information (ESI) available: Spectral data for all new and selected known compounds. See DOI: 10.1039/d0ob00803f
Conflicts of interest
The authors declare no competing financial interest.
References
- 1.Jager M and Minnaard AJ, Chem. Commun, 2016, 52, 656–664. [DOI] [PubMed] [Google Scholar]
- 2.Kulkarni SS, Wang CC, Sabbavarapu NM, Podilapu AR, Liao PH and Hung SC, Chem. Rev, 2018, 118, 8025–8104. [DOI] [PubMed] [Google Scholar]
- 3.Volbeda AG, van der Marel GA and Codée JDC, in Protecting Groups – Strategies and Applications in Carbohydrate Chemistry, ed. Vidal S, Wiley-VCH, Weinheim, 2019, pp. 1–28. [Google Scholar]
- 4.Wang T and Demchenko AV, Org. Biomol. Chem, 2019, 17, 4934–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fraser-Reid B, Jayaprakash KN, López JC, Gómez AM and Uriel C, in ACS Symp. Ser. (Frontiers in Modern Carbohydrate Chemistry), ed. Demchenko AV, Oxford Univ. Press, 2007, vol. 960, pp. 91–117. [Google Scholar]
- 6.Bandara MD, Yasomanee JP and Demchenko AV, in Selective Glycosylations: Synthetic Methods and Catalysts, ed. Bennett CS, Wiley, 2017, pp. 29–58. [Google Scholar]
- 7.Prabhu A, Venot A and Boons GJ, Org. Lett, 2003, 5, 4975–4978. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC and Boons GJ, Science, 2013, 341, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Markad SD and Schmidt RR, Eur. J. Org. Chem, 2009, 5002–5011. [Google Scholar]
- 10.Seeberger PH, Acc. Chem. Res, 2015, 48, 1450–1463. [DOI] [PubMed] [Google Scholar]
- 11.Senf D, Ruprecht C, de Kruijff GHM, Simonetti SO, Schuhmacher F, Seeberger PH and Pfrengle F, Chem. - Eur. J, 2017, 23, 3197–3205. [DOI] [PubMed] [Google Scholar]
- 12.Guberman M and Seeberger PH, J. Am. Chem. Soc, 2019, 141, 5581–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfrengle F and Seeberger PH, in Protecting Groups – Strategies and Applications in Carbohydrate Chemistry, ed. Vidal S, Wiley-VCH, Weinheim, 2019, pp. 451–472. [Google Scholar]
- 14.Ágoston K, Streicher H and Fügedi P, Tetrahedron: Asymmetry, 2016, 27, 707–728. [Google Scholar]
- 15.Panza M, Pistorio SG, Stine KJ and Demchenko AV, Chem. Rev, 2018, 118, 8105–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasomanee JP and Demchenko AV, J. Am. Chem. Soc, 2012, 134, 20097–20102. [DOI] [PubMed] [Google Scholar]
- 17.Yasomanee JP and Demchenko AV, Angew. Chem., Int. Ed, 2014, 53, 10453–10456. [DOI] [PubMed] [Google Scholar]
- 18.Pistorio SG, Yasomanee JP and Demchenko AV, Org. Lett, 2014, 16, 716–719. [DOI] [PubMed] [Google Scholar]
- 19.Yasomanee JP and Demchenko AV, Chem. – Eur. J, 2015, 21, 6572–6581. [DOI] [PubMed] [Google Scholar]
- 20.Kayastha AK, Jia XG, Yasomanee JP and Demchenko AV, Org. Lett, 2015, 17, 4448–4451. [DOI] [PubMed] [Google Scholar]
- 21.Mannino MP, Yasomanee JP, Demchenko AV and Patteti V, in Carbohydrate Chemistry: Proven Synthetic Methods, ed. Murphy P and Vogel P, CRC Press, Boca Raton - London - New York, 2017, vol. 4, pp. 3–10. [Google Scholar]
- 22.Mannino MP, Yasomanee JP and Demchenko AV, Carbohydr. Res, 2018, 470, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pistorio SG, Geringer SA, Stine KJ and Demchenko AV, J. Org. Chem, 2019, 84, 6576–6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mannino MP and Demchenko AV, Chem. – Eur. J, 2020, 26, 2927–2937. [DOI] [PubMed] [Google Scholar]
