

Abstract
The Hippo pathway is an evolutionarily conserved kinase cascade involved in cell growth, apoptosis, development and migration. It is also crucial for stem cell self‐renewal and the maintenance of genomic stability. In addition, this pathway has the unique capacities to sense aspects of tissue architecture, such as cell polarity and mechanical tensions imposed by the surrounding microenvironment, and to control organ size and shape. All of these properties are frequently altered in tumor cells. In this review, we summarize how dysregulation of mammalian Hippo signaling is implicated in cancer.
Overview of the Hippo pathway
Hippo pathway core components
Most components of the Hippo signaling pathway were originally identified in Drosophila by screening for mutations resulting in organomegaly. The Hippo pathway is highly conserved in mammals (Fig. 1, Table 1), and serves to shut down cell proliferation under conditions of high cell density or other stress. Core components include the Mammalian sterile 20‐like kinases (MSTs), Large tumor suppressor kinases (LATSs), and the adaptor proteins Salvador homolog 1 (SAV1; also called WW45) and Mps One Binder kinase activator proteins (MOBs). Under conditions of low cell density, Hippo signaling is inactivated and the mammalian transcriptional activator Yes‐associated protein (YAP) (or its paralog Transcriptional coactivator with PDZ‐binding motif [TAZ]) is activated and free to translocate into the nucleus. Nuclear YAP activates or suppresses transcription factors that regulate target genes involved in cell proliferation, tissue growth, or control of organ size and shape.1, 2 These transcription factors include: TEAD1‐4 (important for growth promotion and the epithelial–mesenchymal transition [EMT]); SMADs (TGF‐β signaling); RUNXs (blood and bone formation); p63/p73 (apoptosis); PAX3 (neural crest formation); PPARγ (adipogenesis); TTF1 (thyroid and lung morphogenesis); and TBX‐5 (WNT/β‐catenin signaling and cardiac and limb development).3, 4 Under conditions of high cell density (or stress stimuli or mechanical compression), MST and LATS are activated. Activated LATS phosphorylates YAP and TAZ, promoting their cytoplasmic retention through binding to 14‐3‐3 protein. This binding prevents activation of YAP/TAZ target transcription factors and facilitates YAP/TAZ proteasomal degradation by the E3 ligase complex SCFβ‐TRCP (Fig. 2).5
Figure 1.
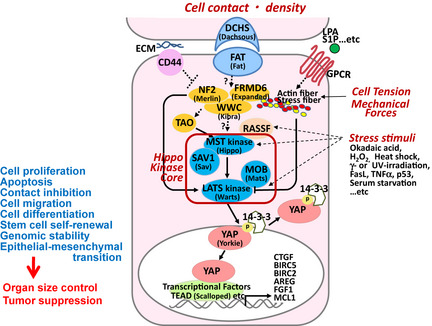
Overview of the mammalian Hippo signaling pathway. Core components of the mammalian Hippo pathway are: MST and LATS kinases, and SAV1 and MOB adaptor proteins. Orthologous Drosophila components are indicated in parentheses. The Hippo pathway is activated in response to increased cell density, tension or stress, and inhibits cell proliferation, and other processes potentially contributing to tumorigenesis. Candidate sensory components upstream of the Hippo kinase core include FAT4, DCHS1/2, NF2, FRMD6 and WWC1. TAO‐1 kinase and RASSF regulate MST phosphorylation. When activation of the Hippo core is triggered, SAV1 allows MST to phosphorylate LATS, and MOB binds to LATS to enhance its catalytic activity. LATS then phosphorylates and inactivates YAP (or its paralog TAZ), promoting their cytoplasmic retention through binding to 14‐3‐3 protein. YAP (or TAZ) is ultimately degraded so that transcription factors (such as TEAD) promoting cell survival are not activated. In contrast, under conditions of low cell density or minimal stress, YAP dissociates from 14‐3‐3, translocates into the nucleus, and activates transcription factors that induce the expression of pro‐survival genes such as CTGF. Solid lines, known direct interactions; dashed lines, unknown mechanisms.
Table 1.
Names of Hippo component molecules in Drosophila and in Human
| Drosophila | Human |
|---|---|
| Dachsous | DCHS1,2 |
| Fat | FAT1‐4 |
| Expanded | FRMD6 (WILLIN) |
| Kibra | WWC1‐3 |
| Merlin | NF2 |
| Rassf | RASSF1‐10 |
| Hippo | MST1 (STK4), MST2 (STK3), MST3, MST4 |
| Salvador | SAV1 (WW45) |
| Warts | LATS1,2 |
| Tricornered | NDR1 (STK38),NDR2(STK38L) |
| Mats | MOB1A (MOBKL1B), MOB1B (MOBKL1A), MOB2, 3A‐C, 4 |
| Yorkie | YAP1,TAZ (WWTR1) |
| Scalloped | TEAD1‐4 |
Figure 2.
