

Abstract
Inhibition of heat shock protein 90 (Hsp90) can lead to degradation of multiple client proteins, which are involved in tumor progression. Epidermal growth factor receptor (EGFR) is one of the most potent oncogenic client proteins of Hsp90. Targeted inhibition of EGFR has shown clinical efficacy in the treatment of patients with non‐small‐cell lung cancer (NSCLC). However, primary and acquired resistance to the existing EGFR inhibitors is a major clinical problem. In the present study, we investigated the effect of the novel Hsp90 inhibitor CH5164840 on the antitumor activity of erlotinib. The NSCLC cell lines and xenograft models were treated with CH5164840 and erlotinib to examine their mechanisms of action and cell growth inhibition. We found that CH5164840 showed remarkable antitumor activity against NSCLC cell lines and xenograft models. The addition of CH5164840 enhanced the antitumor activity of erlotinib against NCI‐H292 EGFR‐overexpressing xenograft models. Phosphorylation of Stat3 increased with erlotinib treatment in NCI‐H292 cells, which was abrogated by Hsp90 inhibition. Furthermore, in a NCI‐H1975 T790M mutation erlotinib‐resistant model, CH5164840 enhanced the antitumor activity of erlotinib despite the low efficacy of erlotinib treatment alone. In addition, ERK signaling was effectively suppressed by combination treatment with erlotinib and CH5164840 in a NCI‐H1975 erlotinib‐resistant model. Taken together, these data indicate that CH5164840 has potent antitumor activity and is highly effective in combination with erlotinib against NSCLC tumors with EGFR overexpression and mutations. Our results support the therapeutic potential of CH5164840 as a Hsp90 inhibitor for combination therapy with EGFR‐targeting agents against EGFR‐addicted NSCLC.
Epidermal growth factor receptor (EGFR) plays a key role in the development and progression of non‐small‐cell lung cancer (NSCLC). Two tyrosine kinase inhibitors (TKI) against EGFR – erlotinib and gefitinib – are currently available for NSCLC therapy. Although targeted inhibition of EGFR has shown promising initial clinical efficacy in the treatment of patients with NSCLC, who have exon 19 (ΔE 746‐A750) and 21 (L858R)‐activating EGFR mutations,1, 2, 3 primary and acquired resistance to these drugs in some patients is an obstacle to treatment efficacy.4 The major mechanisms of resistance to TKI are EGFR mutations,5 MET amplification,6 hepatocyte growth factor (HGF) activation,7 PTEN loss8 and other genomic alterations with wild‐type EGFR, including K‐ras mutations9 and ALK‐fusion proteins.10 A secondary mutation in the EGFR gene (T790M) is the major cause of resistance to gefitinib and erlotinib11, 12 and is found in 50% of clinically resistant patients.13 Due to the potential for resistance to TKI in EGFR‐addicted tumors, identification of an effective treatment using rationalized combinations of agents is particularly promising.
Heat shock protein 90 (Hsp90) is a molecular chaperone that plays an important role in protein folding and stability of client proteins14 and is considered to be an attractive target for cancer therapies.15, 16, 17, 18, 19 Hsp90 is activated by forming multi‐chaperone complexes with co‐chaperones in tumor cells.20 Hsp90 is overexpressed in many tumors, with expression levels correlating with prognosis.21, 22 Inhibition of Hsp90 function leads to the degradation of multiple oncogenic client proteins involved in tumor progression, resulting in a loss of signal transduction, growth inhibition, anti‐angiogenesis and cell death; therefore, multiple signaling pathways are simultaneously blocked by Hsp90 inhibition. Epidermal growth factor receptor is one of the most potent oncogenic client proteins of Hsp90; furthermore, mutated EGFR seems to be more sensitive than wild‐type EGFR to degradation from Hsp90 inhibition.23, 24 These unique features are expected to overcome the problem of resistance to TKI. Stat3 is also a Hsp90 client protein25 and an important signaling mediator for EGFR. Stat3 is activated in approximately 50% of NSCLC primary tumors and lung cancer cell lines26, 27, 28 and this pathway is upregulated in NSCLC cells harboring EGFR mutations.29
First‐generation Hsp90 inhibitors, geldanamycin derivatives, are associated with hepatotoxicity, polymorphic metabolism by NQO1 and efflux by P‐glycoprotein and are therefore not desirable for therapeutic use. Several second‐generation Hsp90 inhibitors have been synthesized and are currently in clinical development.30, 31 Recently, we identified a novel potent Hsp90 inhibitor, CH5164840.32, 33 In the present study, we report the in vitro and in vivo properties of CH5164840 alone and in combination with erlotinib, including its antitumor activity in EGFR‐overexpressing and erlotinib‐resistant NSCLC xenograft models.
Materials and Methods
Compounds
CH5164840 was synthesized by Chugai Pharmaceutical Co., Ltd (Kanagawa, Japan).33 Erlotinib was obtained from Chugai Pharmaceutical/F. Hoffmann‐La Roche (Basel, Switzerland).
