

Abstract
The incidence of esophageal adenocarcinoma has increased in the last 25 years. Columnar metaplasia in Barrett's mucosa is assumed to be a precancerous lesion for esophageal adenocarcinoma. However, the induction process of Barrett's mucosa is still unknown. To analyze the induction of esophageal columnar metaplasia, we established a mouse gastro‐esophageal reflux disease (GERD) model with associated development of columnar metaplasia in the esophagus. C57BL/6 mice received side‐to‐side anastomosis of the esophagogastric junction with the jejunum, and mice were killed 10, 20, and 40 weeks after operation. To analyze the contribution of bone marrow‐derived cells to columnar metaplasia in this surgical GERD model, some mice were transplanted with GFP‐marked bone marrow after the operation. Seventy‐three percent of the mice (16/22) showed thickened mucosa in esophagus and 41% of mice (9/22) developed columnar metaplasia 40 weeks after the operation with a mortality rate of 4%. Bone marrow‐derived cells were not detected in columnar metaplastic epithelia. However, scattered epithelial cells in the thickened squamous epithelia in regions of esophagitis did show bone marrow derivation. The results demonstrate that reflux induced by esophago‐jejunostomy in mice leads to the development of columnar metaplasia in the esophagus. However, bone marrow‐derived cells do not contribute directly to columnar metaplasia in this mouse model.
Approximately half of esophageal cancers in Western countries are esophageal adenocarcinomas (Barrett's cancer),1 and esophageal adenocarcinoma has increased by sixfold in the last 25 years.2 Barrett's mucosa, which was described first by Barrett in 1950,3 is assumed to be a precancerous lesion for esophageal adenocarcinoma.4 Barrett's mucosa is defined as columnar epithelium, replacing esophageal squamous epithelium, initiated by reparative processes in the setting of gastroesophageal reflux disease.5, 6, 7, 8 In 1998, the American College of Gastroenterology (ACG) defined Barrett's esophagus as “a change in the esophageal epithelium of any length that can be recognized at endoscopy and is confirmed to have intestinal metaplasia by biopsy of the tubular esophagus and excludes intestinal metaplasia of the cardia”.9 However, the precise mechanism and genetic changes responsible for initiation of Barrett's mucosa are still not known.
Using rats, a variety of operative models for gastroesophageal reflux disease have been established for eliciting columnar metaplasia.10, 11, 12 It is reported that not only the gastric juice but also the duodenal juice, especially the reflux of bile, play an important role in the generation of columnar metaplasia. Although there are reports of rat gastro‐esophageal reflux disease (GERD) models, there are few reports detailing mouse surgical GERD models.13, 14, 15 The lack of mouse models may relate to the difficulty of operations because of the small size of the animals. If an easy mouse GERD model were established, genetically modified mouse models could be used to analyze the mechanism of columnar metaplasia and esophageal cancer more precisely. A simple operation that anastomoses the esophagogastric junction and intestine, side‐to‐side, induces columnar metaplasia and adenocarcinoma in rats.10 The reflux model with side‐to‐side anastomosis of esophagogastric junction and jejunum appeared easier than other models; however, it had not been tried in mice. In this report, we used this model for mice.
There are at least three reported possible mechanisms for the genesis of columnar metaplasia. The first is extension of the columnar epithelium over the gastroesophageal junction proximally.16, 17, 18 The second is metaplasia of esophageal glands. The third is columnar true metaplasia of squamous epithelium by changes of stem cell differentiation.19, 20 Recently, Spasmolytic Polypeptide (TFF2) Expressing Metaplasia (SPEM), a metaplasia caused by long term Helicobacter infection in the stomach, was reported to support engraftment of bone marrow‐derived cells.21 This prompted us to analyze whether columnar metaplasia in the esophagus is also associated with bone marrow‐derived cells using the mouse reflux model.
Materials and Methods
Animals
Wild type C57BL/6 mice were used for the surgical model. Green fluorescent protein transgenic (GFP Tg) mice (C57BL/6‐Tg [CAG‐EGFP], Japan SLC, Tokyo) were used as donors of bone marrow cells. During the experiments, mice were kept in cages provided with water and regular chow ad libitum until the time of sacrifice. All animal experimental procedures were reviewed and approved by the Institutional Animal Care and Research Advisory Committee of the University of Tokyo.
Study design
The mice were divided into four groups (Group A, B, C and D). Group A, composed of 30 mice, underwent only esophagojejunostomy to cause GERD. Sub‐groups of 10 mice in Group A were killed 10, 20, and 40 weeks after operation (Fig. 1). Group B, composed of 10 mice, received bone marrow transplantation from GFP Tg mice 5 weeks after esophagojejunostomy. Sub‐groups of five mice in Group B were killed 20 and 40 weeks after esophagojejunostomy. Group C (n = 7) received bone marrow cell transplantation from GFP Tg mice 25 weeks after esophagojejunostomy and were killed 40 weeks after esophagojejunostomy. Group D (n = 3) received only bone marrow cell transplantation from GFP Tg mice and were killed 40 weeks after sham‐operation.