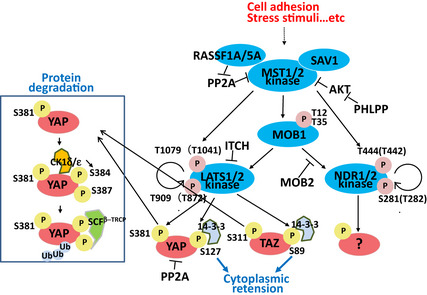
Mechanisms of YAP inactivation. Activation of the Hippo kinase core as described in Figure 1 leads to the indicated phosphorylation events on YAP/TAZ, and their cytoplasmic retention via 14‐3‐3 binding. Alternatively, phosphorylated YAP/TAZ can bind to the E3 ligase complex SCF β‐TRCP, which facilitates YAP/TAZ ubiquitination and proteasomal degradation. RASSF1A activates MST1 by preventing PP2A‐mediated dephosphorylation of MST1/2 and YAP. NDR1/2 are alternative MST substrates.
Hippo pathway upstream components
Candidate upstream sensors activating the mammalian Hippo pathway are FAT tumor suppressor homolog4 (FAT4), Dachsous1/2 (DCHS1/2), Neurofibromin2 (NF2), FERM domain‐containing 6 (FRMD6), and WW and C2 domain‐containing 1 (WWC1; also called KIBRA) (Fig. 1 , Table 1). Mice mutated in FAT4 and/or DCHS1, which regulate apical membrane organization, show unexpectedly small intestines, lungs and kidneys. However, the absence of organomegaly and the presence of normal LATS1 and YAP phosphorylation in these mutants suggest that mammalian Hippo signaling is not regulated by FAT4 or DCHS1, unlike in Drosophila.
CD44 (absent in flies) inactivates NF2 upon cell–cell contact to control Hippo signaling (Fig. 1). NF2 acts as a potent tumor suppressor gene (TSG) by controlling LATS1/2 and YAP.6 Several G protein‐coupled receptor (GPCR) agonists, such as LPA, S1P, and thrombin (Gi‐coupled GPCR agonists), as well as glucagon and epinephrine (Gs‐coupled GPCR agonists), also negatively or positively regulate LATS1/2.7 WWC1(KIBRA) binds to NF2 and FRMD6 to promote Hippo signaling independently of MSTs. While TAO‐1 kinase promotes MST phosphorylation, PP2A dephosphorylates MST1/2 and YAP (Fig. 2). RASSF1A activates MST1 by preventing PP2A‐mediated dephosphorylation.
Apicobasal cell polarity in mammalian epithelial cells is established by the Crumbs, Par, and Scribble complexes, which influence the Hippo pathway (Fig. 3). The Crumbs complex component AMOT co‐localizes with MST1/2, LATS1/2 and YAP in a complex at the tight junction to control cell growth. Zona occludens‐2 (ZO‐2) in the tight junction, and α‐catenin, β‐catenin, or PTPN14 in the adherence junction, also bind to YAP/TAZ8 (Fig. 3). Inhibition of any of cell–cell adhesion; Crumbs, Scribble, or Lgl complex formation; α‐catenin, ZO‐2, or PTPN14 expression; F‐actin polymerization, stress fiber formation, or Rho activity induced by mechanical forces, may decrease YAP phosphorylation in a manner dependent9 or independent10 of Hippo core kinases. The result is increased cell growth.8, 11 Nuclear translocation of YAP is also promoted when the cells are “stretched” or growing on a stiff extracellular matrix (ECM), and repressed when cells are compressed or growing on a soft surface. Importantly, loss of α‐catenin, which occurs in many human cancers, is linked to a poor prognosis. Thus, the Hippo pathway is tightly linked to tissue architecture and regulated by upstream cytoskeletal or tension signaling. For a detailed review of Hippo upstream components, please refer to Yu et al.12
Figure 3.
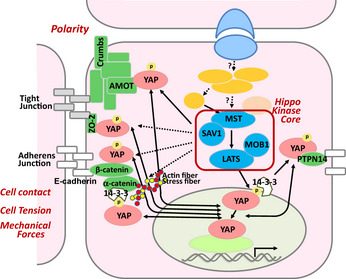
Regulation of YAP by upstream polarization or adhesion protein complexes. Polarization proteins sense cell contact and mechanical forces and transmit signals to the Hippo pathway to inhibit cell proliferation and control organ size and shape. The Crumbs complex component AMOT can co‐localize with MST1/2‐LATS1/2‐YAP complexes at the tight junction to promote cytoplasmic retention of YAP. ZO‐2 in the tight junction, and α‐catenin, β‐catenin, or PTPN14 in the adherence junction, can also bind to YAP and prevent nuclear translocation. YAP nuclear localization can also be reduced by F‐actin polymerization, stress fiber formation or Rho activity induced by mechanical forces.