Cell lines
The human NSCLC cell lines HCC827, NCI‐H292, NCI‐H1781, A549, NCI‐H1650, NCI‐H1975 and NCI‐H441 were obtained from the American Type Culture Collection (Manassas, VA, USA). The human NSCLC cell line EBC‐1 was obtained from RIKEN Cell Bank (Ibaraki, Japan). The human NSCLC cell line PC‐9 was obtained from Immuno‐Biological Laboratories (Fujioka, Japan). All cell lines were maintained according to the supplier's instructions.
Cell culture
For proliferation assays, tumor cells were seeded into microtiter plates containing compounds and incubated at 37°C in 5% CO2. After incubation for 4 days, a Cell Counting Kit‐8 solution (Dojindo Laboratories, Kumamoto, Japan) was added and absorbance was measured at 450 nm with a Microplate‐Reader iMark (Bio‐Rad Laboratories, Hercules, CA, USA). Antiproliferative activity was calculated using the formula (1‐T/C) × 100 (%), where T represents the absorbance of drug‐treated cells and C represents that of untreated control cells at 450 nm. The IC50 values were calculated using Microsoft Excel 2007 (Redmond, WA, USA). Caspase‐3/7 activity and cell viability were measured with the Caspase‐Glo 3/7 Assay Kit (Promega, Madison, WI, USA) and CellTiter‐Glo Luminescent Cell Viability Assay (Promega), respectively, using EnVision High Throughput Screening (PerkinElmer, Waltham, MA, USA).
Western blotting
Western blotting was performed as previously described.34 Primary antibodies were used for pY1068‐EGFR, EGFR (L858R), pS473‐AKT, AKT, pT202/Y204‐ERK, ERK, pY705‐Stat3, Stat3, JAK1, JAK2 and Tyk2 (Cell Signaling, Beverly, MA, USA), EGFR, HER2, MET, Raf1, actin, GAPDH and Histone H1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and Hsp70 and Hsp90 (Stressgen, Victoria, Canada). Signals were developed using ECL Plus (GE Healthcare, Buckinghamshire, UK), followed by LAS‐4000 (Fujifilm, Tokyo, Japan) or the Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE, USA).
RNA interference
Cells were seeded onto a 96‐well plate and transfected with Human STAT3, JAK1, JAK2 and Tyk2 ON‐TARGETplus SMARTpool ORF and ON‐TARGETplus Non‐targeting Pool (Thermo Scientific, Dharmacon, Waltham, MA, USA) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) and treated with 0.2 μM erlotinib. At 96 h after siRNA transfection, cell viability was determined using the CellTiter‐Glo luminescent cell viability assay (Promega).
Xenograft models and efficacy studies
All animal studies were approved by the Chugai Institutional Animal Care and Use Committee. Cancer cells (0.5–1.0 × 107) were implanted subcutaneously into the right flank of athymic nude (BALB/c nu/nu) mice (Charles River Laboratories, Kanagawa, Japan). Tumor volume (TV) was calculated using the formula TV = ab 2/2, where a and b represent tumor length and width, respectively. Once the tumors reached a volume of approximately 200–300 mm3, animals were randomized into each group (n = 4–5) and treatments were initiated. CH5164840 was dissolved in a vehicle of 10% DMSO/10% Cremophor EL/0.02 N HCl in water. CH5164840 and erlotinib were orally administered once daily for 11 days. Tumor growth inhibition (TGI) was calculated using the formula , where T (TV of the treated group, T 0 on day 0 or T t on day t) and C (tumor volume of control group, C 0 on day 0 or C t on day t) represent mean tumor volume. The maximum tolerated dose was defined as the dose that resulted in neither lethality nor more than 20% bodyweight loss. For pharmacodynamic studies, blood was collected 4 h after the last administration of CH5164840 and PBMC were prepared with M‐SMF (JIMRO, Gunma, Japan), lysed and analyzed using western blotting.
Statistical analysis
For in vivo combination studies, sas preclinical package v5 software (SAS Institute, Cary, NC, USA) was used for statistical analyses (Tukey's test). For in vitro experiments, statistical values were defined using the Student's t‐test.
Results
Novel Hsp90 inhibitor CH5164840 has potent growth inhibitory activity against NSCLC in vitro and antitumor activity in vivo
Recently, through virtual screening and structure‐based drug design, we identified CH5164840 as a Hsp90 inhibitor with a novel chemical structure.32, 33 In an initial evaluation, in vitro cell growth inhibition of CH5164840 or erlotinib were examined to determine the sensitivity of NSCLC cell lines with various genotypes, including PC‐9 and HCC827 (EGFR ΔE746‐A750), NCI‐H292 (wild‐type EGFR overexpression), NCI‐H1975 (EGFR T790M and L858R mutant), NCI‐H1650 (EGFR ΔE746‐A750, PTEN null), NCI‐H1781 (HER2 G776insV_G/C mutant) and A549 (K‐ras mutant). The IC50 values of CH5164840 were found to be 140–550 nM in seven NSCLC cell lines, regardless of genetic mutations, although sensitivities to erlotinib were more variable (IC50 values 4.7–13000 nM; Table 1). Next, we examined whether CH5164840 induced the degradation of Hsp90 client proteins in NSCLC cell lines. CH5164840 was found to significantly reduce the protein levels of EGFR, HER2 and MET and to suppress downstream AKT and ERK signaling (Fig. 1). We also confirmed the induction of Hsp70, a marker of Hsp90 inhibition.