Figure 1.
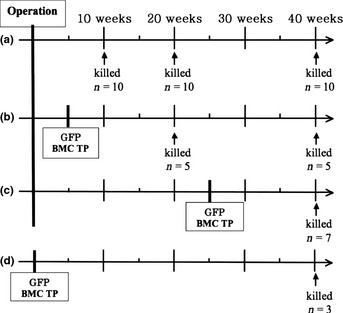
Group A underwent only operation to cause the gastroesophageal reflux disease. Group B received bone marrow cell transplantation from green fluorescent protein (GFP) Tg mice 5 weeks after operation. Group C received bone marrow cell transplantation from GFP Tg mice 25 weeks after operation. Group D received only bone marrow cell transplantation from GFP Tg as control group.
Operation method
The operation was performed as Kumagai et al.10 described in a report of the rat reflux model (Fig. 2a). Briefly, mice were fasted for 24 h before operation. Following induction of anesthesia with intraperitoneal injection of pentobalbital, a 2‐cm midline laparotomy incision was made. An incision of 4 mm was made at the esophago‐gastric junction, and a loop of jejunum, 1 cm distal to the ligament of Treitz, was anastomosed to the esophagogastric junction. All sutures were performed with interrupted 8‐0 silk sutures. The abdominal wall was closed with interrupted 4‐0 silk sutures (Fig. 2b). Animals were allowed to drink water 12 h after the operation and to eat 24 h later. The sham operation group received only laparotomy and closure.
Figure 2.
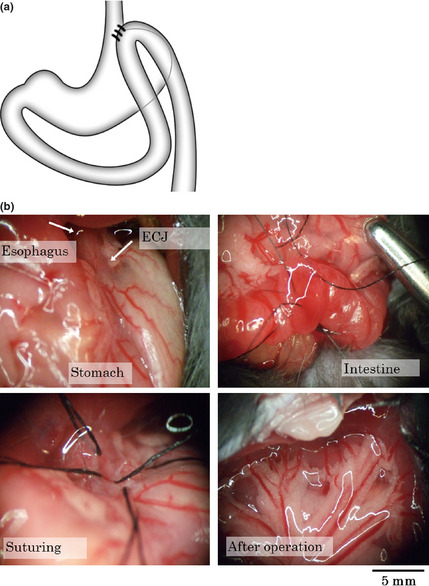
(a) Schema of surgical gastroesophageal reflux disease (GERD) model. The jejunum, 1 cm distal to Treitz's ligament, was anastomosed side‐to‐side to the esophago‐gastric junction. (b) The representative procedure of the operation.
Bone marrow cell transplantation
Bone marrow cell transplantation was performed using male C57BL/6 mice as recipient and female GFP‐Tg mice as donors 5 or 25 weeks after operation in Groups B and C.
For bone marrow cell isolation, female GFP‐Tg mice (8 weeks old) were killed by a high dose of anesthesia and their limbs were removed. Green fluorescent protein‐positive bone marrow cells were flushed with buffer (DMEM with 5% FBS) from the medullary cavities of the tibias and femurs using a 27‐G needle. Bone marrow cells were sieved through 50 μm pore mesh, then washed twice with buffer.
C57BL/6 recipient mice were sublethally irradiated (9 Gy) using an X‐ray generator, and intravenously transplanted with GFP‐positive bone marrow cells (2 × 106 cells in 0.25 mL) within 3 h after irradiation.
Necropsy and tissue processing
Each mouse was anesthetized with pentobarbital and was perfused through the left ventricle with PBS (pH 7.4) for 1 min, followed by 4% buffered paraformaldehyde for 9 min. The esophagus and stomach were excised and opened along the greater curvature. The esophagus and stomach were cut into 2 mm‐wide strips parallel to the lesser curvature. They were placed in 4% paraformaldehyde for 2 h at 4°C. Some tissues were embedded in Tissue‐Tek OCT compound (Sakura Finetek, Tokyo, Japan) and frozen in liquid nitrogen, and the others were embedded in paraffin and 4‐μm sections were prepared.
Histochemistry and immunohistochemistry
All sections were examined with H&E staining and Alcian blue (pH 2.5) stain to detect the presence of the thickening of the mucous membrane, erosion, ulcer, columnar metaplasia, and tumor. Tumor invasion to veins was examined with Elastica van Gieson (EVG) staining. For immunohistochemistry to detect GFP‐positive cells, epithelial cells, leukocyte, proliferating cells, and CDX2 expressing cells, the following primary antibodies were used, respectively: Rabbit polyclonal anti‐GFP antibody (ab6556, Abcam, Tokyo, Japan), mouse monoclonal pan‐cytokeratin antibody (ab6401, Abcam), rat monoclonal anti‐mouse CD45 (AbD Serotec, Oxford, UK), rat monoclonal anti‐mouse Ki67 antibody (M7249, Dako Cytomation, Denmark), and mouse monoclonal anti CDX2 antibody (CDX2‐88, Biogenex, San Francisco, CA, USA).