Tumor‐Related Roles of Downstream Hippo Components
The loss of Hippo signaling is a key prognostic factor in predicting cancer patient survival and sensitivity to chemotherapeutic drugs. Cancer onset and/or development is generally associated with Hippo pathway inactivation or YAP activation, which is caused mainly by epigenetic changes (e.g. methylation of MST or LATS). In addition, genetic mutations (e.g. germline or somatic NF2 mutations) and YAP amplification (somatic) have also been reported. However, mutations in the Hippo pathway genes themselves are uncommon. For a detailed review of Hippo pathway alterations in various human cancers, please refer to Harvey et al.13
Tumor‐related roles of YAP
YAP can act as an oncogene or a TSG, depending on the cellular context and YAP's interacting partners. Conditional YAP activation in the liver reversibly increases liver size, and YAP dysregulation leads to hepatocellular carcinomas (HCCs). Conversely, YAP inactivation leads to loss of hepatocytes and bile duct cells.14, 15
In mice, Camargo et al.14 reported that conditional YAP activation in the intestine expanded multipotent progenitor cells and resulted in early onset of polyps following DSS treatment. However, Barry et al. found that transgenic YAP expression reduced WNT target gene expression and induced rapid destruction of intestinal crypts,16 while loss of YAP led to excessive WNT signaling causing hyperplasia, intestinal stem cell expansion, and formation of ectopic crypts and microadenomas. Barry et al. therefore speculated that cytoplasmic YAP may normally bind to DVL proteins and dampen WNT signaling. The discrepancy between these two reports requires resolution, since either increased nuclear YAP or decreased cytoplasmic YAP heightens the risk of colon cancer. In a third report, Rosenbluh et al.17 showed that YAP and the transcription factor TBX5 form a complex with β‐catenin. Phosphorylation of YAP(Thr357) by the tyrosine kinase YES1 induces localization of this complex to the promoters of the anti‐apoptotic genes BCL2L1 and BIRC5, inducing the transformation and survival of β‐catenin‐driven tumor cells. YAP is thus a key driver of β‐catenin‐related intestinal cancers.
Two transgenic mouse strains overexpressing YAP in the skin have been described.8, 18 Both strains initially show expanded basal epidermal progenitors that fail to undergo normal terminal differentiation, and later develop squamous cell carcinomas (SCCs). These mutants have a severe defect in hair follicle (HF) formation that may be due to impaired planar cell polarity caused by strong expression of YAP before HF morphogenesis.
Crosstalk between Hippo and other tumor‐related pathways
YAP/TAZ binds to phosphorylated R‐SMADs,19, 20 connecting the Hippo pathway to TGF‐β‐ and BMPR‐related signaling (Fig. 4). In general, nuclear YAP/TAZ enhances SMAD activity in the nucleus, while cytoplasmic YAP/TAZ restricts SMAD nuclear accumulation. TGF‐β‐SMAD activation induced by low cell density depends on nuclear YAP/TAZ, while YAP/TAZ‐induced stem cell self‐renewal depends on nuclear SMADs.19, 20 Thus, Hippo signaling may underlie the opposing roles of the TGF‐β pathway in early versus late stages of cancer. TGF‐β inhibits cell‐cycle progression during tumor initiation but subsequently promotes cancer cell proliferation, EMT, and metastasis. Normally‐polarized epithelial cells possess cytoplasmic YAP/TAZ, which restricts TGF‐β‐induced SMAD activity.19 In contrast, cells that have lost Crumbs complex function exhibit impaired cell polarity, elevated nuclear YAP/TAZ, increased nuclear SMADs, and enhanced sensitivity to TGF‐β ligand.19 EMT and the transformed phenotype are then promoted. Crosstalk between Hippo signaling and the TGF‐β‐SMADs pathway may thus explain the differential functions of TGF‐β as a tumor progresses.
Figure 4.
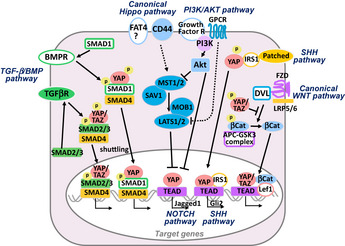
Crosstalk between the Hippo pathway and tumor‐related signaling pathways. The Hippo pathway is influenced by and interacts with components of several tumor‐related pathways, including the transforming growth factor‐β (TGF‐β)/BMP, WNT/β‐catenin, PI3K/Akt, SHH, and NOTCH pathways, as indicated. Please see main text for details.
The Hippo pathway also regulates WNT‐induced signaling.21, 22 YAP/TAZ is recruited to TCF/LEF binding sites together with β‐catenin. Heart‐specific conditional deletion of SAV1, MST1/2 or LATS2 in mice increases nuclear YAP activity, which promotes WNT/β‐catenin signaling and triggers cardiomyocyte proliferation.22 Conversely, WNT signaling enhances YAP mRNA expression. Nuclear accumulation of β‐catenin is augmented by nuclear TAZ via LATS inhibition. Phosphorylated YAP/TAZ also inhibits Disheveled (DVL), promoting β‐catenin degradation and dampening WNT signaling.16, 21 Thus, nuclear YAP/TAZ enhances WNT activity, whereas cytoplasmic YAP/TAZ restricts it.