Table 1.
Antiproliferative activity of CH5164840 in non‐small‐cell lung cancer cell lines
| Cell line | EGFR status | CH5164840 IC50 (μM) | Erlotinib IC50 (μM) |
|---|---|---|---|
| PC‐9 | E746_A750del | 0.16 | 0.0048 |
| HCC827 | E746_A750del | 0.14 | 0.0047 |
| NCI‐H292 | WT overexpression | 0.49 | 0.16 |
| NCI‐H1781 | WT | 0.55 | 1.70 |
| A549 | WT | 0.19 | 3.40 |
| NCI‐H1650 | E746_A750del | 0.16 | 4.10 |
| NCI‐H1975 | L858R and T790M | 0.30 | 13.00 |
EGFR, epidermal growth factor receptor.
Figure 1.
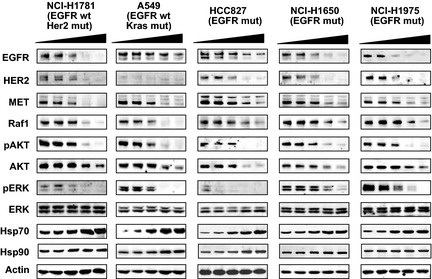
CH5164840 induces degradation of multiple heat shock protein 90 (Hsp90) client proteins in non‐small‐cell lung cancer (NSCLC) cell lines. The NSCLC cell lines were treated with 0, 0.04, 0.2, 1 or 5 μM CH5164840 for 24 h before lysis and analyzed using western blotting. EGFR, epidermal growth factor receptor. HER2, human epidermal growth factor receptor 2; p, phospho.
To evaluate the in vivo antitumor activity of CH5164840, several xenograft studies were conducted using NSCLC cell lines in nude BALB/c mice. Daily oral administration of CH5164840 resulted in a significant level of dose‐dependent antitumor activity in the NCI‐H1650 (EGFR mutant, PTEN null) xenograft models, with a maximum TGI of 131% (Fig. 2a). In addition, we confirmed the dose‐dependent induction of Hsp70 in murine PBMC, which is a marker of Hsp90 inhibition (Fig. 2b). Similar to NCI‐H1650 antitumor activity, CH5164840 showed substantial antitumor activity in NCI‐H292 (wild‐type EGFR overexpression), NCI‐H1975 (EGFR mutant) and NCI‐H441 (wild‐type EGFR, MET overexpression) xenograft models (Table 2). In all in vivo studies, the doses of CH5164840 tested were well tolerated, with no gross toxicity observed in the treated animals (data not shown). Taken together, these data show that CH5164840 inhibited Hsp90, resulting in the inhibition of in vitro cell growth and in vivo antitumor activity against multiple NSCLC cell lines.
Figure 2.
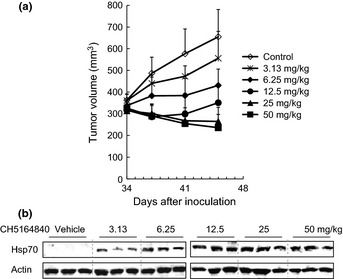
Antitumor activity of CH5164840 in a NCI‐H1650 erlotinib‐resistant xenograft model and pharmacodynamic response. (a) Mice bearing NCI‐H1650 tumors were orally administered the indicated doses of CH5164840 on a daily basis for 11 consecutive days. Mean tumor volume is shown. (b) Four hours after the final administration in (a), murine PBMC were isolated from blood, lysed and analyzed using western blotting.
Table 2.
Antitumor efficacy of CH5164840 in tumor xenograft models at MTD
| Models | Tumor types | Status | Client | MTD (mg/kg) | TGI (%) |
|---|---|---|---|---|---|
| NCI‐H292 | Mucoepidermoid | EGFR overexpression | EGFR | 25 | 92 |
| NCI‐H441 | Papillary adeno | MET overexpression | MET | 50 | 115 |
| NCI‐H1975 | Adenocarcinoma | EGFR mutant |
EGFR L858R and T790M |
50 | 112 |
| NCI‐H1650 | Adenocarcinoma | EGFR mutant |
EGFR E746‐A750del |
50 | 131 |
EGFR, epidermal growth factor receptor; MET, met proto‐oncogene; MTD, maximum tolerated dose; TGI, tumor growth inhibition. CH5164840 was orally administered once daily for 11 days.
CH5164840 enhances the antitumor activity of erlotinib in a NCI‐H292 wild‐type EGFR overexpression, erlotinib‐halfway sensitive model
We then investigated the effect of CH5164840 on erlotinib activity against NCI‐H292 cells, which overexpress wild‐type EGFR, because erlotinib exhibits moderate activity against NCI‐H292 NSCLC cells. To confirm the cellular effects of this combination treatment in vitro, we examined cell growth inhibition and caspase‐3/7 activity. As shown in Figure 3, CH5164840 enhanced the cell growth inhibitory activity of erlotinib. Furthermore, when NCI‐H292 cells were treated with a combination of erlotinib and CH5164840, CH5164840 significantly enhanced the caspase‐3/7 activity of erlotinib (Fig. 3b). To examine the combined antitumor activity of CH5164840 and erlotinib in vivo, 25 mg/kg of erlotinib and/or 12.5 mg/kg of CH5164840 was administered to the NCI‐H292 xenograft models. CH5164840 synergistically enhanced the antitumor activity of erlotinib (Fig. 3c). Combined treatment with erlotinib and CH5164840 produced a statistically significant effect on TGI compared with both monotherapies, causing tumor regression. These data support the effectiveness of a combination strategy using erlotinib and CH5164840 against NSCLC with EGFR overexpression.