For immunohistochemistry with anti‐GFP, deparaffinized sections were blocked with blocking serum provided in the HistoMouse staining kit (Zymed, San Francisco, CA, USA) and with 1.5% goat serum provided in the Vectastain ABC KIT (Vector Laboratories, Burlingame, CA, USA) after blockade of endogenous peroxidase activity with 3% H2O2. Sections were incubated with the primary antibody (1:1000) overnight at 4°C followed by incubation with a biotinylated secondary antibody and HRP‐conjugated streptavidin (Vectastain Elite ABC KIT, Vector Laboratories). Chromogen was developed with diaminobenzidine (Biogenex). For immunohistochemistry with anti‐Ki67, deparaffinized sections were blocked endogenous peroxidase activity with 3% H2O2 after antigen retrieval with Citrate Buffer (pH 6.5) for 20 min at 98°C by microwave. Sections were blocked with blocking serum provided in the HistoMouse staining kit (Zymed) and with 1.5% goat serum provided in the Vectastain ABC KIT (Vector Laboratories) after blockade of endogenous peroxidase activity with 3% H2O2. Sections were incubated with the primary antibody (1:1000) overnight at 4°C followed by incubation with a biotinylated secondary antibody and HRP‐conjugated streptavidin (Vectastain Elite ABC KIT, Vector Laboratories). Chromogen was developed with diaminobenzidine (Biogenex).
For all staining, sections were counterstained with Gill's hematoxylin and mounted. Sections were viewed and photographed on the Olympus BX‐51 bright‐field microscope equipped with a Nikon Digital Sight DS‐L imaging system.
Immunofluorescence
For multiple staining with anti‐CD45, anti‐cytokeratin, anti‐GFP, anti‐E‐cadherin (Santa Cruz DECMA‐1) sections were blocked with blocking serum provided in the M.O.M. staining kit (Vector Laboratories) and incubated with anti‐GFP (1:1000), anti‐Ki67 (1:50), anti‐cytokeratin (1:500) and anti‐E‐cadherin (1:200) at the same time overnight at 4°C, followed by incubation with Alexa Fluor 488 Goat Anti‐rabbit IgG, Alexa Fluor 594 Goat Anti‐rat IgG, Alexa Fluor 350 Goat Anti‐mouse IgG. After being washed with PBS, sections were washed in PBS for 3 min and mounted using Prolong antifade with 4,6‐diamino‐2‐phenylindole (DAPI) (Molecular Probes, Carlsbad, CA, USA). All of the sections were viewed and photographed on a Olympus BX‐51 (Tokyo, Japan) fluorescence microscope, and digital images were captured using a Nikon Digital Sight DS‐L imaging system (Nikon, Tokyo, Japan).
Statistics
Differences in the ratios of mucosal change in histochemistry were evaluated by a chi‐square test using JMP 6.0 software.
Results
Two out of 52 mice died in the early postoperative period. The survival rate at 10 weeks after the operation was 96%. Macroscopically, expansion of esophagus diameter and the thickening of the esophageal wall were seen in mice that underwent esophagojejunostomy (Fig. 3). No formation of either tumor or ulcer was observed macroscopically. The anastomoses were patent in all mice.
Figure 3.
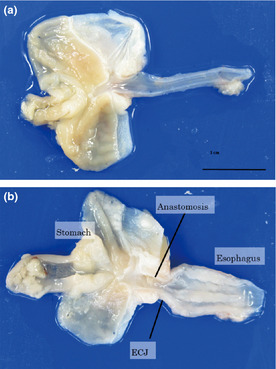
Macroscopically, expansion of esophagus diameter and the thickening of the esophageal wall were seen in operated mice.
Microscopically, thickening of the mucosa, erosion, metaplastic columnar epithelium and duplicated muscularis mucosa were observed (Fig. 4). Proliferating cells, stained positively for Ki67 were also increased in basal regions of the mucosa in thickened squamous epithelium (Fig. 4e,f). Metaplastic columnar epithelium was observed in 28% of mice at 20 weeks after surgery (Figs 4d,5a). Regions of columnar metaplasia contained goblet cells, which stained positively with Alcian blue (pH 2.5) and CDX2 consistent with columnar intestinal metaplasia (Fig. 5b,c). Metaplastic glands showed columnar epithelium with atypia including goblet cells, double muscularis mucosa and proliferating cells that were distributed up to the surface of the mucosa (Fig. 5d). To analyze the geographic distribution of columnar metaplasia, we made serial sections 100 μm apart from an entire block and reconstructed in a bird's eye view. Figure 5e shows the schema of the position of metaplastic columnar epithelium. The metaplastic columnar epithelium was seen in lower esophagus adjacent to an erosion and was distinct from anastomosed intestinal mucosa or stomach.