SHH pathway activation leads to YAP mRNA expression, followed by YAP protein stabilization and nuclear accumulation.23 SHH also increases TEAD1 and IRS1, and induces IRS1 nuclear translocation. YAP, TEAD1 and IRS1 form a nuclear complex that regulates GLI2 and GLI1 expression, enhancing cell‐cycle progression. In mice, YAP overexpression results in hyperactivation of SHH signaling, causing a neuronal differentiation defect in primary cortical progenitors. In humans, increased YAP occurs in medulloblastomas with aberrant SHH signaling, suggesting that therapeutic targeting of YAP might eliminate medulloblastoma recurrence.23
AKT inhibits apoptosis by inactivating MST1/2 via phosphorylation at Thr120/117. In addition, AKT‐mediated MST2 (Thr117) phosphorylation reduces MST2 binding to RASSF1A while increasing its binding to RAF1, again inhibiting MST2 activity. AKT also phosphorylates MST1 (Thr387), which reduces MST1 activity and decreases its cleavage in response to H2O2.24 Reciprocal regulation exists, since MST1/2 overexpression in a prostate cancer cell line inhibits AKT activity. However, AKT‐mediated YAP (Ser127) phosphorylation promotes YAP localization in the cytoplasm and attenuation of p73‐mediated apoptosis.25 Thus, AKT's effects on YAP localization are cell context‐dependent.
MST1/2 deficiency in murine intestinal epithelium decreases YAP phosphorylation and activates NOTCH and β‐catenin signaling.26 Jagged‐1 (NOTCH ligand) is a YAP target in hepatocytes, and Hippo pathway activation in these cells inhibits their proliferation and survival. Conversely, overexpression of constitutively active YAP upregulates Jagged‐1 and increases hepatocyte proliferation. Jagged‐1 induction is required for YAP binding to TEAD4 after MST stimulation. Accordingly, γ‐secretase inhibitors suppress intestinal dysplasia caused by YAP overexpression in mice.14 Thus, YAP and NOTCH may be therapeutic targets for gastrointestinal cancer.
Regulation and Tumor‐Related Roles of Hippo Core Components
MST1/2 kinases
MST activation
Unlike Drosophila Hippo kinase, mammalian MST1/2 contain cleavage sites for caspase‐3 and caspase‐6/7. Cleavage at these sites induced by stresses such as heat shock, tumor necrosis factor‐α (TNF‐α) or UV generate cleaved MST1/2 proteins (∼36 kDa) that have lost the autoinhibitory domain and gained catalytic activity. These shorter MST proteins also localize in the nucleus due to the removal of nuclear export signals. In mouse splenocytes or embryonic fibroblasts, MST1/2 are usually present as 55–60 kDa proteins that are rarely phosphorylated at the autophosphorylation site (Thr183/Thr180) required for full activation. Accordingly, these full‐length MST kinases show little activity and are localized in the cytoplasm. In contrast, in mouse liver, most MST1/2 molecules are 36 kDa and abundantly phosphorylated.26 The mechanism by which MST1/2 are phosphorylated and cleaved in the liver is unclear.
MST1/2 are also controlled by RASSF proteins (Fig. 2). Drosophila RASSF has 10 orthologs in humans (RASSF1–10) but only RASSF1–6 bind to MST1/2. RASSF1 and RASSF5 each have two splice variants. The longer splice variants, RASSF1A and RASSF5A, activate MST1/2 and sustain Hippo signaling. The RASSF1A‐MST1 complex normally localizes at microtubules and regulates the cell‐cycle and microtubule dynamics. The RASSF1A‐MST2 complex promotes MST2 activity by enhancing its autophosphorylation and preventing its degradation. RASSF1A also protects MST1/2 against dephosphorylation and inactivation by PP2A. RASSF6 binds to MST2 but antagonizes Hippo signaling. Interestingly, the RASSF1 gene is localized at 3p21, a chromosomal region frequently deleted in human lung tumors. Notably, RASSF1A is one of the most frequently epigenetically silenced TSGs in human cancers.27
MST1 activity can be enhanced by PH‐domain and leucine‐rich‐repeat protein phosphatase (PHLPP) via a RASSF1A‐independent mechanism (Fig. 2). MST1 cleavage is normally prevented by AKT‐mediated phosphorylation of Thr387,24 but PHLPP reverses this phosphorylation by binding to MST1 in its PP2C domain. Thus, PHLPP functions as a TSG through its effects on AKT. In mice, PHLPP1 loss cooperates with PTEN loss and p53 mutation to promote prostate carcinogenesis.