Figure 3.
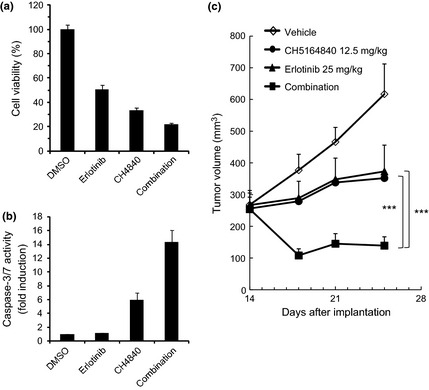
Antitumor activity of CH5164840 in combination with erlotinib on NCI‐H292 EGFR‐overexpressing NSCLC in vitro and in vivo. (a,b) NCI‐H292 cells were treated with 0.2 μM erlotinib and/or 0.5 μM CH5164840 for 48 h (for caspase‐3/7 activity and cell viability) and 96 h (for cell viability). Caspase‐3/7 activity and cell viability were measured with the Caspase‐Glo 3/7 assay and CellTiter‐Glo, respectively, using EnVision High Throughput Screening. Caspase‐3/7 activity was normalized to cell viability. (c) Mice bearing NCI‐H292 tumors were orally administered 12.5 mg/kg CH5164840 daily for 11 consecutive days and/or 25 mg/kg erlotinib. Tukey's test: ***P < 0.001.
We further examined the molecular mechanisms of erlotinib and CH5164840 synergism in NCI‐H292 cells. First, we analyzed the effect of these agents on cell signaling using western blotting. Combination treatment with erlotinib and CH5164840 showed reduced protein levels of EGFR and phospho‐EGFR and suppressed downstream AKT and ERK signaling (Fig. 4a). Additionally, erlotinib produced a dose‐dependent increase in Stat3 phosphorylation compared with DMSO control (Fig. 4b), which was mainly present in the nucleus (Fig. 4c). Interestingly, treatment with CH5164840 in addition to erlotinib reduced the phosphorylation level of Stat3 (Fig. 4c). A previous study has shown that Janus kinase1 (JAK1) phosphorylates Stat3 in NSCLC cells.35 We confirmed that siRNA knockdown of JAK1, but not JAK2 or TyK2, inhibited erlotinib‐induced Stat3 phosphorylation in NCI‐H292 cells (Fig. 4d). In addition, Hsp90 inhibition by CH5164840 induced degradation of the JAK1 protein (Fig. 4a). Furthermore, to explore the effect of JAK1 and Stat3 on cell growth, we examined these knockdowns by siRNA. Inhibition of Stat3 expression by siRNA specifically enhanced cell growth inhibition by erlotinib (Fig. 4e), producing statistically significant results. The combination of erlotinib and JAK1 knockdown showed more effective cell growth inhibition than did Stat3 (Fig. 4e). Taken together, these findings suggest that, in addition to AKT and ERK signaling, suppression of the JAK1‐Stat3 signaling pathway by CH5164840 enhanced the antitumor effects of erlotinib in NSCLC.
Figure 4.
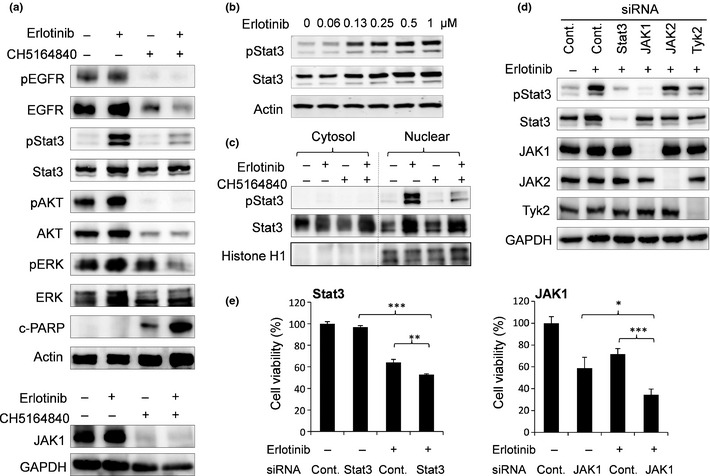
CH5164840 treatment suppressed erlotinib‐induced Stat3 signaling in NCI‐H292 cells. (a) NCI‐H292 cells were treated with 0.2 μM erlotinib and/or 0.5 μM CH5164840 for 48 h, lysed and analyzed using western blotting. (b) NCI‐H292 cells were treated with the indicated concentration of erlotinib for 24 h and analyzed for Stat3 and phospho‐Stat3 protein. (c) NCI‐H292 cells were treated with 0.2 μM erlotinib and/or 0.5 μM CH5164840 for 48 h and cytosol and nuclear fractions were analyzed using western blotting. Histone H1 indicates loading control for nuclear fraction. (d) NCI‐H292 cells were transfected with 10 nM Stat3, JAK1, JAK2, Tyk2 or control siRNA. At 24 h post‐transfection, cells were treated with 0.2 μM erlotinib and cultured for 24 h, lysed and analyzed using western blotting. (e) NCI‐H292 cells were transfected with 10 nM Stat3 siRNA, JAK1 siRNA or control siRNA and treated with DMSO or 0.2 μM erlotinib. At 96 h post‐transfection, cell viability was determined by CellTiter‐Glo. Data are shown as mean ± SD (n = 3). Student's t‐test: *P < 0.05; **P <0.01; ***P < 0.001. c‐PARP, cleaved poly (ADP‐ribose) polymerase; EGFR, epidermal growth factor receptor; p, phospho; −, DMSO; +, compound.