Figure 4.
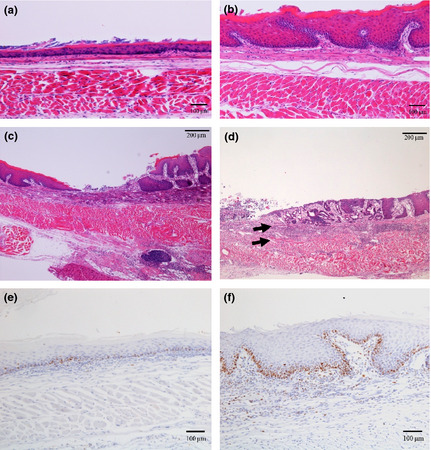
(a–d) Hematoxylin and eosin staining (a) Normal esophagus mucosa (b) 20 weeks after operation: thickening of the mucous membrane was seen. (c) 10 weeks after operation: inflammatory cell infiltration and erosion were seen. (d) 20 weeks after operation: metaplastic columnar epithelium and double muscularis mucosa were seen (e,f) Immunostaining with anti‐Ki67 antibody. (e) Normal esophageal mucosa. (f) 20 weeks after operation: Proliferating cells stained positively for Ki67 were increased in mucosa of mice after surgery.
Figure 5.
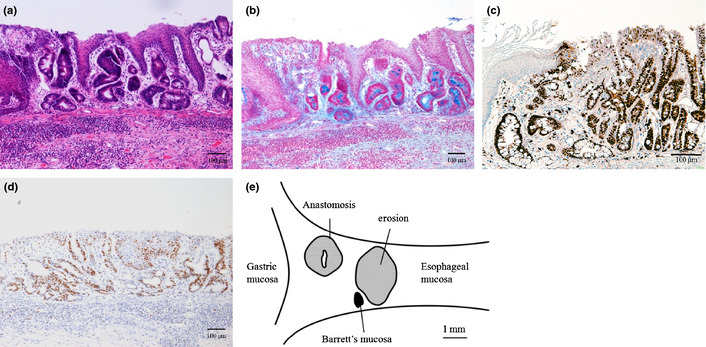
Columnar metaplasia. (a) hematoxylin and eosin staining. (b) Alcian blue (pH 2.5) staining. (c) CDX2 immunostaining for columnar metaplasia. (d) Immunostaining with anti‐Ki67 antibody. (e) Schema of distribution of Barrett's mucosa.
In one mouse killed 40 weeks after surgery, a microscopic tumor was found in the lower esophagus (Fig. 6). The tumor cells invaded into the adventitia and submucosa of esophagus (Fig. 6b,c). Lymphatic ductal invasion was also seen and the duct was negative for EVG staining (Fig. 6d). The tumor contained cornification and many proliferating cells, which stained positively for Ki67 (Fig. 6e). From the histopathological finding, the tumor was characterized as a squamous cell carcinoma. The cancer originated in squamous epithelium and did not connect with columnar metaplasia, although there was columnar metaplasia in the same section.
Figure 6.
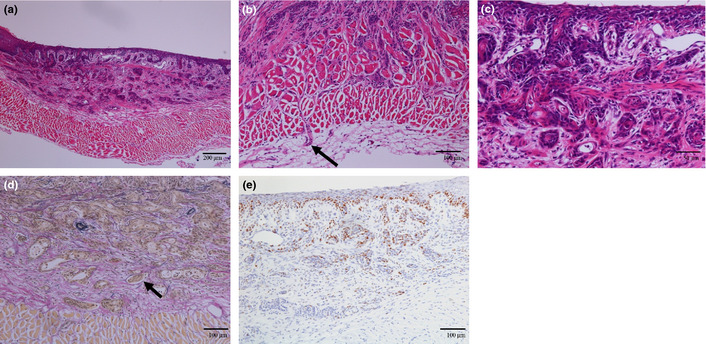
Squamous cell carcinoma observed in a mouse 40 weeks after operation. (a–c) Hematoxylin and eosin staining. (b) The tumor invaded to adventitia of esophagus. (c) The tumor is spreading to submucosal layer. (d) Ductal invasion was also seen. The duct is negative for Elastica van Gieson (EVG) staining diagnosed as lymph vessel invasion. (e) Staining with anti‐Ki67 antibody.