Downstream of MST
The MST/SAV1 complex activates LATS, which phosphorylates YAP/TAZ. MST1 also phosphorylates FOXOs, promoting their nuclear retention and transcriptional activity. Naive T cells from MST1 knockout (KO) mice exhibit increased reactive oxygen species (ROS) and decreased expression of the FOXO targets catalase and SOD2. However, no reduction in either FOXO protein or phosphorylation is seen in MST1 KO T cells or in MST1/2 double KO (DKO) thymocytes.28, 29 Further examination is thus required to determine whether FOXOs are truly regulated by MSTs in vivo. Lastly, MST1/2 overexpression drives JNK/SAPK, p38, and histone H2B phosphorylation in vitro, but in vivo confirmation is lacking.30
Tumor‐related phenotypes in MST mutant mice
Germline inactivating mutations of MST1 result in an immunodeficiency syndrome both in mice and humans.29 However, tumorigenesis has been reported only in mice bearing conditional tissue‐specific MST mutations. For example, liver‐specific MST1/2 DKO mice show decreased YAP phosphorylation and hepatomegaly, and develop liver tumors by age 5–6 months.26, 31 These mutants accumulate oval cells (adult liver stem cells) in their hepatic periportal regions, resulting in enlarged livers and hepatomas. NF2 and SAV1 mutants have a similar phenotype. It is still unclear whether LATS1/2 are essential for tumor suppression by the Hippo pathway, at least in liver. One study showed loss of MST1/2 decreased MOB1 and YAP phosphorylation but had no effect on LATS1/2 phosphorylation,26 while another study found that levels of phosphorylated YAP and LATS1/2 were reduced in MST1/2 DKO liver.31
Mice lacking MST1/2 specifically in the intestinal epithelium show expansion of an undifferentiated cell population, enlarged crypts, and loss of secretory cells. These mutants die about 13 weeks after birth due to colonic adenomas.26 MST1/2 DKO intestinal epithelial cells exhibit reduced YAP phosphorylation but enhanced YAP protein and nuclear localization.
Keratinocyte‐specific MST1/2 DKO mice have no abnormalities and show normal YAP phosphorylation and activation.8 This result was unexpected because MOB1 KO (see below) and YAP transgenic mice both develop skin carcinomas. Thus, YAP may be phosphorylated by an MST‐independent mechanism in keratinocytes.
SAV1
The binding of MST2 to the adaptor protein SAV1 allows MST2 to phosphorylate both SAV1 and LATS1 and enhance LATS1 activity. SAV1 KO mutants develop hyperplasia of epithelial progenitors and are embryonic lethal.32 Skin cells of keratinocyte‐specific SAV1 KO mice show impaired Ca2+‐stimulated MST1/2 phosphorylation and nuclear translocation.32 Liver‐specific SAV1 KO mice exhibit hepatomegaly and expanded oval cells, and eventually develop HCCs and cholangiocellular carcinomas.31, 33 YAP nuclear translocation occurs in SAV1 KO oval cell‐like cholangiocytes but not in hepatocytes, suggesting that SAV1‐regulated MST1/2 activity may be required for YAP nuclear translocation in specific cell types.33
LATS1/2 kinases
LATS1/2 activation
MST1/2‐mediated phosphorylation of LATS1/2 (Thr1079/Thr1041) is necessary but not sufficient to activate these kinases.34 LATS1/2 must also bind to MOB1 to induce autophosphorylation of the serine residues (Ser909/Ser872) critical for complete LATS1/2 activation. LATS2 (Ser83/Ser380) are also phosphorylated by Aurora‐A, leading to LATS2 localization at the mitotic spindle. Similarly, LATS1 (Ser613) is phosphorylated by Cdc2/cyclinB during mitosis. However, whether YAP is involved in LATS1/2‐mediated control of mitotic progression is unknown. PP2A may modulate LATS activity in this context since okadaic acid activates LATS1/2.
At the transcriptional level, p53 induces LATS2 mRNA in response to nocodazole or constitutive H‐Ras expression. LATS2 transcripts are also regulated by microRNAs (e.g. miR‐31, miR‐372, miR‐373) that are oncogenic when they inhibit LATS2 expression in lung, esophageal, gastric, or testicular tumors. At the protein level, WWC1(KIBRA) and/or NF2 stabilizes LATS2 by inhibiting its ubiquitination. Mitotic stress or constitutive H‐Ras activation can also lead to LATS stabilization and nuclear accumulation, whereas LATS1 protein is destabilized by sustained ROS‐PKCδ signaling or activation of the E3 ubiquitin ligase ITCH.
LATS1/2 functions
YAP and TAZ are the only substrates of LATS kinases identified to date. However, LATS1/2 directly or indirectly affect many other molecules involved in cell proliferation, apoptosis, senescence, migration, and/or mitosis, and do so in a YAP/TAZ‐dependent or ‐independent manner. For example: (i) LATS1/2 arrest cells in G1/S or G2/M by inhibiting the kinase activities of the cyclinE/CDK2 and cyclinA/B‐CDC2 complexes in a mechanism that may involve YAP.35 (ii) LATS1 induces apoptosis by upregulating p53 and Bax while downregulating Bcl‐XL and Bcl‐2. (iii) LATS2 is involved in cellular senescence induced by oncogenic H‐Ras‐V12 or p16‐RB‐ROS activation. (iv) LATS1 interacts with zyxin, which is important for actin polymerization, and co‐localizes with F‐actin and LIMK1 at the contractile ring.36 By binding to LIMK1, LATS1 inhibits its kinase activity and thus its effects on actin dynamics, cell migration and mitosis. (v) LATS2 associates with the microtubule‐associated protein Ajuba, which then recruits γ‐tubulin to centrosomes during normal mitosis.