CH5164840 enhances the antitumor activity of erlotinib in an erlotinib‐resistant NCI‐H1975 model
We further explored the combined effect of erlotinib and CH5164840 treatment using NCI‐H1975 NSCLC cells. NCI‐H1975 cells harbor the EGFR T790M gatekeeper mutation, a secondary EGFR mutation that is a major cause of resistance to erlotinib. To investigate the combined effect of erlotinib and CH5164840, we examined the antiproliferative effect of the agents on NCI‐H1975 cells. CH5164840 enhanced the cell growth inhibitory and caspase‐3/7 activity induced by erlotinib (Fig. 5a,b).
Figure 5.
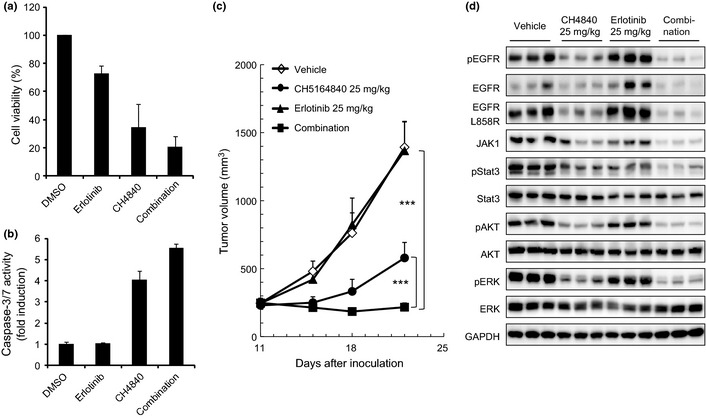
Antitumor activity of CH5164840 in combination with erlotinib in a NCI‐H1975 erlotinib‐resistant model. (a,b) NCI‐H1975 cells were treated with 1 μM erlotinib and/or 1 μM CH5164840 for 48 h (for caspase‐3/7 activity and cell viability) and 96 h (for cell viability). Caspase‐3/7 activity and cell viability were measured with the Caspase‐Glo 3/7 assay and CellTiter‐Glo, respectively, using EnVision High Throughput Screening. Caspase activity was normalized to cell viability. (c) Mice bearing NCI‐H1975 tumors were orally administered 25 mg/kg of CH5164840 and/or 25 mg/kg of erlotinib daily for 11 days. (d) Four hours after the final administration in (c), tumors were resected, lysed and analyzed using western blotting. EGFR, epidermal growth factor receptor; p, phospho. Tukey's test: ***P < 0.001.
We then investigated the in vivo antitumor activity of this combination therapy against human tumor xenograft models in mice. When a combination of 25 mg/kg erlotinib and 25 mg/kg CH5164840 was orally administered to a NCI‐H1975 xenograft model, tumor growth was significantly inhibited, despite erlotinib treatment alone having no effect (Fig. 5c). The enhanced antitumor activity with combination treatment was statistically significant compared with both monotherapies and no gross toxicity was observed in any of the treated animals (data not shown). In addition, combination therapy resulted in reduced protein levels of mutated EGFR, JAK1, phospho‐Stat3, phospho‐AKT and phospho‐ERK (Fig. 5d). These results suggest that CH5164840 potentiates the efficacy of erlotinib in an erlotinib‐resistant model harboring the EGFR T790M gatekeeper mutation.