Table 1 shows the number of histopathological findings in each mouse group. An increased thickness of esophageal mucosa and erosion were already seen in some mice 10 weeks after operation. Columnar metaplasia appeared in two (14%) mice 20 weeks after operation. In mice 40 weeks after operation, nine (41%) mice showed columnar metaplasia and one mouse showed the squamous cell carcinoma in esophagus. The incidence of mucosal thickening, erosion and columnar metaplasia were increased with increasing time after operation. There was no significant difference in the incidence of pathological findings between groups with or without transplantation of bone marrow cell of GTP‐Tg mouse.
Table 1.
Histopathological status of each group of mice
| Thickening | Erosion | Columnar metaplasia | Tumor | |
|---|---|---|---|---|
| Group A (only operation) | 3 (33%) | 1 (11%) | 0 (0%) | |
| 10 weeks (n = 9) | 3 (33%) | 1 (11%) | 0 (0%) | 0 (0%) |
| 20 weeks (n = 9) | 6 (66%) | 4 (44%) | 1 (11%) | 0 (0%) |
| 40 weeks (n = 10) | 8 (80%) | 6 (60%) | 0 (0%) | |
| Group B (BMC TP 5 weeks after operation) | ||||
| 20 weeks (n = 5) | 4 (80%) | 2 (40%) | 1 (20%) | 0 (0%) |
| 40 weeks (n = 5) | 2 (40%) | 1 (20%) | 2 (40%) | 0 (0%) |
| Group C (BMC TP 25 weeks after operation) | ||||
| 40 weeks (n = 7) | 6 (86%) | 4 (57%) | 3 (43%) | 1 (14%) |
| Group D (only BMC TP) | ||||
| 40 weeks (n = 3) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
BMC TP, bone marrow cell transplantation. Only thickening of the esophageal mucosa and erosion were seen in some mice 10 weeks after operation. Barrett's mucosa appeared in two (14%) mice 20 weeks after operation. In mice 40 weeks after operation, 40% of mice showed Barrett's mucosa and one mouse showed carcinoma in esophagus. The ratio of appearance of mucosal thickness, erosion and Barrett's mucosa increased as the time went by after operation.
Transplanted bone marrow‐derived cells were identified as GFP positive cells in the surgical GERD model mice. Ninety‐two percent of the recipient bone marrow cells were GFP positive, and there was no significant difference in the ratio among the groups B, C, and D. Figure 7 shows immunohistochemistry staining with anti‐GFP antibody. In the control group (Group D), a small number of GFP‐positive cells were seen in submucosal layer and no GFP positive epithelial cells were seen (Fig. 7a). In mice treated with esophagojejunostomy, there were many GFP‐positive cells in the thickened esophageal mucosa with esophagitis (Fig. 7b) as well as in carcinoma (Fig. 7d). There were no GFP‐positive cells in columnar metaplasia cells, although many GFP‐positive cells were seen in submucosa and interstitium surrounding columnar metaplasia (Fig. 7c).
Figure 7.
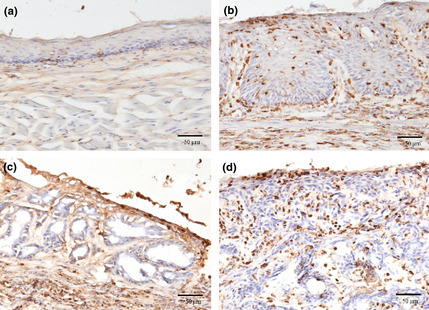
Immunohistochemistry against green fluorescent protein (GFP). (a) Normal mucosa of esophagus. (b) Thickening mucosa with esophagitis. (c) There were no GFP positive cell in Barrett's mucosa. (d) There were many GFP positive cells in carcinoma.
To confirm whether GFP positive cells in the mucosa were leukocytes or epithelial cells, multiple immunofluorescence staining was performed with anti‐CD45, anti‐cytokeratin, anti‐GFP antibodies (Fig. 8). The GFP‐ positive cells in the normal esophagus also showed CD45‐positivity and were identified as leukocytes. While many GFP positive cells in the squamous epithelium in the surgical reflux model mice showed CD45 positivity, a few GFP positive cells were negative for CD45. These cells were also positive for cytokeratin and were identified as epithelial cells derived from transplanted bone marrow cells. There were no GFP‐positive, CD45‐negative and cytokeratin‐positive cells in the columnar metaplasia. The period after operation of bone marrow cell transplantation did not affect the amount or distribution of cells derived from transplanted bone marrow cells. A few cells within the squamous cell carcinoma showed GFP positivity, CD45 negativity and cytokeratin positivity. All GFP positive, CD45 negative and cytokeratin positive cells were isolated as single cells and no clusters of GFP‐positive cells were observed. The GFP positivity in esophagitis mucosa and in squamous cell carcinoma was also confirmed by double‐staining of GFP and pan‐cytokeratin (Fig. 9). To confirm GFP‐stained cells being really squamous epithelial cells, double immunostaining for the molecules that can circumscribe cell boundaries, E‐cadherin, and GFP is shown (Fig. 10).