LATS1/2 are key regulators of checkpoints required to maintain genomic stability. For example: (i) loss of LATS1/2 results in centrosomal amplification and impaired spindle assembly. LAT1/2 DKO cells therefore show loss of diploidy, chromosome misalignment, enlarged nuclei, presence of micronuclei, and defects in cytokinesis.37, 38 (ii) LATS2 KO cells exhibit decreased expression of Aurora‐B and PLK1,37 while loss of LATS1 increases the production of LIMK1‐induced polynucleated cells, leading to cytokinesis failure.36 (iii) In response to DNA damage, LATS1/2 induce p53 by inhibiting MDM2, ensuring proper function of the G1 tetraploidy checkpoint.
In addition to these functions, LATS2 binds to the androgen receptor and inhibits its regulation of target genes, and microarray studies suggest that LATS1/2 participate in the RAS, p53 and WNT pathways.
Tumor‐related phenotypes in LATS1/2 mutant mice
The majority of LATS1 KO mice die during embryogenesis. Survivors develop soft tissue sarcomas and ovarian stromal cell tumors.39 All LATS2 KO mutants die as embryos exhibiting failed cytokinesis and fragmented centrosomes.37, 38
NDR1/2 kinases
NDR1/2 are mammalian orthologs of Tricornered (Trc), which regulates Drosophila morphogenesis. Full activation of mammalian NDR1/2 requires phosphorylation by MST1‐3, which is facilitated by phospho‐MOB1 or phospho‐MOB2 with the assistance of the scaffold protein Furry40 (Fig. 2). NDR1/2 mediate apoptosis downstream of RASSF1A and MST1 by an unknown mechanism. NDR1 also supports centrosome duplication early in S phase, and promotes chromosome attachment to the spindle in M phase.41
NDR1 KO T cells show normal apoptosis in response to proapoptotic stimuli, possibly due to compensation by increased NDR2 protein. However, mice deficient for NDR1 are prone to developing T cell lymphomas.42
Mob1
MOB1 functions
The MOB genes were originally discovered in yeast by searching for mutations interfering with the mitotic exit network and the separation initiation network pathways.43 In mammals, there are four MOB families; MOB1A/1B (orthologs of Drosophila Mats), MOB2, MOB3A/3B/3C, and MOB4. MOB1A/1B are scaffold adaptor proteins that coactivate LATS1/2 and NDR1/2 kinase activities. Interaction with MOB1A/1B also enhances LATS1/2 and NDR1/2 phosphorylation by MSTs, downregulating proliferation.44 MOB2 competes with MOB1 for binding to NDR1/2 and inhibits NDR1/2, but does not bind to LATS1/2. MOB3A/3B/3C and MOB4 proteins do not appear to regulate LATS or NDR kinases.
To interact with LATS1/2 and NDR1/2, MOB1 (Thr12/Thr35) must be phosphorylated by MST1/2. In MST1/2 DKO mutants, MOB1 phosphorylation is abolished in all tissues.26, 29 In vitro, MOB1A overexpression inhibits mitosis, whereas suppression of MOB1A or MOB1B increases proliferation or impairs mitotic exit. MOB1A/1B DKO mice are early embryonic lethal (~E6.5) due to a defect in primitive endoderm formation.45
Tumor‐related phenotypes in MOB1 mutant mice
Mice with a partial loss of MOB1A/1B functions (i.e. homozygous for a MOB1A mutation and heterozygous for a MOB1B mutation, or the reverse), develop a variety of tumors, but trichilemmal carcinomas are most prominent (Fig. 5). All tumors in MOB1‐deficient mutants show loss of the WT MOB1 allele, suggesting increased genomic instability.45 MOB1A/1B DKO keratinocytes exhibit hyperproliferation, impaired contact inhibition, enhanced progenitor self‐renewal, and increased centrosomes. Total LATS1/2, phospho‐LATS1/2 and phospho‐YAP proteins are reduced due to a post‐transcriptional mechanism. Interestingly, MOB1‐deficient mice show the most severe phenotype and develop the broadest range of tumors among strains lacking a Hippo signaling component, suggesting that MOB1s are key molecules in the Hippo pathway that may act on additional targets. Indeed, MOB1 reportedly binds to TSSC1, NUP98, HDAC3, TRAF6, DIPA, and DOCK8.
Figure 5.
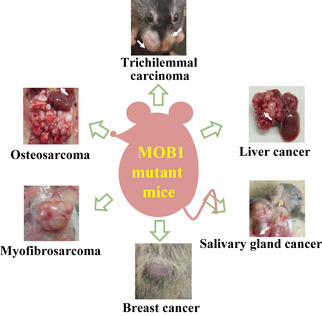
Tumors observed in MOB1 mutant mice. MOB1A/1B DKO mice show one or more of the indicated tumor types.