Discussion
Epidermal growth factor receptor is an attractive target for anticancer therapies because it plays an important role in lung carcinogenesis; moreover, its expression is correlated with poor prognosis36 and EGFR TKI, erlotinib and gefitinib, have shown a clinical response in NSCLC with activating EGFR mutations. However, the development of primary and acquired resistances to these drugs over several years has become a major clinical problem. Recently, many resistant mechanisms of EGFR‐TKI have been investigated37, including EGFR mutations (T790M),5, 38 MET amplification,6 hepatocyte growth factor (HGF) activation,7 PTEN loss8, 39 and AXL activation.40 To overcome TKI resistance, corresponding strategies for each TKI‐resistant mechanism are needed. The combination of a TKI and a Hsp90 inhibitor is considered particularly promising because the inhibition of Hsp90 induces degradation of molecules that are involved in TKI resistance, such as mutant EGFR, MET and EML4‐ALK. Hsp90 inhibitors have shown efficacy against established EGFR‐TKI–resistant cells, EGFR T790M41 and HGF overexpression.42 In fact, the Hsp90 inhibitor IP‐504 has shown clinical activity in patients with ALK rearrangement in a phase 1/2 study.43
We recently identified CH5164840 as a Hsp90 inhibitor with a novel chemical structure.33 CH5164840 can be taken orally and exhibits highly potent antitumor efficacy.34 In the present study, CH5164840 inhibited the cell growth of NSCLC cell lines that have different oncogenic drivers, induced degradation of representative Hsp90 client proteins EGFR, HER2, MET and Raf1 and inhibited phosphorylation of downstream signaling proteins AKT and ERK. Treatment with CH5164840 also induces expression of Hsp70. Generally, studies on Hsp90 inhibitors focus on degradation of client proteins or induction of Hsp70 as a biomarker for the Hsp90 inhibitor. However, induction of Hsp70 by Hsp90 inhibition has been shown to reduce the antitumor effect of Hsp90 inhibitors and knockdown of Hsp70 by siRNA increased the 17‐AAG‐induced apoptosis.44 Therefore, abrogating Hsp70 induction approaches are raised to increasing the sensitivity of Hsp90 inhibitors.45 Furthermore, CH5164840 monotherapy showed antitumor activity against NSCLC xenograft models. CH5164840 showed antitumor activity against a NCI‐H441 (wild‐type EGFR and MET overexpression) model; therefore, it is considered useful for patients who exhibit MET‐associated TKI resistance. In fact, as shown in Figure S1, we confirmed in NCI‐H441 and EBC‐1 (MET amplification) cells, in which erlotinib did not show cell growth inhibition, that CH5164840 enhanced the cell growth inhibitory activity of erlotinib with IC50 values of erlotinib in the individual cell lines of >20 μM (EBC‐1) and 10.0 μM (NCI‐H441) in vitro. Furthermore, combination treatment resulted in degradation of phosphorylated and total EGFR or MET in these cells. Taking these observations together, Hsp90 inhibitor is a potential therapeutic option in combination with erlotinib against TKI‐resistant NSCLC with aberrant c‐MET. Similarly, it may also be effective in patients with PTEN loss, which is one of the mechanisms of TKI resistance, because of the antitumor activity of CH5164840 in the NCI‐H1650 (EGFR ΔE746‐A750, PTEN null) model. These results show that the novel Hsp90 inhibitor CH5164840 has potent antitumor activity in vitro and in vivo against clinically relevant NSCLC, including erlotinib‐resistant models.
The association between EGFR protein expression levels and clinical response to EGFR TKI has been variable46, 47 and, in fact, erlotinib elicits only a moderate response against wild‐type EGFR‐overexpressing NCI‐H292 cells. In the present study, we showed that treatment of NCI‐H292 cells with a combination of EGFR TKI erlotinib and Hsp90 inhibitor CH5164840 led to synergistic cell growth inhibition and induction of apoptosis. These in vitro results were consistent with the regression of antitumor activity by combination treatment in a NCI‐H292 xenograft model. Furthermore, we examined the molecular mechanisms by which CH5164840 enhanced the efficacy of erlotinib. Combination treatment with erlotinib and CH5164840 reduced EGFR and phospho‐EGFR levels, as well as downstream signaling proteins AKT and ERK. Interestingly, we noticed that erlotinib treatment increased phospho‐Stat3 in NCI‐H292 cells, an effect that was strongly suppressed by CH5164840. Erlotinib‐induced phospho‐Stat3 is sensitive to Hsp90 inhibition most likely because Stat3 is a client protein of Hsp90.25 Therefore, in NCI‐H292 cells, the efficacy of erlotinib might be increased through blocking Stat3 signaling by Hsp90 inhibition. Stat3 can be activated through multiple pathways, including EGFR, the IL‐6/gp130 receptor family, platelet‐derived growth factor receptor (PDGFR), Src kinase and JAK. It has been reported that JAK1 causes Stat3 activation in NSCLC cells.35 We confirmed this finding and also that both basal and erlotinib‐induced phospho‐Stat3 were inhibited by JAK1 knockdown with siRNA but not JAK2 and Tyk2 knockdown (Fig. 4d). Although a combination of erlotinib treatment and JAK1 knockdown is more effective for cell growth inhibition than that of erlotinib treatment and stat3 knockdown, the former does not completely inhibit cell growth; therefore, additional blocking of AKT and ERK pathways by a Hsp90 inhibitor might be an important therapeutic approach in NCI‐H292 NSCLC cells. However, the mechanism by which phospho‐Stat3 is increased by erlotinib‐induced EGFR inhibition remains unknown. Further investigation is needed to clarify the upstream pathways of Stat3 activation by EGFR TKI treatment.