Figure 8.
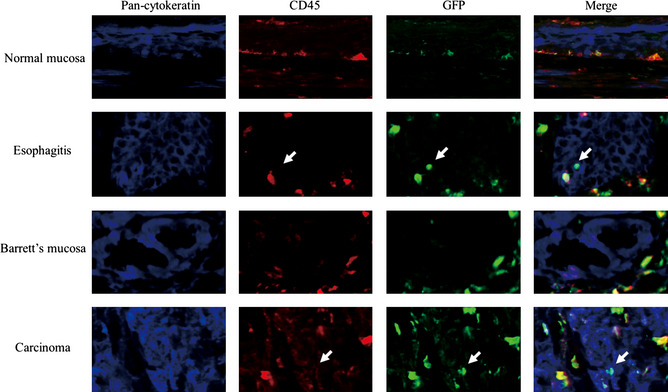
Immunofluorescence multiple staining with anti‐CD45, anti‐cytokeratin, anti‐green fluorescent protein (GFP) antibodies. The GFP positive cells in normal esophagus also showed CD45 positivity and identified as leukocyte. A few GFP positive cells did not show CD45 positivity. These cells also positive for cytokeratin and identified as epithelium cells derived from transplanted bone marrow cells. There was no GFP positive, CD45 negative and cytokeratin positive cells in Barrett's mucosa. A few cells in carcinoma showed GFP positivity, CD45 negativity and cytokeratin positivity.
Figure 9.
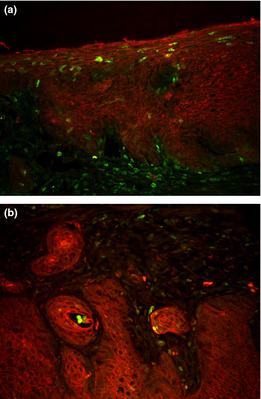
Immunofluorescence double staining with anti‐pan‐cytokeratin and anti‐green fluorescent protein (GFP) antibodies in esophagitis mucosa (a), and in squamous cell carcinoma (b). Pan‐cytokeratin was stained for red and GFP for green. A few cells in esophagitis mucosa and in carcinoma showed GFP positivity and cytokeratin positivity.
Figure 10.
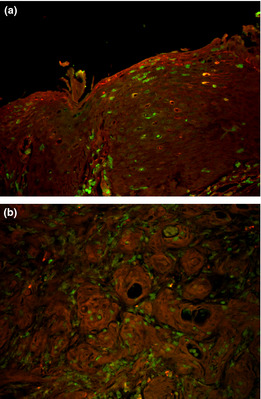
Immunofluorescence double staining with anti‐E‐cadherin and anti‐green fluorescent protein (GFP) antibodies in esophagitis mucosa (a), and in squamous cell carcinoma (b). E‐cadherin was stained for red and GFP for green. A few GFP positive cells in esophagitis mucosa and in carcinoma are circumscribed by E‐cadherin.
Discussion
This study is the first report of mouse surgical reflux model using the operation in which the esophagogastric junction and the jejunum are anastomosed side‐to‐side. The metaplastic columnar epithelium was observed in 14% of mice at 20 weeks, and in 41% at 40 weeks postoperation. The histology showed similarity with human intestinal mucosa in Barrett's mucosa, including an esophagitis background, columnar epithelium with goblet cells, double muscularis mucosa and accentuation of cell proliferation. Thus, the side‐to‐side esophagojejunostomy model in mice provides a strong model of reflux‐induced columnar metaplasia induction in the esophagus. In our preliminary study, esophago‐jejuno end‐to‐side anastomosis with gastrectomy in mice caused only 15.4% columnar metaplasia in esophagus after 40 weeks of operation. Acid reflux should have affected arising columnar metaplasia in esophagus in the model of current study.
In this study, columnar metaplasia appeared in a high percentage of mice 40 weeks after surgery but only a single squamous cell carcinoma was observed. No evidence for adenocarcinoma was found. Xu et al.14 reported 42.4% columnar metaplasia and 12.2% tumors (6.1% adenocarcinoma) 20 weeks after operation in their mouse surgical GERD model with transection of the esophagogastric junction and end‐to‐side esophagojejunostomy. Fein et al.15 showed no occurrence of columnar metaplasia and tumors in normal mice 24 weeks after operation in a mouse surgical reflux model with total gastrectomy and esophagojejunostomy. Thus, while we did not observe adenocarcinoma, the rate of columnar metaplasia was around the same as Xu's model, albeit requiring a longer experimental period. From the lack of columnar metaplasia in Fein's model, backflow of both gastric juice and duodenal juice may be necessary for generating columnar metaplasia in the mouse. The incidence rate of columnar metaplasia and adenocarcinoma in the similar model of rat was 100% and 37.5%.10 We do not know the reason for these differences, however, the volume of reflux through large anastomosis of rat or the period of longer follow‐up, 50 weeks, in rat model may have affected these difference.