Like keratinocyte‐specific YAP transgenic mice,8 keratinocyte‐specific MOB1‐deficient mutants show expanded basal epidermal progenitors. However, while YAP transgenic mice develop SCCs, MOB1‐deficient mice develop trichilemmal carcinomas.8 This difference may be explained by the fact that transgenic YAP expression occurred before HF morphogenesis, whereas the MOB1A/1B genes were deleted after birth (after HF formation). Examination of human tumors has shown that YAP is activated in many human trichilemmal carcinomas, some of which also exhibit MOB1A/1B inactivation.45
Conclusion
Because the Hippo pathway controls cell proliferation, apoptosis, contact inhibition, cell migration, cell differentiation, stem cell self‐renewal, genetic stability, and EMT, impaired Hippo signaling leads to altered organ size and the onset, development, and metastasis of cancers. The Hippo pathway is frequently inactivated in a range of tumor types, emphasizing its non‐redundant and powerful functions in opposing oncogenesis and increasing its attractiveness as a target for new anti‐cancer therapies. However, the kinases of the Hippo pathway are important tumor suppressors, precluding their selection as targets for the development of small‐molecule inhibitor drugs. An alternative therapeutic target may be YAP. YAP overexpression is associated with tumorigenesis, and cancers arising due to inactivation of Hippo signaling can be largely suppressed by heterozygous YAP deletion.6 Certain porphyrin derivatives, such as verteporfin (VP) and protoporphyrin IX (PPIX) reportedly inhibit YAP‐TEAD association and YAP‐induced liver overgrowth.46 Verteporfin is now in clinical use for photocoagulation therapy of age‐related macular degeneration. Further screening to identify compounds blocking the interaction between YAP and its target transcription factors may lead to more effective anti‐tumor drugs targeting the Hippo pathway. In addition, because GPCR signaling affects the Hippo pathway (Fig. 1), antagonists or inhibitors of S1P, LPA or β–adrenergic receptor signaling, or agonists of glucagon, epinephrine, or the dopamine receptor, may accomplish the same goal. For a recent review on Hippo pathway targeting as a therapeutic strategy, please refer to Harvey et al.13
In conclusion, continued molecular exploration of the Hippo pathway is likely to expose additional players or interactions that regulate its functions. We are optimistic that fresh insights will emerge which will both increase our understanding of various human cancers and open new avenues of exploitation for cancer treatment.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
This work was supported by a grant from the Ministry of Education, Sports, and Culture of Japan, and by The Uehara Memorial Foundation.
(Cancer Sci 2013; 104: 1271–1277
References
- 1. Harvey K, Tapon N. The Salvador‐Warts‐Hippo pathway – an emerging tumour‐ suppressor network. Nat Rev Cancer 2007; 7: 182–91. [DOI] [PubMed] [Google Scholar]
- 2. Huang J, Wu S, Barrera J et al The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005; 122: 421–34. [DOI] [PubMed] [Google Scholar]
- 3. Wang K, Degerny C, Xu M et al YAP, TAZ, and Yorkie: a conserved family of signal‐responsive transcriptional coregulators in animal development and human disease. Biochem Cell Biol 2009; 87: 77–91. [DOI] [PubMed] [Google Scholar]
- 4. Hong JH, Hwang ES, McManus MT et al TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 2005; 309: 1074–8. [DOI] [PubMed] [Google Scholar]
- 5. Zhao B, Li L, Tumaneng K et al A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF (beta‐TRCP). Genes Dev 2010; 24: 72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang N, Bai H, David KK et al The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell 2010; 19: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yu FX, Zhao B, Panupinthu N et al Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell 2012; 150: 780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schlegelmilch K, Mohseni M, Kirak O et al Yap1 acts downstream of alpha‐catenin to control epidermal proliferation. Cell 2011; 144: 782–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wada K, Itoga K, Okano T et al Hippo pathway regulation by cell morphology and stress fibers. Development 2011; 138: 3907–14. [DOI] [PubMed] [Google Scholar]
- 10. Dupont S, Morsut L, Aragona M et al Role of YAP/TAZ in mechanotransduction. Nature 2011; 474: 179–83. [DOI] [PubMed] [Google Scholar]
- 11. Cordenonsi M, Zanconato F, Azzolin L et al The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 2011; 147: 759–72. [DOI] [PubMed] [Google Scholar]
- 12. Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev 2013; 27: 355–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer 2013; 13: 246–57. [DOI] [PubMed] [Google Scholar]
- 14. Camargo FD, Gokhale S, Johnnidis JB et al YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 2007; 17: 2054–60. [DOI] [PubMed] [Google Scholar]