Acquired resistance to TKI, which is usually due to the development of a second point mutation (T790M) in EGFR, is also a serious clinical problem. Consistent with other Hsp90 inhibitors,48, 49 CH5164840 monotherapy shows antitumor activity against NCI‐H1975 NSCLC cells harboring the EGFR L858R and T790M mutations. In addition, combination therapy with erlotinib and CH5164840 showed significant synergism, despite the lack of antitumor activity of erlotinib monotherapy in this model; additionally, combination therapy simultaneously induced degradation of mutant EGFR, phospho‐EGFR and downstream AKT, ERK and Stat3 signaling. As such, combination therapy using TKI and a Hsp90 inhibitor might be useful to prevent the development of TKI resistance, because most TKI are at potential risk of encountering TKI resistance and addicted‐oncogenic proteins are degraded by Hsp90 inhibition.15 In conclusion, our data support the clinical development and benefits of using a Hsp90 inhibitor in combination with EGFR‐targeted therapies for the treatment of EGFR‐addicted NSCLC.
Disclosure Statement
All authors are employees of Chugai Pharmaceutical Co., Ltd.
Supporting information
Fig. S1. Antitumor activity of CH5164840 in combination with erlotinib on non‐small‐cell lung cancer with aberrant c‐MET in vitro.
Acknowledgments
The authors thank Dr Neal Rosen (Memorial Sloan‐Kettering Cancer Center), Dr Shigehisa Nagahashi and Dr Hiroshi Sakamoto for their helpful discussion. Thanks also to Ms Fumie Sawamura, Ms Kurisu Honda, Ms Yasue Nagata, Mr Sachiya Yamamoto, Mr Yusuke Ide, Ms Ikuko Matsuo and Ms Yumiko Hashimoto for their technical support.
(Cancer Sci 2013; 104: 1346–1352
References
- 1. Lynch TJ, Bell DW, Sordella R et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 2. Paez JG, Janne PA, Lee JC et al EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497–500. [DOI] [PubMed] [Google Scholar]
- 3. Pao W, Miller V, Zakowski M et al EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101: 13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Costa DB, Kobayashi S, Tenen DG, Huberman MS. Pooled analysis of the prospective trials of gefitinib monotherapy for EGFR‐mutant non‐small cell lung cancers. Lung Cancer 2007; 58: 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7: 169–81. [DOI] [PubMed] [Google Scholar]
- 6. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 7. Yano S, Wang W, Li Q et al Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor‐activating mutations. Cancer Res 2008; 68: 9479–87. [DOI] [PubMed] [Google Scholar]
- 8. Sos ML, Koker M, Weir BA et al PTEN loss contributes to erlotinib resistance in EGFR‐mutant lung cancer by activation of Akt and EGFR. Cancer Res 2009; 69: 3256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pao W, Wang TY, Riely GJ et al KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005; 2: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shaw AT, Yeap BY, Mino‐Kenudson M et al Clinical features and outcome of patients with non‐small‐cell lung cancer who harbor EML4‐ALK. J Clin Oncol 2009; 27: 4247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi S, Boggon TJ, Dayaram T et al EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med 2005; 352: 786–92. [DOI] [PubMed] [Google Scholar]
- 12. Pao W, Miller VA, Politi KA et al Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Balak MN, Gong Y, Riely GJ et al Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor‐mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006; 12: 6494–501. [DOI] [PubMed] [Google Scholar]
- 14. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer 2005; 5: 761–72. [DOI] [PubMed] [Google Scholar]
- 15. Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci 2007; 1113: 202–16. [DOI] [PubMed] [Google Scholar]
- 16. Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther 2002; 2: 3–24. [DOI] [PubMed] [Google Scholar]
- 17. Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003; 3: 213–7. [DOI] [PubMed] [Google Scholar]
- 18. Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res 2009; 15: 9–14. [DOI] [PubMed] [Google Scholar]
- 19. Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 2012; 18: 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kamal A, Thao L, Sensintaffar J et al A high‐affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003; 425: 407–10. [DOI] [PubMed] [Google Scholar]
- 21. Pick E, Kluger Y, Giltnane JM et al High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res 2007; 67: 2932–7. [DOI] [PubMed] [Google Scholar]
- 22. Gallegos Ruiz MI, Floor K, Roepman P et al Integration of gene dosage and gene expression in non‐small cell lung cancer, identification of HSP90 as potential target. PLoS ONE 2008; 3: e0001722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shimamura T, Lowell AM, Engelman JA, Shapiro GI. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res 2005; 65: 6401–8. [DOI] [PubMed] [Google Scholar]
- 24. Sawai A, Chandarlapaty S, Greulich H et al Inhibition of Hsp90 down‐regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res 2008; 68: 589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sato N, Yamamoto T, Sekine Y et al Involvement of heat‐shock protein 90 in the interleukin‐6‐mediated signaling pathway through STAT3. Biochem Biophys Res Commun 2003; 300: 847–52. [DOI] [PubMed] [Google Scholar]