The mouse models seemed to be difficult operatively because of the small size of the animals. The surgical model in this study is technically easy with low motality, because it requires only a single anastomosis without resection of the gastrointestinal tract. We compared this operative model with the other operation models by the same surgeon (S. A.). Operation time is shorter and mortality rate is lower in our model than in other models. As almost all food passes from esophagus to intestine through the stomach in our model, the digestive absorption route is physiological compared to other models. In addition, the structure of the esophago‐gastric junction is retained in this model. Therefore, our surgical model seems to be more similar to human GERD than other models.
At least three possible mechanisms for the genesis of columnar epithelia in Barrett esophagus have been reported. The first is the extension of columnar epithelia at the gastroesophageal junction proximally, representing a contiguous metaplastic change.16, 17, 18 The second is metaplasia arising from esophageal glands. The third is true columnar metaplasia of squamous epithelium by a change in stem cell differentiation.19, 20 In mice, the squamous forestomach lies between columnar cells in the stomach and the esophageal mucosa. As this study and previous studies of rat models that showed columnar metaplasia did not display contiguity with the columnar epithelium of stomach, extension is not likely to be a cause of columnar metaplasia. In our model, the areas of columnar metaplasia were present as islands in squamous epithelia distinct from the esophagojejunal anastomosis. As there are no esophageal glands in mouse or rat, columnar metaplasia cannot originate from esophageal glands in rodents. Therefore, the hypothesis that columnar metaplasia originates from true metaplasia of squamous epithelium appears most likely. By analogy then, columnar metaplasia in this mouse model is very close to that in human Barretts' esophagus.
Multipotential stem cells may contribute to the genesis of Barrett's esophagus.22 Li et al.23 reported glandular‐like cells, originating from bone marrow cells in the submucosa, were observed after bone marrow esophageal implantation along with bile perfusion. Recently, Hutchinson et al.24 reported that epithelial cells of columnar metaplasia in mice are bone marrow‐derived. In our results, however, all columnar metaplasia cells were negative for GFP and did not originate from bone marrow‐derived cells. The reason for this discrepancy may be related to different reflux operation models for mice and that the performance of bone marrow transplant prior to the surgical procedure. Residual embryonic cells are reported to be precursors of a Barrett's‐like metaplasia.25 These cells may be the origin of columnar metaplasia in this mouse model.
In this study, transplanted bone marrow‐derived epithelial cells, stained GFP positive, CD45 negative and cytokeratin positive, were detected in esophageal mucosa with esophagitis and squamous cell carcinoma, but not in normal mucosa or columnar metaplastic mucosa. Okamoto et al.26 showed that bone marrow cells can repopulate the epithelia of the human gastrointestinal tract. Donor‐derived epithelial cells substantially repopulated the gastrointestinal tract during epithelial regeneration after graft‐versus‐host disease or ulcer formation. In this experiment, bone marrow‐derived cells differentiate into squamous cells in esophagitis mucosa with gastroesophageal reflux. Three mechanisms suggest that the bone marrow‐derived cell is taken into the esophagitis mucosa. First, bone marrow‐derived cells differentiate into the squamous cells and replenish the epithelial cells. Second, bone marrow‐derived cells may fuse with damaged epithelial cells. Third, bone marrow‐derived cells may provide cytokines to enhance the epithelial proliferation and contribute to mucosal repair or carcinogenesis. Nevertheless, in the present study, there was no evidence for division or proliferation of bone marrow‐derived cells in mucosa.
In summary, we have found that side‐to‐side esophagojejunostomy elicits columnar metaplasia in the esophagus of mice. While these studies demonstrate that reflux in mice can elicit metaplasia, we find no evidence that this metaplasia is derived from bone marrow‐derived cells.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgment
We thank Ms Miki Furuya for her technical assistance and animal care.