- 15. Dong J, Feldmann G, Huang J et al Elucidation of a universal size‐control mechanism in Drosophila and mammals. Cell 2007; 130: 1120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barry ER, Morikawa T, Butler BL et al Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 2013; 493: 106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rosenbluh J, Nijhawan D, Cox AG et al β‐Catenin‐driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012; 151: 1457–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang H, Pasolli HA, Fuchs E. Yes‐associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc Natl Acad Sci USA 2011; 108: 2270–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Varelas X, Samavarchi‐Tehrani P, Narimatsu M et al The Crumbs complex couples cell density sensing to Hippo‐dependent control of the TGF‐β‐SMAD pathway. Dev Cell 2010; 19: 831–44. [DOI] [PubMed] [Google Scholar]
- 20. Varelas X, Sakuma R, Samavarchi‐Tehrani P et al TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem‐cell self‐renewal. Nat Cell Biol 2008; 10: 837–48. [DOI] [PubMed] [Google Scholar]
- 21. Varelas X, Miller BW, Sopko R et al The Hippo pathway regulates Wnt/beta‐catenin signaling. Dev Cell 2010; 18: 579–91. [DOI] [PubMed] [Google Scholar]
- 22. Heallen T, Zhang M, Wang J et al Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011; 332: 458–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fernandez LA, Northcott PA, Dalton J et al YAP1 is amplified and up‐regulated in hedgehog‐associated medulloblastomas and mediates Sonic hedgehog‐driven neural precursor proliferation. Genes Dev 2009; 23: 2729–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jang SW, Yang SJ, Srinivasan S et al Akt phosphorylates MstI and prevents its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation. J Biol Chem 2007; 282: 30836–44. [DOI] [PubMed] [Google Scholar]
- 25. Basu S, Totty NF, Irwin MS et al Akt phosphorylates the Yes‐associated protein, YAP, to induce interaction with 14‐3‐3 and attenuation of p73‐mediated apoptosis. Mol Cell 2003; 11: 11–23. [DOI] [PubMed] [Google Scholar]
- 26. Zhou D, Conrad C, Xia F et al Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009; 16: 425–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. J Cell Sci 2007; 120: 3163–72. [DOI] [PubMed] [Google Scholar]
- 28. Zhou D, Medoff BD, Chen L et al The Nore1B/Mst1 complex restrains antigen receptor‐induced proliferation of naive T cells. Proc Natl Acad Sci USA 2008; 105: 20321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mou F, Praskova M, Xia F et al The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J Exp Med 2012; 209: 741–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheung WL, Ajiro K, Samejima K et al Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 2003; 113: 507–17. [DOI] [PubMed] [Google Scholar]
- 31. Lu L, Li Y, Kim SM et al Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci USA 2010; 107: 1437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee JH, Kim TS, Yang TH et al A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J 2008; 27: 1231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee KP, Lee JH, Kim TS et al The Hippo‐Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci USA 2010; 107: 8248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Visser S, Yang X. LATS tumor suppressor: a new governor of cellular homeostasis. Cell‐cycle 2010; 9: 3892–903. [DOI] [PubMed] [Google Scholar]
- 35. Tao W, Zhang S, Turenchalk GS et al Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat Genet 1999; 21: 177–81. [DOI] [PubMed] [Google Scholar]
- 36. Yang X, Yu K, Hao Y et al LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol 2004; 6: 609–17. [DOI] [PubMed] [Google Scholar]
- 37. Yabuta N, Okada N, Ito A et al Lats2 is an essential mitotic regulator required for the coordination of cell division. J Biol Chem 2007; 282: 19259–71. [DOI] [PubMed] [Google Scholar]
- 38. McPherson JP, Tamblyn L, Elia A et al Lats2/Kpm is required for embryonic development, proliferation control and genomic integrity. EMBO J 2004; 23: 3677–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. St John MA, Tao W, Fei X et al Mice deficient of Lats1 develop soft‐tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat Genet 1999; 21: 182–6. [DOI] [PubMed] [Google Scholar]
- 40. Chiba S, Ikeda M, Katsunuma K et al MST2‐ and Furry‐mediated activation of NDR1 kinase is critical for precise alignment of mitotic chromosomes. Curr Biol 2009; 19: 675–81. [DOI] [PubMed] [Google Scholar]
- 41. Hergovich A, Lamla S, Nigg EA et al Centrosome‐associated NDR kinase regulates centrosome duplication. Mol Cell 2007; 25: 625–34. [DOI] [PubMed] [Google Scholar]
- 42. Cornils H, Stegert MR, Hergovich A et al Ablation of the kinase NDR1 predisposes mice to the development of T cell lymphoma. Sci Signal 2010; 3: ra47. [DOI] [PubMed] [Google Scholar]
- 43. Hergovich A. MOB control: reviewing a conserved family of kinase regulators. Cell Signal 2011; 23: 1433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lai ZC, Wei X, Shimizu T et al Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 2005; 120: 675–85. [DOI] [PubMed] [Google Scholar]
- 45. Nishio M, Hamada K, Kawahara K et al Cancer susceptibility and embryonic lethality in Mob1a/1b double‐mutant mice. J Clin Invest 2012; 122: 4505–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu‐Chittenden Y, Huang B, Shim JS et al Genetic and pharmacological disruption of the TEAD–YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012; 26: 1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]