- 26. Bromberg JF, Wrzeszczynska MH, Devgan G et al Stat3 as an oncogene. Cell 1999; 98: 295–303. [DOI] [PubMed] [Google Scholar]
- 27. Gao SP, Mark KG, Leslie K et al Mutations in the EGFR kinase domain mediate STAT3 activation via IL‐6 production in human lung adenocarcinomas. J Clin Invest 2007; 117: 3846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mukohara T, Kudoh S, Yamauchi S et al Expression of epidermal growth factor receptor (EGFR) and downstream‐activated peptides in surgically excised non‐small‐cell lung cancer (NSCLC). Lung Cancer 2003; 41: 123–30. [DOI] [PubMed] [Google Scholar]
- 29. Haura EB, Zheng Z, Song L, Cantor A, Bepler G. Activated epidermal growth factor receptor‐Stat‐3 signaling promotes tumor survival in vivo in non‐small cell lung cancer. Clin Cancer Res 2005; 11: 8288–94. [DOI] [PubMed] [Google Scholar]
- 30. Messaoudi S, Peyrat JF, Brion JD, Alami M. Heat‐shock protein 90 inhibitors as antitumor agents: a survey of the literature from 2005 to 2010. Expert Opin Ther Pat 2011; 21: 1501–42. [DOI] [PubMed] [Google Scholar]
- 31. Kim YS, Alarcon SV, Lee S et al Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem 2009; 9: 1479–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miura T, Fukami TA, Hasegawa K et al Lead generation of heat shock protein 90 inhibitors by a combination of fragment‐based approach, virtual screening, and structure‐based drug design. Bioorg Med Chem Lett 2011; 21: 5778–83. [DOI] [PubMed] [Google Scholar]
- 33. Suda A, Koyano H, Hayase T et al Design and synthesis of novel macrocyclic 2‐amino‐6‐arylpyrimidine Hsp90 inhibitors. Bioorg Med Chem Lett 2012; 22: 1136–41. [DOI] [PubMed] [Google Scholar]
- 34. Ono N, Yamazaki T, Nakanishi Y et al Preclinical antitumor activity of the novel heat shock protein 90 inhibitor CH5164840 against human epidermal growth factor receptor 2 (HER2)‐overexpressing cancers. Cancer Sci 2012; 103: 342–9. [DOI] [PubMed] [Google Scholar]
- 35. Song L, Rawal B, Nemeth JA, Haura EB. JAK1 activates STAT3 activity in non‐small‐cell lung cancer cells and IL‐6 neutralizing antibodies can suppress JAK1‐STAT3 signaling. Mol Cancer Ther 2011; 10: 481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirsch FR, Varella‐Garcia M, Cappuzzo F et al Combination of EGFR gene copy number and protein expression predicts outcome for advanced non‐small‐cell lung cancer patients treated with gefitinib. Ann Oncol 2007; 18: 752–60. [DOI] [PubMed] [Google Scholar]
- 37. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR‐mutant non‐small‐cell lung cancer. Nat Rev Cancer 2010; 10: 760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ogino A, Kitao H, Hirano S et al Emergence of epidermal growth factor receptor T790M mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res 2007; 67: 7807–14. [DOI] [PubMed] [Google Scholar]
- 39. Yamamoto C, Basaki Y, Kawahara A et al Loss of PTEN expression by blocking nuclear translocation of EGR1 in gefitinib‐resistant lung cancer cells harboring epidermal growth factor receptor‐activating mutations. Cancer Res 2010; 70: 8715–25. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Z, Lee JC, Lin L et al Activation of the AXL kinase causes resistance to EGFR‐targeted therapy in lung cancer. Nat Genet 2012; 44: 852–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shimamura T, Li D, Ji H et al Hsp90 inhibition suppresses mutant EGFR‐T790M signaling and overcomes kinase inhibitor resistance. Cancer Res 2008; 68: 5827–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Koizumi H, Yamada T, Takeuchi S et al Hsp90 inhibition overcomes HGF‐triggering resistance to EGFR‐TKIs in EGFR‐mutant lung cancer by decreasing client protein expression and angiogenesis. J Thorac Oncol 2012; 7: 1078–85. [DOI] [PubMed] [Google Scholar]
- 43. Sequist LV, Gettinger S, Senzer NN et al Activity of IPI‐504, a novel heat‐shock protein 90 inhibitor, in patients with molecularly defined non‐small‐cell lung cancer. J Clin Oncol 2010; 28: 4953–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo F, Rocha K, Bali P et al Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17‐allylamino‐demethoxy geldanamycin. Cancer Res 2005; 65: 10536–44. [DOI] [PubMed] [Google Scholar]
- 45. Powers MV, Jones K, Barillari C, Westwood I, van Montfort RL, Workman P. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 2010; 9: 1542–50. [DOI] [PubMed] [Google Scholar]
- 46. Tsao MS, Sakurada A, Cutz JC et al Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med 2005; 353: 133–44. [DOI] [PubMed] [Google Scholar]
- 47. Hirsch FR, Varella‐Garcia M, Bunn PA Jr et al Molecular predictors of outcome with gefitinib in a phase III placebo‐controlled study in advanced non‐small‐cell lung cancer. J Clin Oncol 2006; 24: 5034–42. [DOI] [PubMed] [Google Scholar]
- 48. Rice JW, Veal JM, Barabasz A et al Targeting of multiple signaling pathways by the Hsp90 inhibitor SNX‐2112 in EGFR resistance models as a single agent or in combination with erlotinib. Oncol Res 2009; 18: 229–42. [DOI] [PubMed] [Google Scholar]
- 49. Bao R, Lai CJ, Wang DG et al Targeting heat shock protein 90 with CUDC‐305 overcomes erlotinib resistance in non‐small cell lung cancer. Mol Cancer Ther 2009; 8: 3296–306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Antitumor activity of CH5164840 in combination with erlotinib on non‐small‐cell lung cancer with aberrant c‐MET in vitro.