(Cancer Sci, doi: 10.1111/cas.12213, 2013)
References
- 1. Devesa SS, Blot WJ, Fraumeni JF Jr. Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer 1998; 83: 2049–53. [PubMed] [Google Scholar]
- 2. Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 2005; 97: 142–6. [DOI] [PubMed] [Google Scholar]
- 3. Barrett NR. Chronic peptic ulcer of the oesophagus and ‘oesophagitis’. Br J Surg 1950; 38: 175–82. [DOI] [PubMed] [Google Scholar]
- 4. Spechler SJ, Goyal RK. The columnar‐lined esophagus, intestinal metaplasia, and Norman Barrett. Gastroenterology 1996; 110: 614–21. [DOI] [PubMed] [Google Scholar]
- 5. Lagergren J, Bergström R, Lindgren A, Nyren O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med 1999; 340: 825–31. [DOI] [PubMed] [Google Scholar]
- 6. Barrett NR. The lower esophagus lined by columnar epithelium. Surgery 1957; 41: 881–94. [PubMed] [Google Scholar]
- 7. Dixon J, Strugala V, Griffin SM et al Esophageal mucin: an adherent mucus gel barrier is absent in the normal esophagus but present in columnar‐lined Barrett's esophagus. Am J Gastroenterol 2001; 96: 2575–83. [DOI] [PubMed] [Google Scholar]
- 8. Jankowski J. Gene expression in Barrett's mucosa: acute and chronic adaptive responses in the oesophagus. Gut 1993; 34: 1649–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sampliner RE. Practice guidelines on the diagnosis, surveillance, and therapy of Barrett's esophagus. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 1998; 93: 1028–32. [DOI] [PubMed] [Google Scholar]
- 10. Kumagai H, Mukaisho K, Sugihara H et al Cell kinetic study on histogenesis of Barrett's esophagus using rat reflux model. Scand J Gastroenterol 2003; 38: 687–92. [DOI] [PubMed] [Google Scholar]
- 11. Nishijima K, Miwa K, Miyashita T et al Impact of the biliary diversion procedure on carcinogenesis in Barrett's esophagus surgically induced by duodenoesophageal reflux in rats. Ann Surg 2004; 240: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seto Y, Kobori O. Role of reflux oesophagitis and acid in the development of columnar epithelium in the rat oesophagus. Br J Surg 1993; 80: 467–70. [DOI] [PubMed] [Google Scholar]
- 13. Babu A, Meng X, Banerjee AM et al Secretory phospholipase A2 is required to produce histologic changes associated with gastroduodenal reflux in a murine model. J Thorac Cardiovasc Surg 2008; 135: 1220–7. [DOI] [PubMed] [Google Scholar]
- 14. Xu X, LoCicero J III, Macri E, Loda M, Ellis FH Jr. Barrett's esophagus and associated adenocarcinoma in a mouse surgical model. J Surg Res 2000; 88: 120–4. [DOI] [PubMed] [Google Scholar]
- 15. Fein M, Peters JH, Baril N et al Loss of function of Trp53, but not Apc, leads to the development of esophageal adenocarcinoma in mice with jejunoesophageal reflux. J Surg Res 1999; 83: 48–55. [DOI] [PubMed] [Google Scholar]
- 16. Bremner CG, Lynch VP, Ellis FH Jr. Barrett's esophagus: congenital or acquired? An experimental study of esophageal mucosal regeneration in the dog. Surgery 1970; 68: 209–16. [PubMed] [Google Scholar]
- 17. Dixon MF, Neville PM, Mapstone NP, Moayyedi P, Axon AT. Bile reflux gastritis and Barrett's oesophagus: further evidence of a role for duodenogastro‐oesophageal reflux? Gut 2001; 49: 359–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cameron AJ, Lomboy CT, Pera M, Carpenter HA. Adenocarcinoma of the esophagogastric junction and Barrett's esophagus. Gastroenterology 1995; 109: 1541–6. [DOI] [PubMed] [Google Scholar]
- 19. Gillen P, Keeling P, Byrne PJ, West AB, Hennessy TP. Experimental columnar metaplasia in the canine oesophagus. Br J Surg 1988; 75: 113–5. [DOI] [PubMed] [Google Scholar]
- 20. Jankowski JA, Harrison RF, Perry I, Balkwill F, Tselepis C. Barrett's metaplasia. Lancet 2000; 356: 2079–85. [DOI] [PubMed] [Google Scholar]
- 21. Houghton J, Stoicov C, Nomura S et al Gastric cancer originating from bone marrow‐derived cells. Science 2004; 306: 1568–71. [DOI] [PubMed] [Google Scholar]
- 22. Seery JP. Stem cells of the oesophageal epithelium. J Cell Sci 2002; 115: 1783–9. [DOI] [PubMed] [Google Scholar]
- 23. Li Y, Wo JM, Ellis S, Ray MB, Jones W, Martin RC. Morphological transformation in esophageal submucosa by bone marrow cells: esophageal implantation under external esophageal perfusion. Stem Cells Dev 2006; 15: 697–705. [DOI] [PubMed] [Google Scholar]
- 24. Hutchinson L, Stendtrom B, Chen D et al Human barrett's adenocarcinoma of the esophagus, associated myofibroblasts and endothelium can arise from bone marrow derived cells after allogenic stem cell transplant. Stem Cells Dev 2011; 20: 11–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang X, Ouyang H, Yamamoto Y et al Residual embryonic cells as precursors of a Barrett's‐like metaplasia. Cell 2011; 145: 1023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Okamoto R, Yajima T, Yamazaki M et al Damaged epithelia regenerated by bone marrow‐derived cells in the human gastrointestinal tract. Nat Med 2002; 8: 1011–7. [DOI] [PubMed] [Google Scholar]