- 25.Mannino MP and Demchenko AV, Chem. – Eur. J, 2020, 26, 2938–2946. [DOI] [PubMed] [Google Scholar]
- 26.Prasad V, Birzin ET, McVaugh CT, van Rijn RD, Rohrer SP, Chicchi G, Underwood DJ, Thornton ER, Smith III AB and Hirschmann R, J. Med. Chem, 2003, 46, 1858–1869. [DOI] [PubMed] [Google Scholar]
- 27.Baek JY, Shin Y-J, Jeon HB and Kim KS, Tetrahedron Lett, 2005, 46, 5143–5147. [Google Scholar]
- 28.Ruei J-H, Venukumar P, Ingle AB and Mong K-KT, Chem. Commun, 2015, 51, 5394–5397. [DOI] [PubMed] [Google Scholar]
- 29.Xiang S, Hoang KLM, He J, Tan YJ and Liu XW, Angew. Chem., Int. Ed, 2015, 54, 604–607. [DOI] [PubMed] [Google Scholar]
- 30.Lu YL, Ghosh B and Mong KT, Chem. – Eur. J, 2017, 23, 6905–6918. [DOI] [PubMed] [Google Scholar]
- 31.Wu YF and Tsai YF, Org. Lett, 2017, 19, 4171–4174. [DOI] [PubMed] [Google Scholar]
- 32.Escopy S, Geringer SA and De Meo C, Org. Lett, 2017, 19, 2638–2641. [DOI] [PubMed] [Google Scholar]
- 33.Shadrick M, Yu C, Geringer S, Ritter S, Behm A, Cox A, Lohman M and De Meo C, New J. Chem, 2018, 42, 14138–14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones B, Behm A, Shadrick M, Geringer SA, Escopy S, Lohman M and De Meo C, J. Org. Chem, 2019, 84, 15052–15062. [DOI] [PubMed] [Google Scholar]
- 35.Dubey A, Tiwari A and Mandal PK, Carbohydr. Res, 2019, 487, 107887. [DOI] [PubMed] [Google Scholar]
- 36.Shit P, Gucchait A and Misra AK, Tetrahedron, 2019, 75, 130697. [Google Scholar]
- 37.Zemplén G and Pacsu E, Ber. Dtsch. Chem. Ges, 1929, 62, 1613–1614. [Google Scholar]
- 38.Bandara MD, Stine KJ and Demchenko AV, Carbohydr. Res, 2019, 483, 107743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bandara MD, Stine KJ and Demchenko AV, J. Org. Chem, 2019, 84, 16192–16198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bandara MD, Stine KJ and Demchenko AV, Carbohydr. Res, 2019, 486, 107824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thijssen MJL, Halkes KM, Kamerling JP and Vliegenthart JFG, Bioorg. Med. Chem, 1994, 2, 1309–1317. [DOI] [PubMed] [Google Scholar]
- 42.Veeneman GH and van Boom JH, Tetrahedron Lett, 1990, 31, 275–278. [Google Scholar]
- 43.Nagorny P, Fasching B, Li X, Chen G, Aussedat B and Danishefsky SJ, J. Am. Chem. Soc, 2009, 131, 5792–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mende M, Nieger M and Brase S, Chem. – Eur. J, 2017, 23, 12283–12296. [DOI] [PubMed] [Google Scholar]
- 45.Garegg PJ, Kvarnstrom I, Niklasson A, Niklasson G and Svensson SCT, J. Carbohydr. Chem, 1993, 12, 933–953. [Google Scholar]
- 46.Koto S, Uchida T and Zen S, Bull. Chem. Soc. Jpn, 1973, 46, 2520–2523. [Google Scholar]
- 47.Thijssen MJL, Bijkerk MHG, Kamerling JP and Vliegenthart JFG, Carbohydr. Res, 1998, 306, 111–125. [DOI] [PubMed] [Google Scholar]
- 48.Ray AK and Roy N, Carbohydr. Res, 1990, 196, 95–100. [DOI] [PubMed] [Google Scholar]
- 49.Cherif S, Clavel J-M and Monneret C, J. Carbohydr. Chem, 2002, 21, 123–130. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.